

Abstract
The Ah receptor is a transcription factor that modulates gene expression via interactions with multiple protein partners; these are reviewed, including the novel NC-XRE pathway involving KLF6.
Introduction
The Aryl Hydrocarbon Receptor (AHR) is the focus of a classic environmental toxicology research field, which began with the hypothesis for the AHR in 19761,2. While the genesis of AHR research is indeed firmly rooted in environmental toxicology, the AHR has been studied in relation to a diversity of diseases, disorders, and pathophysiologies. This includes immunology3,4, nuclear hormone signaling2,5, metabolic homeostasis and disorders6, cell growth and death7,8, developmental biology9, carcinogenesis10, dioxin toxicity11, cardiotoxicity12, and hepatotoxicity13. While these are all important manifestations of AHR biology, the tremendous body of evidence encompassed within the AHR research area precludes an exhaustive review of the field. With this in mind, the focus of this article is four-fold: 1) to briefly chronicle the AHR field as it relates to the elucidation of the Xenobiotic Response Element (XRE) mediated canonical pathway, 2) to review some classical AHR roles and downstream target genes, 3) to consider AHR function in the context of the newly described non-canonical pathway involving the interaction between the AHR and the tumor suppressor Kruppel-Like Factor 6 (KLF6) at the Non-Consensus Xenobiotic Response Element (NC-XRE) 14, and 4) to speculate on the implications for NC-XRE-driven gene expression, including future research directions. The reader is referred to the excellent reviews cited above, which cover areas not addressed in detail here. For a review of the structurally diverse AHR ligands we refer readers to Denison and Nagy 2003 15 and Nguyen and Bradfield, 200816. The authors wish to note that we have endeavoured to present an accurate chronicle of the archival literature by citing original scientific findings.
History
The earliest work in the Ah Receptor field stemmed from the observation that exposure to various halogenated and polycyclic aromatic hydrocarbon (H/PAHs) compounds (i.e. benzo[a]pyrene, 3-methylcholanthrene) led to the induction of aryl hydrocarbon hydroxylase enzyme activity(AhH, CYP1A1)17-19. This knowledge came from carcinogen studies which showed that coal tar applied to the skin of rabbit ears led to the formation of cancerous lesions20-22. Identification of BaP as the causative agent of those cancerous lesions23,24 and the induction of CYP1A1 by BaP25-27 gave way to broader studies of the PAH family 28,29 and their ability to affect gene expression. Later work identified the much more striking induction of CYP1A1 by the HAH 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD, dioxin)30,31. Although numerous well-documented accidental toxic exposures to TCDD exist11,32-34, TCDD was first identified as the etiological agent of chloracne in a 1957 study of factory workers producing a synthetic auxin35. Subsequent studies of TCDD estimated it to be about 30,000 times as potent as 3-methylcholanthrene in CYP1A1 induction31. This led the field to characterize the perimortem pathology associated with oral acute exposure to TCDD in various species 36,37.
While characterizing the pathophysiology of dioxin toxicity and the induction of CYP1A1 by TCDD, laboratories reported significant differences between species, as well as between different strains of the same species, in both the oral acute LD50 of TCDD and the extent of CYP1A1 enzyme induction by TCDD 38-41. For example, the oral acute LD50 for TCDD in guinea pigs is ~1-2 μg/kg while in the remarkably resistant golden syrian hamster it is ~5 mg/kg41. Interestingly, the dramatically reduced TCDD susceptibility in the golden syrian hamster is not reflected in the relatively modest differences in agonist binding affinities observed amongst species. Nevertheless, the studies of differences in TCDD sensitivity ultimately identified two strains of mice that varied in sensitivity to TCDD exposure, termed the responsive and non-responsive strains, C57BL/6J mice and DBA/2 mice respectively38,42. It was determined that induction of CYP1A1 by these H/PAHs was dose-dependent rather than an altogether non-responsive phenotype, such that the ED50 for CYP1A1 induction in the C57BL/6J strain was approximately 1 nmol/kg while the ED50 for the DBA/2 strain was >10 nmol/kg43. This finding led to the hypothesis that CYP1A1 induction was mediated by a receptor1. Indeed, in acquiring the first evidence for such a receptor, the data supported the hypothesis for a proteinic receptor present in the cytosol, whose agonist binding varied between the B6 (AHRb/b) and D2 (AHRd/d) strains, thus accounting for the large difference in resultant enzyme induction1,44,45. It is noteworthy that later work in studying these two alleles found the difference in sensitivity was attributable 5 amino acids differing between the two variants46. Following the seminal work by Poland, Nebert, and Glover the actual term ‘Ah Receptor’ was first used in a 1980 report by Okey et al., wherein they describe the non-liganded AHR present in the cytoplasm and its’ translocation from the cytosol to the nucleus following dioxin exposure in cell culture47,48.
