

Abstract
The formation of a replication loop on the lagging strand facilitates coordinated synthesis of the leading- and lagging-DNA strands and provides a mechanism for recycling of the lagging-strand DNA polymerase. As an Okazaki fragment is completed, the loop is released, and a new loop is formed as the synthesis of a new Okazaki fragment is initiated. Loop release requires the dissociation of the complex formed by the interactions among helicase, DNA polymerase, and DNA. The completion of the Okazaki fragment may result in either a nick or a single-stranded DNA region. In the replication system of bacteriophage T7, the dissociation of the polymerase from either DNA region is faster than that observed for the dissociation of the helicase from DNA polymerase, implying that the replication loop is released more likely through the dissociation of the lagging-strand DNA from polymerase, retaining the polymerase at replication fork. Both dissociation of DNA polymerase from DNA and that of helicase from a DNA polymerase·DNA complex are much faster at a nick DNA region than the release from a ssDNA region. These results suggest that the replication loop is released as a result of the nick formed when the lagging-strand DNA polymerase encounters the previously synthesized Okazaki fragment, releasing lagging-strand DNA and retaining DNA polymerase at the replication fork for the synthesis of next Okazaki fragment.
Keywords: bacteriophage, DNA replication, DNA-protein interaction, enzyme kinetics, pre-steady-state kinetics
Introduction
Bacteriophage T7 has a simple but efficient DNA replication system (1) consisting of gene 5 DNA polymerase (gp5),3 the processivity factor Escherichia coli thioredoxin (trx), gene 4 helicase-primase (gp4), and gene 2.5 ssDNA-binding protein (gp2.5) (Fig. 1). gp5 forms a high affinity complex with trx (gp5·trx) to increase the processivity of nucleotide polymerization (2, 3). The helicase domain of the hexameric gp4 encircles the lagging-strand DNA and unwinds dsDNA in a reaction fueled by the hydrolysis of dTTP (4, 5). The primase domain in the N-terminal half of gp4 recognizes specific sequences on the lagging-strand template at which it catalyzes the synthesis of tetraribonucleotides that are used as primer for the initiation of the synthesis of Okazaki fragments. Primase maintains contact with the priming sequence during ongoing DNA synthesis, organizing a priming loop that keeps the primer in physical proximity to the replication complex (6). Because of the polarity of DNA synthesis, the leading strand is synthesized continuously whereas the lagging strand is synthesized discontinuously, in the form of Okazaki fragments. Interactions between the lagging-strand DNA polymerase and the helicase domain of gp4 are important in the formation and maintenance of a replication loop (Fig. 1). gp2.5 coats the ssDNA to remove secondary structures and also physically interacts with both gp4 and gp5, interactions essential for coordination of leading- and lagging-strand DNA synthesis (7).
FIGURE 1.
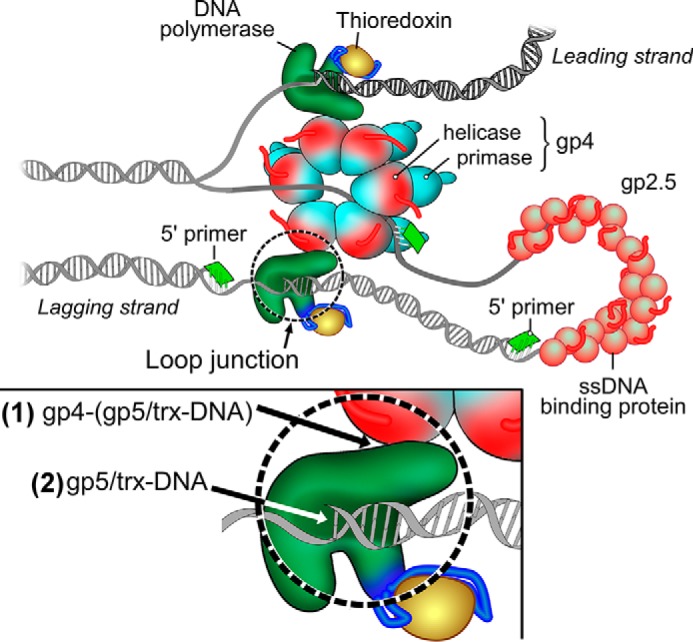
Model of the replisome of bacteriophage T7. The T7 replisome consists of DNA polymerase (gp5), the processivity factor E. coli trx, the hexameric helicase-primase (gp4), and the ssDNA-binding protein (gp2.5). The helicase unwinds dsDNA to generate two ssDNA templates for both leading-strand and lagging-strand DNA synthesis. The primase synthesizes oligoribonucleotides as primers for the lagging-strand DNA polymerase to initiate the synthesis of Okazaki fragments. gp2.5 coats the lagging-strand DNA template formed by the interaction among helicase, polymerase, and DNA. The replication loop consists of ssDNA/dsDNA and the interaction junction of helicase·DNA polymerase·DNA. The release of the loop is dependent on the dissociation of the loop junction by separation of the DNA from polymerase ((2), inset) or the separation of the polymerase from helicase ((1), inset).
