

Abstract
Phosphorothioate (PT) modification of DNA, in which the non-bridging oxygen of the backbone phosphate group is replaced by sulfur, is governed by the DndA-E proteins in prokaryotes. To better understand the biochemical mechanism of PT modification, functional analysis of the recently found PT-modifying enzyme DndEi, which has an additional domain compared with canonical DndE, from Riemerella anatipestifer is performed in this study. The additional domain is identified as a DNA helicase, and functional deletion of this domain in vivo leads to PT modification deficiency, indicating an essential role of helicase activity in PT modification. Subsequent analysis reveals that the additional domain has an ATPase activity. Intriguingly, the ATPase activity is strongly stimulated by DNA substrate containing a GAAC/GTTC motif (i.e. the motif at which PT modifications occur in R. anatipestifer) when the additional domain and the other domain (homologous to canonical DndE) are co-expressed as a full-length DndEi. These results reveal that PT modification is a biochemical process with DNA strand separation and intense ATP hydrolysis.
Keywords: ATPase, bacteria, DNA enzyme, DNA helicase, microbiology
Introduction
Phosphorothioate (PT)3 modification of DNA, in which the non-bridging phosphate is replaced by sulfur (1), is widespread in prokaryotes with diverse sequence contexts (2). In terms of physiological function, PT modifications are found to participate in preventing foreign DNA introduction via a restriction-modification system (3, 4).
PT modifications are governed by a large family of five-gene clusters termed as dndA-E (Fig. 1). DndB is a negative transcriptional regulator for the PT-modifying genes (5), whereas it is not indispensable for PT modification and is absent in some PT-containing bacteria (5–7). DndA and DndC-E are all essential for PT modification identified by in vivo studies (7, 8). DndA is a cysteine desulfurase capable of assembling DndC as an iron-sulfur cluster protein (9, 10). In some cases, DndA is functionally replaced by the cysteine desulfurase IscS (11). DndD was predicted to be an ABC transporter ATP-binding protein (8) and DndE was reported to be a DNA-binding protein (12); nevertheless, their specific functions in PT modification are still obscure. Remarkably, there are two kinds of DndE protein, one is named DndE and the other is named DndEi (Fig. 1). DndEi is a recently found PT-modifying enzyme that has an additional putative helicase domain compared with canonical DndE (13). The putative helicase domain belongs to the AAA+ family of ATPases (13), a large class of ATPases that include several other helicases such as the MCM protein involved in DNA replication and RuvB protein involved in homologous recombination (14). The AAA+ family of proteins also includes dynein, chaperone proteins, transcriptional regulators, vesicular fusion proteins, and other proteins involved in additional diverse functions (14). The additional domain probably endows PT-modifying enzyme with helicase activity that has never been found in PT modification; however, no functional study of DndEi has ever been reported.
FIGURE 1.
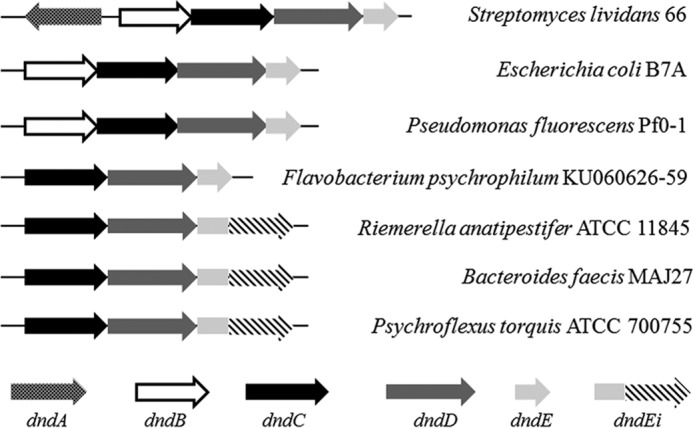
Genomic organization of the PT-modifying genes in 7 members of bacteria. Strains and GenBankTM accession numbers: S. lividans 66 (CM001889.1); E. coli B7A (CP005998.1); Pseudomonas fluorescens Pf0–1 (CP000094.2); Flavobacterium psychrophilum KU060626-59 (KF241852); R. anatipestifer ATCC 11845 (CP003388.1); Bacteroides faecis MAJ27 (NZ_AGDG01000022.1); Psychroflexus torquis ATCC 700755 (CP003879.1).
Bioinformatic analysis has revealed that a dndCDEi gene cluster exist in the genome of Riemerella anatipestifer strain ATCC 11845 (13). In this study, the enzymatic activities of DndEi from this strain were characterized. DndEi has a DNA helicase activity that is proved to be essential for PT modification; meanwhile, it has an ATPase activity that is strongly stimulated by double-stranded DNA containing a GAAC/GTTC motif.
