

Abstract
Ralstonia solanacearum is the causative agent of bacterial wilt of potato. Ralstonia solanacearum strain UY031 belongs to the American phylotype IIB, sequevar 1, also classified as race 3 biovar 2. Here we report the completely sequenced genome of this strain, the first complete genome for phylotype IIB, sequevar 1, and the fourth for the R. solanacearum species complex. In addition to standard genome annotation, we have carried out a curated annotation of type III effector genes, an important pathogenicity-related class of genes for this organism. We identified 60 effector genes, and observed that this effector repertoire is distinct when compared to those from other phylotype IIB strains. Eleven of the effectors appear to be nonfunctional due to disruptive mutations. We also report a methylome analysis of this genome, the first for a R. solanacearum strain. This analysis helped us note the presence of a toxin gene within a region of probable phage origin, raising the hypothesis that this gene may play a role in this strain’s virulence.
Keywords: Short genome report, Bacterial wilt, Ralstonia solanacearum, Bacterial plant pathogen, Methylome, Uruguay
Introduction
Ralstonia solanacearum is the causal agent of bacterial wilt, one of the most devastating plant diseases worldwide [1]. It is a highly diversified bacterial plant pathogen in terms of host range, geographical distribution, pathogenicity, epidemiological relationships, and physiological properties [2]. Strains are divided in four phylotypes, corresponding roughly to their geographic origin: Asia (phylotype I), the Americas (II), Africa (III), and Indonesia (IV) [3]. Strain UY031 belongs to phylotype IIB, sequevar 1 (IIB1), the group considered mainly responsible for bacterial wilt of potato in cold and temperate regions [4]. Phylotype IIB, sequevar 1 is also traditionally classified as race 3 biovar 2.
Strain UY031 was isolated in Uruguay from infected potato tubers in 2003 and displays high aggressiveness both on potato and tomato hosts [5]. This strain is being used as a model in plant-pathogen gene expression studies carried out by our group; having its genome available greatly facilitates the identification of pathogenicity-related genes. Four other IIB1 R. solanacearum strains have been partially sequenced: UW551 [6], IPO1609 [7], NCPPB909 [8], and CFIA906 [8]. This is the first genome of this group to be completely sequenced, and the fourth within the R. solanacearum species complex (the other three are strains GMI1000 [9], Po82 [10] , and PSI07 [11]).
Organism information
Classification and features
Ralstonia solanacearum UY031 strain is classified within the order Burkholderiales of the class Betaproteobacteria. It is an aerobic, non-sporulating, Gram-negative bacterium with rod-shaped cells ranging from 0.5 to 1.5 μm in length (Fig. 1, (a) and (b)). The strain is moderately fast-growing, forming 3–4 mm colonies within 2–3 days at 28 °C. On a general nutrient medium containing tetrazolium chloride and high glucose content, strain UY031 usually produces a diffusible brown pigment and develops pearly cream-white, flat, irregular, and fluidal colonies with characteristic pink whorls in the centre (Fig. 1, (c)). Strain UY031 was isolated from a naturally infected potato tuber showing typical brown rot symptoms (creamy exudates from the vascular rings and eyes of the tuber). This strain is highly pathogenic in different solanaceous hosts including important crops like tomato and potato [5]. Pathogenicity of this strain was also confirmed in several accessions of Solanum commersonii Dunal, a wild species considered as a valuable source of resistance for potato breeding. Due to its great aggressiveness, strain UY031 is being used for selection of resistant germplasm as part of the potato breeding program developed in Uruguay. This strain has been deposited in the CFBP collection of plant-associated bacteria, and has received code CFBP 8401. Minimum Information about the Genome Sequence of R. solanacearum strain UY031 is summarized in Table 1, and a phylogenetic tree is shown in Fig. 2.
Fig. 1.
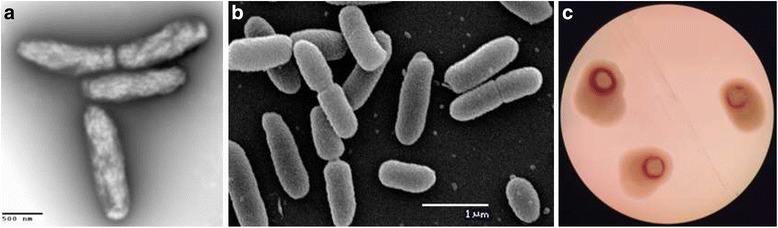
Images of Ralstonia solanacearum strain UY031 using transmission (a) and scanning (b) electron microscopy, as well as light microscopy to visualize colony morphology on solid media (c)
Table 1.
