

Abstract
We characterized DNA methylation quantitative trait loci (mQTLs) in a large collection (n=166) of human fetal brain samples spanning 56–166 days post-conception, identifying >16,000 fetal brain mQTLs. Fetal brain mQTLs are primarily cis-acting, enriched in regulatory chromatin domains and transcription factor binding sites, and show significant overlap with genetic variants also associated with gene expression in the brain. Using tissue from three distinct regions of the adult brain (prefrontal cortex, striatum and cerebellum) we show that most fetal brain mQTLs are developmentally stable, although a subset is characterized by fetal-specific effects. We show that fetal brain mQTLs are enriched amongst risk loci identified in a recent large-scale genome-wide association study (GWAS) of schizophrenia, a severe psychiatric disorder with a hypothesized neurodevelopmental component. Finally, we demonstrate how mQTLs can be used to refine GWAS loci through the identification of discrete sites of variable fetal brain methylation associated with schizophrenia risk variants.
Human brain development is orchestrated by complex transcriptional programs1, which are guided and reinforced by epigenetic modifications to DNA and histone proteins. DNA methylation is the most extensively studied epigenetic modification, playing a key role in many important genomic regulatory processes2. Of note, the establishment and maintenance of cell- and tissue-specific DNA methylation patterns is crucial for normal mammalian development3. Although traditionally regarded as a mechanism of transcriptional repression, DNA methylation can be associated with both increases and decreases in gene expression4, and has recently been implicated in other genomic functions including alternative splicing and promoter usage5.
We recently characterized widespread changes in DNA methylation across human fetal brain development6, although the factors influencing inter-individual methylomic variation during the prenatal period are unknown. Studies in a variety of tissues, including adult human brain7,8, have shown that DNA methylation can be influenced by DNA sequence variation. These methylation quantitative trait loci (mQTLs) have been found to overlap with DNA variants associated with levels of gene expression (expression quantitative trait loci; eQTLs)4,9, and may serve as markers for these as well as other genetic influences on gene regulation. Although mQTLs have previously been assessed in the adult human brain using low-resolution DNA methylation arrays7,8, mQTLs in the developing human brain have not previously been explored.
In this study we combine high-density DNA methylation profiling with genome-wide SNP genotyping in a large (n = 166) collection of human brain samples from the first and second trimester of gestation. Given the growing evidence that many common variants associated with complex diseases act through effects on gene regulation10,11, we subsequently test for enrichment of fetal mQTL amongst risk loci identified in a recent large-scale GWAS of schizophrenia12, a neuropsychiatric disorder with a hypothesized neurodevelopmental component13,14. Finally, we show how mQTL data can be used to refine broad GWAS loci through the identification of discrete sites of variable fetal brain methylation associated with schizophrenia risk variants. As a resource to the wider community we have developed a searchable online database of fetal brain mQTLs that can be accessed at http://epigenetics.essex.ac.uk/mQTL/.
Results
Methylation quantitative trait loci (mQTL) in the developing human brain are widespread and predominantly characterized by cis effects
We performed genome-wide single nucleotide polymorphism (SNP) genotyping and DNA methylation profiling in 166 human fetal brain samples ranging from 56 to 166 days post-conception (see Online Methods and Supplementary Table 1). After stringent quality-control we tested for an additive effect of allele dosage on DNA methylation across all potential pairings of 430,304 SNPs and 314,554 DNA methylation sites to identify fetal brain mQTLs (Supplementary Table 1). We identified 16,809 mQTLs at a conservative Bonferroni-corrected significance threshold of P < 3.69×10−13 (Supplementary Tables 2 and 3). The median DNA methylation change per allele across all identified mQTLs was 6.69% (interquartile range (IQR) = 3.17%–8.96%) for each mQTL SNP (Supplementary Fig. 1), slightly larger than reported in a previous analysis of genome-wide mQTLs in the adult brain (median effect size = 4.11%, IQR = 2.13–6.97%)7. The majority of mQTL SNPs (74.17%) are associated with DNA methylation at only a single probe; in contrast, most DNA methylation sites (69.83%) showing evidence for association do so with multiple mQTL SNPs, presumably as a result of linkage disequilibrium (LD) between SNPs (Supplementary Fig. 2). A searchable database of fetal brain mQTLs is available at: http://epigenetics.essex.ac.uk/mQTL/.
The majority of fetal brain mQTLs (96.3%) involve SNPs and DNA methylation sites on the same chromosome (Fig. 1a and Supplementary Table 2). We defined significant SNP-methylation relationships spanning <500kb as “cis-mQTLs” (n = 15,942, 94.8% of total), with those spanning >500kb or characterized by inter-chromosomal effects (n = 867, 5.16% of total) as representing trans-mQTLs. The strong enrichment of cis-mQTLs concurs with data from other tissues and cell-types7,15–18. Amongst the cis-mQTLs, both effect size (i.e. DNA methylation change per allele, Fig. 1b) and significance (i.e. P-value, Supplementary Fig. 3) are related to the distance between the Illumina 450K array probe and mQTL SNP.
Figure 1. mQTLs in the developing human brain are predominantly cis-acting with effect size related to distance, although notable trans-effects are also present.
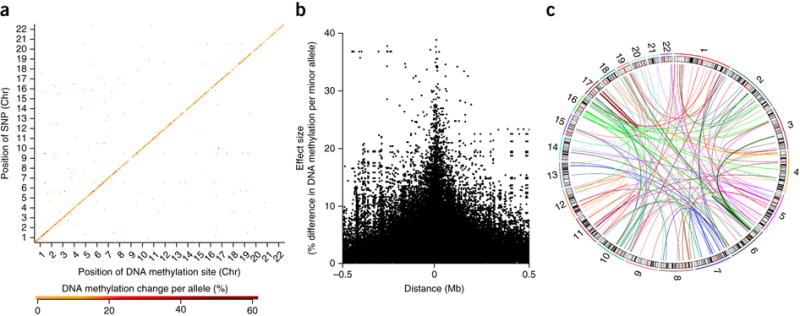
(a) The genomic distribution of Bonferroni significant (P = 3.69 ×10−13) mQTLs in fetal brain, where the position on the x-axis indicates the location of Illumina 450K HumanMethylation array probes and the position on the y-axis indicates the location of informative SNPs. The color of the point corresponds to the difference in DNA methylation per allele compared to the reference allele, with the largest effects plotted in dark red. A clear positive diagonal can be observed demonstrating that the majority of mQTLs in fetal brain are associated with genotype in cis. (b) The relationship between mQTL effect size (DNA methylation change per allele) and distance between the Illumina 450K HumanMethylation array probe and informative SNPs in fetal brain, confirming the predominantly cis nature of mQTLs. A similar relationship is seen between mQTL significance and distance (see Supplementary Fig. 3). (c) Trans-mQTLs in the developing human brain. Shown are all Bonferroni significant (P = 3.69 ×10-13) trans-mQTLs identified in fetal brain samples. The thickness of each line depicts association effect size, with color reflecting the chromosomal location of the mQTL SNP.