Experimental Procedures
Bacterial Strains, Plasmids, and Growth Conditions
Bacterial strains and plasmids used in this study are described in Table 1. Escherichia coli strains were routinely grown in Luria broth (LB) at 37 °C and R. anatipestifer strains were cultured in tryptic soybean broth (Difco Laboratories, Detroit, MI) at 37 °C under 5% CO2.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Characteristics | Source or reference |
|---|---|---|
| E. coli | ||
| DH5α | F− Δlacu169 ( ϕ80lacZΔM15), recA1, endA1, hsdR17 | Biomedal |
| BL21(DE3) | hsdS gal, chromosomal insertion of T7 pol | Novagen |
| S17-1 | lpir hsdR pro thi; chromosomal integrated RP4–2 Tc::Mu Km::Tn7 | Biomedal |
| R. anatipestifer | ||
| ATCC 11845 | Wild-type, PT+ | 15 |
| HXb2 | Wild-type, PT− | 15 |
| Plasmids | ||
| pBluescript SK(+) | Bla, lacZ, orif1, ColE, Apr | Stratagene |
| pET-28a | Expression vector, pBR322 replicon, PT7, His6 tag, Kmr | Novagen |
| pT1 | Based on pET-28a, with DNA region coding DndEi inserted at NdeI-SalI sites | This study |
| pT2 | Based on pET-28a, with DNA region coding DndEih inserted at NdeI-SalI sites | This study |
| pT3 | Based on pT1, with A (the 599th nucletide in dndEi gene) mutated to C | This study |
| pT4 | Based on pT1, with A (the 1232nd nucletide in dndEi gene) mutated to C | This study |
| pReS0 | ColE1 ori, Apr, E. coli-R. anatipestifer shuttle plasmid | 15 |
| pJ1 | Based on pReS0, with dndCD genes and their promoter region inserted at XbaI-XhoI sites | This study |
| pJ2 | Based on pReS0, with dndCDEie genes and their promoter region inserted at XbaI-XhoI sites | This study |
| pJ3 | Based on pReS0, with dndCDEi genes and their promoter region inserted at XbaI-XhoI sites | This study |
| pJ4 | Based on pJ3, with A (the 599th nucletide in dndEi gene) mutated to C | This study |
| pJ5 | Based on pJ3, with A (the 1232nd nucletide in dndEi gene) mutated to C | This study |
Gene Cloning and Plasmid Construction
E. coli strain DH5α was used for plasmid construction.
Construction of plasmids used for overexpression of DndEi, DndEih, DndEi (K200A), and DndEi (D411A)
The gene encoding DndEi (Riean_0555) was amplified from R. anatipestifer ATCC 11845 genomic DNA, using KOD polymerase (Toyobo), with the sense primer A1F (CCGAGACATATGCAAATTAACATAAGAACATC) and the antisense primer A1R (TACTTAGTCGACGTTGTTCTTCTTTATGGGGT). Amplified PCR products were digested with NdeI and SalI (Fermentas), and then ligated into pET-28a, creating the plasmid pT1. The coding region of DndEih was amplified with A2F (GGGTGTCATATGTTCGATTTTTTAACTGAACAT) and A1R primers. Amplified PCR products were digested with NdeI and SalI, and ligated into pET-28a, creating the plasmid pT2. To obtain the DndEi K200A mutant, pT3 was constructed by PCR using pT1 as a template and A3F (AGGAAGTTCAGGAACAGGAGCAACACAATTTGCAT) and A3R (GCTCCTGTTCCTGAACTTCCTGCAACTGCTATG) as primers. To obtain the DndEi D411A mutant, pT4 was constructed by PCR using pT1 as a template and A4F (CGTTATGTTTTGTTAATTGCTGAAGCACATGT) and A4R (GCAATTAACAAAACATAACGCATAGCACGATA) as primers. All constructed plasmids were sequenced completely to ensure that no errors had been introduced.
Construction of Plasmids Used for Heterologous Expression of PT-modifying Genes in R. anatipestifer HXb2
The coding region of DndCD and their promoter region (−715 to −1) were amplified from R. anatipestifer ATCC 11845, with the A5F (AAGAGTCTCGAGTCACGGTAGAAGCGGCAT) and A5R (TCGAGGTCTAGATTAATTTGCATAAAGCTCGT) primers. PCR products were digested with XbaI and XhoI (Fermentas), and then ligated into pBluescript SK(+) to obtain the plasmid SK1 for sequencing. The XbaI/XhoI-digested fragment from SK1 was inserted into the corresponding sites of pReS0 (15), generating pJ1. The coding region of DndCDEie and their promoter region were amplified with the A5F and A6R (CGTCGGTCTAGATTAAGTTTTTTCATCTAACTTATTTCC) primers. The stop codon TAA was introduced to the 3′ end of the dndEie gene after amplification. PCR products were ligated into pBluescript SK(+) to obtain SK2 for sequencing and then inserted into pReS0, generating pJ2. The coding region of DndCDEi and their promoter region were amplified with the A5F and A7R (GACTGCTCTAGACCAGCGGAAGTAAGGTAT) primers. PCR products were ligated into pBluescript SK(+) to obtain SK3 for sequencing and then inserted into pReS0, generating pJ3. SK4 plasmid was constructed by PCR using SK3 as a template and A3F and A3R as primers. The XbaI/XhoI-digested fragment from SK4 was inserted into pReS0, generating pJ4. SK5 plasmid was constructed by PCR using SK3 as a template and A4F and A4R as primers. The XbaI/XhoI-digested fragment from SK5 was inserted into pReS0, generating pJ5.
Expression and Purification of the N-terminal His-tagged DndEi, DndEih, DndEi (K200A), and DndEi (D411A)
E. coli BL21(DE3) harboring the corresponding expression plasmid was first grown at 37 °C to an optical density at 600 nm (A600) of 0.8 and subsequently induced at 22 °C for 10 h with 0.4 mm isopropyl β-d-thiogalactopyranoside. Cells were harvested by centrifugation, and the pellets were re-suspended in buffer A (50 mm Tris-HCl, pH 7.5, 500 mm NaCl). Re-suspended cells were subjected to three freeze-thaw cycles and then sonically lysed. After centrifugation (10,000 × g for 60 min at 4 °C), the supernatant was loaded onto a Ni2+-nitrilotriacetic acid-agarose column (Invitrogen). Elution was carried out with buffer B (50 mm Tris-HCl, pH 7.5, 500 mm NaCl, 200 mm imidazole). Fractions containing targeted protein were pooled and concentrated to 2.5 ml with Amicon Ultra Centrifugal Filter 10,000 MWCO (Millipore). PD-10 columns (GE healthcare) were then used for imidazole elimination and buffer exchange according to the manufacturer's recommendation. Expression and purification of the His-tagged protein were analyzed by 12% SDS-PAGE and the concentration was determined by the Bradford method with bovine serum albumin (BSA) as the standard.
