

Background: Antibody v-domains in scFv format often suffer from aggregation and stability issues that restrict formulation.
Results: Structural and empirical analyses of an optimized scFv revealed that three VL-CDR3 mutations were sufficient to mediate significant stability and affinity improvements.
Conclusion: scFv issues were resolved via removal of side-chain clashes at the VL/VH interface.
Significance: CDR-restricted mutagenesis delivers stability-optimized molecules for high concentration dosing.
Keywords: antibody engineering, chemokine, crystal structure, mutagenesis in vitro, protein stability
Abstract
Fully-human single-chain Fv (scFv) proteins are key potential building blocks of bispecific therapeutic antibodies, but they often suffer from manufacturability and clinical development limitations such as instability and aggregation. The causes of these scFv instability problems, in proteins that should be theoretically stable, remains poorly understood. To inform the future development of such molecules, we carried out a comprehensive structural analysis of the highly stabilized anti-CXCL13 scFv E10. E10 was derived from the parental 3B4 using complementarity-determining region (CDR)-restricted mutagenesis and tailored selection and screening strategies, and carries four mutations in VL-CDR3. High-resolution crystal structures of parental 3B4 and optimized E10 scFvs were solved in the presence and absence of human CXCL13. In parallel, a series of scFv mutants was generated to interrogate the individual contribution of each of the four mutations to stability and affinity improvements. In combination, these analyses demonstrated that the optimization of E10 was primarily mediated by removing clashes between both the VL and the VH, and between the VL and CXCL13. Importantly, a single, germline-encoded VL-CDR3 residue mediated the key difference between the stable and unstable forms of the scFv. This work demonstrates that, aside from being the critical mediators of specificity and affinity, CDRs may also be the primary drivers of biotherapeutic developability.
Introduction
The redundancy in signaling pathways involved in the pathogenesis of many diseases, coupled with advances in antibody engineering technologies, has driven a proliferation of multi-targeting biotherapeutic modalities (1). There are many examples of bispecific molecules that have effective dual targeting capability for synergistic or improved mechanisms of action. One of the most advanced of these is blinatumomab, a bispecific single chain antibody composed of an anti-CD19 scFv4 linked in tandem with an anti-CD3 scFv for T-cell engagement, that is now an approved therapeutic for the treatment of B-cell malignancies (2). Other novel bispecific formats in the clinic include the IgG-scFv MM-141 (3), soluble T-cell receptor-CD3 scFv immune-mobilizing monoclonal TCRs against cancer (ImmTACs) (4), and scFv diabody-based dual affinity retargeting molecules (T-DARTs) (5). The intravenous route of administration, combined with the relatively low dosing regimen for these oncology applications, negates the requirement for high concentration formulation, which is highly desirable for biotherapeutics for chronic inflammatory indications.
On paper, the scFv presents the logical building block for creating multi-specific antibodies from the v-domains of pre-existing IgGs, creating modular, single-polypeptide units. However, it is well established that the behavior of re-appropriated v-domains in the scFv format can vary widely, with many suffering from aggregation, solubility, and stability issues that restrict scalable expression and formulation (6–8). Many strategies aimed at optimizing v-domain pairing and stability have been investigated, including: linker modulation (6, 7), introduction of inter-chain disulfide bonds (9, 10), or structure-guided mutation of frameworks at the domain interface (11, 12). Despite these efforts, the progression of scFv-based bispecific molecules for non-oncology indications has been limited.
We recently described the successful concurrent affinity and stability optimization of an anti-CXCL13 scFv using CDR-restricted mutagenesis and affinity- and stability-driven selection by phage display (13). When reformatted into an scFv-Fc-scFv bispecific molecule, the lead scFv “E10” displayed an exemplary stability and solubility profile and could be formulated at 100 mg/ml in a standard biopharmaceutical platform buffer with the viscosity remaining well below the threshold of 20 centipoise required for subcutaneous administration. The E10 discovery process was particularly intriguing for a number of reasons, including: 1) All functionally improved clones from selection and screening were mutated in VL-CDR3 alone. 2) The v-gene frameworks of E10 remained fully germline and were not modified to improve VL-VH pairing stability. 3) The VH-CDR3 was shown to be highly intolerant to mutation. 4) The lead E10 scFv required only four VL-CDR3 substitutions to mediate significant improvements in both affinity and stability.
Despite these findings, the principles allowing the optimization of highly stable, scFv-based, bispecific therapeutics are not fully understood. It has been shown that even molecules built on v-domain frameworks of high theoretical stability can suffer from poor development characteristics (14). In the study presented here, we have delineated the factors that led to improved target binding affinity versus scFv stability. Empirical analyses of VL-CDR3 point mutants and high-resolution scFv-CXCL13 co-crystal structures were used to elucidate contributing factors. The four VL-CDR3 substitutions were examined in isolation, and subsequently in combination. Each mutant was expressed as scFv-Fc and characterized in comparative kinetic and thermal stability analyses. These empirical data demonstrated that distinct individual residue changes affect binding affinity and/or VH-VL interface stability. In parallel, high-resolution crystal structures of both scFvs were solved in isolation, and in complex with human CXCL13. In addition to providing the first description of the human CXCL13 structure, these analyses show a novel mode of VH-CDR3 engagement that clearly demonstrates why this loop was not amenable to mutation. They also elegantly support the empirical observations from in vitro kinetic and stability analyses and confirm the key residues mediating both affinity and stability improvements.
In summary, we have demonstrated that as few as three amino acid substitutions, confined to the VL-CDR3, are sufficient to mediate the affinity optimization and stability improvements necessary to facilitate high concentration formulation. This study shows that: 1) In vitro Darwinian protein optimization can achieve significant scFv stability and affinity optimization with minimal mutational load. 2) A single CDR amino acid side-chain clash affecting packing at the VH-VL interface can lead to dramatic differences in the biophysical behavior of human scFvs with therapeutic potential. 3) Importantly, these improvements were not directly mediated by new side-chain contacts between the antibody v-domains or with CXCL13, but were significantly impacted through the resolution of subtle repulsive forces in the antibody combining site. These findings are of broad importance in the antibody engineering field as they highlight that the CDRs of human scFvs are not just critical mediators of affinity and specificity but may also be the primary drivers of biotherapeutic developability.