Despite the preponderance of cis-mQTLs, there are some notable trans-mQTL effects (Fig. 1c and Supplementary Table 4), consistent with previous reports of long-range genetic regulation of epigenetic variation in multiple cell-types19. Although the average effect size for trans-mQTLs is significantly lower than that observed for cis-mQTLs (two-sided Wilcoxon rank sum test, P = 6.74×10−7) (Supplementary Fig. 1), there is a higher proportion of larger (DNA methylation change per allele > 25%) effects among trans-mQTLs than cis-mQTLs (1.04% vs 0.715%). Of the 178 DNA methylation sites identified as being associated with trans-acting genetic variation in the fetal brain, 50 (28.09%) and 108 (60.67%) were also identified in studies of pancreatic islet cells16 and lymphocytes19, respectively (Supplementary Table 4 and Supplementary Table 5). These long-range associations between genotype and DNA methylation complement data showing interactions between regulatory elements spanning several Mb20, and even between chromosomes21.
Fetal brain mQTLs are significantly enriched in functional regulatory domains
We used data from ENCODE and the Roadmap Epigenomics Project10,22–24 to assess whether sites characterized by genotype-associated DNA methylation co-localize with genomic regions associated with markers of transcriptional activity. We observed an enrichment of fetal brain mQTLs in genomic regions characterized by ChIP-seq peaks for repressive histone modifications in fetal brain – for example, H3K9me3 (relative enrichment = 1.43, P = 0.00107) and H3K27me3 (relative enrichment = 1.16, P = 0.000144) - and a significant depletion of fetal brain mQTLs in genomic regions defined by ChIP-seq peaks for histone modifications associated with active transcription – for example, H3K4me1 (relative enrichment = 0.828, P = 6.55×10−8) and H3K36me3 (relative enrichment = 0.543, P = 1.39×10−15 (Supplementary Table 6). Fetal brain mQTLs were found to be significantly enriched in regions of open chromatin indicated by DNase1 hypersensitivity sites (DHSs) identified in the adult human brain (Supplementary Table 7), consistent with the observation that intermediately methylated domains, one potential consequence of allele-specific DNA methylation, are enriched in DHSs25. We also identified a significant enrichment of genotype-associated DNA methylation sites overlapping annotated transcription factor binding sites identified by the ENCODE project10,26 (relative enrichment = 1.26, P = 2.96×10−11) (Supplementary Table 8). Of note, there is a highly-significant enrichment (odds ratio = 1.35, P = 1.66×10−9) of fetal brain mQTLs influencing DNA methylation in CCCTC-binding factor (CTCF) motifs (Supplementary Table 8), confirming a finding from a previous study of heritable DNA methylation sites in the human brain27. CTCF is an 11 zinc-finger protein with insulator and chromatin barrier activity whose binding affinity is known to be strongly influenced by DNA methylation28. Given the important role of CTCF in core genomic processes including transcription, chromosomal interactions and chromatin structure29, the enrichment of genetically-mediated DNA methylation at CTCF binding sites highlights an important potential mechanism linking genetic variation to genomic function. In addition to CTCF, a significant enrichment was also observed within binding sites for several other transcription factors, including IRF1, GABP, ELF1, Rad21 and CCNT2, with significant depletion in binding sites for others (e.g. SUZ12, CtBP2) (Supplementary Table 8).
Although the majority of fetal brain mQTLs are conserved in adult brain regions, there are fetal-specific genetic effects on DNA methylation at certain loci
We next generated mQTL data from three adult human brain regions (prefrontal cortex (PFC), striatum (STR) and cerebellum (CER)) dissected from matched donors (n = 83; 21–96 years old, see Online Methods and Supplementary Table 1) to explore the extent to which fetal brain mQTLs are also present in the adult brain. Using a replication mQTL significance threshold of P < 1×10−5, the majority (83.46%) of fetal brain mQTLs are present in at least one of the tested adult brain region (Supplementary Table 9 and Fig. 2a), and there is a highly-significant overall correlation of individual mQTL effect sizes between fetal brain and each of the individual adult brain regions (PFC: r = 0.911, P < 2.2×10−16; STR: r = 0.899, P < 2.2×10−16; CER: r = 0.835, P < 2.2×10−16) (Supplementary Fig. 4) across all Bonferroni-significant fetal brain mQTLs, even in mQTLs that do not meet our replication threshold (Supplementary Fig. 5). Of note, fetal brain mQTLs that do not replicate in adult brain are characterized by significantly lower effect sizes across all brain regions, including the fetal brain discovery sample (P = 3.18×10−141) (Supplementary Fig. 6). Despite the overall strong concordance in the direction of mQTL effects between fetal and adult brain, there are notable examples of heterogeneity between fetal and adult brain tissue. We used a multilevel linear regression model to test the significance of an interaction term and identify differential mQTL effects across our datasets. Of the 10,663 fetal mQTL effects tested, 3,173 (29.76%) were found to be significantly heterogeneous (Bonferroni-corrected P < 4.69 × 10−6) across the fetal and adult datasets (Supplementary Table 10). These include mQTLs that had notably larger or notably smaller effects in the adult brain, and fetal-specific mQTLs showing no significant association with DNA methylation in any adult brain region (see Fig. 2b). We also identified a small number (n = 45) of fetal brain mQTLs that had opposite effects on DNA methylation in fetal and adult brain samples (Fig. 2c and Supplementary Table 11).