Conjugation Assay
E. coli S17-1 was used for conjugative transfer of plasmids into R. anatipestifer. Donor E. coli S17-1 and recipient R. anatipestifer strains were grown to mid-log phase, respectively, and plasmid was transferred into the recipient strains by biparental mating as described previously (16).
Iodine Cleavage of PT-modified Genomic DNA
PT-modified DNA can be cleaved by iodine at the modified sites (17). A 30 mm iodine solution was freshly prepared and reactions (20 μl) were then setup in PCR tubes as follows: genomic DNA (2 μg), 50 mm Na2HPO4, pH 9.0, 3 mm I2. Reactions were heated to 65 °C for 10 min and then slow cooled (0.1 °C/s) to 4 °C using a thermal cycler (Eppendorf Mastercycler pro S). The iodine-cleaved genomic DNA was run on a 0.8% agarose gel in 1× TAE containing 50 μm thiourea.
LC-MS Analysis of PT-containing Dinucleotides
20 μg of genomic DNA was hydrolyzed with 4 units of nuclease P1 (Sigma) and subsequently dephosphorylated by 10 units of alkaline phosphatase (Fermentas), essentially as described elsewhere (2). The digested DNA samples were dried and re-suspended in 50 μl of deionized water for analysis by liquid chromatography-coupled, time-of-flight mass spectrometry. The sample was resolved on an Agilent ZORBAX SB-C18 column (2.1 × 150 mm, 3.5-μm bead size) with a flow rate of 0.3 ml/min and the following parameters: column temperature: 35 °C; solvent A: 0.1% acetic acid in H2O; solvent B: 0.1% acetic acid in acetonitrile; gradient: 4% B for 10 min, 4 to 16% B over 15 min, and 16 to 100% B over 1 min. The HPLC column was coupled to an Agilent 6230 TOF mass spectrometer with an electrospray ionization source in positive mode with the following parameters: gas flow, 10 liters/min; nebulizer pressure, 30 psi; drying gas temperature, 325 °C; and capillary voltage, 3,000 V.
Fluorescent Helicase Assay
DNA substrates (Table 2) used for helicase assay were prepared by annealing an Alexa Fluor 488-labeled oligonucleotide to a black hole quencher-1-labeled oligonucleotide at a 1:1.5 molar ratio. A typical fluorescence helicase assay was performed as described previously (18) with a slight modification. The assay was performed in 30 mm Tris-HCl, pH 7.5, 5 mm MgCl2, 0.075% Triton X-100, 20 nm DNA substrate, 1 mm ATP, and 250 nm capture strand. The unwinding reaction was started by addition of 1 nmol of helicase enzyme and was carried out in a 200-μl volume at 37 °C for up to 60 min. The fluorescent signal increase was registered every 30 s using a Fluorescence Reader (Biotek synergy 2). The fluorophore was excited at 485 nm and the helicase activity was measured at 528 nm. The enzyme activity was calculated as the initial reaction velocity from the linear part of the progress curve using the linear regression method. A linear equation Y = AX + B was fitted to experimental data, where Y is the enzyme activity expressed in fluorescence units, X is the reaction time, and A is the initial velocity.
TABLE 2.
DNA substrates used in helicase assay in this study

Single-stranded DNA Translocation Assay
Translocation activity was investigated by measuring the ability of DndEih to displace streptavidin from a biotinylated probe at the 5′ or 3′ end of ssDNA. A 6-FAM-labeled 50-mer oligodeoxythymidylates with a biotin label on the end was incubated at a final concentration of 30 nm with 1 μm streptavidin in helicase buffer containing 1 mm ATP at 37 °C for 10 min to allow the streptavidin to bind to the DNA. Biotin (30 μm) was then added as a streptavidin trap, and the reaction was initiated by the addition of 5 μm DndEih. After 30 min, 10-μl aliquots of the reaction mixture were removed and added to 2 μl of stop solution (1 m NaCl and 200 mm EDTA, pH 8.0) together with 10 μm non-biotinylated oligodeoxythymidylates to bind to the protein and to reduce band shifting. The samples were analyzed on a 10% native acrylamide:Tris borate-EDTA gel.
ATPase Activity Assay
The ATPase activity was assayed using the EnzCheck pyrophosphate assay kit (Life Technologies) based on a previously reported procedure (19). The standard assay was performed in a 200-μl reaction mixture containing 50 mm Tris-HCl, pH 7.5, 1 mm MgCl2, 0.1 mm sodium azide, 0.2 mm 2-amino-6-mercapto-7-methylpurine ribonucleoside (MESG), 1 unit of purine nucleoside phosphorylase, 1 mm ATP, and 1 nmol of DndEi or DndEih in the absence or presence of 150 nm 34-bp length dsDNA substrates. Inorganic phosphate (Pi) created by ATP hydrolysis activates the enzymatic conversion of MESG to ribose 1-phosphate and 2-amino-6-mercapto-7-methylpurine by purine nucleoside phosphorylase. Conversion of MESG results in a shift in absorbance maximum from 330 to 360 nm, which is monitored by the Biotek synergy 2 plate reader. The absorbance at 360 nm was measured and corrected with a control reaction lacking protein. The relationship between the absorbance at 360 nm and Pi concentration was established by using KH2PO4 as a standard. The enzyme activity was calculated as the initial reaction velocity from the linear part of the progress curve.
