

Abstract
In LeuT, a prokaryotic homolog of neurotransmitter transporters, Na+ stabilizes outward-open conformational states. We examined how each of the two LeuT Na+ binding sites contributes to Na+-dependent closure of the cytoplasmic pathway using biochemical and biophysical assays of conformation. Mutating either of two residues that contribute to the Na2 site completely prevented cytoplasmic closure in response to Na+, suggesting that Na2 is essential for this conformational change, whereas Na1 mutants retained Na+ responsiveness. However, mutation of Na1 residues also influenced the Na+-dependent conformational change in ways that varied depending on the position mutated. Computational analyses suggest those mutants influence the ability of Na1 binding to hydrate the substrate pathway and perturb an interaction network leading to the extracellular gate. Overall, the results demonstrate that occupation of Na2 stabilizes outward-facing conformations presumably through a direct interaction between Na+ and transmembrane helices 1 and 8, whereas Na+ binding at Na1 influences conformational change through a network of intermediary interactions. The results also provide evidence that N-terminal release and helix motions represent distinct steps in cytoplasmic pathway opening.
Keywords: conformational change, membrane transport, molecular dynamics, neurotransmitter, single-molecule biophysics, transport, alternating access, coupling, sodium
Introduction
Ion-coupled transporters utilize the energy stored in transmembrane ion gradients for the energetically uphill transport of solutes. The transporters appear to obey rules that govern when conformational changes are permitted to occur. Coupling between ion and substrate flux depends on the interconversion of inward- and outward-open transporter conformations preferentially when particular binding sites are occupied. For example, when ions and substrate are transported in the same direction (symport), coupling is maximized by only allowing these conformational changes to occur either when the transporter is empty or when it is loaded with both ions and substrate. If partially loaded transporter complexes were to undergo this conformational change, the resulting independent movement of ions or substrate would lead to a loss of coupling. Thus, occupation of ion and substrate sites is expected to influence the dynamics of conformational change in coupled transporters.
Experimental evidence for the influence of binding events on transporter conformational change is sparse. However, in the neurotransmitter:sodium symporter (NSS)2 family, several studies have shown that the Na+-coupled amino acid transporters Tyt1 and LeuT respond conformationally to the binding of Na+ and amino acids (1–5). LeuT and Tyt1 are prokaryotic homologs of metazoan neurotransmitter transporters in the NSS family. LeuT has proven to be a valuable structural model for understanding structure and function of NSS transporters for neurotransmitters such as glycine, γ-aminobutyric acid (GABA), serotonin, norepinephrine, and dopamine. Crystal structures of LeuT show substrate and two Na+ ions bound at sites near the center of the protein (6, 7), with aqueous pathways connecting these sites to the extracellular medium and the cytoplasm in outward- and inward-open conformations, respectively (8). X-ray structures of LeuT have been obtained in outward-open, outward-occluded, and inward-open conformations (6, 7, 9).
Several studies suggest that the openings of extracellular and cytoplasmic permeation pathways are coupled. First, addition of Na+ to LeuT closed the cytoplasmic pathway as measured using FRET (2) and EPR (5); EPR data also indicate that Na+ opened the extracellular pathway (3, 5). Second, an inward-open x-ray structure of LeuT was obtained by mutating the Na+ binding sites and a cytoplasmic gate residue and by binding a Fab fragment to the cytoplasmic surface (9). These changes to the cytoplasmic side of the protein led to closure of the extracellular pathway even though neither the mutations nor the Fab fragment directly affected that pathway. Third, in the human serotonin transporter (SERT), the noncompetitive inhibitor ibogaine increased the accessibility of the cytoplasmic pathway and decreased it in the extracellular pathway (10). Indeed, the expectation is that simultaneous opening of both pathways would lead to a transmembrane leak pathway that would be counter-productive for coupled transport; nevertheless, intermediates in which both pathways are at least partially closed, as in the occluded structures of LeuT (6) and MhsT (11), may facilitate coupling. In other words, although closure of one pathway does not necessarily result in opening of the other pathway, the reverse appears to be true, i.e. the opening of one pathway correlates with closure of the other pathway.
In the absence of Na+ and amino acid substrate, NSS transporters equilibrate between inward- and outward-open conformations (2, 5). Na+, in the absence of substrate amino acids, shifts the distribution between these states in favor of outward-facing (inward-closed) forms, an effect first observed with Tyt1 (1) and subsequently with LeuT (2, 3), GABA transporter (12), and SERT (13). Addition of leucine, a poor substrate for LeuT, stabilizes an intermediate conformation likely resembling crystal structures with substrate bound in an outward-occluded conformation (3, 5), whereas a good substrate, such as alanine in LeuT or tyrosine in Tyt1, overcomes the conformational restriction imposed by Na+, dramatically increasing the prevalence of inward-open conformations (1, 4). Evidence suggests that in Tyt1, the transitions that occur in the apo-state require protonation of an acidic residue that functionally corresponds to Glu-290 in LeuT (14).
The Na+-substrate stoichiometry for LeuT is presently unknown, although there is strong evidence for electrogenic substrate-Na+ symport (6, 15). The Na+ ions found occupying two sites, Na1 and Na2, in outward-open and outward-occluded structures of LeuT (6, 7, 9) may play different functional roles (16), for example in cation selectivity (17–19) and permeability (20). However, it has not been experimentally established whether the roles of the two sites in LeuT in conformational change differ. Na+ is coordinated by the substrate carboxyl group in Na1 (Fig. 1A), and this interaction likely contributes to the strong Na+ dependence of substrate binding in LeuT (6). Na2 is formed by transmembrane helices 1 and 8 (TM1 and TM8), which are close enough to form a coordination shell for Na+ in outward-facing (occluded and open) LeuT structures (6, 7, 9) but not in the inward-open structure where the two helices have separated to open the cytoplasmic permeation pathway (Fig. 1B) (9, 21, 22). This observation suggests that Na+ binding to Na2 may foster interaction between TMs 1 and 8 that stabilizes LeuT in outward-open conformations (9, 16). In contrast, Na+ binding to the Na1 site has been proposed, based on molecular dynamics (MD) simulations, to modulate the transition between occluded and outward-open states (23–25).
FIGURE 1.
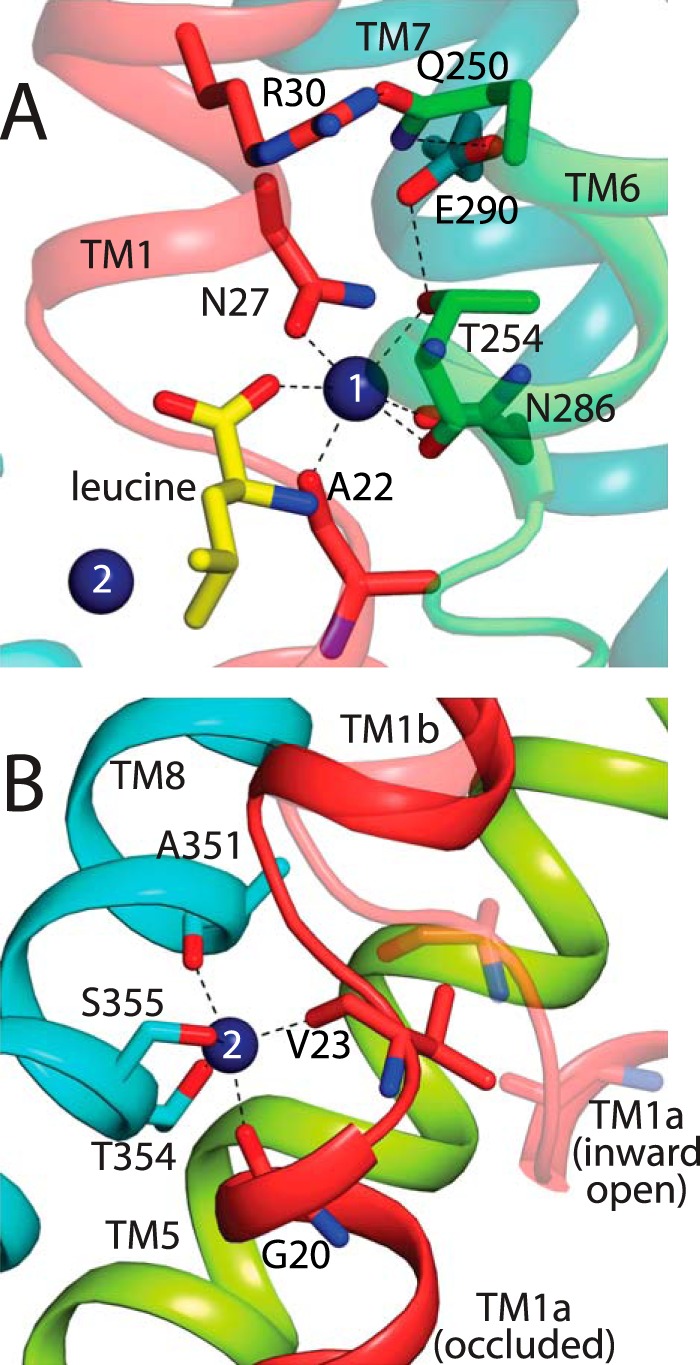
Side chains contributing to Na+ sites in LeuT. A, Na1 site, viewed from the extracellular surface of LeuT, is formed by side chains from Asn-27 in TM1, Thr-254 in TM6, and Asn-286 in TM7. In addition, Na1 includes the substrate carboxyl group and main chain carbonyls from Ala-22 and Thr-254 (6). Proximal residues Glu-290 from TM7, Gln-250 from TM6, and Arg-30 from TM1 are also shown. B, Na2 site, viewed from within the membrane plane, is formed by side chains from Thr-354 and Ser-355 in TM8. In addition, Na2 includes the main chain carbonyls of Gly-20 and Val-23 in TM1 and Ala-351 in TM8 (6). TM1 is shown in two positions, the occluded state in red (6) and in an inward-open conformation in transparent salmon (47). The separation between TMs 1 and 8 found in the inward-open LeuT structure (PDB code 3TT3) (9) is even larger than shown here for an inward-open model, possibly reflecting the detergent/lipid environment in the crystal (21).
Recent EPR studies with the structurally homologous Mhp1, however, were taken as evidence against the role of Na2 in conformational change (26). Although a Na+ site corresponding to the Na2 site of LeuT was found in Mhp1, Na+ had little effect on the conformational equilibrium of this transporter, suggesting that Na2 in Mhp1, and by extension in LeuT, was not responsible for Na+-dependent conformational change. In neither transporter have the individual Na+ sites been directly tested for their involvement in conformational changes.
To explore the roles of the two Na+ binding sites in the conformational dynamics of LeuT, we have monitored the conformation of LeuT Na1 and Na2 mutants in the absence of amino acid substrate. We measured accessibility of a cysteine inserted in the cytoplasmic pathway at the position corresponding to one of the endogenous cysteines of Tyt1 (1) and used single molecule FRET (smFRET) to measure the position of the N terminus. In addition, we carried out MD simulations of wild type and mutant LeuT in outward-open conformations, also in the absence of substrate. Our results show that both the Na+ sites identified in LeuT crystal structures mediate the effect of Na+ on conformation, although the two sites have different modes of action.
Experimental Procedures
Mutagenesis
Site-directed mutagenesis of LeuT was performed using the Stratagene QuikChange protocol. LeuT Na+ site mutants were constructed in the background of the native LeuT, Y265C, and H7C-R86C sequences carrying an His8 tag at the C terminus (6). All mutations were confirmed by DNA sequencing.