Classification and general features of Ralstonia solanacearum strain UY031according to the MIGS recommendations [27]
| MIGS ID | Property | Term | Evidence codea |
|---|---|---|---|
| Classification | Domain Bacteria | TAS [28] | |
| Phylum Proteobacteria | TAS [29] | ||
| Class Betaproteobacteria | TAS [30, 31] | ||
| Order Burkholderiales | TAS [31, 32] | ||
| Family Burkholderiaceae | TAS [31, 33] | ||
| Genus Ralstonia | TAS [34, 35] | ||
| Species Ralstonia solanacearum | TAS [34, 35] | ||
| Strain UY031 | |||
| Gram stain | Negative | IDA | |
| Cell shape | Rod | IDA | |
| Motility | Motile | IDA | |
| Sporulation | Non sporulating | NAS | |
| Temperature range | Mesophile | IDA | |
| Optimum temperature | 27 °C | IDA | |
| pH range; Optimum | 5.5 – 8.0; 6.5 | NAS | |
| Carbon source | Dextrose, lactose, maltose, cellobiose | IDA | |
| MIGS-6 | Habitat | potato plants, soil | TAS [5] |
| MIGS-6.3 | Salinity | <2.0 % | TAS [36] |
| MIGS-22 | Oxygen requirement | Aerobic | IDA |
| MIGS-15 | Biotic relationship | free-living | IDA |
| MIGS-14 | Pathogenicity | Pathogenic | TAS [5] |
| MIGS-4 | Geographic location | Uruguay, San José | TAS [5] |
| MIGS-5 | Sample collection | 2003 | TAS [5] |
| MIGS-4.1 | Latitude | 34°43′58.17”S | NAS |
| MIGS-4.2 | Longitude | 56°32′2.87”W | NAS |
| MIGS-4.4 | Altitude | 116.7 m | NAS |
aEvidence codes - IDA Inferred from direct assay, TAS Traceable author statement (i.e., a direct report exists in the literature), NAS Non-traceable author statement (i.e., not directly observed for the living, isolated sample, but based on a generally accepted property for the species, or anecdotal evidence). These evidence codes are from the Gene Ontology project [37]
Fig. 2.
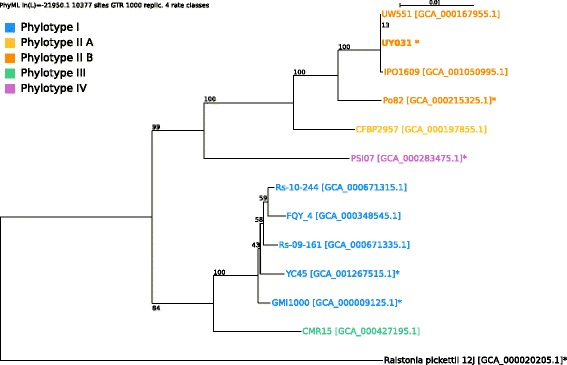
Phylogenetic tree highlighting the position of the Ralstonia solanacearum UY031 (shown in bold) relative to other strains from the same species. The phylogenetic tree was constructed using four conserved prokaryotic marker genes, namely: recA, rpoA, rpoB and rpoC. Each gene was aligned individually with MUSCLE [25]; the resulting multiple alignments were concatenated. PhyML [26] was used to perform tree reconstruction using the GTR model and 1,000 bootstrap replicas. Strain names are colour-coded according to the correspondent phylotype. GenBank accession numbers are displayed within brackets. Strains whose genome was completely sequenced are marked with an asterisk. Ralstonia pickettii 12 J (NCBI accession NC_010682) was used as an outgroup
Genome sequencing information
Genome project history
This sequencing project was carried out in 2015; the result is a complete and finished genome. Project data is available from GenBank (Table 2). Accession codes for reads in the Sequence Read Archive are SRP064191, SRR2518086, and SRZ132405.
Table 3.
Summary of genome: one chromosome and one plasmid
| Label | Size (Mb) | Topology | INSDC identifier | RefSeq ID |
|---|---|---|---|---|
| Chromosome | 3.41 | circular | NA | NA |
| Megaplasmid | 1.99 | circular | NA | NA |
Table 2.