Results
PT Modifications Occur at GpsAAC/GpsTTC Sites in R. anatipestifer ATCC 11845
Previous bioinformatic analysis has revealed that a dndCDEi gene cluster exists in R. anatipestifer ATCC 11845 (13). To confirm the PT phenotype of this strain, we performed the PT-specific iodine cleavage assay as previously described (17). As expected, the genomic DNA (gDNA) from R. anatipestifer ATCC 11845 was cleaved into small fragments by iodine, whereas the gDNA from R. anatipestifer HXb2 (closely related to ATCC 11845, but lacking PT-modifying genes) was not cleaved (Fig. 2A). This result revealed that PT modification indeed existed in R. anatipestifer ATCC 11845, and it also revealed that PT modification occurred on both strands of gDNA, because single strand-modified gDNA cannot be cleaved into small fragments by iodine (17). LC/MS analysis was subsequently performed to investigate the PT sequence context in R. anatipestifer ATCC 11845 by the method of detection of the nuclease P1-resistent PT-containing dinucleotides (2). As shown in Fig. 2B, only d(GpsA) and d(GpsT) dinucleotides were detected in R. anatipestifer ATCC 11845. PT sequencing has revealed that PT modifications occur at GpsAAC/GpsTTC sites in E. coli B7A (17), which has the same iodine-cleaved pattern and the same PT-linked dinucleotides as R. anatipestifer ATCC 11845. Taken together, these results indicated that gDNA is PT modified on both strands of GAAC/GTTC motifs in R. anatipestifer ATCC 11845.
FIGURE 2.

Analysis of the PT modification in R. anatipestifer ATCC 11845. A, electrophoresis pattern of the iodine-cleaved genomic DNA from R. anatipestifer ATCC 11845 (lanes 4 and 5), with R. anatipestifer HXb2 (lacking PT-modifying genes; lanes 2 and 3) as a negative control. M, DNA ladder. B, LC-MS analysis of the PT-linked dinucleotides from R. anatipestifer ATCC 11845.
DndEi Unwinds dsDNA Substrates with a 5′ Single-stranded Region
The dndEi gene from R. anatipestifer ATCC 11845 encodes a hybrid protein that is composed of an E domain (DndEie, homologous to canonical DndE) and an additional putative DNA helicase domain (DndEih) (Fig. 3A). To investigate the helicase activity of DndEi, both the putative helicase domain and the full-length protein were expressed and purified (Fig. 3B). Purification of DndEi mutant proteins, whose use is described later, was also performed as shown in Fig. 3C.
FIGURE 3.
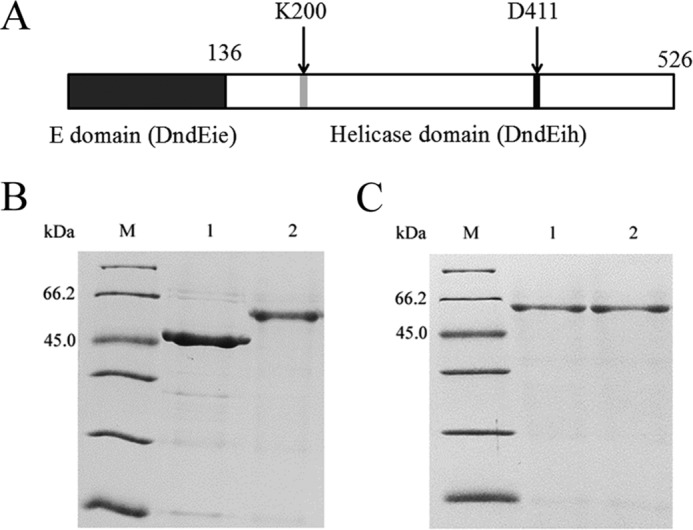
Purification of DndEih, DndEi, and DndEi mutants. A, domain organization of the DndEi protein. The positions of the two conserved motifs are indicated by gray and black boxes, respectively. The positions of the two point mutations are indicated by arrows. B, SDS-PAGE analysis of purified DndEih and DndEi stained by Coomassie Blue. Lanes: M, molecular mass marker; 1, DndEih; 2, DndEi. C, SDS-PAGE analysis of purified DndEi mutants stained by Coomassie Blue. Lanes: M, molecular mass marker; 1, DndEi (K200A); 2, DndEi (D411A).
A previously reported fluorescence resonance energy transfer (FRET) assay (18, 20) was performed to detect the DNA strands separation in real time. In this assay, the fluorescent strand of the duplex DNA was modified with an Alexa Fluor 488 fluorescent dye, whereas the quencher strand was modified with a spectrally paired quencher dye, black hole quencher. Annealing of the two strands brought the two dyes close together, resulting in efficient fluorescence quenching. Duplex unwinding was monitored by observing an increase in Alexa Fluor 488 fluorescence following strand separation. Re-annealing of the fluorescent strand to the quencher strand was suppressed by the presence of a 12-fold molar excess of a capture strand that is complementary to the quencher strand (Fig. 4A). DNA substrates with a 3′-overhang containing a 15-nucleotide ssDNA region and a 25-bp duplex region, a 5′-overhang containing a 15-nucleotide ssDNA region and a 25-bp duplex region, or blunt ends containing a 25-bp duplex region (Table 2) were tested in this assay. DndEih unwound the 5′-overhang but not 3′-overhang or blunt DNA substrates in the presence of ATP (Fig. 4B). The fluorescence intensity had a slight increase in the helicase assay using 3′-overhang DNA substrates, but the increase was not due to the helicase activity of DndEih because the fluorescence curve did not change when the DndEih concentration changed (data not shown). These results revealed that DndEih requires a 5′ single-stranded region to unwind dsDNA, which is consistent with the observation that DndEih translocated along the ssDNA from 5′ to 3′ (Fig. 5). Forked substrates are relevant intermediates in DNA metabolic processes, therefore DNA substrates with single-stranded 5′ and 3′ arms were tested in helicase assay (Fig. 4B). DndEih unwound forked DNA substrates more effectively than 5′-overhang DNA substrates, indicating a preference of DNA substrates with the fork structure. As DndEih is implicated in PT modification that occurs at the GAAC/GTTC sites in R. anatipestifer ATCC 11845, the helicase activities of DndEih to unwind DNA substrates with a GAAC/GTTC site, a PT-modified GpsAAC/GpsTTC site, or no GAAC/GTTC site were tested. As shown in Fig. 6A, the helicase activities (initial velocities) to unwind the three kinds of DNA substrate were at the same level. Subsequently, the full-length DndEi was tested in the helicase assay (Fig. 6B), and it had the same performance as DndEih, indicating that the E domain had no effect on the helicase activity of the helicase domain.