Expression of LeuT Mutants in Escherichia coli and Membrane Preparation
LeuT WT and mutants were expressed in C41 (DE3) E. coli cells as described previously (6, 7). Induction of protein expression was initiated at A600 = 0.4 with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside and continued overnight at 18 °C. Membranes were prepared as described using an Emulsiflex C-3 homogenizer (Avestin, Ottawa, Ontario, Canada) (6, 7) and stored at −80 °C until further use.
Binding Assays
All binding experiments involving LeuT mutants were performed using the scintillation proximity assay described by Quick et al. (27). Membranes prepared from cells expressing LeuT mutants were thawed on ice, resuspended in solubilization buffer (200 mm NaCl containing 20 mm Tris, pH 7.4, and 20% glycerol, with 3 mg of n-dodecyl β-d-maltoside (DDM) per mg of membrane pellet, wet weight), and incubated for 1 h at 4 °C with rotation. The solubilized membranes were bound to Cu2+ His tag YSi SPA beads (PerkinElmer Life Sciences) via the C-terminal His8 tag of the protein for 3 h at 4 °C. For each reaction, 400 μg of SPA beads were used per 40 μg of E. coli membrane protein.
For leucine affinity measurements, the SPA beads were washed three times with 5 volumes of assay buffer (200 mm NaCl containing 20 mm Tris, pH 7.4, 20% glycerol, and 1 mm DDM) to remove non-specifically bound proteins. Aliquots of the bead suspension were transferred to the wells of an Isoplate-96TC (PerkinElmer Life Sciences 6005078), and a range of cold leucine concentrations was added. The reactions were initiated by adding ∼1 μCi of [3H]leucine to a final concentration of 1 nm to 12 μm. For measuring the Na+ dependence of leucine binding, the Cu2+ YSi beads with bound protein were washed three times with 5 volumes of assay buffer free of Na+ (200 mm KCl containing 20 mm Tris, pH 7.4, 20% glycerol, and 1 mm DDM), and the beads were resuspended in the same buffer. Binding of leucine was assayed over a range of Na+ concentrations (0–400 mm), and the concentration of [3H]leucine used to initiate the reactions was, for each mutant being studied, 1/10th the measured KD value for leucine (at 200 mm Na+).
Prior to counting, each plate was incubated for 30 min to allow the beads to settle. Radioactivity from each well was counted using a Wallac Microbeta plate reader using the SPA protocol. Nonspecific radioligand binding was assessed by parallel binding measurements in the presence of 400 mm imidazole.
Cysteine Accessibility Measurements
Membranes from C41 (DE3) E. coli cells expressing LeuT mutants (with Y265C) were suspended in Na+ or K+ buffer (200 mm NaCl or 200 mm KCl, respectively, containing 20 mm Tris, pH 7.4) to a final concentration of 0.2 mg/ml. 40 μg of membranes were loaded per well of a 96-well EMD Millipore MultiScreen-HTS FB filter plate (MSFBN6B10) and washed 10 times with either Na+ or K+ buffer. Membranes were incubated with 0–1 mm 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA) on the filter in either Na+ or K+ buffer for 15 min at room temperature. The reaction was terminated by washing the membranes with ice-cold buffer. These membranes are functionally unsealed toward MTSEA, which is permeant, and Na+. Rates of MTSEA reaction with Y265C in membranes prepared in KCl and assayed in NaCl were unchanged in the presence of 1 μm nigericin (data not shown).
After washing, each filter was transferred to 600 μl of 200 mm NaCl containing 20 mm Tris-Cl, pH 7.0, 3 mm DDM, and 8 m urea and shaken for 30 min at 20 °C to solubilize the membranes and denature the proteins. The extract was added to nickel affinity resin (Ni-NTA-agarose, Qiagen) and labeled for 3 h at 20 °C with Alexa Fluor 750 C5 maleimide (Molecular probes, A30459) using a dye/protein molar ratio of about 20:1. Labeled LeuT was purified batchwise by washing with 40 mm imidazole and elution with 300 mm imidazole in the same buffer. The proteins were separated by SDS-PAGE using a BisTris 4–12% gradient gel, and fluorescence was quantified using an Odyssey CLx Fluorescence Reader (Li-Cor Biosciences, Lincoln, NE).
Preparation of LeuT Proteoliposomes
LeuT from 3 liters of culture was purified by solubilizing with 0.2 g of DDM per g (wet weight) of membrane pellet in 200 mm NaCl containing 20 mm Tris-Cl, pH 8, and 1 mm DDM, to a final DDM concentration of 40 mm. The supernatant fraction after centrifugation at 140,000 × g for 40 min was incubated with 2 ml (settled volume) of nickel affinity resin, Ni-NTA-agarose (Qiagen), in solubilization buffer overnight at 4 °C with gentle agitation. The resin was washed in a column with solubilization buffer containing 40 mm imidazole; LeuT was eluted with the same buffer containing 300 mm imidazole, and imidazole was removed by gel filtration.
Purified LeuT was reconstituted into proteoliposomes at a protein/lipid ratio of 1:100 (w/w) as described previously (6, 15, 28). Reconstitution was carried out in the presence of 20 mm HEPES/Tris, pH 7, 100 mm KCl using Triton X-100-destabilized liposomes that were prepared using E. coli polar lipid extract and phosphatidylcholine (Avanti) at a 3:1 (w/w) ratio. The concentration of protein incorporated in the liposomes was determined by the Amido Black assay method (29). Proteoliposomes were flash-frozen in liquid N2 and stored at −80 °C. Prior to assaying transport, the proteoliposome suspension was thawed, diluted 25-fold into 200 mm KCl containing 20 mm HEPES/Tris, pH 7, frozen, and thawed twice and extruded 21 times using a Mini-Extruder (Avanti Polar Lipids) with a 400-nm polycarbonate filter.
Alanine Transport in Proteoliposomes
The time course of [3H]Ala accumulation was measured at room temperature with proteoliposomes diluted into 200 mm NaCl containing 20 mm HEPES/Tris, pH 7, and as a control, Na+-free KCl buffer. Proteoliposomes were diluted in 66 volumes of assay buffer containing 400 nm [3H]alanine and incubated at 20 °C for intervals of 0–25 min, and the reaction was terminated by rapid dilution into 8 volumes of ice-cold 200 mm KCl containing 20 mm HEPES/Tris, pH 7, followed by rapid filtration through GSWP02500 0.22-μm filters (Millipore) and washing with an additional 8 volumes of buffer. Filters were counted in 4 ml of Optifluor (PerkinElmer Life Sciences).
Single Molecule FRET Measurements
Membranes from cells expressing LeuT mutants (with H7C and R86C) were solubilized with 40 mm DDM, and the protein was incubated overnight at 4 °C in 200 mm NaCl containing 20 mm Tris, pH 7.4, 20% glycerol, 1 mm DDM, and 1 mm tris(2-carboxyethyl)phosphine with Ni-NTA-agarose (Qiagen). The resin was washed with the same buffer containing 40 mm imidazole and then with buffer without imidazole. LeuT was labeled while bound to the resin using 4 μm Alexa Fluor 594 C5-maleimide and 1 μm Alexa Fluor 488 C5-maleimide (acceptor/donor molar ratio = 4:1). After incubation for 3 h at 4 °C with rotation in the same buffer with 100 μm tris(2-carboxyethyl)phosphine, the resin was divided in two parts and loaded in two separate columns. One column was washed with the same Na+-containing buffer and the other with Na+-free buffer (NaCl replaced by KCl). LeuT was eluted with Na+ or K+ buffer containing 300 mm imidazole. The elution fraction with the highest protein concentration was selected for desalting by gel filtration to remove imidazole, eluting with buffer containing either Na+ or K+.
FRET measurements were made on a homebuilt system centered on an inverted IX-71 microscope (Olympus) as described previously (30). A continuous emission 488-nm DPSS 50-milliwatt laser (Spectra-Physics, Santa Clara, CA) was adjusted with neutral density filters to 30 microwatts of power just prior to entering the microscope. Fluorescence was collected through the objective, and donor and acceptor photons were separated using a HQ585LP dichroic mirror. Further selection occurred using an ET525/50M filter on the donor channel and an HQ600LP filter on the acceptor channel (Chroma, Bellows Falls, VT). For each channel, fluorescence was focused onto the aperture of a 100-μm optical fiber (OzOptics, Ottawa, Canada) coupled to an avalanche photodiode (PerkinElmer Life Sciences). Photon traces for the acceptor and donor channels were collected in 1-ms time bins using a digital correlator (Flex03LQ-12, correlator.com, Bridgewater, NJ). Measurements were made in chambered cover glasses (Nunc, Rochester, NY) that were passivated by polylysine-conjugated polyethylene glycol treatment to minimize protein adsorption to the chamber surface. All measurements were at 20.0 ± 1.0 °C.
Protein events were selected from background noise using a threshold determined by comparing photon traces of buffer in the absence and presence of protein. The threshold was for a sum of photons in the donor and acceptor channels and was typically around 30 photons per time bin. Adjacent time bins above the threshold were combined into a single event.
Energy transfer efficiency (ETeff) values were calculated for each event by Equation 1,
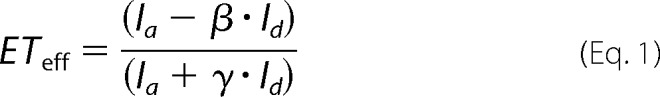 |
where Ia and Id indicate the fluorescence intensity of the acceptor and donor, respectively, β accounts for donor fluorescence bleed through to the acceptor channel (0.06 for Alexa Fluor 488 on our instrument) and γ accounts for differences in the detection efficiency and quantum yield of the fluorophores (1.3 for our system). For each mutant at least three independent photon traces containing several thousand events each were measured.
For each measurement, the ETeff values were compiled into a histogram. The histogram was fit to a sum of three Gaussian peaks, representing the zero peak (resulting from imperfectly labeled protein) and the high and low FRET state of the protein. The positions and widths of the peaks were determined using a global fit to multiple data sets. Areas for each peak were then extracted to determine the population of protein in each conformation.
Data Analysis
Data from binding, transport, accessibility, and smFRET measurements were fit and plotted using Origin (OriginLab, Northampton, MA).
Computational Methods
Continuum Electrostatics Calculations
Electrostatics calculations were carried out using MCCE version 2.9 (31) on crystal structures of the open (9) (PDB entry 3TT1) and occluded states (6) (PDB entry 2A65) in a membrane slab, which was constructed by placing pseudo-atoms uniformly around the protein using an in-house script. The protein and membrane dielectric constant was set to 8.0 or 4.0 for the MCCE calculations, and the dielectric of the water region was set to 80. The Dowser program (79) was used to place additional water molecules in any hydrophilic voids within the protein structure, and these were assigned the same dielectric value as the protein.
Simulation Systems
Molecular dynamics simulations of LeuT were carried out starting with outward open (PDB codes 3TT1) and outward occluded (PDB codes 2A65) conformations. In silico mutations, namely T254A, N286S, and N27S, were made to the outward-open conformation. The WT and the mutant LeuT structures were simulated in the presence of both sodium ions (at Na1 and Na2) or of one ion at Na2. Protonation states were assigned according to calculations performed on the x-ray structures using MCCE as above and in agreement with previous assignments (32). Specifically, Glu-112, Glu-287, and Glu-419 were protonated in all the systems, whereas Glu-290 was deprotonated, except in the outward-open structures in the absence of Na+ at Na1, according to the pKa calculations. Control simulations of the outward-occluded conformation were carried out with both Na+ ions bound, in the presence or absence of substrate.