Experimental Procedures
ScFv-Fc Expression, Purification, and in Vitro Analyses
ScFv-Fc fusion proteins were expressed transiently in Expi293F cells and purified from filtered conditioned medium using ProPlus tips from Phynexus on the automated Phynexus MEA. The resulting proteins were buffer-exchanged into PBS using 40-kDa cut-off Zeba columns (Thermo Scientific) and quantified using a Micro BCA kit (Thermo Scientific). Thermal stability ELISAs and DSC analyses were performed as previously described (13).
Binding Kinetics Analyses
Biacore analysis was performed using a T-200 biosensor, series S CM5 chips, an amine-coupling kit, 10 mm sodium acetate immobilization buffer, pH 5.0, 1× HEPES-buffered saline EDTA-phosphate running buffer containing an additional 250 mm NaCl (final NaCl concentration 400 mm), and 3 m MgCl2 (regeneration solution) (GE Healthcare). Approximately 8000 response units of an anti-human IgG Fc (GE Healthcare) were covalently immobilized to flow cells 1 and 2 of the CM5 chip at pH 5.5. Then 50–100 response units of 3B4/3B4 variant scFv-Fc fusion (diluted in 1× running buffer) were captured on flow cell 2. Human CXCL13 (100–25 nm) diluted in running buffer was flowed across both flow cells at 100 μl/min with a contact phase of 120 s and a dissociation phase of 600 s, followed by a 5-s regeneration pulse with 3 m MgCl2. All experiments were performed at 37 °C. Data were corrected for instrument and bulk artifacts by double referencing (15) a surface-immobilized with capture antibody without scFv-Fc-scFv using Scrubber version 2.0c software (BioLogic Software). The transformed data were fit to a 1:1 binding model in Biacore T200 evaluation software v1.0 (GE Healthcare), which includes a parameter for mass transfer (16).
Production and Purification of 3B4 and 3B4-CXCL13 Complex
2 liters of 0.22 μm filtered 3B4-TEV-Fc conditioned medium was purified on HiTrap rProtein A (GE Healthcare) using low pH elution under standard conditions. The eluted fraction was concentrated and dialyzed into 50 mm Tris, pH 8.0, for His-TEV protease cleavage (Life Technologies). Cleaved Fc was removed with a second HiTrap rProtein A run, and His-TEV protease was removed using Ni2+-nitrilotriacetic acid capture (GE Healthcare). 3B4 was further purified on a HiLoad 26/600 Superdex 200 column (GE Healthcare) pre-equilibrated in 20 mm Tris, pH 7.5, 150 mm NaCl and concentrated to 11 mg/ml for complex formation. The final yield was 5 mg of 3B4/liter of conditioned medium as measured by NanoVue (GE Healthcare).
The 3B4-CXCL13 complex was formed by incubating 1:1.2 (molar ratio) of 3B4:CXCL13 in 20 mm Tris, pH 7.5, and 150 mm NaCl at room temperature for 2 h before passing through a HiLoad 26/600 Superdex 200 column (GE Healthcare). The complex fractions were pooled and concentrated to 11 mg/ml in 20 mm Tris, pH 7.5, and 150 mm NaCl for crystallization. The E10 and E10-CXCL13 complexes were prepared in the same manner.
Crystallization and Data Collection
All crystals were grown at 18 ± 1 °C using the sitting-drop vapor diffusion method. The concentration of E10 was 5.7 mg/ml in TBS buffer (50 mm Tris, 150 mm NaCl, pH 7.5). Each drop contained 0.15 μl of E10 and 0.15 μl of reservoir solution containing 100 mm Tris, pH 8.5, and 2000 mm (NH4)2SO4. The crystals appeared after 2 weeks, reaching a final size of 0.05 × 0.07 × 0.1 mm3. The concentration of E10-CXCL13 was 8.7 mg/ml in TBS buffer. Each drop contained 0.2 μl of E10-CXCL13 and 0.1 μl of reservoir solution containing 100 mm KH2PO4, 100 mm NaH2PO4, 100 mm MES, pH 6.0, and 2000 mm NaCl. The crystals appeared in 3 months, reaching a final size of 0.05 × 0.06 × 0.5 mm3. The concentration of 3B4 was 11 mg/ml in 20 mm Tris, pH 7.5, and 150 mm NaCl. Each drop contained 0.15 μl of 3B4 and 0.15 μl of reservoir solution containing 100 mm HEPES, pH 7.5, and 4300 mm NaCl. The crystals appeared in 2 weeks, reaching a final size of 0.1 × 0.1 × 0.3 mm3. The concentration of 3B4-CXCL13 was 11 mg/ml in 20 mm Tris, pH 7.5, and 150 mm NaCl. Each drop contained 0.2 μl of 3B4-CXCL13 and 0.1 μl of reservoir solution containing 100 mm Tris, pH 8.0, 100 mm NaCl, and 8% PEG 20000. The crystals appeared after 1 month, reaching a final size of 0.03 × 0.05 × 0.4 mm3. X-ray diffraction data were collected remotely at beamline 17-ID of the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) at the Advanced Photon Source (APS), Argonne National Laboratory. Data processing was carried out with the HKL2000 program (17) and autoPROC (18). The final data statistics were from autoPROC. Crystal data and processing statistics are summarized in supplemental Table S1.
Structure Solution and Refinement
All structures were solved by molecular replacement using program Phaser (19). The E10 structure was solved with 3JUY (Protein Data Bank (PDB) ID) as the search model. The E10-CXCL13 complex structure was solved with E10 and an ensemble of 3GV3, 2R3Z, 4HSV, and 3IL8 (PDB ID) for CXCL13 as the search models. E10 and E10-CXCL13 complex structures were used as search models to solve the structures of 3B4 and 3B4-CXCL13 complex, respectively. All structures were refined using PHENIX (20) in the beginning stage and finalized with BUSTER (21). Bulk solvent correction was used. The 2Fo − Fc and Fo − Fc electron density maps were calculated for the inspection and improvement of the structure during refinement. The flexible part of CXCL13 and the CDR regions of 3B4/E10 were built at later stages of the refinement. Solvent molecules, defined as those with peaks greater than or equal to 3σ on the Fo − Fc electron density map with reasonable hydrogen bond networks, were included as water molecules. Graphic work was carried out using Coot (22). The complex structures were verified with annealed omit maps and assessed using PROCHECK (23). Illustrations were prepared with PyMOL (Schrödinger, LLC, New York).