The formation of the replication loops not only allows both leading-strand and lagging-strand DNA polymerases to synthesize DNA in the same direction but also facilitates recycling of the lagging-strand DNA polymerase to the newly synthesized RNA primers at the fork. The lagging-strand polymerase must transiently release from DNA upon termination of each Okazaki fragment. Biochemical studies have revealed a number of molecular scenarios that help to explain how the formation and release of replication loops are regulated (8–11). The visualization of replication intermediates in the T4 and T7 bacteriophage replication systems by electron microscopy demonstrates the existence of replication loops and allows for determination of length distributions (11, 12).
The replication loop consists of single- and double-stranded regions of DNA and a complex of helicase, DNA polymerase, and DNA (Fig. 1). After the synthesis of an Okazaki fragment, the replication loop releases to enable the synthesis of the next Okazaki fragment (1). The release of the loop requires the disruption of this junction by dissociation of DNA polymerase from either helicase or DNA. Because the lagging-strand DNA synthesis is discontinuous, the lagging DNA polymerase must cycle on and off DNA for synthesis of each new Okazaki fragment, an event accompanied by loop release and eventually reformation. Two mechanisms have been proposed to explain the cycling of DNA polymerase. In the collision model, the lagging-strand polymerase collides with the 5′ terminus of an earlier completed fragment and releases from DNA (13). In the signaling model, the lagging-strand polymerase releases from DNA due to one or more distinct macromolecular interaction events involved in repetitive lagging-strand cycles (10). For example, the association of the DnaG primase with the primosome triggers the lagging polymerase recycling in the E. coli replication system (9). The signaling model does not require the collision of DNA polymerase with the previous Okazaki fragment.
In the T7 replication system, loop release was found to occur via both mechanisms using single-molecular technique (14–16). In addition to loop release via the collision mechanism, the synthesis of pAC also triggers loop release before the nascent Okazaki fragment is completed, leaving an ssDNA gap in the lagging DNA strand (14–16). Notably, DNA bound to the polymerase has different structures in both mechanisms. In the collision model, completion of an Okazaki fragment by DNA polymerase results in DNA containing a nick (13, 17). On the other hand, in the signaling model, an ssDNA template for lagging-strand DNA synthesis contains a single-stranded region. We hypothesize that the different intermediate DNA structures formed during DNA synthesis may affect the stability of the loop junction and contribute to loop release via different pathways.
In this study, duplex DNAs containing a nick or short and long ssDNA regions (see Table 1) were used to examine the binding affinity and dissociation among helicase, DNA polymerase, and DNA at a replication fork. Our data show that the binding affinities among helicase, DNA polymerase, and DNA are weaker at a nick than are those at single-stranded regions. We also find that the interaction between helicase and DNA polymerase is stronger than that between DNA and DNA polymerase. Taken together, we propose that the replication loop is more likely to release via the collision pathway by dissociation of polymerase from the DNA; the polymerase is thus retained at the replication fork.
TABLE 1.
DNA containing a nick or short and long single-stranded regions used in this study

Experimental Procedures
Materials
Oligonucleotides were purchased from Integrated DNA Technologies. DNA substrates containing a nick or single-stranded regions were prepared by annealing the designated strands (see Table 1). The dideoxy primer strands contained a dideoxynucleotide at the 3′-end to enable the formation of a stable DNA polymerase·DNA complex in the presence of the next incoming nucleoside 5′-triphosphate (18). Bacteriophage T7 gene 5 DNA polymerase lacking exonuclease activity (gp5 exo−), gp4, and E. coli trx were overproduced and purified as described previously (18). For simplicity, we denote gp5 exo− as gp5 throughout the text.
Binding of DNA Polymerase (gp5·trx) to DNA
SPR analysis of the binding of gp5·trx to three kinds of DNA was performed using a Biacore 3000 instrument (Uppsala, Sweden) as described previously (18). Three annealed DNA substrates (300 response units (RU)) were immobilized to a streptavidin (SA) chip through a biotin at the 5′ terminus of the template strand. In a control flow cell, biotin was used instead of the biotinylated DNA to compensate for background. Binding studies were carried out in buffer A (20 mm HEPES, pH 7.5, 2.5 mm DTT, 200 mm potassium glutamate, 1% glycerol (w/v)) at a flow rate of 10 μl/min at room temperature. A varying concentration of gp5·trx (20-fold molar excess of trx, 0.005–0.6 μm gp5) was flowed over the chip to study the binding of the polymerase to DNA. The binding signal was fitted to Equation 1 using a steady-state model provided by BIAevaluation 3.0.2 computational software (Biacore). The dissociation constants Kd were calculated using the steady-state average RU (18)
where Y is the response signal corresponding to the binding; B is concentration of protein in μm; RUmax is the maximal binding amount in RU; Kd is the dissociation constant in μm. All experiments were carried out three times, and standard errors were derived using Prism software. (The same details apply to Equation 2 below.)
