

Abstract
Phagocytosis is a critical cellular process for innate immune defense against microbial infection. The regulation of phagocytosis process is complex and has not been well defined. An intracellular molecule might regulate cell surface-initiated phagocytosis, but the underlying molecular mechanism is poorly understood (1). In this study, we found that microtubule-associated protein 1S (MAP1S), a protein identified recently that is involved in autophagy (2), is expressed primarily in macrophages. MAP1S-deficient macrophages are impaired in the phagocytosis of bacteria. Furthermore, we demonstrate that MAP1S interacts directly with MyD88, a key adaptor of Toll-like receptors (TLRs), upon TLR activation and affects the TLR signaling pathway. Intriguingly, we also observe that, upon TLR activation, MyD88 participates in autophagy processing in a MAP1S-dependent manner by co-localizing with MAP1 light chain 3 (MAP1-LC3 or LC3). Therefore, we reveal that an intracellular autophagy-related molecule of MAP1S controls bacterial phagocytosis through TLR signaling.
Keywords: autophagy, bacterial adhesion, innate immunity, phagocytosis, Toll-like receptor (TLR)
Introduction
Autophagy is a highly conserved process involved not only in metabolic regulation but also in the innate immune defense against organisms in the cytosol (3–5). An important stage for autophagy is forming an autophagosome, and microtubule-associated protein 1 light chain 3 (MAP1-LC3 or LC3), a mammalian homolog of yeast Atg8, plays an essential role in the formation of the autophagosome (6, 7). It initiates from the elongation of the isolation membrane, and the complex Atg12-Atg5-Atg16 provides docking sites for LC3-II. LC3-II then elongates the isolation membrane until it forms an intact autophagosome. The autophagosome fuses with the lysosome and matures into an autolysosome in which its content is degraded. The lapidated LC3 is the most widely used marker for detection of autophagy (6, 7). Recent studies have shown that TLR4 signaling induced autophagy in macrophages/monocytes (8, 9) and that TLR signaling induced the maturation of phagosomes after exposure to bacteria and promoted the MHC II-dependent presentation of bacterial antigens (10, 11).
Phagocytosis is a conserved form of host defense by which phagocytes internalize large particles such as pathogens or apoptotic cells. Phagocytosis is an essential component of effective innate immunity in mammals (12, 13). Phagocytosis of bacteria is often accompanied by inflammatory responses that are driven by TLRs, suggesting that phagocytosis and TLR signaling may be linked functionally. It has been shown that TLRs regulate phagocytosis in a MyD88-dependent way in which TLR signaling activated p38 to accelerate phagosome maturation. Meanwhile, TLRs might regulate phagocytosis in a MyD88-independent pathway (14, 15). Another possible mechanism of TLR-modulated phagocytosis is that TLR signaling alters gene transcription programs that affect phagocytosis (16, 17). Interestingly, it has been reported that autophagy can lend a hand in phagocytosis (1), but the molecular mechanism is poorly understood.
MAP1S is a newly identified autophagy-related protein (2, 18). This molecule regulates autophagosomal biogenesis and degradation and interacts with autophagosome-associated LC3 (2, 18). MAP1S mediates the aggregation of mitochondria, resulting in cell death and genomic destruction, and plays a role in apoptosis (19, 20). MAP1S is involved in the formation of microtubule bundles (20). Considering that actin and microtubules are responsible for the process of phagocytosis, we hypothesize that this autophagy-related MAP1S protein might play a role in the regulation of phagocytosis. Indeed, we found that MAP1S regulates cell surface TLR activation in macrophages and controls the efficacy of bacterial phagocytosis. Therefore, we uncovered one of the molecular mechanisms detailing how the autophagy-related MAP1S molecule is a significant component of phagocytosis.
Experimental Procedures
Mice
MAP1S knockout mice were generated at the Texas A&M Health Science Center in Houston (18) and then backcrossed with the wild-type BL6 background for more than 15 generations. MyD88−/− was provided by Dr. S. Akira. All mice were maintained in the specific pathogen-free facility at the Texas A&M University Health Science Center. All animal experiments were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee.
Cells and Reagents
Raw264.7 and HEK293T cells were grown in DMEM containing 10% (v/v) heat-inactivated fetal bovine serum (HyClone). Bone marrow-derived macrophages (BMDMs) were obtained from bone marrow cells cultured for 6–8 days in the presence of 10 ng/ml macrophage colony-stimulating factor (R&D Systems). Peritoneal macrophages were isolated by peritoneal lavage after 4 days of intraperitoneal injection of 2 ml of sterile 3% thioglycollate medium (BD Biosciences). Salmonella typhimurium SR11 and Staphylococcus aureus strain Newman were cultured in LB medium.
Antibodies against IκBα, phospho-p38 MAP kinase (Thr-180/Tyr-182), and phospho-Akt (Ser-473) were purchased from Cell Signaling Technology. Antibody against MAP1S (catalog no. 4G1) was purchased from Precision Antibody. Antibody against MyD88 (catalog no. IMG-178) was from Imgenex. Antibodies against LC3 for immunofluorescence staining (catalog no. M115-3B) and Western blot analysis (catalog no. PD014B) and p62/SQSTM1 (catalog no. PM045) were purchased from MBL. Escherichia coli K12 LPS, ODN 2216, and LTA were purchased from InvivoGen and used at the indicated concentrations. Total RNA was isolated from TRIzol reagent (Invitrogen), and cDNA was prepared with SuperScript II reverse transcriptase (Invitrogen). The plasmid construct expressing the NF-κB luciferase reporter has been described previously (21).