FIGURE 4.
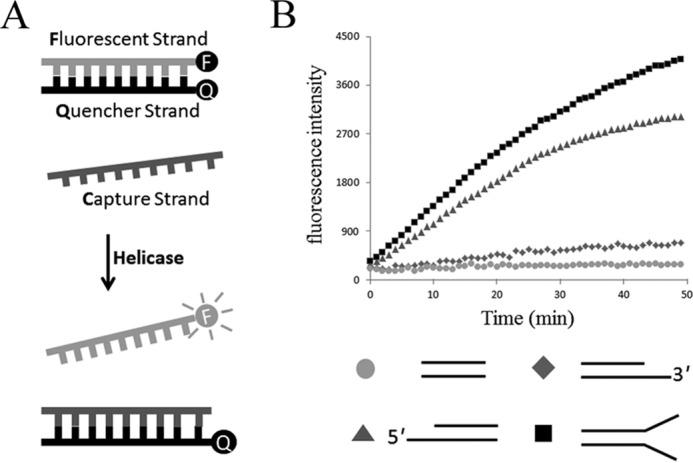
DNA helicase activity of DndEih using various DNA substrates. A, schematic representation of the fluorescence helicase assay based on FRET. The fluorescent strand is labeled with a fluorophore (F) and quencher strand is labeled with a quencher (Q). Annealing of the fluorophore strand to the quencher strand results in the fluorescence signal from the Alexa Fluor 488 dye to be quenched. When the double-stranded DNA substrate is unwound by the helicase, the fluorophore emits light upon its release from the quencher. The capture strand, which is complementary to the quencher strand, prevents the reannealing of the unwound duplex. B, time course of the helicase reaction of DndEih using 3′-overhang (diamonds), 5′-overhang (triangles), blunt-ended (circles), and forked (squares) DNA substrates in the presence of ATP.
FIGURE 5.
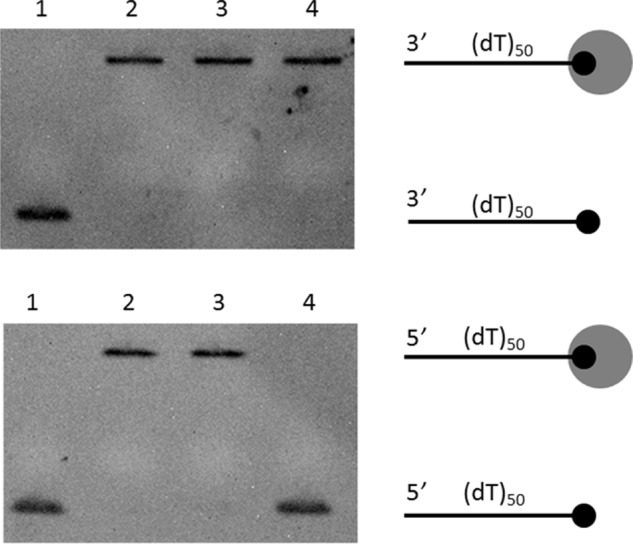
Single-stranded DNA translocation activity of DndEih. Translocation activity was investigated by measuring the ability of DndEih to displace streptavidin from a biotinylated probe at the 5′ or 3′ end of ssDNA. The black circle indicates the biotin-labeled DNA end, with the larger circle representing the streptavidin. Lane 1, biotinylated DNA; lane 2, biotinylated DNA bound to streptavidin; lane 3, streptavidin-bound DNA incubated with DndEih for 30 min in the absence of ATP; lane 4, streptavidin-bound DNA incubated with DndEih for 30 min in the presence of ATP.
FIGURE 6.

DNA helicase activity of DndEih, DndEi, and DndEi mutants. A and B, time course of the helicase reaction of DndEih (A) and DndEi (B) using 5′-overhang DNA substrates with different sequence motif as indicated. C, time course of the helicase reaction of DndEi mutants using 5′-overhang DNA substrates.
Sequence alignment analysis of the helicase domain of bacterial DndEi homologues revealed two conserved motifs (13) that are also conserved in other helicases (21). Motif I (GXXXXGKT, where X is any residue) at the N-terminal is supposed to be involved in nucleotide phosphate binding and hydrolysis (21), and Motif II (DEAH) is supposed to be responsible for coupling of ATPase and helicase activity (21, 22). To elucidate the functional roles of these motifs, we introduced a point mutation in motif I of DndEi, replacing the conserved lysine with alanine (K200A) and a point mutation in motif II, replacing the conserved aspartate with alanine (D411A) (Fig. 3A). K200A and D411A mutant proteins were then overproduced in E. coli and purified (Fig. 3C) using the same procedure as for the wild-type protein. The purified K200A and D411A proteins did not exhibit any detectable helicase activity (Fig. 6C), indicating that both Lys-200 and Asp-411 are essential residues for DNA helicase activity.