In the presence of 5 mm MgCl2 and 0.05 mm ddATP, the active site of DNA polymerase is preferentially located at the 3′-end of primer where ddATP is incorporated opposite thymidine in the template strand (18, 19). The binding affinities of the polymerase to DNA were determined by the same methods described above (18). The chip surface was regenerated by injection of 150 μl of 1 m NaCl at a flow rate of 100 μl/min.
Dissociation Rates of DNA Polymerase from DNA Determined by Trap Assays
The dissociation rates of DNA polymerase from DNA were determined using a RQF-3 KinTek quench flow apparatus (20, 21). One sample syringe contained a preincubated solution of 200 nm gp5·trx and 500 nm unlabeled DNA containing a nick or single-stranded region in the absence or presence of 0.5 mm ddATP and 20 mm MgCl2. This sample was rapidly mixed with 450 nm trap primer/template in the other sample syringe at time intervals ranging from 0.1 to 10 s. The trap DNA consists of a 5′ 32P-radiolabeled 24-mer primer annealed to a 36-mer template. After the sample was mixed at various times, polymerization was initiated by mixing with 500 μm dCTP and 20 mm MgCl2 in the central drive syringe and then incubating for 0.25 s. The 32P-labeled 24-mer primer was extended to 25-mer after incorporation of dCMP. After the sample was expelled from the rapid-quench apparatus, the reaction was rapidly terminated by mixing with 500 μl of 0.3 m EDTA in 50% formamide (v/v). The amount of 32P-labeled extended product (25-mer) was quantified by gel analysis and plotted against time. The graph was fit to a single-exponential equation in Prism software using Equation 2.
where Ef = free enzyme concentration in nm; E0 = concentration of DNA polymerase bound to DNA in nm; k = dissociation rate of polymerase from DNA in s−1; and t is time in seconds (20).
Binding Affinity of Helicase to DNA Polymerase·DNA Complex
The binding of helicase to DNA polymerase·DNA complex was investigated by SPR as described previously (18). DNA substrates (300 RU) were coupled to an SA chip. Binding studies were carried out in buffer A containing 5 mm MgCl2 and 0.05 mm ddATP at a flow rate of 10 μl/min at room temperature. A mixture of 0.3 μm gp5 and 6 μm trx (20-fold molar excess) in buffer A was injected into the flow cell. A varying concentration of wild-type gp4 or gp4-ΔC17 lacking the C-terminal 17 amino acid residues (0.01–0.8 μm in monomeric concentration) was injected in buffer A containing 0.1 mm ATP and 0.1 mm ddATP. In a control flow cell, the same DNA was immobilized without the polymerase to compensate background. The signal was fitted to Equation 1 using the steady-state model provided by BIAevaluation 3.0.2 computational software (Biacore) (18). The dissociation constants Kd were calculated using the steady-state average RU. The chip surface was regenerated as described previously (18).
Results
Binding Affinity of DNA Polymerase to Replication DNA Intermediates
During DNA synthesis, the T7 DNA replisome undergoes changes, not the least of which is the formation, growth, and release of the lagging-strand replication loop. To analyze interactions among the components of the replisome, we first examined binding of DNA polymerase to DNA containing structures that might arise as intermediates during DNA replication.
The binding affinity of DNA polymerase to DNA was studied using SPR (18). In the SPR experiments, three kinds of DNA (300 RU) were immobilized to an SA chip. DNA containing a nick or single-stranded region was prepared by annealing each ssDNA with specific oligonucleotides (Table 1). To compare the binding affinities of DNA polymerase with the various DNAs, varying concentrations of polymerase were flowed over the chip and the response signals were measured. Polymerase lacking the proofreading exonuclease activity was used in these experiments to prevent degradation of the DNA. The binding affinities (Kd) were obtained by fitting the response signal and protein concentration to Equation 1 using the steady-state model (18).
The absence of ddATP and Mg2+ in buffer A led to a random binding of DNA polymerase to DNA to form a binary complex (19). DNA polymerase binding to DNA containing a single-stranded region exhibited higher binding affinities (Kd of 0.29 and 0.37 μm) than that with a nick (0.61 μm) (Fig. 2), in agreement with previous results that DNA polymerase can bind non-specifically to dsDNA and diffuse along the duplex (22). In the presence of ddATP and Mg2+, DNA polymerase, DNA, and ddATP form a ternary complex, in which the polymerase was preferentially positioned and locked at the 3′-end of the primer strand (18, 19). The dissociation constants (Kd) of the polymerase from DNA containing a single-stranded region were about 0.26 μm, lower than that from a nicked DNA (Kd of 0.44 μm) (Fig. 3), indicating that the polymerase bound better to DNA containing a single-stranded region than to a nick.