Project information
| MIGS ID | Property | Term |
|---|---|---|
| MIGS 31 | Finishing quality | Finished |
| MIGS-28 | Libraries used | SMRT library (P5-C3 large insert library) |
| MIGS 29 | Sequencing platforms | PacBio RS II |
| MIGS 31.2 | Fold coverage | 138× |
| MIGS 30 | Assemblers | HGAP.2 workflow |
| MIGS 32 | Gene calling method | Prokka v1.10 (ncRNAs search enabled) |
| Locus tag | RSUY | |
| Genbank ID | CP012687 (chr), CP012688 (pl) | |
| GenBank date of release | September 28, 2015 | |
| GOLD ID | NA | |
| BIOPROJECT | PRJNA278086 | |
| MIGS 13 | Source material identifier | SAMN03402637 |
| Project relevance | Plant pathogen |
Growth conditions and genomic DNA preparation
R. solanacearum strain UY031 was routinely grown in rich B medium (10 g/l bactopeptone, 1 g/l yeast extract and 1 g/l casaminoacids). Genomic DNA was extracted from a bacterial culture grown to stationary phase to avoid over-representation of genomic sequences close to the origin of replication. Twelve ml of a culture grown for 16 h at 30 °C and shaking at 200 rpm (OD600 = 0.87) were used to extract DNA with Blood & Cell Culture DNA Midi kit (Qiagen), following manufacturer’s instructions for gram-negative bacteria. DNA concentration and quality were measured in a Nanodrop (ND-8000 8-sample spectrophotometer).
Genome sequencing and assembly
Whole-genome sequencing was performed on the PacBio RS II platform at the Duke Center for Genomic and Computational Biology (USA). P5-C3 chemistry and a single SMRTcell were used, and quality control was performed with DUGSIM. The number of Pre-Filter Polymerase Read Bases was greater than 749 million (>130x genome coverage). Reads were assembled using RS_HGAP_Assembly.2 protocol from SMRT Analysis 2.3 [12]. This resulted in one circular chromosome (3,412,138 bp) and one circular megaplasmid (1,999,545 bp). These lengths are very similar to those of the corresponding replicons in R. solanacearum Po82, a IIB sequevar 4 strain, also a potato pathogen and which has also been completely sequenced [10]. The origin of replication was defined for both replicons based on the putative origin for reference strain GMI1000 [9].
An assembly quality assessment was performed before all downstream analyses. All reads were mapped back to the assembled sequences using RS_Resequencing.1 protocol from SMRT Analysis 2.3. This analysis revealed that chromosome and megaplasmid sequences had 100 % of bases called (percentage of assembled sequence with coverage > = 1) and 99.9999 % and 99.9992 %, respectively, of consensus concordance.
Genome annotation
Genome annotation was done using Prokka [13] with the option for ncRNA search. Type III effectors of strain UY031 were identified and annotated in three steps: First, 17 of the T3Es from the R. solanacearum species complex [14] were identified based on the Prokka annotations. Second, the 15 T3Es annotated as “Type III Effector Protein”, “Probable Type III Effector Protein” or “Putative Type III Effector Protein” by Prokka were manually annotated using the first BLAST [15] hits (usually 100 % identity) of their DNA sequences against genome sequences of phylotype IIB strains MOLK2 and Po82. Third, the UY031 genome was uploaded to the “Ralstonia T3E” web interface tool [14] to search for additional T3Es not annotated as such with Prokka. The additional 28 T3E genes identified were manually annotated as above. Homologous Gene Group clustering was performed with get_homologues [16] using the orthoMCL program [17] and requiring a minimum sequence identity in BLAST query/subject pairs of 30 %.
The sequencing plataform used to assemble the genome (PacBio RS II) also gives kinectics information about the sequenced genome. The presence of a methylated base in the DNA template delays the incorporation of the complementary nucleotide; such modifications in the kinectics may be used to characterize modified bases by methylation including: 6-mA, 5-mC and 4-mC [18]. The analysis of these modifications in a genome-wide and single-base-resolution scale allowed us to characterize the ‘methylome’ of this strain. These epigenetic marks are commonly used by bacteria, and its implications vary from a defense mechanism, protecting the cell from invading bacteriophages or other foreign DNA, to the bacterial virulence itself [19–21]. We performed methylome analysis and motif detection using RS_Modification_and_Motif_analysis.1 protocol from SMRT Analysis 2.3. Such epigenetic marks arise from DNA methyl-transferases, sometimes coupled with a restriction endonuclease (a Restriction-Modification System). We further characterized which genes give rise to the modified motifs using tools available at REBASE [22].