Simulations were carried out using NAMD (33) with Charmm36 parameters (34–36) using the CMAP correction (37) and TIP3P for waters (38). The dimyristoylphosphocholine bilayer, water, and ions were represented explicitly, with 233 or 234 lipid molecules for the outward-open or occluded proteins, respectively. Dimyristoylphosphocholine was chosen because the hydrophobic matching to LeuT monomers is more optimal than for other pure lipid bilayers (39). The hydrated lipid system was optimized for the LeuT structures using GRIFFIN, as described (40). The system was solvated with around 18,120 water molecules with Na+ and Cl− ions to maintain the system at 100 mm salt concentration (the number of counter ions varied with each system), which resulted in a box size of 94 × 94 × 100 Å.
Simulation Setup
The simulations were performed with constant area in the xy plane for the lipid bilayer. A constant temperature of 310 K was maintained through Langevin dynamics, and a constant pressure of 1 atm was achieved with the Nosé-Hoover Langevin piston algorithm implemented in NAMD (33). The non-bonded interactions were smoothly switched off from 10 to 12 Å, and particle mesh Ewald (PME) summation was used to compute long range electrostatic interactions (41, 42).
The hydrated protein-lipid system was energy-minimized for 100 steps using the steepest descent algorithm (43), followed by 200 steps using the Powell conjugate gradient minimization protocol implemented in CHARMM version 36 (44). Minimization was performed in four stages where initial restraints on all protein atoms were progressively removed from hydrogen atoms, side-chain atoms, and finally all atoms. Initial molecular dynamics equilibration of 8 ns was carried out with harmonic position restraints on the protein atoms, excluding hydrogen atoms; these constraints were gradually released during the equilibration, as follows. For the first 4 ns, the backbone and side-chain atoms were constrained with force constants of 6.58 and 1.64 kcal/mol·Å, respectively. For the subsequent 2 ns, the force constants were reduced to 1.64 and 0.41 kcal/mol·Å, respectively, and in the last 2 ns, they were reduced further to 0.41 and 0.10 kcal/mol·Å, respectively. In the case of N286S, the equilibration included a distance restraint of 50.0 kcal/mol·Å between the ion in Na1 and the binding site oxygen atoms, which increased the subsequent sampling of the Na+-bound state for this mutant. Water, ions, and lipid molecules were thereby allowed to relax around the protein during the equilibration phase; subsequent sampling was considered production.
Simulation Analysis
MD trajectories (n ≥ 3, time scale ∼350 ns per trajectory, or until sodium left the Na1 site, total simulation time ∼1 μs per system) were analyzed every 100 ps with CHARMM c36b2, VMD (45), MDAnalysis (46), and in-house tcl scripts. The extracellular vestibule was defined as conservatively as possible, within a 20 × 15 × 16 Å box centered at (0, −2.5, +2), where the origin of the simulation system was positioned at the center of mass of the membrane phosphate atoms (in the z direction) and the protein (in x and y). This box contains helices TM1b, TM3, TM6a, TM8, and TM10 and was bounded approximately by residues Pro-32, Trp-114, Ala-246, Trp-344, and Ser-399 on the extracellular side, and by Na2 at the bottom of the pathway. The number of waters was averaged over the last 100 ns of all three simulations of each system. Statistical significance was assessed with the unpaired Student's t test. Hydrogen bonds were computed using VMD and were assumed to be formed between two atoms when the donor-acceptor distance was <3.5 Å, and the donor-hydrogen-acceptor angle was 180 ± 30°.
Results
Functional Characterization of Na+ Site Mutants
Na1 is formed by Asn-27, Thr-254, and Asn-286 side chains, main chain carbonyls from Ala-22 and Thr-254, and by the substrate carboxyl when present (Fig. 1A) (6). Na2 is formed by Thr-354 and Ser-355 side chains and main chain carbonyls from Gly-20, Val-23, and Ala-351 (6). We generated LeuT mutants with Thr-254, Thr-354, or Ser-355 replaced with alanine and with Asn-27 or Asn-286 replaced with serine in the background of WT LeuT. The substitutions were intended to diminish the ability of the binding site residues to coordinate Na+ while retaining their chemical characteristics, where possible. In the case of Thr-254, Thr-354, and Ser-355, our only choice was mutation to alanine, but with Asn-27 and Asn-286, we used serine as a replacement to preserve the polar nature of these residues. Each of these single mutants was solubilized, purified, and reconstituted into proteoliposomes and then tested for its ability to transport [3H]alanine in response to an inwardly directed Na+ concentration gradient. All of the mutants were severely compromised in their ability to transport alanine compared with WT LeuT, as shown in Table 1. None of the Na+ site mutants transported alanine at a rate greater than ∼8% of WT LeuT, consistent with the central role of the Na+ sites in LeuT function. We also tested the Y265C mutation used for accessibility measurements, which transported at a rate ∼13% that of WT LeuT. We have noticed that LeuT mutants with unpaired cysteine residues in transmembrane helices are often unstable in detergent, and we suspect that the low activity of this mutant was due to denaturation during the solubilization and reconstitution procedure. Thus, accessibility measurements with mutants containing Y265C were made with protein expressed in native membranes in the absence of detergent.
TABLE 1.
Transport and binding activity of backgrounds and mutants
[3H]Leucine binding and [3H]alanine transport by His-tagged LeuT WT and sodium site mutants (single mutants in WT background) measured by SPA. [3H]Leucine affinity was measured at 200 mm NaCl to determine the KD values. The Na+ dependence of binding was then measured at [3H]leucine concentrations 10% of the measured KD for each mutant. Influx was measured in proteoliposomes containing purified LeuT mutants as described (6, 15).
| Mutant | KD-Leu | K0.5-Na | nNa | Influx |
|---|---|---|---|---|
| nm | mm | pmol/min | ||
| WT | 11.8 ± 1.0 | 59 ± 13 | 1.3 ± 0.1 | 530 ± 160 |
| Y265C | 23.5 ± 2.5 | 98 ± 9 | 1.3 ± 0.1 | 70 ± 25 |
| T354A | 974 ± 18 | >1300 | 1.7 ± 0.1 | 50 ± 5 |
| S355A | 982 ± 382 | >1800 | 1.3 ± 0.1 | 6 ± 16 |
| N27S | 1600 ± 280 | 31 ± 3 | 0.7 ± 0.1 | 5 ± 4 |
| T254A | 1440 ± 96 | >1700 | 1.2 ± 0.1 | 4 ± 6 |
| N286S | 462 ± 23 | 2.6 ± 0.2 | 0.7 ± 0.3 | −2 ± 8 |
Despite the defect in transport activity, all of the mutants retained some affinity for leucine, a poor LeuT substrate that binds tightly to the WT transporter. [3H]Leucine binding was measured by SPA using Cu2+-derivatized beads and His-tagged LeuT WT and mutants solubilized in DDM (27). The KD value for [3H]leucine binding was measured for each mutant in 200 mm NaCl. The Na+ dependence of binding was then measured at [3H]leucine concentrations 10% of the KD value determined for each mutant. All of the mutations altered both leucine affinity and the Na+ dependence of leucine binding (Table 1). Least disruptive was mutation of Asn-286 in Na1 to serine, which increased the leucine KD about 40-fold and decreased EC50 for Na+ (∼20-fold). Mutation of the Na1 residue Asn-27 to serine increased the KD for leucine over 100-fold and decreased the EC50 for Na+ (∼2-fold). The other three mutations, T254A, T354A, and S355A, all increased leucine KD at least 80-fold and increased the EC50 for Na+ to above 1 m. The decrease in leucine binding affinity following mutation of either Na+ site suggests that both Na+ ions are required for optimal binding. In agreement with this finding, the Na+ dependence of leucine binding to WT and several other mutants was noticeably sigmoidal, and the Hill coefficients for fits to those plots were significantly greater than 1 (Table 1). Notable exceptions were the two mutants, N27S and N286S, with K0.5 for Na+ lower than WT, which had Hill coefficients well below 1. These results suggest negative cooperativity between the two Na+ sites in N27S and N286S, in contrast with the positive cooperativity observed with WT and the other mutants.
Measurements of Na+-dependent Conformational Change
To measure the effect of Na+ on LeuT conformation, and how that effect is influenced by Na+ site mutation, we used a biochemical assay based on conformation-dependent accessibility of a cytoplasmic pathway residue (47). In parallel, we examined most of the mutants using smFRET to determine the distribution between outward- and inward-open conformations (2).
The accessibility assay relies on modification of a cysteine residue in the cytoplasmic permeation pathway that becomes more or less accessible when LeuT is in an inward- or outward-open conformation, respectively (Fig. 2A) (47). WT LeuT contains no cysteines. We inserted a single cysteine in TM6, replacing the tyrosine at position 265. This position corresponds to one of the two endogenous cysteines in the cytoplasmic pathway of Tyt1 (Cys-238), which was shown to become less accessible in the presence of Na+ (1). Reactivity toward MTSEA was measured using E. coli membranes from cells expressing each mutant. This preparation of membranes retained binding activity for leucine, which could be measured in a filtration assay. The Y265C mutant retained robust leucine binding activity (Table 1) but, unlike WT LeuT, was sensitive to inactivation upon modification of the inserted cysteine by methane thiosulfonate reagents (data not shown). We followed cysteine modification by fluorescent labeling of the remaining free cysteine after incubating membranes with MTSEA as described under “Experimental Procedures.” Unreacted cysteine was found to decrease with increasing MTSEA concentrations (Fig. 3A). In separate experiments, we verified that the extent of modification was time-dependent at the IC50 value for MTSEA (Fig. 3B) and that the concentration of free MTSEA decreased less than 10% during the incubation as measured using 5-thio-2-nitrobenzoate (48).
FIGURE 2.
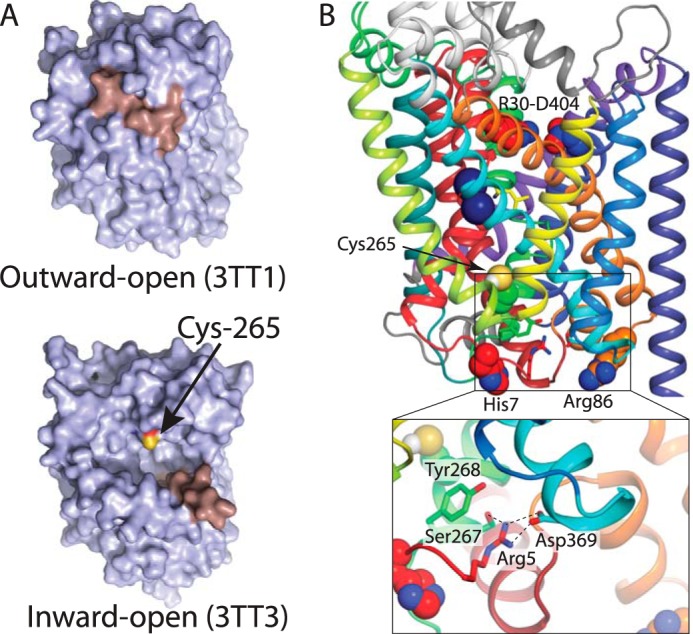
Location of key residues. A, view of the LeuT cytoplasmic surface in the outward-open conformation (PDB code 3TT1) (top) (9) and in an inward-open conformation (PDB code 3TT3) (bottom) (9). The sulfhydryl of Cys-265 is labeled yellow and is accessible to the cytoplasm in the inward-open but not in the occluded structure. TM1 is shown in salmon. B, structure of occluded LeuT (PDB code 2A65) (6), viewed from within the plane of the membrane. The position of Cys-265 in the cytoplasmic pathway is indicated as spheres, below the bound substrate (yellow sticks) and Na+ (blue spheres) and above the cytoplasmic face of LeuT. The position of His-7 and Arg-86 on the N terminus and intracellular loop 1, respectively, are also shown as spheres. The side chains of the intracellular network composed of Ser-267, Tyr-268, Arg-5, and Asp-369 are shown as sticks and in detail in the inset. The color code for transmembrane helices is as follows: TM1, red; TM2, firebrick; TM3, orange; TM4, yellow; TM5, lime; TM6, green; TM7, turquoise; TM8, cyan; TM9, light blue; TM10, deep blue; TM11, purple; TM 12, violet.