FIGURE 2.

Binding of DNA polymerase to various DNAs in the absence of Mg2+ and ddATP. A, scheme for measuring the interaction of gp5·trx with DNA containing a nick or a short or long single-stranded region that was prepared and immobilized onto the SA chip (300 RU). gp5·trx randomly binds to DNA without Mg2+ and ddATP to form a binary complex. nt, nucleotides. B, sensorgrams for the binding of increasing concentrations of gp5·trx (1:20, molar ratio, 0.005–0.6 μm gp5) to the three immobilized DNA in buffer A (see “Experimental Procedures”). C, the binding affinities of gp5·trx to DNA were determined using the steady-state average response at each concentration of gp5·trx shown in B. The solid lines represent the theoretical curve calculated from the steady-state fit model (Biacore). Representative data from multiple experiments are shown.
FIGURE 3.

Binding of DNA polymerase to various DNAs in the presence of Mg2+ and ddATP. A, scheme for measuring the interaction of gp5·trx with DNA containing a nick or a short or long single-stranded region that was prepared and immobilized on to the SA chip (300 RU). gp5·trx is locked at the 3′-end of primer in the presence of Mg2+ and ddATP to form a ternary complex. nt, nucleotides. B, sensorgrams for the binding of increasing concentrations of gp5·trx (1:20, molar ratio, 0.005–0.4 μm gp5) to the three immobilized DNAs in buffer A (see “Experimental Procedures”). C, the binding affinities of gp5·trx to DNA were determined as described in the legend for Fig. 2C. Representative data from multiple experiments are shown.
Comparison of Kd values shows that DNA polymerase is more prone to dissociation from nicked DNA rather than from DNA containing a single-stranded region in both binary and ternary complexes. The addition of ddATP and Mg2+ results in formation of the ternary complex, enhancing the binding of DNA polymerase to DNA. The enhanced binding may arise as a result of the polymerase now being located at the end of primer, thus leading to a stronger binding mode than that of the random binding in the binary complex. DNA structures obviously affect the binding affinity of DNA polymerase, giving a higher binding affinity to DNA containing a single-stranded region than to the nicked DNA in both binary and ternary complexes.
Dissociation Rates of Polymerase from DNA Determined by Trap Assays
The effects of DNA structures on DNA polymerase binding was further examined by measuring the dissociation rates of DNA polymerase in the binary or ternary complexes by a DNA trapping experiment using a RQF-3 KinTek quench flow apparatus (Fig. 4A). In the absence of ddATP and Mg2+, DNA polymerase binds to DNA to form a binary complex. Once the polymerase dissociates from the binary complex, it will bind to the trap DNA composed of a radiolabeled 24-mer primer and 36-mer template, and then extend the 24-mer primer to a 25-mer by incorporating deoxycytidine monophosphate. Measuring the amount of extended 25-mer product determines the dissociation rate of polymerase from DNA. The dissociation rates determined in this assay indicate that dissociation from nicked DNA (1.0 s−1) is faster than that from DNA containing a short or long single-stranded region (0.51 s−1 or 0.42 s−1, respectively) (Fig. 4). In the ternary complex formed in the presence of ddATP and Mg2+, dissociation of the polymerase from all DNA structures was slower than from the binary complex in the absence of ddATP and Mg2+ (Fig. 5). Dissociation of the polymerase from the ternary complex occurred at a rate of 0.54 s−1 for a nicked DNA and 0.069 or 0.064 s−1 for DNA containing a short or long single-stranded region, respectively.
FIGURE 4.

Dissociation rates (koff) of DNA polymerase from DNA containing a nick or a short or long single-stranded region in the absence of ddATP and Mg2+. A, scheme for measuring the dissociation of polymerase from DNA containing a nick or a short or long single-stranded region. Dissociated polymerase binds to 24-mer/36-mer and catalyzes the extension of a 24-mer to a 25-mer product. B, gp5·trx (200 nm) was preincubated with 500 nm unlabeled DNA containing a nick or a short or long single-stranded region in the absence of ddATP and Mg2+ to form a binary complex. This solution was mixed with 450 nm 32P-labeled 24-mer/36-mer at time intervals ranging from 0.1 to 10 s. Then polymerization of nucleotides was initiated with 500 μm dCTP and 20 mm Mg2+ for a constant time of 0.25 s, during which dCMP was incorporated and 32P-labeled primer was extended from a 24-mer to a 25-mer. The dissociation rates of polymerase from DNA were obtained by fitting the 25-mer product and time according to Equation 2. The solid lines represent the theoretical curves calculated from the steady-state fit model. Representative data from multiple experiments are shown.