Cell Sorting and FACS Analysis
Various cell types were sorted from single-cell suspensions of C57BL/6 mouse spleens. Cells were stained with combinations of fluorescence-conjugated monoclonal antibodies (BD Biosciences) and were sorted by a FACSAria (BD Biosciences) with the following sorting criteria for each cell type: B cell, CD19+; CD4 T cell, CD3+CD4+; natural killer cell, NK1.1+; macrophage, CD11b+F4/80+; and myeloid dendritic cell, CD11c+CD11b+. Dendritic cells were sorted further with CD11c+CD45RAhighCD11blow for plasmatocytoid dendritic cells and CD11c+CD45RAnegCD11bhigh for conventional dendritic cells. The purity of all sorted cell types was greater than 96%, as determined by post-sorting flow cytometry with a FACSCalibur (BD Biosciences). FACS analysis for TNF-α was performed as described in our previous report (21).
Transient Transfections and Luciferase Assay
All transfections were performed in triplicate in 24-well plates. Approximately 2 × 105 cells/well were seeded 24 h before transfection. Plasmids were transfected into cells using Lipofectamine 2000 (Invitrogen) following the instructions of the manufacturer. Briefly, 0.8 μg of reporter plasmids plus 0.05 μg of pCMV-LacZ vector were diluted with Opti-MEM and then mixed with diluted Lipofectamine 2000. After 20 min of incubation at room temperature, the mixtures were added to each well. 24 h post-transfection, cells were either analyzed for luciferase activity or challenged further with different TLR agonists or other stimuli at the specified treatment times. Luciferase assays were performed using a luciferase assay system (Promega). β-Galactosidase activity was used as an internal control. Each experiment was conducted a minimum of three times.
Immunofluorescence Staining
Cells were seeded on glass coverslips overnight, fixed in 4% paraformaldehyde for 10 min, and permeabilized with 0.5% Triton X-100 for 15 min. The cells were washed with PBS three times and blocked with 5% BSA in PBS for 1 h at room temperature. The cells were then incubated with primary antibodies (1:200) against MyD88 in blocking buffer at 4 °C overnight. The cells were washed three times with phosphate-buffered saline with Tween 20 for 5 min each and incubated with primary antibodies (1:150) against LC3 for 1 h at room temperature. Then cells were washed three times with phosphate-buffered saline with Tween 20 and incubated with FITC-conjugated anti-rabbit secondary antibody (1:200) for 1 h. After washing with PBS, the specifically bound antibodies were detected using Tyramide Signal Amplification Plus fluorescence systems from PerkinElmer Life Sciences. The coverslips were then rinsed and mounted with Aqua mounting solution. Images were analyzed with a Zeiss Axioskop-2 microscope.
Immunoprecipitation and Immunoblotting
For immunoprecipitation, cell lysates were incubated with the indicated antibody plus protein G-Sepharose at 4 °C overnight to form immunocomplexes. After extensive washing with lysis buffer, the immunocomplexes were analyzed by immunoblotting. For immunoblotting, cells with or without treatment were collected and lysed in lysis buffer containing 150 mm NaCl, 50 mm Tris (pH 8.0), 0.5 mm EDTA, and 1% Nonidet P-40 plus complete protease inhibitor mixture (Roche Applied Science). Following brief vortexing and rotation, cell lysates were separated by SDS-PAGE and transferred to PVDF membranes. These membranes were blocked with 5% fat-free milk in PBS for 30 min and incubated with the indicated antibody in PBS with 1% fat-free milk for 2 h. The membranes were then washed in phosphate-buffered saline with Tween 20 and incubated for 1 h with HRP-conjugated secondary antibody. After subsequent washes, the immunoreactive bands were detected with ECL plus immunoblotting detection reagents (Amersham Biosciences).
Phagocytosis
Monolayers of primary macrophages (peritoneal macrophages and BMDMs) were infected with S. aureus Newman strain and S. typhimurium SR11 at a multiplicity of infection of 10:1. We synchronized the infection of monolayers by centrifuging the infected tissue culture plates at 1000 rpm for 2 min. Following a 30-min incubation at 37 °C, 5% CO2, the cells were washed three times with PBS, and then we added fresh DMEM supplemented with 100 μg/ml of gentamicin. Macrophages were incubated in DMEM with gentamicin for 60 min, washed with PBS, and lysed in sterile water for 10 min. The cell lysate was diluted serially and replated on the LB plate to determine the colony-forming unit count (bacterial colonies are enumerated). We presented the percentage of phagocytosis relative to the average of phagocytosis by wild-type control cells, set as 100%.
ELISA
Levels of cytokine proteins in the primary macrophages supernatants were determined by the specific ELISA kits (R&D Systems).