The Helicase Domain of DndEi Is Essential for PT Modification
Because the DNA helicase domain is not found in all of the DndE homologues, we next identified whether the helicase domain is required for PT modification in R. anatipestifer. Five plasmids (pJ1–5, Fig. 7A) containing the PT-modifying genes or its derivatives from R. anatipestifer ATCC 11845 were constructed based on the E. coli-R. anatipestifer shuttle plasmid pReS0. The recombinant plasmids were transferred into R. anatipestifer HXb2 (lacking PT-modifying genes) by conjugation. Genomic DNA of the transformants was then extracted and analyzed by a iodine cleavage assay (Fig. 7B) and LC/MS (Fig. 7C). The gDNA from HXb2 with dndCDEi (pJ3) was cleaved into small fragments (Fig. 7B) and PT-linked dinucleotides were detected in HXb2 (pJ3) by LC/MS (Fig. 7C), indicating that the intact dndCDEi conferred PT modification in the heterologous host HXb2. Meanwhile, absence of the DNA helicase domain (pJ2) resulted in loss of its PT modification activity (Fig. 7, B and C), indicating that the helicase domain of DndEi is essential for PT modification. Furthermore, HXb2 (pJ4, DndEi K200A) and HXb2 (pJ5, DndEi D411A), in which DndEi was not deleted but the helicase activity was inactivated, did not have any detectable PT phenotype (Fig. 7, B and C), suggesting that the helicase activity is implicated in PT modification.
FIGURE 7.

Analysis of the PT modification in R. anatipestifer HXb2 harboring a pReS0-based plasmid (pJ1–5), which has the PT-modifying genes from R. anatipestifer ATCC 11845. A, schematic diagram of the PT-modifying genes in pReS0-based plasmids (Table 1). B, electrophoresis pattern of the iodine-cleaved genomic DNA from R. anatipestifer HXb2 (pJ1) (lanes 4 and 5), HXb2 (pJ2) (lanes 6 and 7), HXb2 (pJ3) (lanes 8 and 9), HXb2 (pJ4) (lanes 10 and 11), HXb2 (pJ5) (lanes 12 and 13), and HXb2 (pReS0 empty vector) (lanes 2 and 3). M, DNA ladder. C, LC-MS analysis of the PT-linked dinucleotides from R. anatipestifer HXb2 harboring a pReS0-based plasmid. Electrospray ionization: m/z [M + H]+ modes for LC-MS: d(GpsA), 597.1388; d(GpsT), 588.1272.
DndEi Has an ATPase Activity Strongly Stimulated by the GAAC/GTTC Motif
Helicases disrupt the hydrogen bonds that hold the two strands of duplex DNA together in a reaction that is coupled with the hydrolysis of a nucleoside 5′-triphosphate, and thus nearly all helicases are also DNA-dependent nucleoside 5′-triphosphatases (23). We therefore examined the ATPase activity of the helicase domain of DndEi by using the pyrophosphate assay kit. It was assayed by measuring an increase in inorganic phosphate as a result of ATP hydrolysis as described previously (19). As shown in Fig. 8A, DndEih exhibited an ATPase activity that was stimulated by double-stranded DNA, and DNA substrates with different sequence context had the same effect on the stimulation of the ATPase activity (Fig. 8B).
FIGURE 8.

ATPase activity of DndEih (A and B) and DndEi (C and D). A and C, representative time courses of ATP hydrolysis by DndEih and DndEi. The ATPase assay was performed in the absence of DNA or in the presence of different kinds of dsDNA substrate (with no GAAC/GTTC motif; with a GAAC/GTTC motif; with a PT-modified GpsAAC/GpsTTC motif). The amount of Pi produced was calculated based on a standard KH2PO4 curve. Each data point represents the mean of triplicate experiments. B, rates of Pi accumulation in the ATPase assay performed in A. The data are presented as mean ± S.D. D, rates of Pi accumulation in the ATPase assay performed in C. The data are presented as mean ± S.D.
Because the helicase domain and the E domain (had no ATPase activity; data not shown) form a hybrid protein in vivo, the ATPase activity of the full-length DndEi was subsequently detected (Fig. 8C) to see whether the E domain had an effect on ATPase activity of the helicase domain. DndEi also exhibited an ATPase activity that was stimulated by double-stranded DNA as DndEih. However, DNA fragments containing a GAAC/GTTC motif stimulated ATPase (5.4-fold) greater than DNA fragments without this motif (3.9-fold) and DNA fragments containing a PT-modified GpsAAC/GpsTTC motif (3.2-fold) (Fig. 8D). These results indicated that the E domain can affect the ATPase activity of the helicase domain by recognizing different DNA substrates. Considering the DNA fragment with a GAAC/GTTC motif is the substrate of PT-modifying enzymes, the strongly stimulated ATPase activity is probably relevant to DNA PT modification.
Discussion
DNA helicases are a class of enzymes vital to all living organisms (23). They are molecular motor enzymes that use the energy of ATP hydrolysis to separate duplex DNA into single strands (21). Nearly all DNA metabolic processes, such as DNA replication (24), recombination (25), and repair (26), involve the separation of DNA strands that necessitates the use of DNA helicases. In this study, DNA helicase was shown to have a novel biological function (i.e. participating in DNA PT modification). The PT-modifying enzyme DndEi from R. anatipestifer was identified as a 5′ to 3′ DNA helicase with a preference of forked DNA substrates. Both deletion of the helicase domain and inactivation of the helicase activity of DndEi can lead to PT modification deficiency, indicating that DNA helicase activity is necessary for PT modification in R. anatipestifer.
As other DNA helicases, DndEi has an ATPase activity. It has been reported that the activity of ATPase is strongly stimulated by specific motifs in some nucleic acid metabolic processes (27, 28). For example, transcription factor δ has an associated DNA-dependent ATPase activity that is strongly stimulated by the TATA region of promoters, and the stimulated ATPase activity is required for activation of run-off transcription (27). This site-specific stimulation of ATPase activity was also found in DndEi by the GAAC/GTTC motif, which is consistent with the fact that GpsAAC/GpsTTC is the PT modification site in R. anatipestifer. Interestingly, more ATP was hydrolyzed by DndEi in the presence of DNA substrates with a GAAC/GTTC motif than in the presence of DNA substrates with a PT-modified GpsAAC/GpsTTC motif; meanwhile, the two kinds of DNA substrates were unwound by DndEi at the same speed. In contrast, DndEi without the E domain unwound the two kinds of DNA substrates consuming equal amounts of ATP. Taken together, these results indicate that, except for helicase working, additional ATP hydrolysis is catalyzed by DndEi in PT modification. The additional ATP hydrolysis is predicted to be used for sulfur incorporation into the phosphate backbone, which presents an energetically uphill proposition (29).