AHR nuclear translocation follows ligand binding 45,49,50 and is a critical step in the induction of CYP1A151,52. Furthermore, it was demonstrated that induction of CYP1A1 by TCDD was regulated by cis-DNA elements in the target gene promoter52-54. These motifs were rigorously studied to elucidate the core recognition motif 5'-GCGTG-3’, which was dubbed the Xenobiotic Response Element (XRE)55-59. While characterizing the induction of CYP1A1 by the AHR, many labs identified proteins associated with the cytosolic receptor which were not associated with the nuclear form of the receptor60,61. It was ultimately shown that several proteins were necessary for proper translocation and transcriptional activation by the AHR62-64. In the case of the AHR protein partner Aryl hydrocarbon receptor nuclear translocator (Arnt), this discovery began with mutagen studies65 identifying BaP resistant Hepa-1 cell clones with decreased CYP1A1 induction following exposure to a number of different mutagens. The frequency with which these mutagenic clones developed increased with higher doses of the mutagen and the BaP resistant phenotype persisted in successive cell generations. These observations suggested a genetic aberration rather than an acquired resistance to explain the BaP resistant phenotype66. Subsequent studies of several of these clones found that AHR expression as well as AHR nuclear translocation dynamics following TCDD exposure were similar to that of the parent Hepa1 line67. This was followed by studies assessing the biochemical characteristics of the AHR which hypothesized a smaller form of the receptor to exist following ligand binding, as assayed by sucrose density gradient and gel filtration analyses68, and a larger form of the receptor to be present in the nuclear fraction 60.
The suggestion that the nuclear receptor might interact with other protein partners led several labs to the hypothesis that the additional proteins facilitated DNA-binding and more specifically that the DNA-bound form of nuclear AHR was heteromeric in structure69,70. Ultimately, this heteromer was shown to include the protein identified as the now well known Arnt, through studies of the hepa1 mutants discussed previously71. The Arnt cDNA was cloned and the protein found to contain a basic helix-loop-helix region and 2 domains with homology to the drosophila proteins PERiod and SIngle-Minded (Per and Sim)71,72, a structural feature that unites all three in a protein family together with the AHR—the PAS family of bHLH proteins. The AHR protein was first purified in 1991 by Bradfield and coworkers73; the mouse and human AHR ORFs were subsequently cloned shortly thereafter74,75. Based on studies of the glucocorticoid receptor76, another group identified the HSP90 protein in an interaction with the AHR77. The HSP90/AHR interaction was later shown to have a functional role in maintaining the cytosolic receptor in a conformation capable of binding to TCDD78.
The Arnt protein was so named as it was thought to aid in translocation of the AHR from the cytoplasm to the nucleus upon ligand binding71,79. While not immediately clear at the time, later studies by the Poland lab and the Hankinson lab tenably cemented the framework for what we know as the canonical AHR signaling pathway (AHR/Arnt pathway, XRE pathway) with data showing: 1) the unliganded AHR is cytosolic, 2) upon ligand binding the AHR translocates to the nucleus, 3) Arnt is constitutively located in the nucleus—a finding not anticipated by the genetic studies, 4) the nuclear form of the AHR complex does not include HSP90, but does include Arnt and 5) AHR-DNA binding requires Arnt 80,81.