Statistical Analysis
Results are expressed as mean ± S.D. and are representative of at least three separate experiments. The values are shown as mean ± S.E. of six independent experiments for phagocytosis, and each was performed in triplicate. Unpaired two-tailed Student's t test was used to determine statistical differences in the means of two columns. A p value of less than 0.05 was considered to be statistically significant.
Results
MAP1S Is Expressed Primarily in Macrophages and Controls the Phagocytosis of Bacteria
MAP1S plays an important role in the regulation of autophagy and liver cancer development (2). We found that MAP1S is also highly expressed in the spleen, in which macrophages, members of the phagocytic lineage, are the specific cell type that expresses MAP1S at high levels (Fig. 1). To investigate the role of MAP1S in the phagocytosis of bacteria, we utilized our Map1s knockout mouse (2). We performed traditional phagocytosis assays by infecting primary peritoneal macrophages derived from C57BL/6 mice and Map1s knockout mice with different bacteria, including S. aureus, S. typhimurium, and E. coli. We found that the Map1s-deleted macrophages exhibited impaired phagocytosis of each bacterial strain compared with macrophages derived from the wild-type macrophages (Fig. 2A and data not shown). We obtained similar results by assessing phagocytosis of an E. coli strain expressing GFP (22) using both confocal microscopy (Fig. 2B) and flow cytometry (Fig. 2C) with both peritoneal macrophages and bone marrow-derived macrophages. Taken together, these data demonstrate that the MAP1S autophagy protein plays a critical role in controlling the phagocytosis of bacteria.
FIGURE 1.
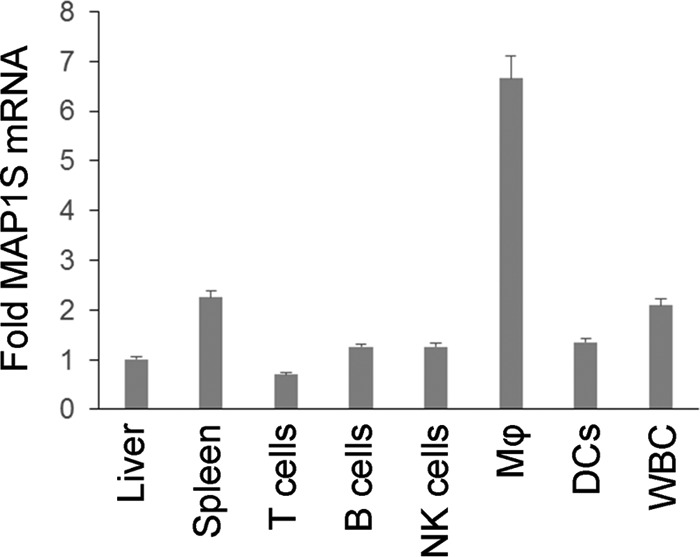
Characterization of the MAP1S expression profile. Real-time RT-PCR was performed to analyze the MAP1S mRNA expression in various cell types in the spleen, including B and CD4+ T lymphocytes, natural killer (NK) cells, dendritic cells (DCs), and macrophages (Mφ), as well as white blood cells (WBC). -Fold expression is shown by comparison with the expression level in the liver. Single-cell suspensions of splenocytes were sorted by FACS. β-Actin was used as a control. Data are shown as mean ± S.D. (n = 3 technical replicates).
FIGURE 2.
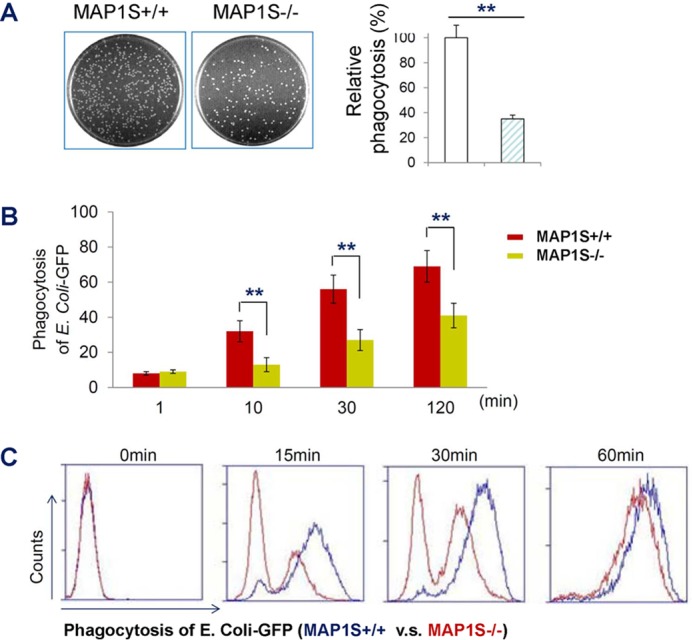
Autophagy MAP1S controls phagocytosis of bacteria. A, phagocytosis assay with standard plate count Salmonella. Peritoneal macrophages from the MAP1S wild type and MAP1S KO mice were infected with S. typhimurium strain SR11 at a multiplicity of infection of 10. Right panel, quantitative results and percentage of phagocytosis relative to the average of phagocytosis of wild-type control cells, set as 100%. B, confocal microscopy-based phagocytosis assay. Peritoneal macrophages from wild-type and MAP1S KO mice were infected with a multiplicity of infection of 1:10 or 1:100 of E. coli-GFP for different times. The number of phagocytosed bacteria was determined by counting E. coli-GFP in 200 macrophages. C, flow cytometry-based phagocytosis assay. BMDMs from MAP1S wild-type and MAP1S KO mice infected with E. coli-GFP (multiplicity of infection of 1:10 or 1:100). The values in A and B are shown as mean ± S.E. of six independent experiments for phagocytosis, and each was performed in triplicate. **, p < 0.01 by unpaired two-tailed Student's t test.