Results
Comparative Biophysical Analysis of 3B4- and E10-scFv-Fc Fusion Proteins
ScFv-Fc fusion proteins constructed with either parental 3B4 scFv or optimized E10 scFv at the N terminus were expressed and purified for comparative biophysical analysis. Fig. 1A is a schematic representation of these fusion proteins highlighting the four VL-CDR3 amino acid residues that differ between both scFvs. DSC analysis revealed a 5 °C increase in thermal stability for the optimized E10 scFv-Fc (Fig. 1B). Protein solubility and aggregation potential of a 100 mg/ml solution of each fusion protein, stored at 4 °C, were monitored over 7 weeks by periodic sampling and analytical size-exclusion chromatography analysis. Fig. 1C shows that the 3B4-containing scFv-Fc had high aggregation, with almost 20% HMMS in the starting preparation, increasing to >30% after only 7 days and continuing to rise. In contrast, the E10-scFv-Fc had a low starting percentage of high molecular mass species, which remained at ∼1% HMMS up to 50 days.
FIGURE 1.
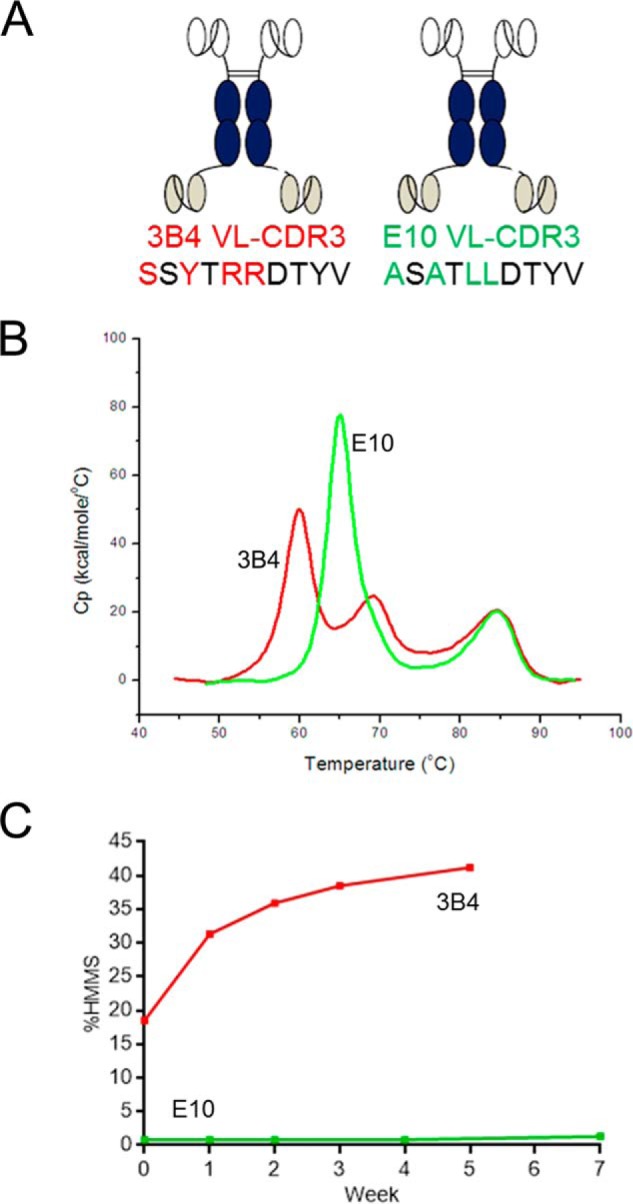
Comparative biophysical analysis of parental 3B4 scFv-Fc-scFv and optimized E10 scFv-Fc-scFv. A, schematic representation of the two bispecific proteins highlighting the four amino acid differences between the two clones in VL-CDR3. B, DSC analysis showing the 5 °C increase in Tm1 between E10 (green) and 3B4 (red). C, comparative size-exclusion chromatography analysis showing time-dependent increases in formation of HMMS after storage of a 100 mg/ml solution of 3B4 (red) and E10 (green) bispecific molecules at 4 °C.
Crystal Structure of Optimized E10 scFv in Complex with CXCL13
To better understand the influence of these mutations, we generated crystals of 3B4 and E10 scFvs, either alone or complexed with CXCL13, and determined the structure to a resolution of 2.1 Å by x-ray crystallography (Protein Data Bank codes for 3B4, E10, 3B4-CXCL13, and E10-CXCL13 are 5C2B, 5C6W, 5CBA and 5CBE, respectively). Fig. 2A illustrates a unique mode of interaction between the VH-CDR3 of E10 scFv and CXCL13, generating an intermolecular four-stranded antiparallel β-sheet composed of the extended VH-CDR3 loop of E10 and the β1-sheet of CXCL13. This interaction was shown to be mediated exclusively through backbone interactions (Fig. 2B), and is conserved in the 3B4-CXCL13 complex structure. A network of charge-charge interactions between Arg21 and Arg22 of the 310-helix and Leu46 from the 40S loop of CXCL13 and a triad of negatively charged residues at the base of the VH-CDR3 loop (Glu95, Asp97, Asp100), supported by Asp95 from VL, further contributes to strong target engagement by E10 (Fig. 2C). Conservation of these interactions provides structural rationale for the failure to retrieve any affinity-optimized mutants from the 3B4 VH-CDR3 mutagenesis libraries (13).
FIGURE 2.