Following the discovery that the unliganded AHR and Arnt are found in the cytosol and nucleus respectively, data from the Hankinson lab functionally characterized the domains of the AHR and Arnt proteins. Extensive mutagenesis experiments have ultimately characterized the regions necessary for AHR ligand binding, HSP90 interaction, dimerization, DNA-binding, and transcriptional activation82-87 (Figure 1). Previous work had reported the unliganded cytosolic AHR to interact with HSP90 and other as yet unidentified protein partners77,88,89. These proteins were subsequently identified as 1) a protein previously shown to interact with a hepatitis B virus protein known as X-associated protein 2 (XAP2)90—and independently identified as the AHR interacting protein AIP91 or ARA992)—and 2) p2393,94.
Fig. 1. Schematic representation of the functional domains in the AHR, Arnt and KLF6 proteins.

The figure depicts the location of the functional domains within the proteins. The blue text refers regions important in the AHR/KLF6 NC-XRE pathway, the red text refers to the AHR/Arnt XRE pathway, and black text is not specific to either. The regions in this figure are based on previous studies for the AHR14,83 Arnt82,84,86,87, and KLF6219,259,260.
This body of research in the AHR field (including many other important studies not cited here) led to a functional characterization of the molecular events that constitute what is collectively dubbed the canonical AHR signaling pathway. While it took many years to delineate the cell signaling events precedent to CYP1A1 enzyme induction following TCDD exposure, the advent of microarray and next-generation sequencing technologies has identified multifarious AHR target genes that are regulated via the classic XRE. Indeed, this new research has also identified novel protein partners and cis DNA motifs for the AHR; these target genes and protein interactions are the focus of the sections below.
Section II: The canonical pathway (XRE)
As depicted in Figure 2, in the canonical pathway the unliganded AHR resides in the cytosol, bound by chaperone proteins HSP90, p23, and ARA9 (XAP/AIP). Upon ligand binding, the AHR is presumed to undergo a conformational change in the PAS A domain which facilitates nuclear translocation and AHR/Arnt dimerization through the PAS A region concomitant with dissociation from HSP9095. This change in structure following ligand binding presumably allows a conserved nuclear localization sequence in the N-terminal 42 amino acids to stimulate nuclear translocation96, which is facilitated by importins2,84. Once in the nucleus, the AHR dissociates from chaperone proteins and heterodimerizes with Arnt. AHR/Arnt dimerization is facilitated by the ligand-induced conformational change in the PAS A region95. As shown in Figure 2, the HLH domain of the AHR, and to a lesser extent the PAS A and B domains, are necessary for AHR/Arnt dimerization, while the basic region located in the N-terminus of the protein is necessary for DNA-binding83. Similar to the AHR, the basic region of Arnt is required for XRE DNA-binding, and the HLH and PAS A domain of Arnt mediate protein binding with the AHR82,86. Both of these proteins contain a nuclear localization signal in the N-terminus of the protein, while the nuclear export signal of the AHR is located between amino acids 62-7284,97. Although the precise mechanisms remain unclear98, it has been suggested that phosphorylation of the AHR and Arnt is necessary for DNA binding by the heterodimer, possibly through a mechanism involving protein kinase c99-101.
Fig. 2. The existence of multiple mechanisms to regulate AHR activity.