MAP1S Regulates Cell Surface TLR Activation in Macrophages
A link between autophagy and phagocytosis has been noticed recently in which Atg7 controls the phagocytosis of mycobacteria through regulating scavenger receptor expression (23). Then we checked whether MAP1S utilized the same mechanism. However, we found that MAP1S deficiency did not affect the expression of scavenger receptors in macrophages (data not shown). Then we assessed the effect of the Map1s deletion on Toll-like receptor activation because phagocytosis is a receptor-mediated process. We exposed bone marrow-derived macrophages isolated from parent C57BL/6 mice and isogenic Map1s−/− mice to TLR agonists and heat-killed S. typhimurium LT2 (SR11) for 36 h. We found that Map1s-deleted primary macrophages treated with LTA, LPS, the outer surface lipoprotein A (OspA), and heat-killed S. typhimurium secreted significantly less IL-6 and TNF-α than C57Bl/6 macrophages (Fig. 3, A, D, and E). Notably, the LTA-treated group exhibited the most dramatic changes, whereas poly(I:C) yielded similar results from both cell lineages (Fig. 3, A, D, and F). We further examined the LTA-induced cytokine secretion of Map1s−/− and C57Bl/6-derived macrophages at different time points using a variety of doses (Fig. 3, B and C). We noted that the TLRs that exhibited impaired activation (TLR1, TLR2, and TLR4) are also expressed at the cell surface. Furthermore, TLR3, an intracellular TLR and the only TLR that utilizes a MyD88-independent pathway, exhibited similar activation in both cell types by Poly(I:C) (Fig. 3F), and Flagellin failed to induce strong cytokine secretion in our assays (24, 25). MAP1S has been shown to be important for mitochondrial stress, which can affect cell viability, but the cell viability of wild-type and knockout cells after different stimulations was not different (Fig. 3G). Overall, the results suggest that MAP1S regulates cell surface TLR activation and the MyD88-dependent TLR signaling pathway.
FIGURE 3.

MAP1S regulates TLR signaling. A and D, BMDMs from MAP1S+/+ (WT) and MAP1S−/− (KO) C57BL/6 mice were seeded into 24-well plates and treated with different TLR agonists, including LPS (100 ng/ml), LTA (10 μg/ml), OspA (50 ng/ml), Flagellin (100 ng/ml), poly(I:C) (100 ng/ml), ODN 2216 (10 μg/ml), and heat-killed S. typhimurium (HK-ST, 105 cells/ml), or left untreated (UT). IL-6 (A) in the supernatant of cell-cultured medium was measured 36 h after the challenge with TLR agonists. B and C, IL-6 was measured after BMDMs from MAP1S+/+ and MAP1S−/− C57BL/6 mice were treated with indicated amount of LTA for 36 h (B) or 10 μg/ml LTA for the indicated time (C). E, BMDMs were treated with LTA (10 μg/ml) for 24 h. The production of TNF-α was detected by flow cytometry. The filled region indicates TNF-α expression in KO cells, whereas the line represents TNF-α produced in WT cells. Data shown are from an experiment that is representative of three separate experiments. F, BMDMs were stimulated with poly(I:C) (1 μg/ml) for 4 h, and IFNβ mRNA was measured by quantitative PCR. G, cell viability assay for BMDMs after 24-h treatment with HK-ST (105 cells/ml), HK-E. coli (105 cells/ml), and LTA (10 μg/ml) or left untreated. All the experiments were repeated three times, and results with bar graphs are shown as mean ± S.D. *, p < 0.05; **, p < 0.01 (by unpaired two-tailed Student's t test).
MAP1S Interacts with MyD88
MAP1S is an intracellular protein involved in autophagy (2). To determine the mechanism of how this intracellular molecule regulates cell surface TLR activation, we reasoned that MAP1S might associate with components in the TLR signaling pathway (26–28). We then investigated the association between MAP1S and molecules involved in downstream intracellular TLR signaling. In co-immunoprecipitation experiments, either with anti-MAP1S antibody or antibodies against TLR signaling, including anti-MyD88, anti-MAL (MyD88-adaptor-like), anti-TRIF [TIR (Toll/interleukin-1 receptor) domain-containing adaptor protein inducing interferon β], and anti-TRAF6 antibodies, we found that MAP1S associated only with MyD88, a key adapter in TLR signaling pathway in RAW264.7 cells upon LTA stimulation (Fig. 4, A—C, and data not shown). MAP1S and MyD88 immunoprecipitated from cell lysates were revealed by immunoblots with specific antibodies (Fig. 4, A–C). Furthermore, LTA induced accumulation of MAP1S in parallel with MyD88 in the cytosol of macrophages (Fig. 4, D and E). Collectively, these data indicated that MAP1S interacted with MyD88 to regulate TLR signaling and to control bacterial phagocytosis.