Figure 2. Despite a highly-significant overall correlation of individual mQTL effects between fetal brain and adult brain regions, a subset of loci are characterized by fetal-specific mQTLs.
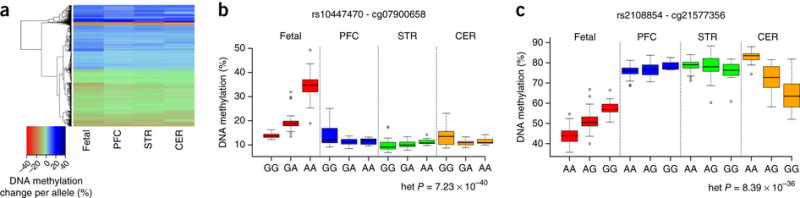
(a) Heatmap showing effect sizes in adult brain for all fetal brain mQTLs tested. Using a replication mQTL significance threshold of P < 1.00E-5, the majority (83.46%) of fetal brain mQTLs are present in at least one of the tested adult brain region. Despite the overall strong concordance in the direction of mQTL between fetal and adult brain, there are notable examples of heterogeneity in mQTL effects between fetal and adult brain tissue. Shown are examples of (b) a fetal-specific mQTL between rs10447470 and cg07900658 (heterogeneity P = 7.23E-40) and (c) an mQTL between rs2108854 and cg21577356 showing an opposite direction of effect in fetal brain and cerebellum (heterogeneity P = 8.39E-36).
Fetal brain mQTLs overlap with genetic variants associated with RNA transcript abundance in the brain
We used eQTL data from ten adult brain regions30 to test whether identified fetal brain mQTLs overlap with genetic variants associated with RNA transcript abundance in cis. We compared the distribution of the minimum brain eQTL P-value of all interrogated SNPs split into the subsets of those identified as fetal brain mQTLs and those not (Supplementary Fig. 7), finding that variants associated with DNA methylation are indeed more likely to be associated with gene expression in cis (Wilcoxon rank sum test P < 2.2×10−16). Of the 414,172 SNPs tested in both the mQTL and eQTL datasets, 9,869 were identified as being Bonferroni-significant cis mQTLs and 2,674 as Bonferroni-significant (P < 5.99×10−9) eQTLs, with an overlap of 750 variants associated with 227 DNA methylation probes and 127 transcript probes (Supplementary Table 12). At a more relaxed eQTL threshold (P < 1.00×10−7), there is an overlap of 1,042 variants associated with 315 DNA methylation probes and 183 transcript probes. A list of all variants associated with both DNA methylation and gene expression in cis is given in Supplementary Table 13. This overlapping set of variants likely includes multiple SNPs in LD that are associated with one gene expression transcript and DNA methylation site. Because the extent and magnitude of LD varies across the genome, we established an LD-independent set of SNPs associated with DNA methylation and tested the overlap of these with the sentinelized subsignals – i.e. the most associated marker from a set in high LD (r2 > 0.8) - from the brain eQTL dataset30. Compared to 1 million simulated mQTL SNP sets matched for allele frequency, this overlap was significantly greater than expected (relative enrichment = 4.23, P < 1.00×10−6 after 1 million simulations; Supplementary Table 14 and Supplementary Fig. 8).
There is a significant enrichment of schizophrenia-associated GWAS variants in fetal brain mQTLs
Our catalogue of fetal brain mQTLs provides a unique resource for investigating putative functional consequences of genetic variation associated with postulated neurodevelopmental disorders such as schizophrenia. A recent large-scale GWAS identified 108 independent genomic loci exhibiting genome-wide significant association with the disorder (P < 5×10−8), with evidence for a substantial polygenic component within signals that fall below this stringent level of significance12. Because the majority of these variants reside in regions of strong LD and do not index coding variants affecting protein structure, there remains considerable uncertainty about the causal genes involved in pathogenesis and the way in which they are functionally regulated by schizophrenia risk variants. We used PLINK31 to ‘clump’ our list of significant (P < 3.69×10−13) fetal brain mQTL variants into a set of quasi-independent SNPs (SNP pairwise r2 < 0.25 within 250kb (non-major histocompatibility complex (MHC)) or 10000kb (MHC)), see Online Methods) and tested for enrichment of schizophrenia-associated variants across a range of GWAS significance-thresholds, using up to 1,000,000 simulated SNP sets to generate empirical P values (see Online Methods). We observed a highly-significant enrichment (relative enrichment = 4.11, P = 3.0×10−6) of genome-wide significant schizophrenia risk variants amongst fetal brain mQTLs, with a trend for stronger enrichment at more stringent levels of GWAS significance (Supplementary Fig. 9 and Table 1). To examine the specificity of any enrichment, these analyses were repeated using large GWAS datasets from i) a non-neurodevelopmental brain disorder (Alzheimer’s disease (AD)32) and ii) two non-neurological phenotypes (body mass index (BMI)33 and type 2 diabetes (T2D)34. Although our confidence in the enrichment of fetal brain mQTL in these datasets is limited by the smaller number of semi-independent GWAS SNPs, levels of enrichment were found to be notably lower for all other tested phenotypes (Table 1). Variants associated with AD are, however, nominally significantly enriched at the most relaxed GWAS threshold (GWAS threshold P < 5×10−5: relative enrichment = 3.18, P = 0.022) and several individual GWAS variants identified for this and the other tested phenotypes are also significant mQTLs in fetal brain mQTLs (Supplementary Table 15).
Table 1. Fetal brain mQTLs are significantly enriched for schizophrenia genetic risk variants.