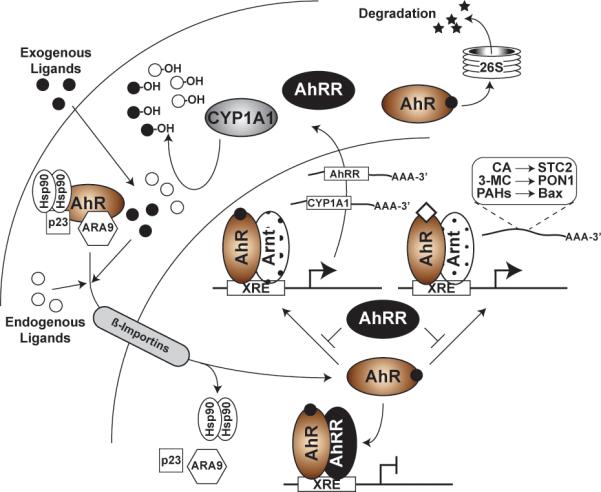
The diagram depicts the different strategies by which AHR function is controlled. In the canonical pathway, AHR activation is induced by exogenous or endogenous ligand binding, nuclear translocation, and recruitment to XREs in partnership with the Arnt protein. AHR signaling is silenced by one of several mechanisms, including 1) induction of CYP1A1 and subsequent metabolic clearance of the ligand pool, 2) expression of the AHR repressor (AHRR), and 3) AHR degradation through the 26S proteosome. AHR activity is also influenced by the distinct properties of the bound agonists (depicted by the open diamond) which can alter the repertoire of responsive target genes.
The liganded AHR/Arnt heterodimer then binds to XREs within target gene promoters, a process that alters DNA topology102 and concomitant stability of the protein-DNA complex. The classic and most well characterized DNA-binding motif for the AHR/Arnt heterodimer is the XRE. It was first defined through experiments analyzing CYP1A1 induction by the AHR103, which identified a number of dioxin responsive domains bound by protein in an AHR-dependent manner, without the requirement of nascent protein synthesis55,56. An assessment of the critical nucleotides in the XRE needed to facilitate AHR binding revealed a core consensus motif of 5'-GCGTG-3’57. In the case of transcriptional activation, flanking residues in the XRE (5'-T/GnGCGTGA/C-3’) confer the greatest AHR transactivation capability57,59, such that nucleotide substitutions within 4 bp 5’ and/or 3’ of the core motif decrease transactivation potential for the AHR59,104.
AHR-XRE target genes
The cytochrome p450 enzyme CYP1A1 is the archetypal XRE-mediated target gene for the AHR. The Ah receptor Repressor (AHRR) gene is another XRE-mediated AHR target gene up-regulated following AHR activation. The AHRR functions as a dominant negative protein, competitively inhibiting AHR transactivation possibly by binding to and sequestering Arnt to form a transcriptionally inactive heterodimer. However, Evans et al. presented evidence favoring a mechanism of AHRR action involving “transrepression” of AHR signaling that does not depend on competitively inhibiting the formation of an AHR/Arnt complex or its DNA binding. The discrepancies notwithstanding, the AHRR confers a type of negative feedback inhibition on the AHR signaling pathway105 (Figure 2). Following transcriptional activation, the AHR releases from DNA and is rapidly exported from the nucleus and degraded via the ubiquitin-mediated 26S proteosome pathway106. The half-life of the cytosolic AHR is 28 hours, while the half-life of the AHR following TCDD exposure is 3 hours. Not surprisingly, inhibiting proteosomal degradation of the AHR significantly increases the amount of nuclear AHR, which “superinduces” CYP1A1 gene expression106. This suggests that rapid AHR turnover following its activation and DNA binding serves as a key regulatory step in the AHR signaling pathway. In fact, it was recently suggested that the TCDD-inducible poly-(ADP-ribose) polymerase (TiPARP), an established AHR target gene, suppresses ongoing AHR activity by TiPARP directly interacting with the AHR to disrupt receptor activity, and by promoting AHR turnover via proteosomal degradation107,108.
In addition to CYP1A1 and the AHRR gene, other XRE mediated AHR target genes include additional phase I heme monooxygenases in the cytochrome p450 family such as cyp1a2109, cyp2a5110, and cyp1b1111 as well as the phase II enzymes GSTA1112 and ALDH3113. Likewise, the AHR and Arnt transactivate NAD(P)H dehydrogenase quinone 1 (NQO1) expression114 through a mechanism involving the erythroid 2-related nuclear factor protein (NRF2) following exposure to either the classic AHR ligands or any of a number of different NRF2 activators115 (Figure 3). The increased expression of these enzymes (including CYP1A1) serves as a negative feedback loop for Ah receptor signaling whereby the enzymes attenuate AHR activation through metabolic depletion of the ligand pool116 (Figure 2).