Structure of the anti-CXCL13 scFv E10 in complex with CXCL13. A, graphic view showing the complex. scFv heavy chain (VH) and light chain (VL) are shown in magenta and yellow, respectively. CXCL13 is shown in gray. B, detail showing the four-stranded antiparallel β-sheet formed between the ascending and descending strands of VH-CDR3 and β1-sheet of CXCL13. Hydrogen bonds between side chains and backbone are shown as dashed lines. C, detail illustrating the charge-charge interactions between negatively charged residues in VH-CDR3 (Glu95, Asp97, D100H shown in magenta) and positively charged residues in the 310-helix and 40S loop of CXCL13 (Arg21, Arg23, Lys46).
Systematic Empirical Analysis of VL-CDR3 Mutations Individually and in Combination
ScFv-Fc mutants interrogating each of the four VL-CDR3 positions in isolation, and in combination, were expressed and purified for comparative in vitro analysis. All clones were subjected to thermal stability analysis via thermal ELISA, DSC, and CXCL13 binding kinetics via Biacore. All data are summarized in Table 1. In general, all mutants tended to display either parental 3B4 behavior or optimized E10 behavior, with a small number of mutants exhibiting intermediate stability or affinity characteristics. We observed a strong correlation in stability ranking of clones using the thermal stability ELISA or DSC (p < 0.0001). This initial comparative analysis revealed that the optimized properties of E10 were conferred by three of the four amino acid substitutions in VL-CDR3 generated using random mutagenesis. The single point mutation S89A (E10.1) in isolation was enough to almost completely recapitulate the improved stability properties of E10, without having much effect on binding affinity for CXCL13 (Fig. 3, A–D, Table 1). In contrast, Y91A (E10.2) had the suboptimal Tm of 3B4 but an identical off-rate when compared with optimized E10 (Fig. 4, A–D, Table 1). Remarkably, in each of these cases, a single point mutation was sufficient to mediate significant stability and affinity changes. Interestingly, R93L (E10.3) had intermediate stability and affinity characteristics (Fig. 5, A–D, Table 1). The fourth position, R94L (E10.4), did not seem to make any additional contributions to stability or affinity and performed nearly identically to 3B4 in all in vitro assays (Table 1). This finding suggested that R94L was simply a non-deleterious “passenger” mutation resulting from the random mutagenesis approach used in the initial library construction.
TABLE 1.
Affinity and stability characterization of scFv-Fc variants
ScFv variants with mutations in the VL-CDR3 are shown, with residue changes from parental 3B4 sequence in bold and underlined. In some cases, alternative amino acids (italicized) have been introduced that are different from both the parental 3B4 and optimized E10.
| VL-CDR3 sequence | Tm | ΔTm | Thermal ELISA EC50 | Biacore koff | |
|---|---|---|---|---|---|
| °C | °C | nm | s−1 | ||
| 3B4 | SSYTRRDTYV | 59.8 | 0 | 14.28 | 0.0108 |
| E10 | ASATLLDTYV | 64.92 | 5.12 | 2.89 | 0.0034 |
| E10.1 | ASYTRRDTYV | 63.13 | 3.33 | 1.76 | 0.0094 |
| E10.2 | SSATRRDTYV | 59.08 | −0.72 | 18.98 | 0.0034 |
| E10.3 | SSYTLRDTYV | 62.5 | 2.7 | 2.51 | 0.0053 |
| E10.4 | SSYTRLDTYV | 60.53 | 0.73 | 15.92 | 0.0082 |
| E10.5 | ASATRRDTYV | 62.85 | 3.05 | 1.93 | 0.0029 |
| E10.6 | ASYTLRDTYV | 65.9 | 6.1 | 2.35 | 0.004 |
| E10.7 | ASYTRLDTYV | 63.67 | 3.87 | 1.87 | 0.0076 |
| E10.8 | ASYTLLDTYV | 65.8 | 6 | 1.77 | 0.0037 |
| E10.9 | SSATLRDTYV | 61.43 | 1.63 | 4.59 | 0.0029 |
| E10.10 | SSATRLDTYV | 59.92 | 0.12 | 18.72 | 0.0046 |
| E10.11 | SSATLLDTYV | 62 | 2.2 | 1.85 | 0.0021 |
| E10.12 | SSYTLLDTYV | 62.75 | 2.95 | 4.55 | 0.0061 |
| E10.13 | ASATLRDTYV | 64.97 | 5.17 | 1.97 | 0.0019 |
| E10.14 | ASATRLDTYV | 63.19 | 3.39 | 1.31 | 0.0028 |
| E10.15 | SSYTKKDTYV | 61.18 | 1.38 | 8.27 | 0.0079 |
| E10.16 | ASATFFDTYV | 62.9 | 3.05 | 2.2 | 0.0023 |
FIGURE 3.

Characterization of mutant E10.1, S89A. A, graphic representation of 3B4, E10, and E10.1 scFv-Fc fusion proteins highlighting VL-CDR3 amino acid content (red, 3B4-specific residues; green, E10-specific residues). B, off-rate analysis as measured by Biacore comparing 3B4 (red), E10 (green), and E10.1 (gray). C, thermal stability ELISA comparing behavior of 3B4 (red), E10 (green), and 10.1 (gray) fusion proteins before (light color) and after (dark color) incubation for 60 min at 60 °C. OD450, optical density at 450 nm; RT, room temperature. Error bars indicate ± S.E. D, DSC analysis comparing 3B4 (red), E10 (green), and E10.1 (gray). E, superposition of VH-VL interface of 3B4 (yellow for VL and magenta for VH) and E10 (gray) in the region of VL residue 89 (shown in “green” for 3B4). Waters are shown as red spheres. The hydrogen-bonding network for 3B4 is shown with dashed yellow lines. The side chain-OH of 3B4 VL Ser89 has no hydrogen-bonding partners.
FIGURE 4.