FIGURE 4.

MAP1S interacts with MyD88. A–C, endogenous co-immunoprecipitation showing the interaction between MAP1S and MyD88 upon LTA stimulation. Raw264.7 was treated with LTA for 0.5 or 24 h. Cell lysates of Raw264.7 were immunoprecipitated with control IgG, antibody against MAP1S (A and C), or antibody against MyD88 (C), followed by immunoblotting for the presence of MyD88 and MAP1S in the immunocomplex. D and E, Western blotting demonstrating that LTA induced the accumulation of MAP1S as well as MyD88 in wild-type macrophages in a similar time frame. FL, full-length; HC, heavy chain; SC, short chain.
MAP1S Regulates p38 Activation in the TLR Signaling Pathway
We elucidated the downstream TLR signaling pathway, which is a part of the phenotypic response because of the MAP1S deficiency. We knew that the cytokines induced by LTA were attenuated by the MAP1S deficiency (Fig. 4A). Therefore, we assessed the activation of molecules in the TLR signaling pathway upon LTA stimulation. There was no difference in IκBα degradation in primary macrophages from MAP1S WT and MAP1S KO mice. In contrast, the phosphorylation level of p38 decreased dramatically in MAP1S-deficient primary macrophages. Moreover, the Akt phosphorylation level was opposite to that of the pattern observed with p-p38 (Fig. 5). The results are consistent with previous reports showing that the expression of cytokines is decreased by inhibition of p38 activity and that p38 inhibitors impair phagocytosis and inhibit phagosome maturation (10, 11). Therefore, these data indicate that MAP1S is involved in the regulation of TLR signaling through the activation of the p38-Akt pathway but not through that of the NF-κB pathway.
FIGURE 5.
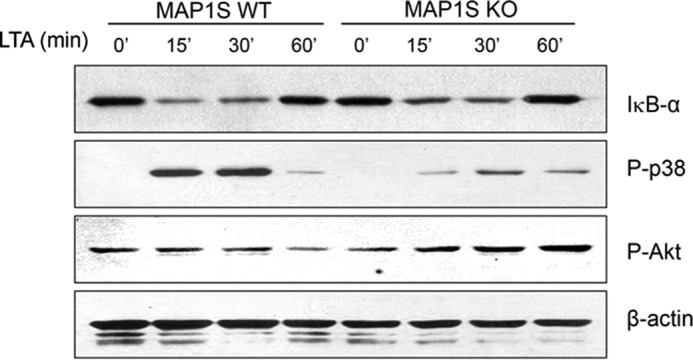
MAP1S regulates p38 but not NF-κB activation in the TLR signaling pathway. BMDMs from MAP1S wild-type and MAP1S KO mice treated with 10 μg/ml LTA for the indicated times. Immunoblots of IκBα, phospho-p38, and phospho-Akt demonstrate their expression levels. β-Actin was used as a loading control.
MyD88 Is Recruited into the Processing of TLR-induced Autophagy by MAP1S
A recent hot research topic in innate immunity is the induction of autophagy by TLR activation (1, 29). To investigate whether activated TLR-induced autophagy was altered by MAP1S deficiency, we stimulated wild-type and knockout mouse macrophages with LTA and LPS for 24 h. We found that, upon LTA and LPS stimulation, the MAP1S deficit did not attenuate the conversion of LC3I to LC3II in primary macrophages, but it led to an accumulation of LC3II and p62 (Fig. 6A). This indicated that regulation of MAP1S in activated TLR-induced autophagy did not occur at the initiation of phagosomes but, instead, during phagosome maturation.
FIGURE 6.

MyD88 is recruited into the processing of TLR-induced autophagy by MAP1S. A, BMDMs from WT and MAP1S KO mice were stimulated with LPS or LTA for 24 h. Cell lysates were harvested to detect the LC3I/II ratio and p62 level by immunoblotting. UT, untreated. B, confocal images of cellular localization of MyD88 in peritoneal primary macrophages from wild-type and MAP1S KO mice stimulated with LTA (10 μg/ml) for 24 h. C, quantification of punctate foci is the average of 10 randomly selected images in the field of 512 × 512 pixels (2). Data are shown as mean ± S.E. of three independent experiments, and each was performed in triplicate. **, p < 0.01 by unpaired two-tailed Student's t test. D, BMDMs from WT and MAP1S KO mice were stimulated with LTA for 24 h. Localization of MyD88 (red) and LC3 (green) in WT and MAP1S KO BMDM were analyzed by confocal microscopy, and colocalization resulted in yellow. Scale bar = 10 μm. E, simplified working model. An autophagy-related protein, MAP1S, regulates bacterial phagocytosis and TLR signaling.