Genome properties
The genome of R. solanacearum strain UY031 has one chromosome (3,412,138 bp) and one circular megaplasmid (1,999,545 bp) (Table 3). The average GC content of the chromosome is 66.5 % while that of the megaplasmid is 66.7 %. A total of 4,778 genes (4,683 CDSs and 95 RNAs) were predicted. Of the protein-coding genes, 3,566 (76.1 %) had functions assigned while 1,212 were considered hypothetical (Table 4). Of all CDSs, 76.6 % could be assigned to one COG functional category and for 83.1 % one or more conserved PFAM-A domains were identified (Table 5).
Fig. 3.
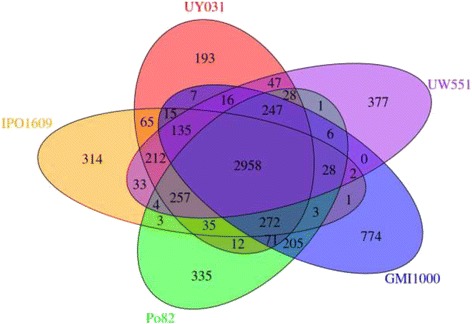
Venn diagram of the Ralstonia solanacearum homologous gene groups. The R. solanacearum genomes compared were as follows: strains Po82, GMI1000, IPO1609, UW551, and UY031
Table 4.
Genome statistics
| Attribute | Value | % of total |
|---|---|---|
| Genome size (bp) | 5,411,683 | 100.00 |
| DNA coding (bp) | 4,737,274 | 87.5 |
| DNA G + C (bp) | 3,604,487 | 66.6 |
| DNA scaffolds | 2 | 100.00 |
| Total genes | 4,778 | 100.00 |
| Protein coding genes | 4,683 | 98.0 |
| RNA genes | 95 | 1.9 |
| Pseudo genes | NA | NA |
| Genes in internal clusters | NA | NA |
| Genes with function prediction | 3,566 | 74.6 |
| Genes assigned to COGs | 3,586 | 76.6 |
| Genes with Pfam domains | 3,892 | 83.1 |
| Genes with signal peptides | 501 | 10.6 |
| Genes with transmembrane helices | 1132 | 24.1 |
| CRISPR repeats | 0 | - |
Table 5.
Number of genes associated with general COG functional categories
| Code | Value | % | Description |
|---|---|---|---|
| J | 160 | 3.4 | Translation, ribosomal structure and biogenesis |
| A | 2 | <0.1 | RNA processing and modification |
| K | 273 | 5.8 | Transcription |
| L | 240 | 5.1 | Replication, recombination and repair |
| B | 3 | <0.1 | Chromatin structure and dynamics |
| D | 28 | 0.6 | Cell cycle control, Cell division, chromosome partitioning |
| V | 45 | 1.0 | Defense mechanisms |
| T | 162 | 3.5 | Signal transduction mechanisms |
| M | 237 | 5.1 | Cell wall/membrane biogenesis |
| N | 119 | 2.5 | Cell motility |
| U | 61 | 1.3 | Intracellular trafficking and secretion |
| O | 154 | 3.3 | Posttranslational modification, protein turnover, chaperones |
| C | 226 | 4.8 | Energy production and conversion |
| G | 165 | 3.5 | Carbohydrate transport and metabolism |
| E | 342 | 7.3 | Amino acid transport and metabolism |
| F | 75 | 1.6 | Nucleotide transport and metabolism |
| H | 154 | 3.3 | Coenzyme transport and metabolism |
| I | 177 | 3.8 | Lipid transport and metabolism |
| P | 176 | 3.8 | Inorganic ion transport and metabolism |
| Q | 73 | 1.6 | Secondary metabolites biosynthesis, transport and catabolism |
| R | 352 | 7.5 | General function prediction only |
| S | 362 | 7.7 | Function unknown |
| - | 1097 | 23.4 | Not in COGs |
The total is based on the total number of protein coding genes in the genome
Insights from the genome sequence
We performed a pan-genome analysis of the R. solanacearum UY031 genome, comparing it to four other genomes: two closely-related R. solanacearum strains (UW551 and IPO1609) and two others with complete genome sequences available (GMI1000 and Po82). The pan-genome consists of 7,594 HGGs while the core genome consists of 2,958 HGGs; the variable genome consists of 2,643 HGGs, and the number of strain-specific HGGs ranges from 193 to 774 (Fig. 3). We identified 193 HGGs that are UY031-specific; 75.1 % of them were annotated as hypothetical proteins.