Different significance thresholds were used to classify SNP associations from a recent large GWAS analysis of schizophrenia12. Empirical P-values were derived by comparing a million simulated SNP sets, matched for allele frequency. There is an enrichment of schizophrenia-associated variants amongst fetal brain mQTLs at all tested GWAS significance thresholds. No enrichment of fetal brain mQTLs was observed in similar-sized GWAS analyses of Alzheimer’s disease32, body mass index33 and type 2 diabetes (T2D)34.
| Schizophrenia (n = 79,845) Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) |
Alzheimer’s disease (n = 54,162) Lambert et al. (2013) |
Body mass index (n = 322,154) Locke et al. (2015) |
Type 2 diabetes (n = 69,033) Morris et al. (2012) |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GWAS P-value threshold | 5.00E-05 | 5.00E-06 | 5.00E-07 | 5.00E-08 | 5.00E-05 | 5.00E-06 | 5.00E-07 | 5.00E-08 | 5.00E-05 | 5.00E-06 | 5.00E-07 | 5.00E-08 | 5.00E-05 | 5.00E-06 | 5.00E-07 | 5.00E-08 | |
| Number of associated SNPs | 6178 | 2838 | 982 | 636 | 260 | 132 | 101 | 80 | 1228 | 706 | 482 | 357 | 229 | 116 | 79 | 58 | |
| Number of mQTL SNPs | 51 | 38 | 28 | 16 | 5 | 2 | 1 | 0 | 13 | 7 | 5 | 4 | 3 | 2 | 1 | 0 | |
| Simulated overlap | Mean | 17.5 | 9.62 | 6.06 | 3.89 | 1.58 | 0.8 | 0.623 | 0.5 | 7.81 | 4.51 | 3.07 | 2.29 | 1.44 | 0.729 | 0.504 | 0.378 |
| SD | 4.16 | 3.08 | 2.45 | 1.97 | 1.24 | 0.884 | 0.779 | 0.696 | 2.8 | 2.13 | 1.75 | 1.5 | 1.2 | 0.856 | 0.705 | 0.613 | |
| Relative enrichment | 2.91 | 3.95 | 4.62 | 4.11 | 3.17 | 2.5 | 1.61 | 0 | 1.66 | 1.55 | 1.63 | 1.75 | 2.09 | 2.74 | 1.99 | 0 | |
| Confidence interval (95%) for enrichment | Lower | 1.96 | 2.38 | 2.55 | 2 | 1.25 | 0.667 | 0.5 | - | 0.929 | 0.778 | 0.714 | 0.667 | 0.75 | 0.667 | 0.5 | - |
| Upper | 5.1 | 9.5 | 14 | 16 | Inf | Inf | Inf | - | 4.33 | 7 | Inf | Inf | Inf | Inf | Inf | - | |
| Empirical P-value for enrichment of mQTLs amongst GWAS SNPs | < 1.00E-06 | < 1.00E-06 | < 1.00E-06 | 3.00E-06 | 0.022 | 0.1895 | 0.4662 | 1 | 0.0553 | 0.1727 | 0.1919 | 0.1997 | 0.1754 | 0.1672 | 0.3965 | 1 | |
The identified mQTLs residing in schizophrenia-associated GWAS regions do not necessarily represent the actual causal risk variants; in many instances we are likely to be capturing ‘passenger’ effects whereby the variant influencing DNA methylation and the schizophrenia-associated SNP are instead co-segregating in the same LD block. Therefore we sought to identify instances where a likely causal risk variant for schizophrenia was an mQTL SNP. We used 1000 Genomes Project data (http://www.1000genomes.org/) to identify all variants in strong LD (r2 > 0.8) with the 125 autosomal index SNPs associated with schizophrenia12. Of note, two of the actual schizophrenia GWAS index SNPs represent Bonferroni-significant fetal brain mQTLs: rs2535627 (associated with DNA methylation at cg11645453, P = 3.05×10−13, Supplementary Fig. 10) and rs4648845 (associated with DNA methylation at cg02275930, P = 4.54×10−15, Supplementary Fig. 11). 46 additional mQTL variants that are in strong LD with another six index SNPs are part of 86 highly-significant fetal brain mQTL pairs (Supplementary Table 16).
mQTLs can be used to localize putative causal loci within large genomic regions associated with schizophrenia
To generate a more comprehensive database of mQTLs in the fetal brain and identify more examples where the same SNP is associated with both DNA methylation and disease, we imputed our genotype data using the most recent panel downloaded from the 1000 Genome Project (see Online Methods). Using an imputed set of 5,177,320 variants we identified an additional 256,040 mQTLs, which reflected the non-imputed dataset in terms of genomic distribution and observed effect sizes (Supplementary Table 2 and Supplementary Fig. 12). The full list of fetal brain mQTLs after imputation can be downloaded from: http://epigenetics.essex.ac.uk/mQTL//All_Imputed_BonfSignificant_mQTLs.csv.gz. Our imputed data enabled us to identify 1,067 instances where the same SNP is associated with both DNA methylation and schizophrenia, with a comprehensive list available for download from: http://epigenetics.essex.ac.uk/mQTL//PGC_IndexSNPs_QTLs_Inc2PCs_AllTissues_MatchS NPPosition.csv). Because they could be biased by the LD structure at associated loci the imputed mQTL data were not used for subsequent enrichment analyses, but they enabled us to further refine schizophrenia candidate regions and undertake co-localization analyses in order to identify variants associated with both DNA methylation and schizophrenia. We performed a Bayesian co-localization analysis35 across the 105 autosomal regions associated with schizophrenia12, spanning 19,378 DNA methylation sites included in our analysis. Instead of focusing only on the intersection of significant variants associated with two phenotypes independently, this approach compares the pattern of association results from the schizophrenia GWAS and mQTL analyses across a region, combining the summary statistics into posterior probabilities for five hypotheses (see Online Methods). As this methodology assumes that the causal variant is present, or at least very well tagged, in the dataset, these co-localization analyses were performed using our imputed fetal brain mQTL dataset. The posterior probabilities for 65 regions, involving 296 DNA methylation sites in 306 pairs, were supportive of a co-localized association signal for both schizophrenia and DNA methylation in that region (PP3+PP4 > 0.99; Supplementary Table 17). Twenty-six of these pairs (covering 15 regions associated with schizophrenia) had a higher posterior probability for both schizophrenia and DNA methylation being associated with the same causal variant (PP4/PP3 > 1), with 16 (10 regions) of these having sufficient support for them to be considered as ‘convincing’ (PP4/PP3 > 5) according to the criteria of Giambartolomei and colleagues35. Of note, three of these top-ranked pairs are annotated to the AS3MT locus within a robust schizophrenia-associated region on chromosome 10 (Supplementary Table 17). The utility of mQTL mapping for localizing putative causal loci associated with disease within this region is shown in Fig. 3, with additional examples for other schizophrenia-associated regions available on our website (http://epigenetics.essex.ac.uk/mQTL/).