Because MAP1S can interact with MyD88 upon LTA stimulation, we wanted to determine whether the MyD88 expression pattern was altered in the MAP1S knockout macrophages. We performed immunofluorescent staining on MAP1S WT and KO primary macrophages using anti-MyD88 antibody. Intriguingly, we observed that LTA could induce MyD88 accumulation with the punctate structures characteristic of autophagy dependent on MAP1S (Fig. 6, B and C). Remarkably, we also found that, upon TLR activation, MyD88 co-localizes with LC3 in a MAP1S-dependent mode (Fig. 6D). These results indicated that MAP1S, in response to TLR activation, promotes the recruitment and accumulation of MyD88 in the cytoplasm and may be involved in the autophagy process.
Discussion
In the immune system, phagocytosis of bacteria plays a critical role in cleaning bacterial pathogens. In this case, phagocytosis requires receptor-mediated recognition of the molecular patterns expressed on the surface of infectious agents. The receptors are collectively called pattern recognition receptors. The toll-like receptor is one of well characterized pattern recognition receptors. TLRs and TLR signaling play an important role in the regulation of phagocytosis. However, the regulation of phagocytosis is a complex issue.
Autophagy (or autophagocytosis) is a cellular process by which a cell degrades its own components through the lysosomal machinery and maintains a balance between the synthesis, degradation, and subsequent recycling of cellular products. However, recent evidence redefines this concept and finds that autophagy degrades not only its own components but also intracellular bacteria and viruses (foreign bodies). Furthermore, it can aid phagocytosis induced by infection. TLR signaling activates a variety of defense mechanisms, including phagocytes, phagosome maturation, and autophagy. Sanjuan et al. (1) have provided evidence that TLR signaling engaged traditional autophagocytic components to promote phagocytosis. It is also supported by our results showing that MAP1S is involved in TLR signaling and promotes the phagocytosis of bacteria.
TLRs detect the invasion of microbial pathogens and induce the expression of inflammatory cytokines. Phagocytosis triggers the degradation of pathogens and the presentation of pathogen-derived peptide antigens. These subsequent reactions of TLRs and phagocytosis instruct the development of antigen-specific acquired immunity (10). Therefore, it is essential to study the relationship between TLRs and phagocytosis. Phagocytosis of bacteria has been shown to be impaired because of defective phagosome maturation in the absence of TLR2, TLR4, or MyD88 (11). In agreement with this study, our data show that a defect of MAP1S also attenuates phagocytosis and bacterial ingestion by macrophages. This strongly suggests that cooperation of MAP1S and TLR-MyD88 enhances maturation of the phagosome in TLR-activated cells.
Compared with the LTA stimulation group, the difference in the LPS and OspA stimulation groups was mild. The underlying mechanism is not fully understood. We only showed that MAP1s interacts with MyD88. There are four adaptors, and LPS utilizes all of them. OspA activates the signaling pathway through TLR1/2, whereas LTA is recognized by TLR2/6. We are currently investigating whether MAP1s interacts with the other three adaptors after different stimulations. This might provide the answer regarding the difference. Another hypothesis is that MAP1S may play a role in TLR2 trafficking (30, 31), which is an intriguing concept regarding the TLR2 signaling pathway. This hypothesis needs to be investigated further.
TLRs and autophagy play an important role in innate immunity. Recent evidence suggests that TLRs are involved in the regulation of autophagy (10). In this study, we investigated the function of MAP1S in TLR signaling-induced autophagy. We found that MAP1S promoted the recruitment of MyD88 to its functional site in the cytoplasm and enhanced the expression of inflammatory cytokines in response to MyD88-dependent TLR signaling (Fig. 6E). Surprisingly, we also found that it was MAP1S that led MyD88 to the autophagosome in MyD88-dependent, TLR-activated cells. Consistent with other reports of TLR signaling-induced autophagy, TLRs promoted phagosome maturation through the TLR-MyD88-p38 dependent pathway (10, 11). Our results further indicated that MAP1S promoted the recruitment of MyD88 and the activation of p38 by signals from TLRs to enhance maturation of the phagosome. The underlying mechanism through p38, but not through NF-κB, is not clear at this moment, although this phenomenon has been reported (10, 11). The current knowledge to explain this phenomenon may be at the MAPK/IKK level (32, 33), which distinguishes p38 and NF-kB pathway. However, we do not yet have evidence showing that MAP1s can interact with a MAPK (e.g. MEKK3/6). This will be investigated further. Another possibility is that p38 signaling, but not NF-κB signaling, regulates phagosome maturation. MAP1S has been suggested to be an autophagy-promoting protein. Loss of MAP1S leads to the accumulation of p-AKT, which has been shown to suppressed autophagy. This is consistent with a previous report showing that loss of MAP1S dampened autophagy (2).
Moreover, MAP1S associates with microtubules and autophagic components to affect autophagosomal maturation (2), and our results show that MAP1S is involved in TLR signaling-induced autophagy through association with MyD88. These findings may indicate that MAP1S recruits MyD88, autophagic components, or even endocytic organelles through association with microtubules. This conclusion is also supported by another potential mechanism: that TLRs trigger phagosome maturation by increasing the rate of phagosome motility along microtubules (11).