Type III effector genes are among the most important for virulence determinants in bacterial plant pathogens such as R. solanacearum [14]. Based on comparisons with effector gene sequences in public databases (see above) we have identified 60 T3Es (Table 6), of which 11 appear to be nonfunctional due to frameshifts or other mutations that disrupt the coding sequence. For example, the effector RipS5 is encoded by a gene that has been clearly interrupted by a 34 kbp prophage. Table 6 also shows the orthologs of these genes in the related strains GMI1000, Po82, IPO1609, and UW551. In the table it can be seen that the genes that code for RipAA and RipAR have frameshifts or truncations in strain UY031 only. The absence of a particular effector may be enough for a pathogen to avoid host defenses, and therefore cause disease. These two genes are therefore a good starting point for additional investigations of phenotypic differences between these strains. Other effector genes of interest are those that are present and do not have disrupting mutations in UY031 but are absent or appear to be nonfunctional in other strains. We have found several such cases (Table 6), but in all cases there is at least one other strain that also has the same gene in what appears to be a functional state.
Table 6.
List of T3E genes identified in R. solanacearum UY031 genome and their orthologs
| Former effector name | New effector namea | UY031 (RSUY_) |
GM1000 (RS) |
Po82 (RSPO_) |
IPO1609 (RSIPO_) |
UW551 (RRSL_) |
|---|---|---|---|---|---|---|
| AWR2 | RipA2 | 32720 | p0099 | m00080 | 03169 | 03418 |
| AWR3 | RipA3 | 40320 | p0846 | m01165 | 03901 + 05027b | - |
| AWR4 | RipA4 | 40330/40b | p0847 | m01166b | 03902/3b | - |
| AWR5 | RipA5_1 | 41860 | p1024 | m01289/90b | 04049 | 01071 |
| AWR5 | RipA5_2 | 19780 | - | c01821 | 01281 | 00546 |
| Rip2 | RipB | 30390 | c0245 | c03161 | 00263 | 02573 |
| Rip62 | RipC1 | 42590 | p1239 | m01371 | 04123 | 03371 |
| Rip34 | RipD | 33840 | p0304 | m01520 | 04484 | 00947 |
| Rip26 | RipE1 | 01190 | c3369 | c00070 | 03083 | 00852 |
| - | RipE2 | 35100 | - | c02513 | 04353 | 03923 |
| PopF1 | RipF1_1 | 45370 | p1555 | m01541 | 03403 | 04777 |
| PopF2 | RipF2 | 45510 | - | m01557 | 05028/9b | 04764 |
| Gala2 | RipG2 | 38790 | p0672 | m01007 | 04892 | 02264 |
| Gala3 | RipG3 | 32420 | p0028 | m00035 | 03202 | 00752 |
| Gala4 | RipG4 | 19910 | c1800 | c01835 | 01266/68b | 00532 |
| Gala5 | RipG5 | 19920 | c1801 | c01836 | 01264 | 00531 |
| Gala6 | RipG6 | 17940 | c1356 | c01999 | 01463 | 01561 |
| Gala7 | RipG7 | 17950 | c1357 | c01998 | 01462 | 01562 |
| HLK1 | RipH1 | 19380 | c1386 | c01846 | 01319 | 00426 |
| HLK2 | RipH2 | 35470 | p0215 | m00201/2c | 04317 | 03559 |
| HLK3 | RipH3 | 33320 | p0160 | m00157 | 03105 | 00041b |
| Rip1 | RipI | 00490 + 32050b | c0041 | c03319 | 00098b | 02976 + 02040b |
| Rip22 | RipJ | 24610b | c2132 | c02749 | - | - |
| Rip16 | RipM | 19180 | c1475 | c01871/2/3 | 01339 + 05024b | 00705 |
| Rip58 | RipN | 43290 | p1130 | m00869 | 04184 | 04736 |
| Rip35 | RipO1 | 34050 | p0323 | m01496 | 04463 | 00926 |
| Rip63 | RipQ | 44390b | p1277 | m00717 | 04287b | 02855b |
| PopS | RipR | 42640 | p1281 | m01376 | 04127 | 03375 |
| SKWP1 | RipS1 | 00860 | c3401 | c00036 | 00017 | 04182 |
| SKWP2 | RipS2 | 44630 | p1374 | m00690 | 04310 | - |
| SKWP3 | RipS3 | 41210 | p0930 | m01229 | 03993/4b | 00237b |
| SKWP5 | RipS5 | 10370 + 10840b | p0296 | c02546b | - | - |
| SKWP7 | RipS7 | 35110b | - | m00383 | 04352b | 03921 |
| Rip59 | RipU | 43920 | p1212 | m00805 | 04243 | 04660 |