FIGURE 3.
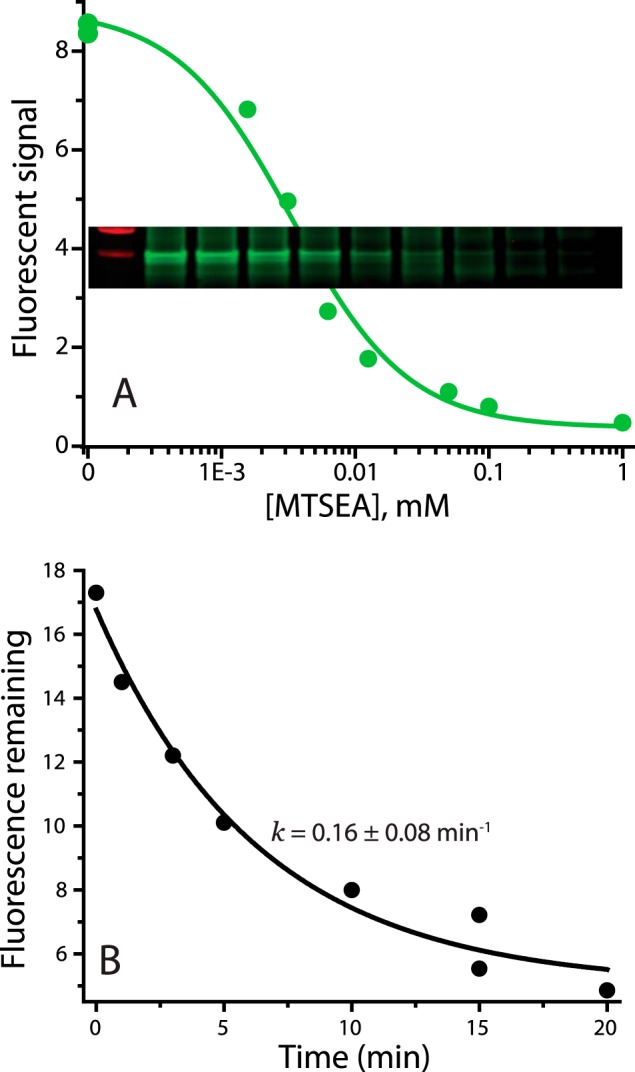
Accessibility assay. A, accessibility assay, raw data. Pretreatment of membranes from E. coli expressing LeuT Y265C with the indicated concentrations of MTSEA for 15 min decreased the extent of subsequent labeling with Alexa Fluor 750 C5 maleimide as described under “Experimental Procedures.” Inset, fluorescence results from gel scan on Li-Cor Odyssey imager. Green circles, line, quantification of band intensities. B, decrease in Alexa Fluor 750 labeling of LeuT Y265C resulting from incubation with 25 μm MTSEA for the indicated times. This MTSEA concentration leads to inactivation of most of the labeling capacity within 15 min. The data were fit as an exponential decay plus a constant for background fluorescence.
smFRET has been used to demonstrate LeuT conformational changes in response to Na+ and substrate (2, 4). Cysteine residues had been introduced at position 7 in the N terminus and residue 86 at the cytoplasmic end of TM3 and labeled with fluorescent dyes (2). Changes in the smFRET signal were initially interpreted as movement of TM1a (2), which is widely believed to move away from TMs 3 and 8 in the process that opens the cytoplasmic permeation pathway (2, 9, 47). However, recent EPR studies of LeuT conformational dynamics show that despite the large Na+-dependent movement of a probe attached to position 7, Na+ induced little or no change in position for a probe placed at position 12, at the beginning of TM1 (5). Arg-5 in the N terminus forms an ion pair with Asp-369 in TM8 that is stabilized by interactions with Ser-267 and Tyr-268 from TM6 (Fig. 2B) (6, 49). Thus, distance changes between residues 7 and 86 in LeuT may reflect a step in cytoplasmic pathway opening that disrupts this ion pair, before any movement of TM1 away from TM3.
To measure Na+-dependent conformational change in LeuT Na1 and Na2 mutants with smFRET, we replaced residues in the N-terminal region (His-7) and the cytoplasmic end of TM3 (Arg-86) with cysteine, as in the previous studies, and reacted them with Alexa Fluor 488 (donor) and Alexa Fluor 594 (acceptor) maleimides. LeuT molecules labeled with both dyes show low FRET efficiency between the fluorophores in inward-open conformations when the N terminus is separated from TM3 and a high FRET efficiency in the outward-open state when the cytoplasmic pathway is closed.
Na2 Site Mutation Blocks the Influence of Na+ on Conformation
Reactivity of Cys-265 in a construct with unaltered Na+ sites is shown in Fig. 4A, which displays results of a typical experiment with the symbols representing individual measurements (combined results from multiple experiments are shown in Fig. 6A). The results show that addition of Na+ to apo-LeuT increased the MTSEA concentration required for half-maximal cysteine modification, consistent with decreased Cys-265 accessibility in Na+. This result agrees with previous observations that, in the absence of substrate, Na+ stabilizes the outward-open conformation and closes the cytoplasmic pathway (1–3).
FIGURE 4.

Effect of Na+ on conformation of LeuT WT and Na+ site mutants as measured by cytoplasmic pathway accessibility. Membranes from E. coli cells expressing LeuT Na+ mutants were treated with a range of MTSEA concentrations in the presence (open circles) or absence (filled circles) of Na+. The membranes were then solubilized, and denatured and unreacted cysteine residues were labeled with Alexa Fluor 750 C5 maleimide as described in detail under “Experimental Procedures.” The cysteine accessibility results are expressed as percentage of maximal labeling with Alexa Fluor 750 C5 maleimide (without MTSEA) for the Y265C background construct (A); Na2 mutant T354A (B); Na1 mutant T254A (C); and Na1 mutant N286S (D). These are representative experiments that were repeated 5–8 times with similar results.
FIGURE 6.
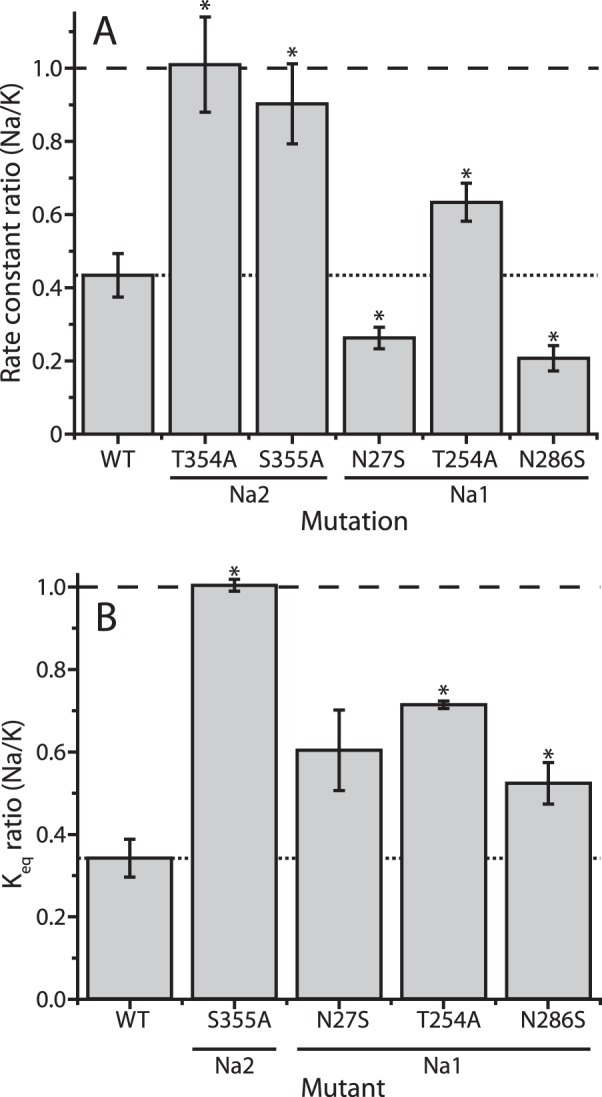
Summarized Na+-dependent conformational changes. A, accessibility results. Cysteine accessibility measurements were used to calculate pseudo-first order rate constants for the MTSEA reaction from the IC50 values in Na+ and K+ and the incubation time. The rate constant in Na+ as a fraction of the rate constant in K+ was calculated for each experiment, and the results presented are the means of 5–8 experiments, with error bars showing the standard errors. Values significantly different from WT (p < 0.05) are indicated with asterisks. B, smFRET results. From equilibrium constants for the distribution between low and high FRET efficiency states (representing open and closed N-terminal position, respectively) in NaCl versus KCl, the ratio (KeqNa/KeqK) was calculated for each of 3–5 experiments with each mutant. A ratio of 1 indicates that Na+ did not change the Keq, and a ratio of <1 indicates that Na+ shifted the distribution toward the high efficiency peak representing the closed N-terminal position. Each bar represents the mean ± S.E. of ratios from 3 to 5 experiments using at least two independent LeuT preparations, and the asterisks represent values that are significantly different from WT (p < 0.05). For the difference between WT and N27S, the unpaired two-sample t test indicated a p value of 0.083.
To investigate whether Na2 was responsible for the ability of Na+ to stabilize LeuT in an outward-open conformation, we tested the mutants T354A and S355A in the Y265C background. Fig. 4B shows that Na+ failed to shift the conformational equilibrium in the Na2 mutant T354A, and similar results were found with S355A (see Fig. 6A). These results represent the first experimental evidence that Na+ binding in Na2 is responsible for closing the cytoplasmic pathway as required for stabilizing LeuT in an outward-open conformation.
We also examined Na2 mutants using smFRET. The distribution of energy transfer efficiency values for LeuT with unmodified Na+ sites is shown in Fig. 5A in the absence (open bars) and presence (filled bars) of Na+. The effect of Na+ was to shift the distribution of LeuT (labeled at Cys-7 and Cys-86) from the low efficiency peak representing an open cytoplasmic pathway to the high efficiency peak associated with cytoplasmic pathway closure. Note that even in the presence of Na+, there remains a small population in which the cytoplasmic pathway is open, suggesting transitions between the inward- and outward-open proton-loaded forms. The Na2 mutant T354A proved unstable in detergent when labeled with FRET fluorophores. However, S355A was stable under these conditions and gave distinct peaks for high and low efficiency populations, suggesting that it could exist in outward- and inward-open conformations (Fig. 5B). Addition of Na+ had no detectable effect on this mutant, in agreement with the accessibility results (Fig. 4); and reaffirming that mutation of Na2 residues abrogated the Na+-dependent conformational shift.
FIGURE 5.

Effect of Na+ on conformation of LeuT WT and Na+ site mutants as measured by a shift in distribution between low and high FRET efficiency populations. Na1 and Na2 mutations were made in a LeuT background construct H7C-R86C and labeled with Alexa Fluor 488 and 594 maleimides. A, H7C-R86C (WT), a histogram showing the distribution of FRET states in the presence (filled bars) and absence (empty bars) of Na+. The peak centered around 0.8 represents inward-closed conformations in which the fluorophore on Cys-7 is close to the one on Cys-86, and the peak near 0.4 represents inward-open conformations in which the two fluorophores are separated. B, distribution of the Na2 mutant S355A between conformational states was not shifted by Na+. C and D, Na+ induced a shift in distribution in the Na1 site mutants T254A and N286S that was smaller than the shift induced in WT LeuT. A larger proportion of molecules was found in inward-open (low FRET efficiency) conformations in S355A and T254A (B and C) relative to WT and N286S (A and D). The distributions were fitted to three peaks at high, low, and zero efficiency (from imperfectly labeled protein) in the presence (dashed line) and absence (solid line) of Na+.