Figure 3. mQTL mapping can localize putative causal loci associated with disease.
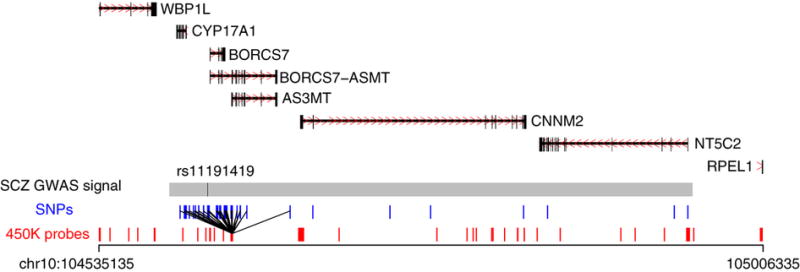
Co-localization analyses yielded strong support for variants annotated to the AS3MT gene being associated with both schizophrenia and DNA methylation within a broad genomic region (chr10:104535135-105006335) identified in a recent GWAS analysis of schizophrenia12. All potential causal variants (r2 > 0.8 with index variant) present in the imputed mQTL dataset are indicated by vertical blue lines and all DNA methylation probes within each region by vertical red lines. Bonferroni-significant mQTLs are indicated by black lines between the respective variant and DNA methylation probe, where the line width reflects the magnitude of the effect. Additional examples of fetal brain mQTLs in genomic regions showing genome-wide significant association with schizophrenia are available on our website (http://epigenetics.essex.ac.uk/mQTL/).
Schizophrenia-associated genomic regions are enriched for fetal-specific mQTLs
Given the hypothesized neurodevelopmental component to schizophrenia, we examined the extent to which the mQTLs overlapping with schizophrenia-associated variants are characterized by fetal-specific effects (Supplementary Table 16). Across the 78 mQTLs also tested in adult brain samples, overall effect sizes were significantly larger in fetal brain than all adult brain regions tested (Wilcoxon rank-sum test: PFC P = 0.0420, STR P = 0.00226, CER P = 0.00998) (Fig. 4). Our heterogeneity analysis highlighted 16 (20.5%) instances where significantly different relationships between genotype and DNA methylation are found across the adult and fetal datasets, with eight classed as fetal-specific variants (i.e. those not reaching our replication threshold (P < 1.00×10−5) in any adult brain region) and the remaining eight demonstrating smaller effects across the adult brain.
Figure 4. Fetal mQTLs in schizophrenia-associated regions have larger effects on DNA methylation during neurodevelopment than the in adult brain.

For robustly-associated schizophrenia GWAS variants characterized by human fetal brain mQTLs (Supplementary Table 16), we compared effect sizes between fetal and adult brain. Effect sizes (red to blue) for the corresponding mQTLs in adult brain are significantly lower across all three adult brain regions tested (Wilcoxon rank-sum test: PFC P = 0.0420, STR P = 0.00226, CER P = 0.00998). The heterogeneity P-value for each mQTL is depicted by the green (high heterogeneity) to yellow (low heterogeneity) column. White indicates that the mQTL was not tested in the adult brain samples.
Discussion
To explore the functional consequences of genetic variation in the developing human brain, we characterized mQTLs in human fetal brain samples (spanning 56–166 days post-conception), identifying over 16,000 associated pairs of SNPs and DNA methylation sites. We find that fetal brain mQTLs are significantly enriched in functional regulatory domains including DHSs, regions of repressive histone modifications, and specific transcription factor binding sites across the genome, and show significant overlap with genetic variants influencing gene expression in the brain. Although the majority of fetal brain mQTLs appear to be conserved across adult brain regions, we find evidence for fetal-specific genetic effects at certain loci. Our data concur with findings from an independent study of cortical mQTLs across development [Jaffe et al, Nat Neuro, in press]; mQTL effects are highly consistent across both analyses (Supplementary Fig. 13) and largely conserved between fetal and adult brain.
There is growing evidence that the majority of common variants associated with complex traits act through effects on gene regulation10,11. Our data add to a growing literature showing that DNA methylation is genetically influenced27, with mQTLs representing a potential mechanism linking genetic variation to complex phenotypes4,9,36,37. We find a significant enrichment of schizophrenia-associated GWAS variants in fetal brain mQTLs, indicating that common genetic variants conferring risk for schizophrenia are associated with altered DNA methylation in fetal human brain. The hypothesis that schizophrenia has an early neurodevelopmental component is supported by several lines of epidemiological and neuropathological evidence13,14. However, direct molecular evidence of schizophrenia risk factors operating in the fetal brain is scarce38,39. We have recently found that genomic loci that are differentially methylated between schizophrenia patients and unaffected controls in the adult brain are enriched at those undergoing dynamic changes in DNA methylation during human fetal brain development6,40. In the present study, we find that genetic variants exhibiting genome-wide significant association with schizophrenia12 show a four-fold enrichment amongst fetal brain mQTLs, directly implicating altered gene regulation during fetal brain development in the etiology of the disorder.
To conclude, we report the first systematic analysis of genetically-mediated DNA methylation in the developing human brain. Our data support the hypothesis that a significant proportion of the genetic variants conferring schizophrenia risk have regulatory effects that become manifest early in the prenatal period, and demonstrate the utility of mQTL mapping for localizing putative causal loci associated with complex disease phenotypes within large genomic regions. As a resource to the wider community, we have developed a searchable online database of fetal brain mQTLs that can be accessed at http://epigenetics.essex.ac.uk/mQTL/.