| Rip12 | RipV1 | 17880 | c1349 | c02006 | 01470 | 01554 |
| - | RipV2 | 19160b | - | c01875/76b | 01341 | 00703 |
| PopW | RipW | 07010 | c2775 | c00735 | 02524 | 02682 |
| PopA | RipX | 40640 | p0877 | m01196 | 03933 | 02443 |
| Rip3 | RipY | 30260 | c0257 | c03153 | 00276 | 01439 |
| Rip57 | RipZ | 42040 | p1031 | m01312 | 04067 | 00271b |
| AvrA | RipAA | 26380b | c0608 | c02748 | 00659 | 01581 |
| PopB | RipAB | 40630 | p0876 | m01195 | 03932 | 02442 |
| PopC | RipAC | 40620 | p0875 | m01194 | 03931 | 02441 |
| Rip72 | ripAD | 45790 | p1601 | m01585 | 03364 | 02518 |
| Rip4 | RipAE | 29570 | c0321 | c03085 | 00343 | 01625 |
| Rip41 | RipAI | 40230 | p0838 | m01156 | 03894 | 01021 |
| Rip21 | RipAJ | 13300 | c2101 | c01332 | 04893 | 01260 |
| Rip38 | RipAL | 39210b | - | m01053 | - | 02221 |
| Brg40 | RipAM | 02270 | c3272 | c00191 | 02968 | 02810 |
| Rip43 | RipAN | 40310 | p0845 | m01164 | 03900 | 01013 |
| Rip50 | RipAO | 40750 | p0879 | m01206 | 03944 | 03105 |
| Rip60 | RipAP | 43960 | p1215b | m00800 | 04247 | 04655 |
| Rip51 | RipAQ | 40810 | p0885 | - | 03951 | 03113 |
| Rip61 | RipAR | 44220b | p1236 | m00770 | 04270 | 01136 |
| Rip39 | RipAV | 39280 | p0732 | m01061 | - | 02213 |
| Brg13 | RipAX1 | 02040 | c3290 | m01221 | 02991 | - |
| Rip55 | RipAY | 41810 | p1022 | m01283 | 04046 | 01066 |
| - | RipBH | 45880 | - | m01600 | 03355 | 00782 |
| - | RipBI | 45200b | - | m00718 | 03419 | 00326 |
| - | RipTPS | 39290 | p0731 | m01062b | - | 02212 |
aAccording to Peeters et al. [14]; b: these genes appear to be nonfunctional due to various reasons (frameshift, truncation, etc.); genes in other columns that appear in the form locus tag x + locus tag y are genes which also appear to be nonfunctional due to frameshifts. c:this gene is duplicated
Our modification analysis revealed two motifs that are essentially always methylated, namely: CAACRAC and GTWWAC. Both are fairly frequent in the genome, occurring respectively 2144 and 716 times. Motif CAACRAC is associated with the product of gene RSUY_11320 (R. Roberts, personal communication), which is hypothesized to be an enzyme of the Restriction-Modification System, with a restriction nuclease and a DNA methyltransferase role. This gene does not have homologs in other R. solanacearum strains and is located close to a region containing phage-related genes. This region contains gene RSUY_11410, which has been annotated as encoding a zonular occludens toxin. The provenance of this annotation is an enterotoxin gene found in Vibriocholera [23]; in R. solanacearum the role of this toxin gene is still unclear [24]. Motif GTWWAC is probably associated with the product of gene RSUY_22890 (R. Roberts, personal communication), which is hypothesized to be a solitary DNA methyltransferase (no restriction endonuclease linked). This gene does have homologs in other R. solanacearum strains (GMI1000, IPO1609, Po82 and PSI07). To our knowledge this is the first R. solanacearum genome with a methylome profile available.
Conclusions
The complete sequence of R. solanacearum UY031 strain presented here should provide a rich platform upon which additional plant-pathogen studies can be carried out. Even though this is the fifth phylotype IIB1 sequenced, we found many differences with respect to the genomes of the other strains. In particular, the repertoire of T3E genes has many variations among these strains, and this may help explain some of the most relevant pathogenicity-related phenotypes described in the literature, opening the way to new control methods for bacterial wilt.