Na1 Mutation Alters, but Does Not Abolish, the Conformational Response to Na+
LeuT mutated at Na1 retained conformational responsiveness to Na+, as measured by Cys-265 accessibility (Figs. 4 and 6A). However, mutation of Na1 altered the extent of the response in a manner that varied depending on the specific mutation. Fig. 4C shows that, relative to the Y265C background mutant, Na+ had a decreased effect on T254A (also Fig. 6A). Remarkably, when either Asn-27 or Asn-286 was mutated to serine, LeuT responded to Na+ with an enhanced conformational shift toward outward-open conformations, revealed by a greater rightward shift of the Na+ curve (shown in Fig. 4D for N286S and in Fig. 6A for both mutants). smFRET measurements also demonstrated Na+-induced shifts in distribution between conformational states for the LeuT Na1 mutants (Figs. 5, C and D, and 6B). However, each of these Na+-induced shifts was smaller than that seen in the LeuT construct with intact Na+ sites.
Fig. 6 summarizes the effects of Na+ on the conformational equilibrium in LeuT WT and Na1 and Na2 mutants. For Cys-265 accessibility measurements (Fig. 6A), we used the IC50 values for MTSEA and the time of incubation to calculate a pseudo-first order rate constant for the MTSEA reaction (see Fig. 7A). For smFRET experiments (Fig. 6B), we used the equilibrium constant (Keq) between low and high efficiency FRET populations (Fig. 7B). In Fig. 6, we express the effect of Na+ as the ratio of the values in the presence and absence of Na+. We found that the ratio of rate constants was more reproducible between experiments than were the absolute values for those rate constants (Fig. 7A). In LeuT constructs with unmodified Na+ sites (Y265C and labeled H7C-R86C), Na+ decreased the fraction of inward-open conformations to less than half of the fraction in K+ controls (dotted versus dashed lines in Fig. 6), and this decrease was eliminated for Na2 mutants T354A and S355A. For the Na1 mutant T254A, Na+ suppressed the fraction significantly, but not as much as in WT, using both Cys-265 accessibility and smFRET to measure conformation. For Na1 mutants N27S and N286S, however, the two methods diverged in their assessment of the conformational response to Na+. Cys-265 accessibility indicated that Na+ had a greater effect on conformation in these mutants relative to the construct with unmodified Na+ sites (Fig. 6A). In contrast, smFRET measurements indicated that the same Na1 mutations decreased the influence of Na+ on LeuT conformation, again relative to the corresponding construct with intact Na+ sites (Fig. 6B).
FIGURE 7.

Rate constants and equilibrium constants (absolute values) summarized over all experiments. A, rates of cytoplasmic pathway modification for LeuT WT and sodium site mutants in Na+ and K+. We used the IC50 value from cysteine accessibility data (as in Fig. 4) and the time of incubation to calculate a pseudo-first order rate constant for the MTSEA reaction. For LeuT mutants in which Na+ decreased accessibility, such as Y265C (WT), the modification rate in Na+ (light gray bars) was less than the rate in K+ (dark gray bars). The data represent the mean of rate constants determined in 5–8 individual experiments, with error bars representing the standard errors. Asterisks indicate mutants for which the rate with Na+ was significantly lower than that with K+ (p > 0.05). The p value for T254A was 0.06. The variability in the individual rates was greater than for the Na+/K+ ratios (Fig. 6A) because of variations between experiments that affected both rates. There was a high degree of correlation between the Na+ and K+ rates in different experiments as evidenced by Pearson product-moment correlation coefficients of 0.80, 0.76, 0.78, 0.36, 0.81, and 0.57 for WT, T354A, S355A, N27S, T254A, and N286S, respectively. B, conformational equilibria determined from smFRET measurements in LeuT WT and sodium site mutants in Na+ and K+. From the areas (A) under the fitted peaks for low and high FRET efficiency as shown in Fig. 5, equilibrium constants were calculated as Alow/Ahigh. The ability of Na+ to decrease the Keq for LeuT WT (Y265C) was completely blocked by mutation of the Na2 residue Ser-355 and decreased by mutation of Na1 residues Asn-27, Thr-254, and Asn-286. The data presented are means of equilibrium constants determined in 3–5 individual experiments with error bars showing the S.E. Asterisks indicate mutants for which the Keq with Na+ was significantly lower than that with K+ (p < 0.001). The p value for N27S was 0.128. Na+ site mutant Keq values in the absence of Na+ all differed significantly from WT (p < 0.002).
We note that, in addition to the Na+-dependent changes in conformation for WT and the Na1 mutants, these data also show variations between mutants in the absence of Na+. Using Cys-265 accessibility, S355A, N27S, and T254A were more reactive than WT, even in the absence of Na+, suggesting that inward-open conformations were more prevalent in these mutants (Fig. 7A). Fig. 5, B and C (and Fig. 7B), shows that the equilibrium between open and closed conformational states determined by smFRET contained a higher proportion of inward-open states in S355A and T254A.
Effects of Na1 Mutation on Ion Coordination and on the Extracellular Gate
An interesting observation from the above results is that in the Na2 mutants (when Na1 is intact), Na+ binding has no effect on the conformational equilibrium of the transporter, and yet, when Na2 is intact, mutations in Na1 are able to modulate that equilibrium, both in the presence and absence of Na+. Previous simulation studies have suggested that Na1 binding to a pre-existing outward-facing conformation modulates the opening of the extracellular pathway in the transition between outward-open and outward-occluded states (23–25). To gain insight into whether the Na1 mutations in LeuT affect this transition, we carried out molecular dynamics simulations initiated with the substrate-free Na+-bound outward-open structure of LeuT (PDB ID 3TT1 (9)). The Tyr-108 residue that had been replaced by Phe in the crystallographic construct was restored, and the protein was embedded in a hydrated lipid bilayer. Multiple simulations were carried out for WT and for the three Na1 mutants, with a total of ∼1 μs simulation time for each construct.
As expected, in the simulations of WT LeuT, ion coordination at Na1 maintained the interaction distances present in the crystal structure of the outward-open state, with the exception of a water molecule that supplements the coordination (Fig. 8A) in place of the substrate carboxyl group. Comparison of simulations of the outward-open WT protein in the presence and absence of an ion at Na1 (with Na2 occupied) revealed an increase (∼19%) in the number of water molecules in the pathway in the presence of Na+ (Table 2), which is comparable with earlier reports (23–25). This increased hydration occurs with only small (∼0.05 Å) differences in conformation, as measured by the root-mean-squared deviation (r.m.s.d.) of TM1b and TM6a relative to the crystal structures (Table 2). These results are consistent with earlier work indicating that Na1 mediates hydration of the pathway, a process that may reduce the ability of the pathway to close, and thereby aid in substrate binding to the Na+-loaded transporter (23–25).
FIGURE 8.

Na+ coordination at WT and mutant Na1 sites in outward-open LeuT. Distances between Na+ and coordinating oxygen atoms in Na1 were computed from MD simulations of WT (A), N27S (B), T254A (C), and N286S (D) LeuT and are plotted as a normalized distribution. Distances were measured to water oxygen (purple), the backbone oxygen atom of Ala-22 (black), or Thr-254 (green) and the side chain oxygen atom of Asn-27/Ser-27 (red) or Asn-286/Ser-286 (cyan). For N286S, sections of the trajectories in which the ion was no longer bound (all oxygen-Na+ distances >3.2 Å) were excluded from the analysis. The distance to the Glu-290 side chain was determined using the minimum distance to either carboxyl O atom (orange). Insets show representative snapshots of the side chains that contribute to Na1 in WT LeuT, as well as the network of interactions connecting Na1 to the Arg-30–Asp-404 salt bridge in the extracellular pathway.
TABLE 2.
Properties of the extracellular pathway during MD simulations of outward-open LeuT
Properties were computed for the last 100 ns of simulations in the presence (+Na1) or absence (−Na1) of Na+ at Na1. In the latter case, Glu-290 was protonated, and the ion had been replaced by a water molecule by Dowser. The number of waters was counted in the extracellular vestibule, as defined under “Experimental Procedures.” The r.m.s.d. (Å) of the Cα atoms of helices TM1a (residues 24–39) and TM6b (residues 241–256), after superposing the backbone atoms of helices TM3, -4, -8, and -10 from the scaffold, with respect to those of the outward-open (“open”, PDB code 3TT1) or occluded (“occ,” PDB code 2A65) energy-minimized structures, was averaged over the last 150 ns of three >300-ns-long trajectories. ± indicates the standard deviation over the three trajectories. The occluded state with substrate and without a Na+ ion at Na1 was not simulated (NS).
| Water count |
r.m.s.d. from occ |
r.m.s.d. from outward-open |
||||
|---|---|---|---|---|---|---|
| −Na1 | +Na1 | −Na1 | +Na1 | −Na1 | +Na1 | |
| Å | Å | |||||
| WT occ | NS | 11.2 ± 0.02 | NS | 0.72 ± 0.01 | NS | 2.80 ± 0.01 |
| WT open | 22.2 ± 0.1 | 27.4 ± 0.02 | 2.60 ± 0.01 | 2.79 ± 0.01 | 1.37 ± 0.01 | 1.32 ± 0.01 |
| N27S | 22.6 ± 0.1 | 21.0 ± 0.14 | 2.25 ± 0.03 | 2.60 ± 0.02 | 1.70 ± 0.02 | 1.53 ± 0.01 |
| T254A | 23.0 ± 0.1 | 25.7 ± 0.02 | 2.69 ± 0.02 | 2.79 ± 0.01 | 1.43 ± 0.02 | 1.26 ± 0.01 |
| N286S | 23.8 ± 0.1 | 25.4 ± 0.02 | 2.40 ± 0.02 | 2.75 ± 0.01 | 1.43 ± 0.02 | 1.35 ± 0.01 |
With the WT simulations for reference, we then made in silico mutations of Na1 mimicking the experimental substitutions in the outward-open structure of LeuT. Simulations were carried out with Na2 occupied and either with or without an ion at Na1. On the timescale of these simulations (∼350-ns-long trajectories), the Na+ ion remained coordinated to the WT and to the T254A and N27S mutant binding sites (Fig. 8, A–C). However, the binding site adapted in the two mutants, so that the ion interacted directly with the carboxyl oxygen atoms of Glu-290 in addition to the oxygen atoms of the other binding site residues, apparently to compensate for the interactions lost upon mutation. In simulations of the N286S mutant, the ion did not interact significantly with Glu-290, despite the fact that the ion lost coordination from the side chain at position 286 (Fig. 8D). Presumably because of this reduced number of interactions in the N286S mutant, the Na+ ion escaped from the Na1 binding pocket within the first 60 ns in 6 out of 10 simulation trajectories computed for this mutant, suggesting a significantly weaker affinity than in the WT and the other Na1 mutants, at least for the outward-open state. Note that the experimental K0.5-Na values in Table 1 cannot be directly compared with these simulation results because the binding experiments were carried out in the presence of amino acid substrate.