Characterization of mutant E10.2, Y91A. A, graphic representation of 3B4, E10, and E10.2 scFv-Fc fusion proteins highlighting VL-CDR3 amino acid content (red, 3B4-specific residues; green, E10-specific residues). B, off-rate analysis as measured by Biacore comparing 3B4 (red), E10 (green), and E10.2 (gray). C, thermal stability ELISA comparing behavior of 3B4 (red), E10 (green), and 10.2 (gray) fusion proteins before (light color) and after (dark color) incubation for 60 min at 60 °C. OD450, optical density at 450 nm; RT, room temperature. Error bars indicate ± S.E. D, DSC analysis comparing 3B4 (red), E10 (green), and E10.2 (gray). E, juxtaposition of 3B4 VL-CDR3 (yellow) and E10 VL-CDR3 (blue) in complex with the 310-helix of CXCL13 (gray). The position 91 side chain is marked in red for clarity in both cases. F, the impact of the Y91A mutation is made clearer with a superposition of the 3B4-CXCL13 (yellow) and E10-CXCL13 (blue) complexes. Mutation at this position to alanine removes steric occlusion at the interface increasing the surface area of interaction by ∼200 Å2.
FIGURE 5.

Characterization of mutant E10.3, R93L. A, schematic representation of 3B4, E10, and E10.3 scFv-Fc fusion proteins highlighting VL-CDR3 amino acid content (red, 3B4-specific residues; green, E10-specific residues). B, off-rate analysis as measured by Biacore comparing 3B4 (red), E10 (green), and E10.3 (gray). C, thermal stability ELISA comparing the behavior of 3B4 (red), E10 (green), and 10.1 (gray) fusion proteins before (light color) and after (dark color) incubation for 60 min at 60 °C. OD450, optical density at 450 nm; RT, room temperature. Error bars indicate ± S.E. D, DSC analysis comparing 3B4 (red), E10 (green), and E10.3 (gray). E, surface charge representation shows that the R93L mutation removes a repulsive charge-charge interaction between Arg93 in 3B4 and Lys46 in the 40S loop of CXCL13. F, juxtaposition of 3B4-CXCL13 (yellow) and E10-CXCL13 (blue) complexes highlighting the impact of R93L on both stability and affinity. Leu93 in E10 VL CDR3 interacts with Tyr30 and Trp32 in Vl-CDR1, as indicated by dashed lines. It also facilitates a more favorable interaction between Tyr30, which can adopt an alternative rotamer in E10, and Lys46 in the 40S loop of CXCL13.
S89A is the Primary Driver of Stability Increases in E10 scFv
Subsequent resolution of the parental 3B4 scFv-CXCL13 complex further supported these observations and elegantly illustrates how the critical VL-CDR3 mutations in E10 mediate their functional effects. Fig. 3A shows a schematic representation of 3B4, E10, and E10.1 (S89A) scFv-Fc with VL-CDR3 amino acid differences highlighted. These clones were compared in affinity and stability analyses. S89A had no effect on affinity with its measured off-rate being practically identical to parental 3B4 (0.0094 versus 0.0108 s−1; Fig. 3B). However, it had a significant effect on stability, showing similar activity to optimized E10 in both thermal ELISA and DSC. Stability data are shown in Fig. 3, C and D, and summarized in Table 1. Comparison of the co-crystal structures for both E10 and 3B4 scFv with CXCL13 (Fig. 3E) shows that Ser89 sits at the VH-VL interface and presents a polar side chain in a hydrophobic environment proximal to Phe98 in VL- framework 4 and Phe100J in VH-CDR3. Replacement with Ala89 removes the unsatisfied hydrogen bond donor/acceptor pair, thus optimizing the interface. Data obtained with additional combination mutants support that S89A is sufficient to significantly improve scFv stability. Data summarized in Table 1 demonstrate that any of the 16 mutants with Ala89 in VL-CDR3 exhibit E10-like behavior with respect to stability as measured by both thermal ELISA and DSC.
Y91A Is Critical in Driving Affinity Improvements in E10 scFv
Fig. 4A shows a schematic representation of 3B4, E10, and E10.2 (Y91A) scFv-Fc with VL-CDR3 amino acid differences highlighted. These clones were compared as described above. In this case, Y91A had a significant effect on affinity for CXCL13 with an identical off-rate to E10 (Fig. 4B; 0.0034 s−1). However, unlike S89A, Y91A had no measurable impact on scFv stability with performance comparable with parental 3B4 in both thermal ELISA (Fig. 4C) and DSC (Fig. 4D). The data are summarized in supplemental Table S1. We again sought some structural insight into the major functional impact of this single point mutation. Fig. 4E shows a juxtaposition of the co-complexes of 3B4-CXCL13 (left panel) and E10-CXCL13. Tyr91 makes contact with residues in the 310 helix of CXCL13. Removal of the bulky tyrosine side chain by replacement with Ala91 allows a substantial shift in the interface with CXCL13, bringing additional antibody residues into contact with the antigen (Fig. 4E). This is further highlighted in Fig. 4F, which shows a superposition of both co-complexes. In comparison with 3B4, the distance between E10 VL-CDR3 and CXCL13 is significantly reduced, resulting in a 23% (200 Å2) increase in the buried surface area for the antibody-antigen complex.
R93L Contributes to Changes in Both Affinity and Stability
Fig. 5A shows a schematic representation of 3B4, E10, and E10.3 (R93L) scFv-Fc with VL-CDR3 amino acid differences highlighted. These clones were compared as described above. In this case, R93L impacted both affinity and stability. Fig. 5B shows that the R93L mutant had an off-rate intermediate to those of 3B4 and E10 at 0.0053 s−1, suggesting that it is mediating some direct interaction with CXCL13. Furthermore, thermal ELISA (Fig. 5C) and DSC analysis (Fig. 5D) indicate that this residue also has a role to play in stabilization, showing little change in EC50 after thermal incubation and a 2.7 °C increase in Tm1. These data are summarized in supplemental Table S1. The 3B4/E10-CXCL13 complex structures once again provided structural support for our empirical observations. The surface charge representation in Fig. 5E shows that R93L removes a positively charged patch in the paratope, which may dilute the nearby attractive electrostatic interactions described above and introduce a repulsive interaction with Lys46 of CXCL13. Leu93 also makes new intra-domain interactions with both Trp32 and Tyr30 in VL-CDR1, leading to increased scFv stability (Fig. 5F).