Recently, there has been increasing interest in the connection between TLR signaling and autophagy (1, 29, 34). Studies have mainly focused on the induction of autophagy by TLR ligands. By knocking out autophagy-associated genes, it has been demonstrated that autophagy plays a critical role in both innate and adaptive immunity (9, 35). However, it is unclear whether this critical role is via the classical autophagy pathway or whether it results from autophagy-independent functions of the autophagy genes. We identified an autophagy-associated protein, MAP1S, that may help to clarify the relationship between autophagy and phagocytosis. Our results indicate that the function of MAP1S in TLR signaling may not be solely through its role in the classical autophagy pathway but an autophagy-independent function of the gene in addition to its function in classical autophagy. This protein, normally involved in autophagy (2), also interfaces with the cell surface-initiated TLR-MyD88-p38 pathway to regulate bacterial phagocytosis. Unveiling the interaction between MAP1S and MyD88 represents a potential target to concurrently manipulate autophagy and bacterial phagocytosis to block phagolysosomal fusion and prevent a deadly overshoot of immune responses, such as septic shock.
Author Contributions
D. Z. and Y. L designed the experiments, which were performed by M. S., Y. Z., T. Z., F. H., and J. C. L. L., F. W., and W. L. M. generated the MAP1S knockout mice. Data were analyzed by D. Z., Y. L., M. S., and Y. Z. D. Z. and M. S. wrote the paper.
Acknowledgments
We thank Margie Moczygemba for help with the FACS analysis and Dr. William Margolin for GFP-E. coli (13).
This work was supported in part by National Institutes of Health Grant CA176698 and the Texas A&M Health Science Center (to D. Z.), the National Natural Science Foundation of China (to Y. L.), National Institutes of Health Grant CA142862 and DOD New Investigator Award W81XWH (to L. L.), and the John S. Dunn Foundation (to W. M. and F. W.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TLR
- Toll-like receptor
- LTA
- lipoteichoic acid
- BMDM
- bone marrow-derived macrophage.
References
- 1.Sanjuan M. A., Dillon C. P., Tait S. W., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J. L., Withoff S., and Green D. R. (2007) Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257 [DOI] [PubMed] [Google Scholar]
- 2.Xie R., Nguyen S., McKeehan K., Wang F., McKeehan. W. L., and Liu L. (2011) Microtubule-associated protein 1S (MAP1S) bridges autophagic components with microtubules and mitochondria to affect autophagosomal biogenesis and degradation. J. Biol. Chem. 286, 10367–10377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levine B. (2005) Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell 120, 159–162 [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa I., Amano A., Mizushima N., Yamamoto A., Yamaguchi H., Kamimoto T., Nara A., Funao J., Nakata M., Tsuda K., Hamada S., and Yoshimori T. (2004) Autophagy defends cells against invading group A Streptococcus. Science 306, 1037–1040 [DOI] [PubMed] [Google Scholar]
- 5.He C., and Klionsky D. J. (2009) Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suzuki K., and Ohsumi Y. (2007) Molecular machinery of autophagosome formation in yeast, Saccharomyces cerevisiae. FEBS Lett. 581, 2156–2161 [DOI] [PubMed] [Google Scholar]
- 7.Deretic V. (2010) Autophagy in infection. Curr. Opin. Cell Biol. 22, 252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delgado M. A., Elmaoued R. A., Davis A. S., Kyei G., and Deretic V. (2008) Toll-like receptors control autophagy. EMBO J. 27, 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saitoh T., and Akira S. (2010) Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 189, 925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blander J. M., and Medzhitov R. (2004) Regulation of phagosome maturation by signals from toll-like receptors. Science 304, 1014–1018 [DOI] [PubMed] [Google Scholar]
- 11.Blander J. M., and Medzhitov R. (2006) On regulation of phagosome maturation and antigen presentation. Nat. Immunol. 7, 1029–1035 [DOI] [PubMed] [Google Scholar]
- 12.Stuart L. M., and Ezekowitz R. A. (2008) Phagocytosis and comparative innate immunity: learning on the fly. Nat. Rev. Immunol. 8, 131–141 [DOI] [PubMed] [Google Scholar]
- 13.Greenberg S., and Grinstein S. (2002) Phagocytosis and innate immunity. Curr. Opin. Immunol. 14, 136–145 [DOI] [PubMed] [Google Scholar]
- 14.Kong L., and Ge B. X. (2008) MyD88-independent activation of a novel actin-Cdc42/Rac pathway is required for Toll-like receptor-stimulated phagocytosis. Cell Res. 18, 745–755 [DOI] [PubMed] [Google Scholar]
- 15.Kong L., Sun L., Zhang H., Liu Q., Liu Y., Qin L., Shi G., Hu J. H., Xu A., Sun Y. P., Li D., Shi Y. F., Zang J. W., Zhu J., Chen Z., Wang Z. G., and Ge B. X. (2009) An essential role for RIG-I in toll-like receptor-stimulated phagocytosis. Cell Host Microbe 6, 150–161 [DOI] [PubMed] [Google Scholar]