Interestingly, in the simulations of LeuT Na1 site mutants, even with Na+ bound at Na1, there were fewer waters in the extracellular pathway than in WT LeuT (Table 2; p < 10−5). In particular, the pathway in N27S contained a similar number of waters to the WT protein in the absence of Na+ at Na1, reflecting the fact that N27S tended to adopt a conformation that was more occluded than WT under equivalent conditions; in the presence of Na+, the r.m.s.d. of N27S to the occluded state was similar to that of the WT protein without Na+ (2.6 Å; Table 2), whereas in the absence of Na+ N27S is even more similar to the occluded state structure than the WT under the same conditions (r.m.s.d. 2.3 Å; Table 2). This tendency of the N27S mutant to become more closed on the extracellular side might be related to the high accessibility of its cytoplasmic pathway in the absence of Na+ (Fig. 7A).
To understand in molecular detail how N27S and T254A affect the extracellular pathway closure even though Na+ remains bound to Na1 during the simulations (Fig. 8, B and C), we analyzed a network of polar interactions that connects the residues comprising Na1 with a salt bridge between Arg-30 in TM1b and Asp-404 in TM10 spanning the extracellular pathway (Figs. 1A, 2B, and 8A). The Arg-30–Asp-404 salt bridge is observed in structures of the inward-open and outward-occluded states (where it constitutes part of the so-called “thin gate” that prevents bound substrate from dissociating back to the extracellular side (8)) but not when the extracellular pathway is open (6, 7, 9). Consistent with this, and with published simulation studies (50), the Arg-30–Asp-404 salt bridge remained formed during simulations of the outward-occluded WT protein in the presence of substrate (Fig. 9A). In contrast, in the outward-open conformation, in the absence of substrate but with Na+ bound at both Na1 and Na2, the Arg-30–Asp-404 bridge was formed ∼45% of the simulation time (Fig. 9A) (25). Notably, mutations in Na1 altered the propensity for this salt bridge to form, even in the absence of substrate. The N27S mutant, for example, formed the salt bridge more frequently than WT LeuT (∼60% of the simulation time), consistent with its more occluded conformation (Table 2). In contrast, the N286S mutant formed the salt bridge less frequently (<25% of the simulation time). These observed changes in the extracellular pathway behavior appear to be consistent with accessibility measurements showing more inward-closed conformations in N286S (decreased rate constant) and more inward-open conformations for T254A and N27S (increased rate constant) compared with WT (Y265C) in the presence of Na+ (Fig. 7A).
FIGURE 9.
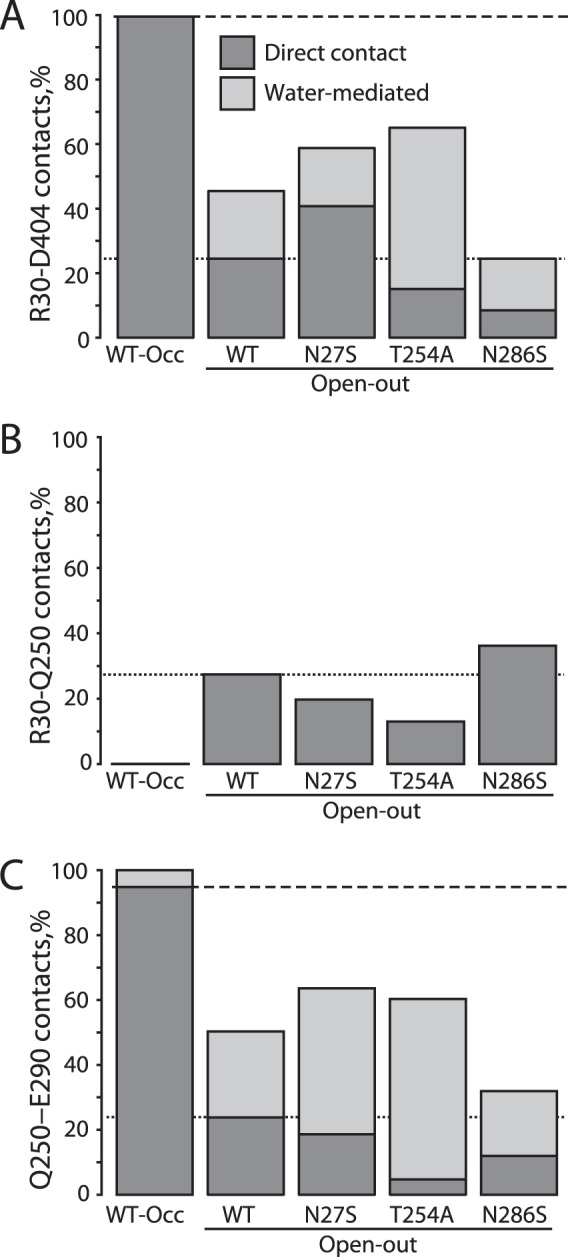
Changes in salt bridge and hydrogen bond propensity in the extracellular pathway of LeuT WT and Na1 mutants. The prevalence of specific side-chain interactions is shown as the percentage of the simulation time that atoms are within a cutoff distance for the following: A, Arg-30–Asp-404 salt bridge, defined as terminal side-chain C atoms <5.5 Å (for direct interactions) and the presence of a common water molecule that hydrogen-bonds with both Arg-30 and Asp-404 simultaneously (for water-mediated interactions). The dashed and dotted lines in A–C represent direct (non-water-mediated) interactions for the WT-occluded and -open simulations, respectively. B, Arg-30–Gln-250 hydrogen bonds, defined as the minimum amine hydrogen to amide oxygen distance <2.8 Å, and C, Gln-250–Glu-290 hydrogen bonds, defined as the minimum amide hydrogen to terminal oxygen distance <3 Å. The same definition of water-mediated contacts was used as for Arg-30–Asp-404. For N286S, sections of the trajectories in which the ion was no longer bound (all oxygen-Na+ distances >3.2 Å), were excluded from the analysis.
The connection between this salt bridge and Na1 involved Gln-250 in TM6 and Glu-290 in TM7 (Fig. 9, B and C), as described previously (51). In simulations of WT outward-occluded LeuT, for example, Gln-250 formed negligible direct interactions with Arg-30 (Fig. 9B) but instead interacted directly with Glu-290 (Fig. 9C). Because Glu-290 interacted with Gln-250, Arg-30 was free to form a salt bridge with Asp-404, as described previously (Fig. 9A). In contrast, in the substrate-free outward-open state with Na1 occupied, Gln-250 alternated between Glu-290 contacts (Fig. 9C) and interactions with Arg-30 (Fig. 9B), consequently sequestering Arg-30 away from Asp-404 (Fig. 9A) (51).
Similar patterns of reciprocity could be seen for the Na1 mutants (Fig. 9). For example, N286S formed the salt bridge directly less often than WT, and thus Arg-30 interacted with Gln-250 more frequently, although the opposite was true for N27S. A slightly different pattern could be seen in T254A, in which water-mediated contacts became more prevalent between Arg-30 and Asp-404, presumably because of the direct interaction between Glu-290 and the ion in that mutant (Fig. 8C).
Overall, these mutations of Na1 led to subtle changes in the local interaction network that may affect the influence of Na+ on the extracellular pathway, and this in turn may impact the propensity of the cytoplasmic pathway to open or close. Similar disruptions in the extracellular pathway, as well as longer range effects, were reported recently upon in silico mutation of cytoplasmic pathway gating residues, or when Li+ was placed in the Na2 and Na1 binding sites (24).
We also considered the possibility that the mutation of Na1 affects the protonation state of Glu-290, which is essential for the proton-dependent antiport step of the LeuT transport cycle. Calculations of the pKa value of Glu-290 using a continuum electrostatic approach under the Poisson framework (31) confirmed that in the outward-open WT LeuT, Glu-290 is likely to be deprotonated when Na+ is bound to Na1 (Table 2 and Fig. 1A) (32), whereas when Na1 is empty, the predicted pKa was close to 7. In the Na1 mutants, however, only small shifts were seen in the calculated pKa values of Glu-290 relative to WT, particularly for N286S (Table 2), suggesting that Glu-290 protonation is less likely than other factors, described above, to mediate the effects of Na1 mutation.
Discussion
Here, we provide the first experimental evidence that Na+ binding at Na2 stabilizes outward-facing conformations of LeuT. Mutation of either of the two side chains (Thr-354 and Ser-355) contributing to Na2 blocked the influence of Na+ on the LeuT conformational equilibrium (Figs. 4 and 5). This result demonstrates the importance of the close interaction between TMs 1 and 8 at the point where the two helices cross, near the substrate binding site, observed in outward-facing structures of LeuT (Fig. 1B) (6, 9). Those regions of TMs 1 and 8 are separated in an inward-open x-ray structure of LeuT (9), in a model of LeuT in the inward-open state (47), and in inward-open crystal structures of the related transporters Mhp1 (52), BetP (53), and vSGLT (54), suggesting that Na+ binding in Na2 fosters close interaction between TMs 1 and 8 and that this interaction stabilizes outward-facing conformations in LeuT (9, 16). As a corollary, Na2 should have a higher affinity for Na+ in the outward-facing states than in the inward-facing states. That the conformational response to Na+ requires an intact Na2 site suggests that the outward-open conformation is also required for high affinity leucine binding, accounting for low substrate and ion affinity in Na2 mutants (Table 1).
Aside from the NSS family, several other transporter families also adopt the five-transmembrane helix inverted-repeat, LeuT-like fold (55). These include the APC (56), BCCT (57), SSS (54), NCS1 (58), and Nramp (59) families. Crystal structures of the Na+-dependent members of these families support a conserved Na+ site homologous to the Na2 site of LeuT (54, 57, 58), although the Na1 site of LeuT is apparently not conserved across families. Measurements of conformational dynamics in the structurally similar Na+-dependent hydantoin transporter Mhp1 demonstrated no significant conformational response to Na+ despite the presence of a Na+ binding site corresponding to Na2 in LeuT (26). This led to the proposal that it was the non-conserved Na1 site that was responsible for the effect on LeuT conformation (26). Our results clearly show the importance of Na2 for the response of LeuT to Na+ and highlight the limits to comparisons between families, even those of the same structural fold.
Within the larger five TM-inverted repeat, LeuT-like fold superfamily (55) containing NSS, there are transporters with diverse coupling strategies, including amino acid exchangers and transporters coupled by symport and antiport to diverse ions, including Na+, H+, Cl−, and K+ (60–64). Although the common overall protein fold suggests a similar conformational mechanism of transport for these proteins, the diversity of coupling processes presages a corresponding variety in the way that substrates and ions influence conformational change. Even within the NSS family, there are some transporters coupled to Cl− and others not (32, 65–67). However, the conformational response to Na+ is conserved from Tyt1 to SERT (1, 13), and we anticipate that the Na2 site will be found critical for this response throughout the family. Mutations of the Na1 and Na2 sites in SERT have been shown to affect transport, ion selectivity, and reactivity of an endogenous cysteine residue (68–70). Nevertheless, we caution against extending our conclusions beyond the NSS transporters.
Interestingly, we also observed that mutation of Ser-355 in Na2 altered the conformational equilibrium even in the absence of Na+ (Fig. 7). This effect may be explained in the light of observations of a hydrogen bond between Ser-355 and Val-23 during simulations of outward-facing LeuT in the absence of Na+ at the Na2 site (71). The loss of this hydrogen bond in S355A could therefore lead to a higher population of inward-facing conformations even in the absence of Na+ (Fig. 7).