FIGURE 5.

Dissociation rates (koff) of DNA polymerase from DNA containing a nick or a short or long single-stranded region in the presence of ddATP and Mg2+. A and B, the same as described in the legend for Fig. 4 except that 200 nm gp5·trx and 500 nm unlabeled DNA were preincubated in the presence of 0.5 mm ddATP and 20 mm Mg2+ to form a ternary complex. Representative data from multiple experiments are shown.
The dissociation rates of DNA polymerase were slower from DNA containing a single-stranded region than that from nicked DNA in both binary and ternary complexes, suggesting that the binding of the polymerase to DNA is favorable in the presence of a single-stranded region when compared with a nick. In the ternary complex, the dissociation rate of the polymerase from DNA containing a single-stranded region is about 8-fold lower than that from a nick. The decreased rate could arise from an inability of the nicked DNA to form a stable ternary complex with polymerase and ddATP, thus leading to a fast dissociation of the polymerase from the nicked DNA. Dissociation of DNA polymerase was significantly slower in the ternary complex than from the binary complex, indicating that the presence of ddATP and Mg2+ stabilizes the complex of DNA polymerase and DNA.
Binding Affinity of Helicase to DNA Polymerase·DNA Complex
Helicase (gp4) interacts with DNA polymerase (gp5) that is bound to DNA (18). Whether DNA structures affect the binding affinity of helicase to polymerase bound to DNA is unknown. Therefore, the binding affinity between helicase and DNA polymerase to which DNA containing a nick or a short or long single-stranded region is bound was determined. In the SPR experiments, each of three DNAs was immobilized onto the chip; DNA polymerase was flowed over the DNA in the presence of ddATP and Mg2+ to form a stable complex.
The C-terminal tail of helicase plays an important role in the interaction with DNA polymerase (18, 23). To determine the contribution of the C-terminal tail of helicase to the interaction between helicase and DNA-bound polymerase, both helicase (gp4) and helicase lacking the C-terminal tail (gp4-ΔC17) were examined. Varied concentrations of gp4-ΔC17 were flowed over the chip, and the interactions between gp4-ΔC17 and polymerase were detected. In a control experiment where polymerase was omitted, no detectable binding of gp4-ΔC17 to the immobilized DNA was observed (18). The binding affinities of gp4-ΔC17 to DNA polymerase bound to DNA containing a short or long single-stranded region were similar to each other (Kd of 0.28 μm) but were 2-fold higher than the binding to a nick (Kd of 0.44 μm) (Fig. 6).
FIGURE 6.

Binding affinity of helicase lacking the C-terminal tail (gp4-ΔC17) with DNA polymerase bound to DNA containing a nick or a short or long single-stranded region. A, scheme for measuring the interaction of gp4-ΔC17 with DNA polymerase binding to DNA containing a nick or short or a long single-stranded region in the presence of Mg2+ and ddATP. Three kinds of DNA were immobilized on the SA chip (300 RU). A mixture of 0.3 μm gp5 and 6 μm trx (1:20, molar ratio) was flowed over the chip to form a stable gp5·trx·DNA·ddATP ternary complex. gp4-ΔC17 was injected into the flow cell. B, increasing concentrations of gp4-ΔC17 monomer (0.01–0.8 μm) in the buffer containing 0.1 mm ATP and 0.1 mm ddATP were injected into the flow cell, and the response binding signal was recorded. The arrows indicate the beginning of injecting proteins into chip cell. C, the binding affinities of gp4-ΔC17 to gp5·trx were determined using the steady-state average response at each concentration of gp4-ΔC17. The solid lines represent the theoretical curves calculated from the steady-state fit model. Representative data from multiple experiments are shown.
Similarly, the binding affinities of helicase to DNA polymerase bound to various DNA were also investigated. The DNA structures (nick or short or long single-stranded region) obviously affected the binding affinity between helicase and DNA polymerase. The binding affinities between helicase and polymerase bound to DNA containing either short or long single-stranded region were similar (Kd of 0.11 μm) but were 2-fold tighter than that at a nick (Kd of 0.26 μm) (Fig. 7). The higher affinity could arise from more exposed surface for the interaction with helicase when polymerase is bound to DNA containing a single-stranded region than when DNA contains a nick.
FIGURE 7.