The E domain can affect the ATPase activity of the helicase domain by recognizing different DNA substrates. Therefore, the E domain of DndEi may play an important role in sequence recognition in DNA PT modification. Previous study has revealed that DndCDE proteins from Salmonella enterica form a stable complex in vitro (30). Taken together, it is possible that DndEi recognizes the modification sites and unwind DNA duplex at the initiation of PT modification, and then recruits other PT-modifying proteins to incorporate sulfur into the DNA backbone. Nevertheless, more functional studies need to be done to further understand the role of helicase in DNA PT modification.
Bacteria obtain PT-modifying genes via horizontal gene transfer facilitated by genomic islands (31). However, PT-modifying enzymes lack the helicase domain in some bacteria, such as Prevotella histicola. Phylogenetic analysis revealed that DndE from P. histicola had a closer relationship with the E domain of DndEi from Capnocytophaga canimorsus than with DndE from Stanieria cyanosphaera (Fig. 9), implying that DndE and DndEi have the same ancestor. Presumably, DndE (P. histicola) had the helicase domain initially and lost it in evolution because there was another DNA helicase having the same function. Similarly, there is no dndA gene in S. enterica, and the iscS gene encoding a similar cysteine desulfurase plays the role as dndA in DNA PT modification (11). Therefore, it is possible that DNA helicase is also indispensable for PT modification in bacteria lacking the helicase domain. DNA helicase encoded by another gene, which is not adjacent to PT-modifying genes, may participate in DNA PT modification.
FIGURE 9.

Phylogenetic analysis of DndE homologues, using MEGA v5.0 with 500 bootstrap replicates. Ampersand: DndE; asterisk: the E domain of DndEi (DndEie). GenBankTM accession numbers of the DndE homologues are: C. canimorsus (WP_041913577.1); R. anatipestifer ATCC 11845 (ADQ81723.1); Vibrio cholerae BJG-01 (EGS71244.1); Gelidibacter mesophilus (WP_027127205); Methylobacterium extorquens (WP_012754233); Klebsiella oxytoca (WP_046878204.1); C. perfringens (WP_003471805); Desulfovibrio africanus (PCS EMG37157); Methanosarcina mazei (KKI02757.1); S. cyanosphaera (WP_041620172.1); P. histicola (WP_036868676.1); Flexibacter litoralis (WP_041264540); and Anabaena cylindrica PCC 7122 (AFZ60204.1).
Author Contributions
D. Y., Q. H., B. C., and T. Z. designed the study and wrote the paper; T. Z. purified the wild-type and mutant DndEi protein; T. Z. performed the helicase assay and ATPase assay; P. J. performed the conjugation assay; P. J. extracted the genomic DNA used for PT analysis; T. Z. performed the iodine cleavage assay; T. Z., L. K., and X. Z. performed the LC/MS analysis; T. Z., L. K., and X. Z. performed the bioinformatic analysis of the DndE homologues.
Acknowledgment
We thank Dr. Wei Zhang for analyzing LC/MS data.
This work was supported by grants from the National Natural Science Foundation of China (30570400, 31170085, 31070058, 31470183, 31472224, 31272590, and 31400029), the Ministry of Science and Technology (2006AA02Z224, 2012CB721004, and 2009ZX09501-008), and the Shanghai Pujiang Program from the Science and Technology Commission of Shanghai Municipality (12PJD021). The authors declare that they have no conflicts of interest with the contents of this article.
- PT
- phosphorothioate
- gDNA
- genomic DNA
- MESG
- 2-amino-6-mercapto-7-methylpurine ribonucleoside.
References
- 1.Wang L., Chen S., Xu T., Taghizadeh K., Wishnok J. S., Zhou X., You D., Deng Z., and Dedon P. C. (2007) Phosphorothioation of DNA in bacteria by dnd genes. Nat. Chem. Biol. 3, 709–710 [DOI] [PubMed] [Google Scholar]
- 2.Wang L., Chen S., Vergin K. L., Giovannoni S. J., Chan S. W., DeMott M. S., Taghizadeh K., Cordero O. X., Cutler M., Timberlake S., Alm E. J., Polz M. F., Pinhassi J., Deng Z., and Dedon P. C. (2011) DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc. Natl. Acad. Sci. U.S.A. 108, 2963–2968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu T., Yao F., Zhou X., Deng Z., and You D. (2010) A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 38, 7133–7141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao B., Cheng Q., Gu C., Yao F., DeMott M. S., Zheng X., Deng Z., Dedon P. C., and You D. (2014) Pathological phenotypes and in vivo DNA cleavage by unrestrained activity of a phosphorothioate-based restriction system in Salmonella. Mol. Microbiol. 93, 776–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng Q., Cao B., Yao F., Li J., Deng Z., and You D. (2015) Regulation of DNA phosphorothioate modifications by the transcriptional regulator DptB in Salmonella. Mol. Microbiol. 97, 1186–1194 [DOI] [PubMed] [Google Scholar]
- 6.Ou H. Y., He X., Shao Y., Tai C., Rajakumar K., and Deng Z. (2009) dndDB: a database focused on phosphorothioation of the DNA backbone. Plos One 4, e5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu T., Liang J., Chen S., Wang L., He X., You D., Wang Z., Li A., Xu Z., Zhou X., and Deng Z. (2009) DNA phosphorothioation in Streptomyces lividans: mutational analysis of the dnd locus. BMC Microbiol. 9, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou X., He X., Liang J., Li A., Xu T., Kieser T., Helmann J. D., and Deng Z. (2005) A novel DNA modification by sulphur. Mol. Microbiol. 57, 1428–1438 [DOI] [PubMed] [Google Scholar]