Although the possibility that Na2 might participate in conformational change had been noted previously (9, 16, 71), participation of Na1 in this process had not been experimentally tested for LeuT. None of the Na1 mutations studied here completely blocked the conformational effect of Na+, but two of the mutations (N27S and N286S) actually enhanced the Na+-induced cytoplasmic pathway closure. These effects are unlikely to result from modulation of Na2 itself, because all the Na1 positions mutated are separated from Na2 by >7.7 Å (distance to Asn-27 Cγ atom in simulations of occluded state). Thus, it appears that Na1 is not required for the Na+-dependent conformational change but modulates the process by a mechanism distinct from the Na2-mediated interaction between TMs 1and 8.
By simulating the Na1 site mutants in the absence of substrate, we could clearly show changes in the Na+ binding site configuration, with the protein adapting to the new coordination (Fig. 8). These modifications led to differences in propensity for extracellular Arg-30–Asp-404 salt bridge formation in the Na1 mutants, as the network of interactions connecting Na1 and the salt bridge shifted (Fig. 9). Thus, the influence of Na1 on conformational change appears likely to reflect the ability of the ion to organize local interactions, rather than global changes in helix-helix distance, as Na2 does. A recent simulation study suggests that an allosteric interaction network involving specific residues in TM1, TM6, and TM8 connect the Na1 and Na2 binding sites to the cytoplasmic gate (24), although the relative contributions of Na2 and Na1 sites to the network were not delineated. Taken together, the results also raise the possibility that Na1 fosters interactions required for extracellular pathway closure upon substrate binding, thereby allowing the cytoplasmic pathway to open.
The results presented here provide insight into the mechanisms that underlie the control of conformational change by ligand binding. The effect of Na+ on LeuT conformation (Figs. 4 and 5) (2–5) and extracellular pathway hydration (Table 2) (23, 25) and the Na+ requirement for substrate binding (Table 1) (72) together serve to limit the probability of uncoupled transport. The strong decrease in the frequency of outward- to inward-open conformational transitions that accompany Na+ binding (4) lowers the possibility that Na+ will be transported alone. Furthermore, the Na+ requirement for substrate binding reduces the chances of unaccompanied substrate flux. Although these properties minimize uncoupled flux, efficient coupled transport requires that substrate binding is able to overcome the conformational restriction imposed by Na+. This phenomenon has been observed (1, 4), and although the molecular determinants underlying the action of substrate have yet to be identified, the participation of the substrate carboxyl in Na1 suggests that this site is critical for the outward- to inward-open transition. Ongoing studies are addressing the role of Na+ binding sites in the substrate-mediated outward- to inward-open conformational transition. We propose that these interactions hold the key to understanding how LeuT achieves coupling of substrate and ion fluxes.
The accessibility assay used here to measure LeuT conformational change is based on previous studies using mammalian and prokaryotic transporters in the NSS family and others (1, 13, 47, 73–76). It has several advantages for measurements of conformational change in these proteins. The assay measures the ability of an aqueous reagent to penetrate into a pathway that is closed in outward-open or occluded LeuT structures (6, 9). It depends on mutation of only one position in that pathway to cysteine (a mutation that has minimal effect on binding activity), and it measures the rate at which that cysteine reacts with an aqueous cysteine reagent. The reactive Cys-265 is over 10 Å from the cytoplasmic end of the pathway and over 11 Å from the binding sites. Its accessibility results from conformational changes that open the cytoplasmic pathway, and it is unlikely to be influenced directly by ion or ligand binding (Fig. 2B). The accessibility assay avoids attachment of fluorescent or spin-labeled probes that could obstruct or alter conformational changes. Importantly, the cysteine reactivity assay can be performed on native membrane fragments (or intact cells (1, 10)), avoiding the use of detergents that might disrupt or modify conformational responses (77, 78).
Our results identified some differences between the conformational changes measured by cytoplasmic pathway accessibility and by N-terminal movement. For example, the enhanced influence of Na+ on conformational change observed in LeuT mutants N27S and N286S was revealed by accessibility measurements (Fig. 4) but not by measurements of N-terminal opening, which indicated these mutants to be less responsive to Na+ than WT (labeled H7C-R86C) LeuT (Fig. 5). Although we cannot rule out the possibility that the presence of detergent or the fluorescent probes attached to LeuT might have distorted the conformational responses measured by smFRET, the general broad agreement between the techniques suggests that each technique reliably reports on a different aspect of conformational change. These differences suggest that the conformational change that converts LeuT from outward- to inward-open states consists of several steps that may not occur synchronously. The divergent response to Na+ measured by the two techniques could result if Na+ had a stronger effect on closing the cytoplasmic pathway in these mutants relative to WT LeuT but had a weaker effect on interactions between the N terminus and TMs 6 and 8 in the mutants. This suggests the existence of conformations with a closed cytoplasmic pathway and an open N terminus, as in Refs. 5, 22.
Assuming that the differences between smFRET and accessibility measurements result from different responses of the N terminus and the cytoplasmic pathway rather than an artifact, the results suggest that opening the cytoplasmic pathway involves several distinct processes. These include release of the N terminus from its interactions with TMs 6 and 8, here detected by smFRET and also consistent with predictions based on molecular simulations (22), helix movements that allow aqueous reagents to react with Cys-265 in the cytoplasmic pathway, and possibly other motions (24), which may all be part of the larger conformational change that closes the extracellular pathway and opens the cytoplasmic pathway. In the future, it may be possible to take advantage of techniques, such as these, that isolate the individual motions to understand how Na+ and substrate influence the individual steps in conformational change.
Author Contributions
S. T. generated most of the mutants, performed most of the binding and cysteine accessibility experiments, and prepared purified, fluorescently labeled LeuT for FRET analysis. S. T. and Y.-W. Z. developed methods for measuring conformational change. L. R. F. and A. N. carried out and analyzed molecular dynamics simulations. D. C. D. measured single molecule FRET distributions. E. M. made some of the cysteine accessibility measurements and determined transport activity in reconstituted liposomes. E. Rosado measured leucine binding by SPA in some of the mutants. S. R. generated some of the mutants used. L. R. F. and G. R. conceived the approach, and S. T., E. Rhoades, L. R. F., and G. R. designed the experiments. S. T., E. Rhoades, L. R. F., and G. R. wrote the manuscript, and all authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Satinder Singh and Joseph Mindell for helpful advice for LeuT purification and reconstitution and Dr. José Faraldo-Gómez for helpful discussions. This study used the computational resources of the Biowulf system at the National Institutes of Health, Bethesda, MD (biowulf.nih.gov).
This work was supported in part by the Division of Intramural Research of the National Institutes of Health, NINDS (to L. R. F.), National Institute on Drug Abuse Grants DA007259 and DA008213 (to G. R.), and NINDS Grant NS079955 (to E. Rhoads). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NSS
- neurotransmitter:sodium symporter
- smFRET
- single molecule FRET
- SERT
- serotonin transporter
- MD
- molecular dynamics
- DDM
- dodecyl maltoside
- MTSEA
- 2-aminoethyl methanethiosulfonate hydrobromide
- SPA
- scintillation proximity assay
- TM
- transmembrane helix
- r.m.s.d.
- root-mean squared deviation
- PDB
- Protein Data Bank
- Ni-NTA
- nickel-nitrilotriacetic acid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1.Quick M., Yano H., Goldberg N. R., Duan L., Beuming T., Shi L., Weinstein H., and Javitch J. A. (2006) State-dependent conformations of the translocation pathway in the tyrosine transporter Tyt1, a novel neurotransmitter:sodium symporter from Fusobacterium nucleatum. J. Biol. Chem. 281, 26444–26454 [DOI] [PubMed] [Google Scholar]
- 2.Zhao Y., Terry D., Shi L., Weinstein H., Blanchard S. C., and Javitch J. A. (2010) Single-molecule dynamics of gating in a neurotransmitter transporter homologue. Nature 465, 188–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Claxton D. P., Quick M., Shi L., de Carvalho F. D., Weinstein H., Javitch J. A., and McHaourab H. S. (2010) Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat. Struct. Mol. Biol. 17, 822–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao Y., Terry D. S., Shi L., Quick M., Weinstein H., Blanchard S. C., and Javitch J. A. (2011) Substrate-modulated gating dynamics in a Na+-coupled neurotransmitter transporter homologue. Nature 474, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kazmier K., Sharma S., Quick M., Islam S. M., Roux B., Weinstein H., Javitch J. A., and McHaourab H. S. (2014) Conformational dynamics of ligand-dependent alternating access in LeuT. Nat. Struct. Mol. Biol. 21, 472–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamashita A., Singh S. K., Kawate T., Jin Y., and Gouaux E. (2005) Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 7.Singh S. K., Piscitelli C. L., Yamashita A., and Gouaux E. (2008) A competitive inhibitor traps LeuT in an open-to-out conformation. Science 322, 1655–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krishnamurthy H., Piscitelli C. L., and Gouaux E. (2009) Unlocking the molecular secrets of sodium-coupled transporters. Nature 459, 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krishnamurthy H., and Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacobs M. T., Zhang Y.-W., Campbell S. D., and Rudnick G. (2007) Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J. Biol. Chem. 282, 29441–29447 [DOI] [PubMed] [Google Scholar]
- 11.Malinauskaite L., Quick M., Reinhard L., Lyons J. A., Yano H., Javitch J. A., and Nissen P. (2014) A mechanism for intracellular release of Na+ by neurotransmitter/sodium symporters. Nat. Struct. Mol. Biol. 21, 1006–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ben-Yona A., and Kanner B. I. (2009) Transmembrane domain 8 of the γ-aminobutyric acid transporter GAT-1 lines a cytoplasmic accessibility pathway into its binding pocket. J. Biol. Chem. 284, 9727–9732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fenollar-Ferrer C., Stockner T., Schwarz T. C., Pal A., Gotovina J., Hofmaier T., Jayaraman K., Adhikary S., Kudlacek O., Mehdipour A. R., Tavoulari S., Rudnick G., Singh S. K., Konrat R., Sitte H. H., and Forrest L. R. (2014) Structure and regulatory interactions of the cytoplasmic terminal domains of serotonin transporter. Biochemistry 53, 5444–5460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao Y., Quick M., Shi L., Mehler E. L., Weinstein H., and Javitch J. A. (2010) Substrate-dependent proton antiport in neurotransmitter:sodium symporters. Nat. Chem. Biol. 6, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ryan R. M., and Mindell J. A. (2007) The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat. Struct. Mol. Biol. 14, 365–371 [DOI] [PubMed] [Google Scholar]
- 16.Rudnick G. (2013) How do transporters couple solute movements? Mol. Membr. Biol. 30, 355–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noskov S. Y., and Roux B. (2008) Control of ion selectivity in LeuT: two Na+ binding sites with two different mechanisms. J. Mol. Biol. 377, 804–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu H., Noskov S. Y., and Roux B. (2010) Two mechanisms of ion selectivity in protein binding sites. Proc. Natl. Acad. Sci. U.S.A. 107, 20329–20334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Y., Zomot E., and Kanner B. I. (2006) Identification of a lithium interaction site in the γ-aminobutyric acid (GABA) transporter GAT-1. J. Biol. Chem. 281, 22092–22099 [DOI] [PubMed] [Google Scholar]