Acknowledgements
We thank Carlos Balsalobre and Cristina Madrid for their helpful advice and for kindly providing materials and protocols; and Carlos Morais for help with NCBI submission. We also thank COST action Sustain from the European Union for funding and Nemo Peeters and Stéphane Genin for hosting MP for a short stay to carry out UY031 effector annotation. RGS has a Ph.D. fellowship from FAPESP, Brazil. JCS has an investigator fellowship from the Conselho Nacional de Desenvolvimento Cientifico e Tecnologico, Brazil.
Abbreviations
- IIB1
Phylotype IIB, sequevar 1
- T3E
Type III effectors
- HGG
Homologous gene groups
Footnotes
Competing interests
The authors declare that they have followed all local, national and international guidelines and legislation and obtained the required permissions and/or licenses for this study.
The authors declare that they do not have any financial and non-financial competing interests.
Authors’ contributions
Conceived the project: MV, JCS, RGS. Provided strains and metadata: MIS, MJP. Assembled and annotated the genome: RGS. Performed effector gene annotation: MP, NSC. Analyzed and interpreted results: JCS, MV, MP, NSC, RGS, MIS, MJP. Wrote the manuscript: JCS, MV, MP, RGS, MIS, MJP. All authors read and approved the final manuscript.
References
- 1.Mansfield J, Genin S, Magori S, Citovsky V, Sriariyanum M, Ronald P, et al. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol Plant Pathol. 2012;13(6):614–29. doi: 10.1111/j.1364-3703.2012.00804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Genin S, Denny TP. Pathogenomics of the Ralstonia solanacearum species complex. Annu Rev Phytopathol. 2012;50:67–89. doi: 10.1146/annurev-phyto-081211-173000. [DOI] [PubMed] [Google Scholar]
- 3.Fegan M, Prior P. How complex is the ‘Ralstonia solanacearum’ species complex? In: Allen CP, editor. Bacterial wilt: The disease and the ralstonia solanacearum species complex. Prior, Hayward AC. St. Paul, MN: American Phytopathological Society; 2005. pp. 449–61. [Google Scholar]
- 4.Janse JD, van den Beld HE, Elphinstone J, Simpkins S, Tjou-Tam-Sin NNA, van Vaerenbergh J. Introduction to Europe of Ralstonia solanacearum biovar 2, race 3 in Pelargonium zonale cuttings. J Plant Pathol. 2004;86(2):147–55. [Google Scholar]
- 5.Siri MI, Sanabria A, Pianzolla MJ. Genetic diversity and aggressiveness of Ralstonia solanacearum strains causing bacterial wilt of potato in Uruguay. Plant Dis. 2011;95(10):1292–301. doi: 10.1094/PDIS-09-10-0626. [DOI] [PubMed] [Google Scholar]
- 6.Gabriel DW, Allen C, Schell M, Denny TP, Greenberg JT, Duan YP, et al. Identification of open reading frames unique to a select agent: Ralstonia solanacearum race 3 biovar 2. MPMI. 2006;19(1):69–79. doi: 10.1094/MPMI-19-0069. [DOI] [PubMed] [Google Scholar]
- 7.Guidot A, Elbaz M, Carrere S, Siri MI, Pianzzola MJ, Prior P, et al. Specific genes from the potato brown rot strains of Ralstonia solanacearum and their potential use for strain detection. Phytopathology. 2009;99(9):1105–12. doi: 10.1094/PHYTO-99-9-1105. [DOI] [PubMed] [Google Scholar]
- 8.Yuan KX, Cullis J, Levesque CA, Tambong J, Chen W, Lewis CT, et al. Draft genome sequences of Ralstonia solanacearum race 3 biovar 2 strains with different temperature adaptations. Genome Announc. 2015;3(4). [DOI] [PMC free article] [PubMed]
- 9.Salanoubat M, Genin S, Artiguenave F, Gouzy J, Mangenot S, Arlat M, et al. Genome sequence of the plant pathogen Ralstonia solanacearum. Nature. 2002;415(6871):497–502. doi: 10.1038/415497a. [DOI] [PubMed] [Google Scholar]
- 10.Xu J, Zheng HJ, Liu L, Pan ZC, Prior P, Tang B, et al. Complete genome sequence of the plant pathogen Ralstonia solanacearum strain Po82. J Bacteriol. 2011;193(16):4261–2. doi: 10.1128/JB.05384-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C, et al. Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genomics. 2010;11:379. doi: 10.1186/1471-2164-11-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods. 2013;10(6):563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 13.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 14.Peeters N, Carrere S, Anisimova M, Plener L, Cazale AC, Genin S. Repertoire, unified nomenclature and evolution of the type III effector gene set in the Ralstonia solanacearum species complex. BMC Genomics. 