Discussion
In vitro affinity maturation of antibodies is now commonly reported in the literature. Accurate structural investigation of affinity and/or stability-improving structural changes before and after maturation, however, is comparatively rare. As a result, the mechanisms that drive affinity improvements for antigen binding are still poorly understood. The development of the highly stable, pm potency, fully human, anti-CXCL13 scFv E10 presented a rare opportunity to study a molecule that had exhibited multi-parameter improvements via only four mutations in a single CDR loop (13). In the study presented here, we used both empirical analyses of point mutations and co-crystal structural analyses to investigate the mutations that enabled the generation of the affinity- and stability-optimized scFv E10.
A commonly held assumption in antibody engineering is that affinity-improving mutations likely directly mediate new contacts with antigen through newly presented amino acid side chains. Indeed, in silico design of mutations based on co-crystal structural analyses have shown that such new contacts can significantly enhance binding affinity in protein-protein (24) and protein-hapten interactions (25). In contrast, post hoc structural analyses of mutations generated naturally during B-cell development (26) or by Darwinian display-based evolution (27), have shown that considerable changes in binding energy can be achieved not by adding significant new contacts with antigen, but rather by “fine tuning” or “rigidification” of the antibody-antigen interface (28).
In the study reported here, we have shown that in E10, none of the affinity-improving mutations mediated new contacts themselves. Surprisingly, the affinity-enhancing mutations instead removed repulsive obstacles to tighter binding between the VL-CDR3 and CXCL13, thereby improving the fitness of the interface. The resulting refinements in existing contacts led to dramatic improvements in kDa and increased buried surface area between E10 and CXCL13, in comparison with the parental clone 3B4. Our findings echo those of Garcia-Rodriguez et al. (29), who demonstrated that CDR-based mutations improving the affinity of a BoNT/A1-A2 cross-reactive antibody for the A2 form eliminated an electrostatic clash while also generating a new energetic interaction.
Another classical assumption is that if an antibody contains somatic mutations in the CDRs, then those deviations are probably important for antigen binding affinity or specificity. This study shows that assumption to be flawed, as Arg93 and Arg94 in the VL-CDR3 of 3B4 are the only residues that deviate from the germline in this CDR, but the presence of Arg93 is actually deleterious to CXCL13 binding function. Arg93 and Arg94 are somatic mutations that were most likely generated in the context of a different VH partner in the human B-cell background of the naive scFv library from which 3B4 was derived (13). In the original v-gene pairing, these two mutations could indeed have been beneficial for binding to an unknown antigen, but when isolated as part of 3B4, Arg93 is a hindrance to high affinity binding of CXCL13. Similarly, not all mutations that are found in in vitro affinity-matured antibodies need to mediate functional improvements to survive selective pressure. Non-deleterious “passenger mutations” may be carried through, even in molecules such as E10 that were put under harsh selective pressure. This phenomenon is exemplified by R94L, where its contribution to affinity and stability improvements was minimal.
Importantly, it has been shown that although in vivo-generated somatic mutations can improve the potency of antibodies, they can also destabilize the v-domains (30). In the case of 3B4, however, the unique VH-CDR3 interaction with CXCL13 was resistant to mutagenesis (13), and so the interface instability had to be repaired by mutation of a germline residue in the VL-CDR3. The parental anti-CXCL13 antibody 3B4 is composed of the VH1–69 and Vλ2–14 germlines (13). Domains from the VH1 and Vλ2 families have been associated with stable scFv v-domain pairing (31, 32), and both VH1–69 and Vλ2–14 are highly represented in the human antibody repertoire (33, 34). Indeed, an extensive study of VH-VL pairing found Vλ2–14 to be the most highly represented Vλ germline in the repertoire, where it is often paired with VH1–69 (34). Despite this, 3B4 was found to suffer from very low stability and solubility in an scFv-based bispecific format (13).
In the present study, we address a “real world example” molecule whose investigation supports early theoretical studies that used v-family consensus sequences to establish trends for stability across common VH-VL pairings and would suggest that the pairing of both VH1–69 and Vλ2–14 could potentially be very stable (31, 35). Our study shows that the surprisingly unstable v-gene pairing observed in 3B4 can be essentially ameliorated by only a single mutation in the VL-CDR3. Like the affinity-enhancing mutations described above, this S89A mutation was found to repair an intrinsic flaw in the packing between the two v-domains, rather than creating a new VH-VL contact. This observation is complementary to previous studies that employed multiple framework-based mutations on the basis of structural analyses and did not attempt to achieve improvements via CDR mutagenesis (11, 12). This study shows that the low stability of scFvs built on theoretically stable v-domain scaffolds (14), whether fully human or humanized, can be driven by as little as a single germline side chain clashing with the VH-CDR3. As VH-CDR3 structures are still problematic to model, particularly for human antibodies, which are poorly represented in the PDB database (36), such clashes in CDRs can be rapidly identified and ameliorated by CDR mutagenesis, and subsequently rationalized by structural biology.
Our previous findings highlighted the utility of “hammer-hug” and thermal selection and screening principles as effective practice for isolating optimized scFvs (13). This study further suggests that, in the future, human synthetic libraries designed for high stability and solubility (32, 37) might be further improved by adding empirically discovered CDR mutations that bias toward overall scFv stability characteristics.
Author Contributions
O. C., B. J. F., and W. J. J. F. conceived the study and O. C., L. L., E. R. L., M. S., L. M., J. B., and W. S. designed and coordinated the experiments. C. T., V. T., A. S. P. T., W. S., and J. B. performed and analyzed the experiments. O. C., W. J. J. F., C. T., and J. B. wrote the paper.
Supplementary Material
Acknowledgments
Use of the IMCA-CAT beamline 17-ID (or 17-BM) at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract Number DE-AC02-06CH11357.
This work was supported by the Pfizer Post-Doctoral Research program. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table S1.
The atomic coordinates and structure factors (codes 5C2B, 5C6W, 5CBA, and 5CBE) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- scFv
- single-chain Fv
- CDR
- complementarity-determining region
- DSC
- differential scanning calorimetry
- TCR
- T-cell receptor
- TEV
- tobacco etch virus.