- 20.Borre L., Andreassen T. F., Shi L., Weinstein H., and Gether U. (2014) The second sodium site in the dopamine transporter controls cation permeation and is regulated by chloride. J. Biol. Chem. 289, 25764–25773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grouleff J., Søndergaard S., Koldsø H., and Schiøtt B. (2015) Properties of an inward-facing state of LeuT: conformational stability and substrate release. Biophys. J. 108, 1390–1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng M. H., and Bahar I. (2014) Complete mapping of substrate translocation highlights the role of LeuT N-terminal segment in regulating transport cycle. PLoS Comput. Biol. 10, e1003879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao C., Stolzenberg S., Gracia L., Weinstein H., Noskov S., and Shi L. (2012) Ion-controlled conformational dynamics in the outward-open transition from an occluded state of LeuT. Biophys. J. 103, 878–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stolzenberg S., Quick M., Zhao C., Gotfryd K., Khelashvili G., Gether U., Loland C. J., Javitch J. A., Noskov S., Weinstein H., and Shi L. (2015) Mechanism of the association between Na+ binding and conformations at the intracellular gate in neurotransmitter:sodium symporters. J. Biol. Chem. 290, 13992–14003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zomot E., Gur M., and Bahar I. (2015) Microseconds simulations reveal a new sodium-binding site and the mechanism of sodium-coupled substrate uptake by LeuT. J. Biol. Chem. 290, 544–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kazmier K., Sharma S., Islam S. M., Roux B., and Mchaourab H. S. (2014) Conformational cycle and ion-coupling mechanism of the Na+/hydantoin transporter Mhp1. Proc. Natl. Acad. Sci. U.S.A. 111, 14752–14757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quick M., and Javitch J. A. (2007) Monitoring the function of membrane transport proteins in detergent-solubilized form. Proc. Natl. Acad. Sci. U.S.A. 104, 3603–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh S. K., Yamashita A., and Gouaux E. (2007) Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature 448, 952–956 [DOI] [PubMed] [Google Scholar]
- 29.Kaplan R. S., and Pedersen P. L. (1985) Determination of microgram quantities of protein in the presence of milligram levels of lipid with amido black 10B. Anal. Biochem. 150, 97–104 [DOI] [PubMed] [Google Scholar]
- 30.Trexler A. J., and Rhoades E. (2009) α-Synuclein binds large unilamellar vesicles as an extended helix. Biochemistry 48, 2304–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Georgescu R. E., Alexov E. G., and Gunner M. R. (2002) Combining conformational flexibility and continuum electrostatics for calculating pK(a)s in proteins. Biophys. J. 83, 1731–1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Forrest L. R., Tavoulari S., Zhang Y.-W., Rudnick G., and Honig B. (2007) Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., and Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 35.MacKerell A. D. Jr., Feig M., and Brooks C. L. 3rd. (2004) Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699 [DOI] [PubMed] [Google Scholar]
- 36.Klauda J. B., Venable R. M., Freites J. A., O'Connor J. W., Tobias D. J., Mondragon-Ramirez C., Vorobyov I., MacKerell A. D. Jr., and Pastor R. W. (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Best R. B., Zhu X., Shim J., Lopes P. E., Mittal J., Feig M., and Mackerell A. D. Jr. (2012) Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 39.Pantano D. A., and Klein M. L. (2009) Characterization of membrane-protein interactions for the leucine transporter from Aquifex aeolicus by molecular dynamics calculations. J. Phys. Chem. B 113, 13715–13722 [DOI] [PubMed] [Google Scholar]
- 40.Staritzbichler R., Anselmi C., Forrest L. R., and Faraldo-Gómez J. D. (2011) GRIFFIN: A versatile methodology for optimization of protein-lipid interfaces for membrane protein simulations. J. Chem. Theory Comput. 7, 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 42.Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., and Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 43.Bixon M., and Lifson S. (1967) Potential functions and conformations in cycloalkanes. Tetrahedron 23, 769–784 [Google Scholar]
- 44.Brooks B. R., Brooks C. L. 3rd, Mackerell A. D. Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., et al. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Humphrey W., Dalke A., and Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 46.Michaud-Agrawal N., Denning E. J., Woolf T. B., and Beckstein O. (2011) MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 32, 2319–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forrest L. R., Zhang Y. W., Jacobs M. T., Gesmonde J., Xie L., Honig B. H., and Rudnick G. (2008) Mechanism for alternating access in neurotransmitter transporters. Proc. Natl. Acad. Sci. U.S.A. 105, 10338–10343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stauffer D. A., and Karlin A. (1994) Electrostatic potential of the acetylcholine binding sites in the nicotinic receptor probed by reactions of binding-site cysteines with charged methanethiosulfonates. Biochemistry 33, 6840–6849 [DOI] [PubMed] [Google Scholar]
- 49.Kniazeff J., Shi L., Loland C. J., Javitch J. A., Weinstein H., and Gether U. (2008) An intracellular interaction network regulates conformational transitions in the dopamine transporter. J. Biol. Chem. 283, 17691–17701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Celik L., Schiøtt B., and Tajkhorshid E. (2008) Substrate binding and formation of an occluded state in the leucine transporter. Biophys. J. 94, 1600–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kantcheva A. K., Quick M., Shi L., Winther A.-M., Stolzenberg S., Weinstein H., Javitch J. A., and Nissen P. (2013) Chloride binding site of neurotransmitter sodium symporters. Proc. Natl. Acad. Sci. U.S.A. 110, 8489–8494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shimamura T., Weyand S., Beckstein O., Rutherford N. G., Hadden J. M., Sharples D., Sansom M. S., Iwata S., Henderson P. J., and Cameron A. D. (2010) Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science 328, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perez C., Koshy C., Yildiz O., and Ziegler C. (2012) Alternating-access mechanism in conformationally asymmetric trimers of the betaine transporter BetP. Nature 490, 126–130 [DOI] [PubMed] [Google Scholar]
- 54.Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., and Abramson J. (2008) The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science 321, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khafizov K., Staritzbichler R., Stamm M., and Forrest L. R. (2010) A study of the evolution of inverted-topology repeats from LeuT-fold transporters using AlignMe. Biochemistry 49, 10702–10713 [DOI] [PubMed] [Google Scholar]
- 56.Shaffer P. L., Goehring A., Shankaranarayanan A., and Gouaux E. (2009) Structure and mechanism of a Na+ independent amino acid transporter. Science 325, 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ressl S., Terwisscha van Scheltinga A. C., Vonrhein C., Ott V., and Ziegler C. (2009) Molecular basis of transport and regulation in the Na+/betaine symporter BetP. Nature 458, 47–52 [DOI] [PubMed] [Google Scholar]
- 58.Weyand S., Shimamura T., Yajima S., Suzuki S., Mirza O., Krusong K., Carpenter E. P., Rutherford N. G., Hadden J. M., O'Reilly J., Ma P., Saidijam M., Patching S. G., Hope R. J., Norbertczak H. T., et al. (2008) Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science 322, 709–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ehrnstorfer I. A., Geertsma E. R., Pardon E., Steyaert J., and Dutzler R. (2014) Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nat. Struct. Mol. Biol. 21, 990–996 [DOI] [PubMed] [Google Scholar]
- 60.Talvenheimo J., Fishkes H., Nelson P. J., and Rudnick G. (1983) The serotonin transporter-imipramine “receptor”: different sodium requirements for imipramine binding and serotonin translocation. J. Biol. Chem. 258, 6115–6119 [PubMed] [Google Scholar]
- 61.Jung H., Buchholz M., Clausen J., Nietschke M., Revermann A., Schmid R., and Jung K. (2002) CaiT of Escherichia coli, a new transporter catalyzing l-carnitine/γ-butyrobetaine exchange. J. Biol. Chem. 277, 39251–39258 [DOI] [PubMed] [Google Scholar]
- 62.Chaudhry F. A., Krizaj D., Larsson P., Reimer R. J., Wreden C., Storm-Mathisen J., Copenhagen D., Kavanaugh M., and Edwards R. H. (2001) Coupled and uncoupled proton movement by amino acid transport system N. EMBO J. 20, 7041–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lauf P. K., McManus T. J., Haas M., Forbush B. 3rd, Duhm J., Flatman P. W., Saier M. H. Jr., and Russell J. M. (1987) Physiology and biophysics of chloride and cation cotransport across cell membranes. Fed. Proc. 46, 2377–2394 [PubMed] [Google Scholar]
- 64.Chen X. Z., Peng J. B., Cohen A., Nelson H., Nelson N., and Hediger M. A. (1999) Yeast SMF1 mediates H+-coupled iron uptake with concomitant uncoupled cation currents. J. Biol. Chem. 274, 35089–35094 [DOI] [PubMed] [Google Scholar]
- 65.Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J. A., and Kanner B. I. (2007) Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature 449, 726–730 [DOI] [PubMed] [Google Scholar]
- 66.Tavoulari S., Rizwan A. N., Forrest L. R., and Rudnick G. (2011) Reconstructing a chloride-binding site in a bacterial neurotransmitter transporter homologue. J. Biol. Chem. 286, 2834–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ben-Yona A., Bendahan A., and Kanner B. (2011) A glutamine residue conserved in the neurotransmitter:sodium:symporters is essential for the interaction of chloride with the GABA transporter GAT-1. J. Biol. Chem. 286, 2826–2833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Henry L. K., Iwamoto H., Field J. R., Kaufmann K., Dawson E. S., Jacobs M. T., Adams C., Felts B., Zdravkovic I., Armstrong V., Combs S., Solis E., Rudnick G., Noskov S. Y., DeFelice L. J., et al. (2011) A conserved asparagine residue in transmembrane segment 1 (TM1) of serotonin transporter dictates chloride-coupled neurotransmitter transport. J. Biol. Chem. 286, 30823–30836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Felts B., Pramod A. B., Sandtner W., Burbach N., Bulling S., Sitte H. H., and Henry L. K. (2014) The two Na+ sites in the human serotonin transporter play distinct roles in the ion coupling and electrogenicity of transport. J. Biol. Chem. 289, 1825–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Koldsø H., Noer P., Grouleff J., Autzen H. E., Sinning S., and Schiøtt B. (2011) Unbiased simulations reveal the inward-facing conformation of the human serotonin transporter and Na(+) ion release. PLoS Comput. Biol. 7, e1002246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao C., and Noskov S. Y. (2011) The role of local hydration and hydrogen-bonding dynamics in ion and solute release from ion-coupled secondary transporters. Biochemistry 50, 1848–1856 [DOI] [PubMed] [Google Scholar]
- 72.Shi L., Quick M., Zhao Y., Weinstein H., and Javitch J. A. (2008) The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol. Cell 30, 667–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ermolova N., Madhvani R. V., and Kaback H. R. (2006) Site-directed alkylation of cysteine replacements in the lactose permease of Escherichia coli: helices I, III, VI, and XI. Biochemistry 45, 4182–4189 [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y. W., and Rudnick G. (2006) The cytoplasmic substrate permeation pathway of serotonin transporter. J. Biol. Chem. 281, 36213–36220 [DOI] [PubMed] [Google Scholar]
- 75.Bulling S., Schicker K., Zhang Y.-W., Steinkellner T., Stockner T., Gruber C. W., Boehm S., Freissmuth M., Rudnick G., Sitte H. H., and Sandtner W. (2012) The mechanistic basis of non-competitive ibogaine inhibition in serotonin and dopamine transporters. J. Biol. Chem. 287, 18524–18534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wilson G., and Karlin A. (2001) Acetylcholine receptor channel structure in the resting, open, and desensitized states probed with the substituted-cysteine-accessibility method. Proc. Natl. Acad. Sci. U.S.A. 98, 1241–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Quick M., Shi L., Zehnpfennig B., Weinstein H., and Javitch J. A. (2012) Experimental conditions can obscure the second high affinity site in LeuT. Nat. Struct. Mol. Biol. 19, 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Quick M., Winther A. M., Shi L., Nissen P., Weinstein H., and Javitch J. A. (2009) Binding of an octylglucoside detergent molecule in the second substrate (S2) site of LeuT establishes an inhibitor-bound conformation. Proc. Natl. Acad. Sci. U.S.A. 106, 5563–5568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang L., and Hermans J. (1996) Hydrophilicity of cavities in proteins. Proteins 24, 433–438 [DOI] [PubMed] [Google Scholar]