Binding affinity of helicase (gp4) with DNA polymerase bound to DNA containing a nick or a short or long single-stranded region. A–C, the same as described in the legend for Fig. 6 except that the full-length helicase was used instead of the helicase lacking the C-terminal tail. Representative data from multiple experiments are shown.
gp4 or gp4-ΔC17 forms a complex with DNA polymerase bound to DNA containing a single-stranded region better than that at a nick. Comparison of gp4 with gp4-ΔC17 indicates that the presence of the C-terminal tail of helicase significantly increases the binding affinity between helicase and DNA polymerase that was bound to any of the three DNAs, in agreement with previous results (18). When compared with the binding affinity between polymerase and DNA (Table 2), helicase binding to polymerase bound to DNA is much tighter, suggesting that the dissociation of DNA polymerase from DNA is more favorable than the dissociation of DNA polymerase from helicase.
TABLE 2.
The dissociation constants (Kd) among helicase, DNA polymerase, and DNA
Discussion
Formation of a replication loop during lagging-strand DNA synthesis allows both leading-strand and lagging-strand DNA synthesis to occur within a single complex of proteins, the replisome. Consequently, the synthesis of both strands proceeds in the same overall direction and the lagging-strand polymerase can be recycled to a newly synthesized RNA primer. After synthesis of an Okazaki fragment, the replication loop is released for the synthesis of a new Okazaki fragment, an event that triggers the reformation of the replication loop. The release of the replication loop requires the dissolution of the junction that tethers the lagging strand to the replisome. This junction is composed of helicase, DNA polymerase, and DNA. Dissociation of the polymerase from DNA or dissociation of polymerase from helicase can disrupt the junction.
Three interactions between helicase and DNA polymerase are known to exist during leading-strand DNA synthesis in the replisome of the replication system of bacteriophage T7 (18). The C-terminal tail of the helicase interacts with a basic patch on the polymerase for its loading onto DNA. A second interaction involves the acidic C-terminal tail of the helicase and a basic patch on the thioredoxin binding domain of the polymerase to enable recycling of the polymerase during DNA synthesis. DNA polymerase that transiently dissociates during leading-strand DNA synthesis is captured by one of the acidic C-terminal tails of the hexameric helicase and can then be recycled back to the leading strand to continue DNA synthesis (18, 23). After dissociation from DNA, the polymerase is still attached to helicase (18, 23). A third interaction that does not involve the acidic C-terminal tail of the helicase occurs during leading-strand synthesis.
On the lagging-strand DNA, protein interactions determine the stability of the replication loop. Two mechanisms have been proposed to explain how the replication loop is released (14, 15). In the collision mechanism, the encounter of lagging-strand DNA polymerase with the 5′ terminus of the previously synthesized Okazaki fragment triggers loop release. In this model, the lagging-strand DNA polymerase dissociates from the nick that is formed upon closure of the lagging-strand gap. In the signaling model, the condensation of the first two nucleotides of the primer to yield pppAC triggers loop release before the nascent Okazaki fragment is completed. This mechanism results in an ssDNA gap in the lagging-strand DNA to which DNA polymerase binds. The use of both mechanisms provides an elegant solution to cope with the stochastic nature of the primase activity (14). The appearance of either nicks or gaps in the DNA in these two models led us to examine the affinities of the replication proteins with similar structures. Differences in affinity of the proteins for these structures could help to identify the trigger that leads to loop release.
Affinities represented by dissociation constants (Kd) of DNA polymerase from DNA, as well as helicase from a polymerase·DNA complex, as determined by SPR, are lower for nicked DNA than those for DNA containing a single-stranded region of DNA. The trap assays also show that the dissociation of polymerase from nicked DNA (0.54 s−1) is considerably faster than that from DNA containing a single-stranded region (0.064–0.069 s−1). All the data show that the polymerase·DNA complex and helicase·polymerase·DNA complex are less stable at nicked DNA than at DNA containing a single-stranded region, suggesting that the replication loop is more likely to release at a nick after polymerase encounters the previously synthesized Okazaki fragment via the collision mode (Fig. 8) (14).
FIGURE 8.
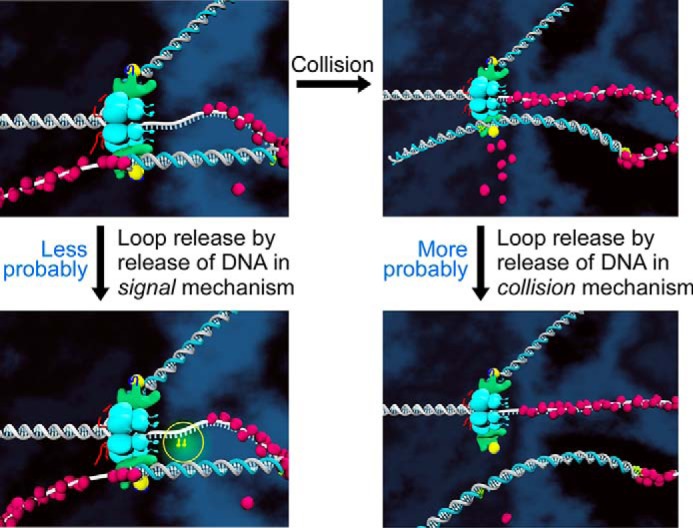
Model for the release of the replication loop. Upon finishing the synthesis of an Okazaki fragment, the loop is released by dissociation of DNA polymerase from DNA rather than from the helicase. The lagging-strand DNA polymerase is retained at the replication fork for the synthesis of the next Okazaki fragment. The dissociation of DNA polymerase from a nick in DNA is easier than that from a single-stranded DNA gap, suggesting that loop release is triggered upon DNA synthesis reaching the 5′ terminus of the previously synthesized Okazaki fragment (collision model).