Online Methods
Human brain samples
Human fetal brain tissue was acquired from the Human Developmental Biology Resource (HDBR) (http://www.hdbr.org) and MRC Brain Banks network (http://www.mrc.ac.uk/research/facilities/brain-banks/access-for-research). Ethical approval for the HDBR was granted by the Royal Free Hospital research ethics committee under reference 08/H0712/34 and Human Tissue Authority (HTA) material storage licence 12220; ethical approval for MRC Brain Bank was granted under reference 08/MRE09/38. A detailed description of these samples can be found in Spiers et al (2015)6. Briefly 173 fetal brain samples (94 male, 79 female) ranging from 56 to 169 days post-conception were used for DNA methylation and SNP profiling. Brain tissue was obtained frozen and had not been dissected into regions. Half of the brain tissue from each individual fetus was homogenized for subsequent genomic DNA extraction. Postnatal prefrontal cortex (PFC), striatum (STR) and cerebellum (CER) samples were obtained from the MRC London Neurodegenerative Disease Brain Bank and the Douglas Bell-Canada Brain Bank (DBCBB) (www.douglasbrainbank.ca) and included both schizophrenia and controls. Postmortem brain specimens were collected postmortem following consent obtained with next of kin, dissected by neuropathology technicians, snap-frozen and stored at −80°C. Genomic DNA was isolated from all brain samples using a standard phenol-chloroform extraction protocol. DNA was tested for degradation and purity using spectrophotometry and gel electrophoresis.
Genome-wide quantification of DNA methylation
500ng of DNA from each sample was treated with sodium bisulfite in duplicate, using the EZ-96 DNA Methylation kit (Zymo Research, CA, USA). DNA methylation was quantified using the Illumina Infinium HumanMethylation450 BeadChip (Illumina Inc, CA, USA) run on an Illumina iScan System (Illumina, CA, USA) using the manufacturers’ standard protocol. Signal intensities for each probe were extracted using Illumina GenomeStudio software (Illumina, CA, USA) and imported into the R statistical programme using the methylumi and minfi packages41,42. Multidimensional scaling (MDS) plots of variable probes on the sex chromosomes were used to check that the predicted gender corresponded with the reported gender for each individual. Further data quality control and processing steps were conducted using the wateRmelon package43 in R. The pfilter function was used to filter firstly samples with >1% probes with a detection P value > 0.05 were removed and probes with a detection P value > 0.05 in at least 1% samples or/and a beadcount < 3 in 5% of samples were removed across all samples to control for poor quality probes. The dasen function was used to normalize the data as previously described43. Cross-hybridizing probes44,45, probes with any SNP within 10bp of the CpG site or single base extension44 and probes on the sex chromosomes were excluded from the QTL analysis. These data are publically available through GEO and can be found under accession numbers: GSE58885, GSE61431, GSE61380. Genotype data is available to access from dbSNP. All fetal brain mQTL data are also available via an online database at http://epigenetics.essex.ac.uk/mQTL/.
Genome-wide SNP genotyping
200ng of genomic DNA from each sample was genotyped using the Illumina HumanOmniExpress BeadChip (Illumina Inc, CA, USA). Following scanning, Illumina GenomeStudio software was used for genotype calling and the data were exported as ped and map files. PLINK31 was used to remove samples with > 5% missing values, and SNPs with > 1% missing values, Hardy-Weinburg equilibrium P < 0.001, and a minor allele frequency of < 5%. Subsequently, SNPs were also filtered so that each of the three genotype groups with 0, 1, or 2 minor alleles (or two genotype groups in the case of rare SNPs with 0 or 1 minor allele) had a minimum of 5 observations.
Methylation QTL (mQTL) analyses
Before commencing QTL analyses, genotypes at the polymorphic SNP probes on the HumanMethylation 450K array were compared to calls from the HumanOmniExpress genotyping array to confirm sample identity. All genome-wide SNP-methylation probe pairs were tested using the R package MatrixEQTL46. This package enables fast computation of QTLs by only saving those more significant than a pre-defined threshold (set to P = 0.0001 for these analyses). An additive linear model was fitted to test if the number of alleles (coded 0,1,2) predicted DNA methylation (beta value 0–100) at each site, including covariates for age, sex and the first two principal components from the genotype data to control for ethnicity differences. In addition, a brain bank covariate was also included for the adult datasets.
Identifying overlap and testing for enrichment of expression QTLs (eQTLs) among fetal brain mQTLs
P values for all cis eQTLs (within 1Mb) were supplied by the authors of a recent manuscript documenting eQTLs in the human brain30 to enable a more thorough examination of the overlap of eQTL and mQTL. To identify all variants associated with DNA methylation and gene expression in cis, our definition of cis mQTL was relaxed to match that used in the eQTL study. Chromosome and base position of the SNPs were used to map between the two datasets. A Bonferroni significance threshold was established for the eQTL results (P from aveALL analysis < 5.99×10−9), based on the number of cis eQTLs tested across all SNPs overlapping with those tested in the mQTL dataset (414,172), in addition to two more relaxed exploratory thresholds (P < 10−9; 10−7).
Prior to testing for a significant overlap with SNPs associated with brain eQTLs all SNPs associated with at least one DNA methylation site in the fetal brain were ‘clumped’ based on their best mQTL P-value using PLINK31 to create a list of quasi-independent SNPs (r2 < 0.25 for all pairs of SNPs within 250kb) and prevent LD between SNPs in the set biasing the results. Given the extensive correlation between variants in the major histocompatibility complex (MHC) region, a more stringent clumping procedure was used for SNPs located in chr6:25000000–35000000, where the window for pairwise SNP comparisons was extended to 10000kb. To test for a larger overlap than expected by chance, up to 1 million simulated sets, matched for allele frequency, were drawn to calculate the expected overlap and generate empirical P-values. SNPs were categorised into MAF bins split at intervals of 2%, and SNPs from each bin were selected to match the distribution in the test set. Empirical significance for enrichment of eQTLs in mQTLs was ascertained by counting the number of simulations with at least as many SNPs overlapping the set of sentinelized subsignals from the aveALL analysis in Ramasamy et al (2014)30, as the true ‘clumped’ Bonferroni significant mQTL SNP set and dividing by the number of simulations performed. Fold change statistics were calculated as the true overlap divided by the mean overlap of these simulations, and 95% confidence intervals as the true overlap divided by the 2.5th and 97.5th quantiles of the distribution of overlaps.