References
- 1.Chan A. C., and Carter P. J. (2010) Therapeutic antibodies for autoimmunity and inflammation. Nat. Rev. Immunol. 10, 301–316 [DOI] [PubMed] [Google Scholar]
- 2.Bargou R., Leo E., Zugmaier G., Klinger M., Goebeler M., Knop S., Noppeney R., Viardot A., Hess G., Schuler M., Einsele H., Brandl C., Wolf A., Kirchinger P., Klappers P., Schmidt M., Riethmüller G., Reinhardt C., Baeuerle P. A., and Kufer P. (2008) Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science 321, 974–977 [DOI] [PubMed] [Google Scholar]
- 3.Fitzgerald J. B., Johnson B. W., Baum J., Adams S., Iadevaia S., Tang J., Rimkunas V., Xu L., Kohli N., Rennard R., Razlog M., Jiao Y., Harms B. D., Olivier K. J. Jr., Schoeberl B., Nielsen U. B., and Lugovskoy A. A. (2014) MM-141, an IGF-IR- and ErbB3-directed bispecific antibody, overcomes network adaptations that limit activity of IGF-IR inhibitors. Mol. Cancer Ther. 13, 410–425 [DOI] [PubMed] [Google Scholar]
- 4.Liddy N., Bossi G., Adams K. J., Lissina A., Mahon T. M., Hassan N. J., Gavarret J., Bianchi F. C., Pumphrey N. J., Ladell K., Gostick E., Sewell A. K., Lissin N. M., Harwood N. E., Molloy P. E., Li Y., Cameron B. J., Sami M., Baston E. E., Todorov P. T., Paston S. J., Dennis R. E., Harper J. V., Dunn S. M., Ashfield R., Johnson A., McGrath Y., Plesa G., June C. H., Kalos M., Price D. A., Vuidepot A., Williams D. D., Sutton D. H., and Jakobsen B. K. (2012) Monoclonal TCR-redirected tumor cell killing. Nat. Med. 18, 980–987 [DOI] [PubMed] [Google Scholar]
- 5.Moore P. A., Zhang W., Rainey G. J., Burke S., Li H., Huang L., Gorlatov S., Veri M. C., Aggarwal S., Yang Y., Shah K., Jin L., Zhang S., He L., Zhang T., Ciccarone V., Koenig S., Bonvini E., and Johnson S. (2011) Application of dual affinity retargeting molecules to achieve optimal redirected T-cell killing of B-cell lymphoma. Blood 117, 4542–4551 [DOI] [PubMed] [Google Scholar]
- 6.Mabry R., Gilbertson D. G., Frank A., Vu T., Ardourel D., Ostrander C., Stevens B., Julien S., Franke S., Meengs B., Brody J., Presnell S., Hamacher N. B., Lantry M., Wolf A., Bukowski T., Rosler R., Yen C., Anderson-Haley M., Brasel K., Pan Q., Franklin H., Thompson P., Dodds M., Underwood S., Peterson S., Sivakumar P. V., and Snavely M. (2010) A dual-targeting PDGFRβ/VEGF-A molecule assembled from stable antibody fragments demonstrates anti-angiogenic activity in vitro and in vivo. MAbs 2, 20–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mabry R., Lewis K. E., Moore M., McKernan P. A., Bukowski T. R., Bontadelli K., Brender T., Okada S., Lum K., West J., Kuijper J. L., Ardourel D., Franke S., Lockwood L., Vu T., Frank A., Appleby M. W., Wolf A., Reardon B., Hamacher N. B., Stevens B., Lewis P., Lewis K. B., Gilbertson D. G., Lantry M., Julien S. H., Ostrander C., Chan C., Byrnes-Blake K., Brody J., Presnell S., Meengs B., Levin S. D., and Snavely M. (2010) Engineering of stable bispecific antibodies targeting IL-17A and IL-23. Protein Eng. Des. Sel. 23, 115–127 [DOI] [PubMed] [Google Scholar]
- 8.Mabry R., and Snavely M. (2010) Therapeutic bispecific antibodies: The selection of stable single-chain fragments to overcome engineering obstacles. IDrugs 13, 543–549 [PubMed] [Google Scholar]
- 9.Michaelson J. S., Demarest S. J., Miller B., Amatucci A., Snyder W. B., Wu X., Huang F., Phan S., Gao S., Doern A., Farrington G. K., Lugovskoy A., Joseph I., Bailly V., Wang X., Garber E., Browning J., and Glaser S. M. (2009) Anti-tumor activity of stability-engineered IgG-like bispecific antibodies targeting TRAIL-R2 and LTβR. MAbs 1, 128–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orcutt K. D., Ackerman M. E., Cieslewicz M., Quiroz E., Slusarczyk A. L., Frangioni J. V., and Wittrup K. D. (2010) A modular IgG-scFv bispecific antibody topology. Protein Eng. Des. Sel. 23, 221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jordan J. L., Arndt J. W., Hanf K., Li G., Hall J., Demarest S., Huang F., Wu X., Miller B., Glaser S., Fernandez E. J., Wang D., and Lugovskoy A. (2009) Structural understanding of stabilization patterns in engineered bispecific Ig-like antibody molecules. Proteins 77, 832–841 [DOI] [PubMed] [Google Scholar]
- 12.Miller B. R., Demarest S. J., Lugovskoy A., Huang F., Wu X., Snyder W. B., Croner L. J., Wang N., Amatucci A., Michaelson J. S., and Glaser S. M. (2010) Stability engineering of scFvs for the development of bispecific and multivalent antibodies. Protein Eng. Des. Sel. 23, 549–557 [DOI] [PubMed] [Google Scholar]
- 13.Fennell B. J., McDonnell B., Tam A. S., Chang L., Steven J., Broadbent I. D., Gao H., Kieras E., Alley J., Luxenberg D., Edmonds J., Fitz L. J., Miao W., Whitters M. J., Medley Q. G., Guo Y. J., Darmanin-Sheehan A., Autin B., Shúilleabháin D. N., Cummins E., King A., Krebs M. R., Grace C., Hickling T. P., Boisvert A., Zhong X., McKenna M., Francis C., Olland S., Bloom L., Paulsen J., Somers W., Jensen A., Lin L., Finlay W. J., and Cunningham O. (2013) CDR-restricted engineering of native human scFvs creates highly stable and soluble bifunctional antibodies for subcutaneous delivery. MAbs 5, 882–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang K., Geddie M. L., Kohli N., Kornaga T., Kirpotin D. B., Jiao Y., Rennard R., Drummond D. C., Nielsen U. B., Xu L., and Lugovskoy A. A. (2015) Comprehensive optimization of a single-chain variable domain antibody fragment as a targeting ligand for a cytotoxic nanoparticle. MAbs 7, 42–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Myszka D. G. (1999) Improving biosensor analysis. J. Mol. Recognit. 12, 279–284 [DOI] [PubMed] [Google Scholar]