- 16.Underhill D. M., and Gantner B. (2004) Integration of Toll-like receptor and phagocytic signaling for tailored immunity. Microbes Infect. 6, 1368–1373 [DOI] [PubMed] [Google Scholar]
- 17.Doyle S. E., O'Connell R. M., Miranda G. A., Vaidya S. A., Chow E. K., Liu P. T., Suzuki S., Suzuki N., Modlin R. L., Yeh W. C., Lane T. F., and Cheng G. (2004) Toll-like receptors induce a phagocytic gene program through p38. J. Exp. Med. 199, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie R., Wang F., McKeehan W. L., and Liu L. (2011) Autophagy enhanced by microtubule- and mitochondrion-associated MAP1S suppresses genome instability and hepatocarcinogenesis. Cancer Res. 71, 7537–7546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L., Vo A., and McKeehan W. L. (2005) Specificity of the methylation-suppressed A isoform of candidate tumor suppressor RASSF1 for microtubule hyperstabilization is determined by cell death inducer C19ORF5. Cancer Res. 65, 1830–1838 [DOI] [PubMed] [Google Scholar]
- 20.Dallol A., Cooper W. N., Al-Mulla F., Agathanggelou A., Maher E. R., and Latif F. (2007) Depletion of the Ras association domain family 1, isoform A-associated novel microtubule-associated protein, C19ORF5/MAP1S, causes mitotic abnormalities. Cancer Res. 67, 492–500 [DOI] [PubMed] [Google Scholar]
- 21.Zhang D., Zhang G., Hayden M. S., Greenblatt M. B., Bussey C., Flavell R. A., and Ghosh S. (2004) A toll-like receptor that prevents infection by uropathogenic bacteria. Science 303, 1522–1526 [DOI] [PubMed] [Google Scholar]
- 22.Liu J., Hu B., Morado D. R., Jani S., Manson M. D., and Margolin W. (2012) Molecular architecture of chemoreceptor arrays revealed by cryoelectron tomography of Escherichia coli minicells. Proc. Natl. Acad. Sci. U.S.A. 109, E1481–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonilla D. L., Bhattacharya A., Sha Y., Xu Y., Xiang Q., Kan A., Jagannath C., Komatsu M., and Eissa N. T. (2013) Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 39, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uematsu S., Fujimoto K., Jang M. H., Yang B. G., Jung Y. J., Nishiyama M., Sato S., Tsujimura T., Yamamoto M., Yokota Y., Kiyono H., Miyasaka M., Ishii K. J., and Akira S. (2008) Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 9, 769–776 [DOI] [PubMed] [Google Scholar]
- 25.Mathur R., Oh H., Zhang D., Park S. G., Seo J., Koblansky A., Hayden M. S., and Ghosh S. (2012) A mouse model of Salmonella typhi infection. Cell 151, 590–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beutler B. A. (2009) TLRs and innate immunity. Blood 113, 1399–1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takeda K., and Akira S. (2007) Toll-like receptors. Curr. Protoc. Immunol. Chapter 14, Unit 14.12 [DOI] [PubMed] [Google Scholar]
- 28.Medzhitov R. (2007) Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 [DOI] [PubMed] [Google Scholar]
- 29.Yano T., Mita S., Ohmori H., Oshima Y., Fujimoto Y., Ueda R., Takada H., Goldman W. E., Fukase K., Silverman N., Yoshimori T., and Kurata S. (2008) Autophagic control of listeria through intracellular innate immune recognition in Drosophila. Nat. Immunol. 9, 908–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nilsen N. J., Deininger S., Nonstad U., Skjeldal F., Husebye H., Rodionov D., von Aulock S., Hartung T., Lien E., Bakke O., and Espevik T. (2008) Cellular trafficking of lipoteichoic acid and Toll-like receptor 2 in relation to signaling: role of CD14 and CD36. J. Leukocyte Biol. 84, 280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Granick J. L., Falahee P. C., Dahmubed D., Borjesson D. L., Miller L. S., and Simon S. I. (2013) Staphylococcus aureus recognition by hematopoietic stem and progenitor cells via TLR2/MyD88/PGE2 stimulates granulopoiesis in wounds. Blood 122, 1770–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuzawa A., Saegusa K., Noguchi T., Sadamitsu C., Nishitoh H., Nagai S., Koyasu S., Matsumoto K., Takeda K., and Ichijo H. (2005) ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6, 587–592 [DOI] [PubMed] [Google Scholar]
- 33.Uhlik M. T., Abell A. N., Johnson N. L., Sun W., Cuevas B. D., Lobel-Rice K. E., Horne E. A., Dell'Acqua M. L., and Johnson G. L. (2003) Rac-MEKK3-MKK3 scaffolding for p38 MAPK activation during hyperosmotic shock. Nat. Cell Biol. 5, 1104–1110 [DOI] [PubMed] [Google Scholar]
- 34.Xu Y., Jagannath C., Liu X. D., Sharafkhaneh A., Kolodziejska K. E., and Eissa N. T. (2007) Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saitoh T., Fujita N., Jang M. H., Uematsu S., Yang B. G., Satoh T., Omori H., Noda T., Yamamoto N., Komatsu M., Tanaka K., Kawai T., Tsujimura T., Takeuchi O., Yoshimori T., Akira S. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 456, 264–268 [DOI] [PubMed] [Google Scholar]