Enrichment of regulatory regions
Published 450K array probe annotations23 were used to identify probes located in transcription factor binding sites (TFBSs) or DNase1 hypersensitivity sites (DHSs) based on data made publically available as part of the ENCODE project10,26. In addition, brain specific DHSs were downloaded from the UCSC (University of California, Santa Cruz) Genome Browser for ‘Frontal_cortex_OC’, ‘Cerebellum_OC’ and ‘Cerebrum_frontal_OC’ and used to annotate DNA methylation sites in the same manner. Peaks associated with 5 histone modifications identified separately in two fetal brain samples (17 weeks gestation; 1 male, 1 female; sample IDs E081 and E082) were downloaded from the Epigenomics Roadmap project24. Due to the heterogeneity in the Chip-seq profiles, presumed due to experimental differences rather than biological differences47, DNA methylation sites had to be located within peaks generated from both brain samples to be classed as overlapping any of the histone marks. The overlap between regulatory features and the DNA methylation sites identified from the set of Bonferroni significant mQTLs in the fetal brain dataset was tested for enrichment using a two sided Fisher’s 2×2 exact test. The significance level for enrichment of overlap with transcription factor binding sites was calculated using a Bonferroni correction for the 149 different transcription factor binding sites tested.
Heterogeneity model
All Bonferroni-significant mQTLs (P < 3.69×10−13) identified in the fetal brain, for which corresponding mQTL data was available from all three adult brain regions, were tested for heterogeneous relationships between DNA methylation and genetic variation across the datasets (n = 10,663). A null model of no heterogeneity was fitted in line with the linear model fitted to test for mQTL effects between the number of alleles (coded 0,1,2) and DNA methylation (beta value 0–100) with fixed effect covariates for sex, age and the first two genetic principal components. As the adult brain regions were dissected from the same set of individuals, we expect their DNA methylation values to be correlated. In addition, we expect DNA methylation values within a brain region to be correlated, and therefore both of these covariates were included as random effects in addition to an indicator variable discriminating fetal from adult samples to control for absolute differences in DNA methylation level associated with age/development stage. This was compared to a heterogeneity model which included an interaction between genotype and development stage indicator with an ANOVA to calculate the heterogeneity P-value.
Enrichment of disease-associated variants among fetal brain mQTLs
A similar simulation procedure to that used to test the overlap of mQTLs and eQTLs was used to test for a larger overlap between fetal brain mQTL SNPs and those identified in GWAS of complex disorders including: schizophrenia12, Alzheimer’s disease32, body mass index (BMI)33, and type 2 diabetes34. The clumping procedure as described for the eQTL enrichment analysis was repeated separately for each phenotype to ensure that the best mQTL SNP present in those analysed in the GWAS was retained. Up to 1 million simulations were performed to generate the expected overlap between the set of mQTL SNPs and variants associated with each disorder at four GWAS significance thresholds (P < 5×10−5, 5×10−6, 5×10−7, 5×10−8) and derive fold change statistics and empirical P values.
Imputation
Prior to imputation PLINK31 was used to remove samples with >5% missing data. We also excluded SNPs characterized by >1% missing values, a Hardy-Weinberg equilibrium P < 0.001 and a minor allele frequency of <5%. These were recoded as vcf files using PLINK1.948 and VCFtools49 before uploading to the Michigan Imputation Server (https://imputationserver.sph.umich.edu/start.html#!pages/home) which uses SHAPEIT50,51 to phase haplotypes, and Minimac352 with the most recent 1000 Genomes reference panel (phase 3, version 5). Imputed genotypes were then filtered and recoded with PLINK1.948 removing samples with >5% missing values, and SNPs with >2 alleles, those indicated as a fail in the FILTER columns using the flag ‘--vcf-filter’, in addition to those characterized by >1% missing values, a Hardy-Weinberg equilibrium P < 0.001, a minor allele frequency of < 5%, or < 5 observations for any genotype group in line with the SNP filtering for the raw genotype groups. This resulted in 5,177,320 variants in the imputed set of genotypes. MatrixEQTL46 was used to test genome-wide mQTLs as previously described, except only mQTL with P < 1×10−8 were recorded.
Colocalization analyses
Schizophrenia associated genomic loci were taken as the 105 autosomal regions published as part of the PGC mega-analysis12. Given our definition of cis mQTLs (i.e. associations between SNPs and DNA methylation probes within 500kb), all DNA methylation sites located within 500kb of these regions were identified and cis mQTL analysis was repeated using the imputed genotypes using MatrixEQTL46 and recording all mQTL results. Colocalization analysis was performed as previously described35 using the R coloc package (http://cran.r-project.org/web/packages/coloc) for each DNA methylation site within each region. In total 19,607 possible mQTLs were tested. From both the PGC schizophrenia GWAS data and our mQTL results we inputted the regression coefficients, their variances and SNP minor allele frequencies, and the prior probabilities were left as their default values. This methodology quantifies the support across the results of each GWAS for 5 hypotheses by calculating the posterior probabilities, denoted as PPi for hypothesis Hi.
H0: there exist no causal variants for either trait;
H1: there exists a causal variant for one trait only, schizophrenia;
H2: there exists a causal variant for one trait only, DNA methylation;
H3: there exist two distinct causal variants, one for each trait;
H4: there exists a single causal variant common to both traits.
Code Availability
Annotated analysis scripts for the analyses used in this study are available for download at http://epigenetics.essex.ac.uk/mQTL/.
Supplementary Material
Acknowledgments
This work was supported by grants from the UK Medical Research Council (MRC) grant numbers MR/K013807/1 (to J.M.) and MR/L010674/1 (to N.J.B.), and US National Institutes of Health (grant number AG036039) to J.M. R.P. and H.S. were funded by MRC PhD studentships. The human embryonic and fetal material was provided by the Joint MRC (grant number G0700089)/Wellcome Trust (grant number GR082557) Human Developmental Biology Resource. We thank Dr Mike Weale for providing eQTL data from the BRAINEAC database.
Footnotes
Author contributions
J.M. and N.J.B. conceived and supervised the study, and obtained funding. E.H. undertook primary data analysis and bioinformatics. L.S. provided analytical support. H.S., J.V., R.P., T.M. and J.B. performed laboratory work. C.T. and G.T. provided samples for analysis. M.O’D. provided support fot GWAS enrichment analyses. E.H., N.J.B. and J.M. drafted the manuscript. All of the authors read and approved the final submission.
Competing financial interests
The authors declare no competing financial interests.
Contributor Information
Eilis Hannon, Email: e.j.hannon@exeter.ac.uk.