2013;14:859. doi: 10.1186/1471-2164-14-859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79(24):7696–701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–5. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanchez-Romero MA, Cota I, Casadesus J. DNA methylation in bacteria: from the methyl group to the methylome. Curr Opin Microbiol. 2015;25:9–16. doi: 10.1016/j.mib.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 20.Garcia-Del Portillo F, Pucciarelli MG, Casadesus J. DNA adenine methylase mutants of Salmonella typhimurium show defects in protein secretion, cell invasion, and M cell cytotoxicity. Proc Natl Acad Sci U S A. 1999;96(20):11578–83. doi: 10.1073/pnas.96.20.11578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heithoff DM, Sinsheimer RL, Low DA, Mahan MJ. An essential role for DNA adenine methylation in bacterial virulence. Science. 1999;284(5416):967–70. doi: 10.1126/science.284.5416.967. [DOI] [PubMed] [Google Scholar]
- 22.Roberts RJ, Vincze T, Posfai J, Macelis D. REBASE--a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 2015;43(Database issue):D298–9. doi: 10.1093/nar/gku1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Di Pierro M, Lu R, Uzzau S, Wang W, Margaretten K, Pazzani C, et al. Zonula occludens toxin structure-function analysis. Identification of the fragment biologically active on tight junctions and of the zonulin receptor binding domain. J Biol Chem. 2001;276(22):19160–5. doi: 10.1074/jbc.M009674200. [DOI] [PubMed] [Google Scholar]
- 24.Murugaiyan S, Bae JY, Wu J, Lee SD, Um HY, Choi HK, et al. Characterization of filamentous bacteriophage PE226 infecting Ralstonia solanacearum strains. J Appl Microbiol. 2011;110(1):296–303. doi: 10.1111/j.1365-2672.2010.04882.x. [DOI] [PubMed] [Google Scholar]
- 25.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 27.Field D, Garrity G, Gray T, Morrison N, Selengut J, Sterk P, et al. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26(5):541–7. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci U S A. 1990;87(12):4576–9. doi: 10.1073/pnas.87.12.4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Garrity GM, Bell JA, Lilburn T. Phylum XIV. Proteobacteria phyl. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. Second. New York: Springer; 2005. p. Part B:1. [Google Scholar]
- 30.Garrity GM, Bell JA, Lilburn T. Class II. Betaproteobacteria class. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. Second. New York: Springer; 2005. p. 575. [Google Scholar]
- 31.List Editor. Validation List Number 107. List of new names and new combinations previously effectively, but not validly, published. Int J Syst Evol Microbiol. 2006,56:1–6 [DOI] [PubMed]
- 32.Garrity GM, Bell JA, Lilburn T. Order I. Burkholderiales ord. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. Second. New York: Springer; 2005. p. 575. [Google Scholar]
- 33.Garrity GM, Bell JA, Lilburn T. Family I. Burkholderiaceae fam. nov. In: Garrity GM, Brenner DJ, Krieg NR, Staley JT, editors. Bergey’s manual of systematic bacteriology. Second. New York: Springer; 2005. p. 575. [Google Scholar]
- 34.Yabuuchi E, Kosako Y, Yano I, Hotta H, Nishiuchi Y. Transfer of two Burkholderia and an Alcaligenes species to Ralstonia gen. Nov.: proposal of Ralstonia pickettii (Ralston, Palleroni and Doudoroff 1973) comb. nov., Ralstonia solanacearum (Smith 1896) comb. nov. and Ralstonia eutropha (Davis 1969) comb. Nov. Microbiol Immunol. 1995;39(11):897–904. doi: 10.1111/j.1348-0421.1995.tb03275.x. [DOI] [PubMed] [Google Scholar]
- 35.List Editor. Validation List No. 57. Validation of the publication of new names and new combinations previously effectively published outside the IJSB. Int J Syst Bacteriol. 1996,46:625–626 [DOI] [PubMed]
- 36.Denny TP, Hayward AC, Schaad NW, Jones JB, Chun W. Laboratory guide for identification of plant pathogenic bacteria. Third. St. Paul, MN, USA: American Phytopathological Society Press; 2001. II. Gram negative bacteria. F. Ralstonia. [Google Scholar]
- 37.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]