- 9.Chen F., Zhang Z., Lin K., Qian T., Zhang Y., You D., He X., Wang Z., Liang J., Deng Z., and Wu G. (2012) Crystal structure of the cysteine desulfurase DndA from Streptomyces lividans which is involved in DNA phosphorothioation. Plos One 7, e36635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.You D., Wang L., Yao F., Zhou X., and Deng Z. (2007) A novel DNA modification by sulfur: DndA is a NifS-like cysteine desulfurase capable of assembling DndC as an iron-sulfur cluster protein in Streptomyces lividans. Biochemistry 46, 6126–6133 [DOI] [PubMed] [Google Scholar]
- 11.An X., Xiong W., Yang Y., Li F., Zhou X., Wang Z., Deng Z., and Liang J. (2012) A novel target of IscS in Escherichia coli: participating in DNA phosphorothioation. Plos One 7, e51265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu W., Wang C., Liang J., Zhang T., Hu Z., Wang Z., Lan W., Li F., Wu H., Ding J., Wu G., Deng Z., and Cao C. (2012) Structural insights into DndE from Escherichia coli B7A involved in DNA phosphorothioation modification. Cell Res. 22, 1203–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbier P., Lunazzi A., Fujiwara-Nagata E., Avendaño-Herrera R., Bernardet J. F., Touchon M., and Duchaud E. (2013) From the Flavobacterium genus to the phylum Bacteroidetes: genomic analysis of dnd gene clusters. FEMS Microbiol. Lett. 348, 26–35 [DOI] [PubMed] [Google Scholar]
- 14.Enemark E. J., and Joshua-Tor L. (2008) On helicases and other motor proteins. Curr. Opin. Struct. Biol. 18, 243–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Q., Miao S., Ni X., Lu F., Yu H., Xing L., and Jiang P. (2013) Construction of a shuttle vector for use in Riemerella anatipestifer. J. Microbiol. Methods 95, 262–267 [DOI] [PubMed] [Google Scholar]
- 16.Hu Q., Han X., Zhou X., Ding C., Zhu Y., and Yu S. (2011) OmpA is a virulence factor of Riemerella anatipestifer. Vet. Microbiol. 150, 278–283 [DOI] [PubMed] [Google Scholar]
- 17.Cao B., Chen C., DeMott M. S., Cheng Q., Clark T. A., Xiong X., Zheng X., Butty V., Levine S. S., Yuan G., Boitano M., Luong K., Song Y., Zhou X., Deng Z., Turner S. W., Korlach J., You D., Wang L., Chen S., and Dedon P. C. (2014) Genomic mapping of phosphorothioates reveals partial modification of short consensus sequences. Nat. Commun. 5, 3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boguszewska-Chachulska A. M., Krawczyk M., Stankiewicz A., Gozdek A., Haenni A. L., and Strokovskaya L. (2004) Direct fluorometric measurement of hepatitis C virus helicase activity. FEBS Lett. 567, 253–258 [DOI] [PubMed] [Google Scholar]
- 19.Yao F., Xu T., Zhou X., Deng Z., and You D. (2009) Functional analysis of spfD gene involved in DNA phosphorothioation in Pseudomonas fluorescens Pf0–1. FEBS Lett. 583, 729–733 [DOI] [PubMed] [Google Scholar]
- 20.Tani H., Fujita O., Furuta A., Matsuda Y., Miyata R., Akimitsu N., Tanaka J., Tsuneda S., Sekiguchi Y., and Noda N. (2010) Real-time monitoring of RNA helicase activity using fluorescence resonance energy transfer in vitro. Biochem. Biophys. Res. Commun. 393, 131–136 [DOI] [PubMed] [Google Scholar]
- 21.Tuteja N., and Tuteja R. (2004) Unraveling DNA helicases: motif, structure, mechanism and function. Eur. J. Biochem. 271, 1849–1863 [DOI] [PubMed] [Google Scholar]
- 22.Tanner N. K., and Linder P. (2001) DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell 8, 251–262 [DOI] [PubMed] [Google Scholar]
- 23.Matson S. W., and Kaiser-Rogers K. A. (1990) DNA helicases. Annu. Rev. Biochem. 59, 289–329 [DOI] [PubMed] [Google Scholar]
- 24.Waga S., and Stillman B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem. 67, 721–751 [DOI] [PubMed] [Google Scholar]
- 25.Sung P., and Klein H. (2006) Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 7, 739–750 [DOI] [PubMed] [Google Scholar]
- 26.Brosh R. M. (2013) DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer 13, 542–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conaway R. C., and Conaway J. W. (1989) An RNA polymerase II transcription factor has an associated DNA-dependent ATPase (dATPase) activity strongly stimulated by the TATA region of promoters. Proc. Natl. Acad. Sci. U.S.A. 86, 7356–7360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okanami M., Meshi T., and Iwabuchi M. (1998) Characterization of a DEAD box ATPase/RNA helicase protein of Arabidopsis thaliana. Nucleic Acids Res. 26, 2638–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckstein F. (2007) Phosphorothioation of DNA in bacteria. Nat. Chem. Biol. 3, 689–690 [DOI] [PubMed] [Google Scholar]
- 30.Cao B., Zheng X., Cheng Q., Yao F., Zheng T., Ramesh Babu I., Zhou H., Dedon P., and You D. (2015) In vitro analysis of phosphorothioate modification of DNA reveals substrate recognition by a multiprotein complex. Sci. Rep. 5, 12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He X., Ou H. Y., Yu Q., Zhou X., Wu J., Liang J., Zhang W., Rajakumar K., and Deng Z. (2007) Analysis of a genomic island housing genes for DNA S-modification system in Streptomyces lividans 66 and its counterparts in other distantly related bacteria. Mol. Microbiol. 65, 1034–1048 [DOI] [PubMed] [Google Scholar]