Helen Spiers, Email: helen.h.spiers@kcl.ac.uk.
Joana Viana, Email: j.viana@exeter.ac.uk.
Ruth Pidsley, Email: r.pidsley@garvan.org.au.
Joe Burrage, Email: J.Burrage@exeter.ac.uk.
Therese M Murphy, Email: T.Murphy@exeter.ac.uk.
Claire Troakes, Email: Claire.troakes@kcl.ac.uk.
Gustavo Turecki, Email: gustavo.turecki@mcgill.ca.
Michael C. O’Donovan, Email: ODonovanMC@cardiff.ac.uk.
Leonard C. Schalkwyk, Email: lschal@essex.ac.uk.
Nicholas J. Bray, Email: BrayN3@cardiff.ac.uk.
Jonathan Mill, Email: j.mill@exeter.ac.uk.
References for main text
- 1.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–9. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 3.Ziller MJ, et al. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013;500:477–81. doi: 10.1038/nature12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner JR, et al. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–7. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spiers H, et al. Methylomic trajectories across human fetal brain development. Genome Res. 2015;25:338–52. doi: 10.1101/gr.180273.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gibbs JR, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gamazon ER, et al. Enrichment of cis-regulatory gene expression SNPs and methylation quantitative trait loci among bipolar disorder susceptibility variants. Mol Psychiatry. 2013;18:340–6. doi: 10.1038/mp.2011.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gutierrez-Arcelus M, et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife. 2013;2:e00523. doi: 10.7554/eLife.00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurano MT, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–5. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicolae DL, et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schizophrenia Working Group of the PGC et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421-+. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fatemi SH, Folsom TD. The neurodevelopmental hypothesis of schizophrenia, revisited. Schizophr Bull. 2009;35:528–48. doi: 10.1093/schbul/sbn187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberger DR. From neuropathology to neurodevelopment. Lancet. 1995;346:552–7. doi: 10.1016/s0140-6736(95)91386-6. [DOI] [PubMed] [Google Scholar]
- 15.Teh AL, et al. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res. 2014;24:1064–74. doi: 10.1101/gr.171439.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olsson AH, et al. Genome-wide associations between genetic and epigenetic variation influence mRNA expression and insulin secretion in human pancreatic islets. PLoS Genet. 2014;10:e1004735. doi: 10.1371/journal.pgen.1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drong AW, et al. The presence of methylation quantitative trait loci indicates a direct genetic influence on the level of DNA methylation in adipose tissue. PLoS One. 2013;8:e55923. doi: 10.1371/journal.pone.0055923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutierrez-Arcelus M, et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015;11:e1004958. doi: 10.1371/journal.pgen.1004958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemire M, et al. Long-range epigenetic regulation is conferred by genetic variation located at thousands of independent loci. Nat Commun. 2015;6:6326. doi: 10.1038/ncomms7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–45. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 22.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slieker RC, et al. Identification and systematic annotation of tissue-specific differentially methylated regions using the Illumina 450k array. Epigenetics Chromatin. 2013;6:26. doi: 10.1186/1756-8935-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roadmap Epigenomics Consortium et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliott G, et al. Intermediate DNA methylation is a conserved signature of genome regulation. Nat Commun. 2015;6:6363. doi: 10.1038/ncomms7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McRae AF, et al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 2014;15:R73. doi: 10.1186/gb-2014-15-5-r73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H, et al. Widespread plasticity in CTCF occupancy linked to DNA methylation. Genome Res. 2012;22:1680–8. doi: 10.1101/gr.136101.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nat Rev Genet. 2014;15:234–46. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramasamy A, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17:1418–28. doi: 10.1038/nn.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lambert JC, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Locke AE, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. doi: 10.1038/nature14177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Morris AP, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–90. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giambartolomei C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383. doi: 10.1371/journal.pgen.1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meaburn EL, Schalkwyk LC, Mill J. Allele-specific methylation in the human genome: implications for genetic studies of complex disease. Epigenetics. 2010;5:578–82. doi: 10.4161/epi.5.7.12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Eijk KR, et al. Identification of schizophrenia-associated loci by combining DNA methylation and gene expression data from whole blood. Eur J Hum Genet. 2014 doi: 10.1038/ejhg.2014.245. [DOI] [PMC free article] [PubMed]
- 38.Hill MJ, Bray NJ. Evidence that schizophrenia risk variation in the ZNF804A gene exerts its effects during fetal brain development. Am J Psychiatry. 2012;169:1301–8. doi: 10.1176/appi.ajp.2012.11121845. [DOI] [PubMed] [Google Scholar]
- 39.Tao R, et al. Expression of ZNF804A in human brain and alterations in schizophrenia, bipolar disorder, and major depressive disorder: a novel transcript fetally regulated by the psychosis risk variant rs1344706. JAMA Psychiatry. 2014;71:1112–20. doi: 10.1001/jamapsychiatry.2014.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pidsley R, et al. Methylomic profiling of human brain tissue supports a neurodevelopmental origin for schizophrenia. Genome Biol. 2014;15:483. doi: 10.1186/s13059-014-0483-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis S, Du P, Bilke S, Triche J, Bootwalla M. methylumi: Handle Illumina methylation data. R package version 2.14.0. 2015 [Google Scholar]
- 42.Aryee MJ, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30:1363–9. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pidsley R, et al. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen YA, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price ME, et al. Additional annotation enhances potential for biologically-relevant analysis of the Illumina Infinium HumanMethylation450 BeadChip array. Epigenetics Chromatin. 2013;6:4. doi: 10.1186/1756-8935-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012;28:1353–8. doi: 10.1093/bioinformatics/bts163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ernst J, Kellis M. Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues. Nat Biotechnol. 2015;33:364–76. doi: 10.1038/nbt.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danecek P, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2012;9:179–81. doi: 10.1038/nmeth.1785. [DOI] [PubMed] [Google Scholar]
- 51.Delaneau O, Zagury JF, Marchini J. Improved whole-chromosome phasing for disease and population genetic studies. Nat Methods. 2013;10:5–6. doi: 10.1038/nmeth.2307. [DOI] [PubMed] [Google Scholar]
- 52.Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet. 2012;44:955–9. doi: 10.1038/ng.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.