We also compared the binding affinity of DNA polymerase·helicase with that of DNA polymerase·DNA. The dissociation constant (Kd) between helicase and DNA polymerase was 0.089 μm (18). gp4E, which has the helicase domain but lacks the primase domain, binds to DNA polymerase with a dissociation constant of 0.09 μm (19), indicating that the helicase domain alone contributes to the interaction with the polymerase. The dissociation constants (Kd) between helicase and DNA-bound polymerase were 0.11–0.26 μm (Table 2). Thus, the binding of DNA to the polymerase weakens its interaction with the helicase. In addition, binding of the polymerase to the various DNA structures (0.25–0.44 μm) was weaker than that to the helicase (Table 2). Therefore, the loop is most likely released by dissociation of the polymerase from DNA rather than dissociation from the helicase. During loop release, the lagging-strand DNA will release from the replication fork, whereas the lagging-strand DNA polymerase, still attached to helicase at the replication fork, can initiate the synthesis of the next Okazaki fragment by extending a new primer.
The retention of DNA polymerase within the replisome has been observed in other replication systems. In the T4 replication system, after loop release, the lagging-strand polymerase remains associated with the replisome (10). In the E. coli replication system, Pol III core and the τ subunit of the clamp loader are tightly bound (24). The τ subunit interacts with the Pol III core and the helicase even after release of the replication loop (25). The β sliding clamp, which is reserved on an Okazaki fragment, attracts a new Pol III core in preference over other polymerases from solution after Pol III dissociates (25, 26).
Author Contributions
H. Z. and Y. T. designed the study and collected the data. H. Z., S. J. L., Z. W., and J. C. analyzed the data. H. Z., S. J. L., and C. C. R. wrote the paper. H. Z., S. J. L., and C. C. R. approved the final version of the manuscript.
Acknowledgment
We thank Steven Moskowitz (Advanced Medical Graphics) for illustrations.
This work was supported by the Natural Science Foundation of China Grants 31370793 and 81422041 (to H. Z.). The authors declare that they have no conflicts of interest with the contents of this article.
- gp5
- gene 5 DNA polymerase
- gp4
- gene 4 helicase-primase
- gp2.5
- gene 2.5 ssDNA-binding protein
- trx
- thioredoxin
- RU
- response unit(s)
- SA
- streptavidin
- ddATP
- 2′,3′-dideoxyadenosine 5′-triphosphate
- Pol III
- DNA polymerase III.
References
- 1.Hamdan S. M., and Richardson C. C. (2009) Motors, switches, and contacts in the replisome. Ann. Rev. Biochem. 78, 205–243 [DOI] [PubMed] [Google Scholar]
- 2.Tabor S., Huber H. E., and Richardson C. C. (1987) Escherichia coli thioredoxin confers processivity on the DNA-polymerase-activity of the gene-5 protein of bacteriophage-T7. J. Biol. Chem. 262, 16212–16223 [PubMed] [Google Scholar]
- 3.Wuite G. J. L., Smith S. B., Young M., Keller D., and Bustamante C. (2000) Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404, 103–106 [DOI] [PubMed] [Google Scholar]
- 4.Ahnert P., and Patel S. S. (1997) Asymmetric interactions of hexameric bacteriophage T7 DNA helicase with the 5′- and 3′-tails of the forked DNA substrate. J. Biol. Chem. 272, 32267–32273 [DOI] [PubMed] [Google Scholar]
- 5.Satapathy A. K., Kochaniak A. B., Mukherjee S., Crampton D. J., van Oijen A., and Richardson C. C. (2010) Residues in the central β-hairpin of the DNA helicase of bacteriophage T7 are important in DNA unwinding. Proc. Natl. Acad. Sci. U.S.A. 107, 6782–6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandey M., Syed S., Donmez I., Patel G., Ha T., and Patel S. S. (2009) Coordinating DNA replication by means of priming loop and differential synthesis rate. Nature 462, 940–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marintcheva B., Hamdan S. M., Lee S. J., and Richardson C. C. (2006) Essential residues in the C terminus of the bacteriophage T7 gene 2.5 single-stranded DNA-binding protein. J. Biol. Chem. 281, 25831–25840 [DOI] [PubMed] [Google Scholar]