- 16.Kortt A. A., Gruen L. C., and Oddie G. W. (1997) Influence of mass transfer and surface ligand heterogeneity on quantitative BIAcore binding data: analysis of the interaction of NC10 Fab with an anti-idiotype Fab′. J. Mol. Recognit. 10, 148–158 [DOI] [PubMed] [Google Scholar]
- 17.Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 18.Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., and Bricogne G. (2011) Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 21.Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER version 2.11.1, Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 22.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 23.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993) PROCHECK: A program to check stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291 [Google Scholar]
- 24.Clark L. A., Boriack-Sjodin P. A., Eldredge J., Fitch C., Friedman B., Hanf K. J., Jarpe M., Liparoto S. F., Li Y., Lugovskoy A., Miller S., Rushe M., Sherman W., Simon K., and Van Vlijmen H. (2006) Affinity enhancement of an in vivo matured therapeutic antibody using structure-based computational design. Protein Sci. 15, 949–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tinberg C. E., Khare S. D., Dou J., Doyle L., Nelson J. W., Schena A., Jankowski W., Kalodimos C. G., Johnsson K., Stoddard B. L., and Baker D. (2013) Computational design of ligand-binding proteins with high affinity and selectivity. Nature 501, 212–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cauerhff A., Goldbaum F. A., and Braden B. C. (2004) Structural mechanism for affinity maturation of an anti-lysozyme antibody. Proc. Natl. Acad. Sci. U.S.A. 101, 3539–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Y., Wiesmann C., Fuh G., Li B., Christinger H. W., McKay P., de Vos A. M., and Lowman H. B. (1999) Selection and analysis of an optimized anti-VEGF antibody: crystal structure of an affinity-matured Fab in complex with antigen. J. Mol. Biol. 293, 865–881 [DOI] [PubMed] [Google Scholar]
- 28.Corrada D., and Colombo G. (2013) Energetic and dynamic aspects of the affinity maturation process: characterizing improved variants from the bevacizumab antibody with molecular simulations. J. Chem. Inf. Model. 53, 2937–2950 [DOI] [PubMed] [Google Scholar]
- 29.Garcia-Rodriguez C., Levy R., Arndt J. W., Forsyth C. M., Razai A., Lou J., Geren I., Stevens R. C., and Marks J. D. (2007) Molecular evolution of antibody cross-reactivity for two subtypes of type A botulinum neurotoxin. Nat. Biotechnol. 25, 107–116 [DOI] [PubMed] [Google Scholar]
- 30.Sun S. B., Sen S., Kim N. J., Magliery T. J., Schultz P. G., and Wang F. (2013) Mutational analysis of 48G7 reveals that somatic hypermutation affects both antibody stability and binding affinity. J. Am. Chem. Soc. 135, 9980–9983 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Ewert S., Huber T., Honegger A., and Plückthun A. (2003) Biophysical properties of human antibody variable domains. J. Mol. Biol. 325, 531–553 [DOI] [PubMed] [Google Scholar]
- 32.Zhai W., Glanville J., Fuhrmann M., Mei L., Ni I., Sundar P. D., Van Blarcom T., Abdiche Y., Lindquist K., Strohner R., Telman D., Cappuccilli G., Finlay W. J., Van den Brulle J., Cox D. R., Pons J., and Rajpal A. (2011) Synthetic antibodies designed on natural sequence landscapes. J. Mol. Biol. 412, 55–71 [DOI] [PubMed] [Google Scholar]
- 33.Glanville J., Zhai W., Berka J., Telman D., Huerta G., Mehta G. R., Ni I., Mei L., Sundar P. D., Day G. M., Cox D., Rajpal A., and Pons J. (2009) Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. U.S.A. 106, 20216–20221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiller T., Schuster I., Deppe D., Siegers K., Strohner R., Herrmann T., Berenguer M., Poujol D., Stehle J., Stark Y., Heßling M., Daubert D., Felderer K., Kaden S., Kölln J., Enzelberger M., and Urlinger S. (2013) A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs 5, 445–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Röthlisberger D., Honegger A., and Plückthun A. (2005) Domain interactions in the Fab fragment: a comparative evaluation of the single-chain Fv and Fab format engineered with variable domains of different stability. J. Mol. Biol. 347, 773–789 [DOI] [PubMed] [Google Scholar]
- 36.Almagro J. C., Teplyakov A., Luo J., Sweet R. W., Kodangattil S., Hernandez-Guzman F., and Gilliland G. L. (2014) Second antibody modeling assessment (AMA-II). Proteins 82, 1553–1562 [DOI] [PubMed] [Google Scholar]
- 37.Mahon C. M., Lambert M. A., Glanville J., Wade J. M., Fennell B. J., Krebs M. R., Armellino D., Yang S., Liu X., O'Sullivan C. M., Autin B., Oficjalska K., Bloom L., Paulsen J., Gill D., Damelin M., Cunningham O., and Finlay W. J. (2013) Comprehensive interrogation of a minimalist synthetic CDR-H3 library and its ability to generate antibodies with therapeutic potential. J. Mol. Biol. 425, 1712–1730 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.