- 8.López de Saro F. J., Georgescu R. E., O'Donnell M. (2003) A peptide switch regulates DNA polymerase processivity. Proc. Natl. Acad. Sci. U.S.A. 100, 14689–14694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X., and Marians K. J. (2000) Two distinct triggers for cycling of the lagging strand polymerase at the replication fork. J. Biol. Chem. 275, 34757–34765 [DOI] [PubMed] [Google Scholar]
- 10.Yang J., Nelson S. W., and Benkovic S. J. (2006) The control mechanism for lagging strand polymerase recycling during bacteriophage T4 DNA replication. Mol. Cell 21, 153–164 [DOI] [PubMed] [Google Scholar]
- 11.Lee J., Chastain P. D. 2nd, Griffith J. D., and Richardson C. C. (2002) Lagging strand synthesis in coordinated DNA synthesis by bacteriophage T7 replication proteins. J. Mol. Biol. 316, 19–34 [DOI] [PubMed] [Google Scholar]
- 12.Park K., Debyser Z., Tabor S., Richardson C. C., and Griffith J. D. (1998) Formation of a DNA loop at the replication fork generated by bacteriophage T7 replication proteins. J. Biol. Chem. 273, 5260–5270 [DOI] [PubMed] [Google Scholar]
- 13.Georgescu R. E., Kurth I., Yao N. Y., Stewart J., Yurieva O., and O'Donnell M. (2009) Mechanism of polymerase collision release from sliding clamps on the lagging strand. EMBO J. 28, 2981–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamdan S. M., Loparo J. J., Takahashi M., Richardson C. C., and van Oijen A. M. (2009) Dynamics of DNA replication loops reveal temporal control of lagging-strand synthesis. Nature 457, 336–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stratmann S. A., and van Oijen A. M. (2014) DNA replication at the single-molecule level. Chem. Soc. Rev. 43, 1201–1220 [DOI] [PubMed] [Google Scholar]
- 16.Geertsema H. J., and van Oijen A. M. (2013) A single-molecule view of DNA replication: the dynamic nature of multi-protein complexes revealed. Curr. Opin. Struct. Biol. 23, 788–793 [DOI] [PubMed] [Google Scholar]
- 17.Benkovic S. J., Valentine A. M., and Salinas F. (2001) Replisome-mediated DNA replication. Ann. Rev. Biochem. 70, 181–208 [DOI] [PubMed] [Google Scholar]
- 18.Zhang H., Lee S. J., Zhu B., Tran N. Q., Tabor S., and Richardson C. C. (2011) Helicase-DNA polymerase interaction is critical to initiate leading-strand DNA synthesis. Proc. Natl. Acad. Sci. U.S.A. 108, 9372–9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamdan S. M., Marintcheva B., Cook T., Lee S. J., Tabor S., and Richardson C. C. (2005) A unique loop in T7 DNA polymerase mediates the binding of helicase-primase, DNA binding protein, and processivity factor. Proc. Natl. Acad. Sci. U.S.A. 102, 5096–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodside A. M., and Guengerich F. P. (2002) Misincorporation and stalling at O6-methylguanine and O6-benzylguanine: Evidence for inactive polymerase complexes. Biochemistry 41, 1039–1050 [DOI] [PubMed] [Google Scholar]
- 21.Patel S. S., Wong I., and Johnson K. A. (1991) Pre-steady-state kinetic-analysis of processive DNA-replication including complete characterization of an exonuclease-deficient mutant. Biochemistry 30, 511–525 [DOI] [PubMed] [Google Scholar]
- 22.Etson C. M., Hamdan S. M., Richardson C. C., and van Oijen A. M. (2010) Thioredoxin suppresses microscopic hopping of T7 DNA polymerase on duplex DNA. Proc. Natl. Acad. Sci. U.S.A. 107, 1900–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamdan S. M., Johnson D. E., Tanner N. A., Lee J. B., Qimron U., Tabor S., van Oijen A. M., and Richardson C. C. (2007) Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 27, 539–549 [DOI] [PubMed] [Google Scholar]
- 24.Downey C. D., and McHenry C. S. (2010) Chaperoning of a replicative polymerase onto a newly assembled DNA-bound sliding clamp by the clamp loader. Mol. Cell 37, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kurth I., and O'Donnell M. (2013) New insights into replisome fluidity during chromosome replication. Trends in Biochem. Sci. 38, 195–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Indiani C., McInerney P., Georgescu R., Goodman M. F., and O'Donnell M. (2005) A sliding-clamp toolbelt binds high-and low-fidelity DNA polymerases simultaneously. Mol. Cell 19, 805–815 [DOI] [PubMed] [Google Scholar]