Fig. 3. Schematic representation of the various AHR partnerships affecting transcriptional control.
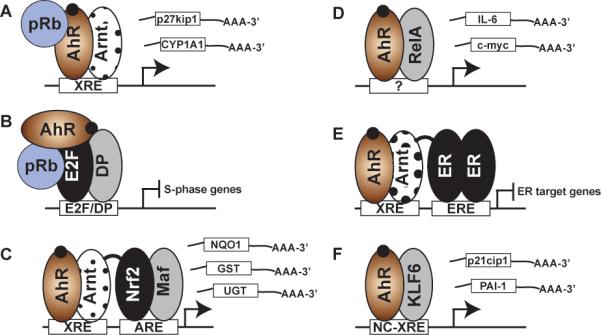
A) The canonical pathway involves AHR/Arnt heterodimer binding to the XREs flanking target gene promoters and functioning as a scaffold for a number of cofactors (including pRb) necessary for maximal transcriptional activation. B) The AHR forms a co-repressor complex with pRb to suppress E2F/DP mediated expression of S-phase genes. C) XRE binding by the AHR/Arnt heterodimerin close proximity to an antioxidant response element (ARE) in phase II metabolic gene promoters facilitates an interaction with the Nrf2/Maf heterodimer to induce gene expression. The prototypical target gene for this interaction is NQO1. D) Through an interaction with RelA, the AHR binds to NFκB target gene promoters including c-Myc and IL-6, to induce gene expression. The precise nature of the DNA binding site remains unresolved—that is, a RelA driven interaction at a NFκB response element or binding to a novel AHR/RelA response element. E) Modulation of estrogen receptor (ER) signaling by direct AHR/Arnt heterodimer interactions suppressing ER mediated gene expression, or by steric hinderance of ER DNA binding due to overlapping XRE and ERE sites (not depicted). F) The novel NC-XRE pathway involves an AHR/KLF6 interaction and DNA binding to NC-XRE motifs flanking target genes such as PAI-1,p21cip1 and E-cadherin (see Fig. 4).
It has been suggested that the induction of metabolic enzymes by the AHR is a critical component of its role as a a high fidelity biosensor, which prevents prolonged receptor activation117,118. With respect to the endogenous ligands, their rapid turn-over protects against prolonged AHR activation which can lead to transcriptional changes that perturb the physiological equilibrium normally maintained by the AHR. Evidence of such perturbations under conditions of prolonged AHR activation affecting cell cycle control have been reported119,120. While it may be a property associated with endogenous ligands, the resistance of certain exogenous agonists such as TCDD to metabolic degradation undermines the benefits of this negative feedback mechanism resulting in deleterious consequences. Agonist induced AHR signaling is highly conserved across the vertebrate lineage121 suggesting that the clearance of agonists provides a general mechanism for controlling AHR biology. Although AHR and Arnt orthologs are present in several invertebrates122, the inability to demonstrate ligand binding by the invertebrate forms123 imputes that AHR agonist binding and the metabolic clearance of agonists are more recent evolutionary events. Given that many of the exogenous AHR ligands are toxic (through a mechanism requiring the AHR), it is not immediately clear what evolutionary benefit the AHR might confer as a sensor of exogenous ligands. This is especially true since many exogenous ligands that activate the AHR are 1) only relatively recently present in the environment in large quantities, and 2) some such as TCDD are poorly metabolized by AHR-induced biotransformation enzymes. This would suggest that activation of the receptor by exogenous ligands constitutes AHR ‘hijacking’ diverting it away from its innate physiological role. Paradoxically, CYP1A1 knockout mice are more sensitive to toxicity from oral BaP exposure, compared with wild-type mice124, suggesting that the receptor's response to certain exogenous agonists is beneficial. However, the lack of BaP genotoxicity in AHR knock out mice suggests that the protective role of CYP1A1 is contingent on the presence of a functional receptor. These observations serve to illustrate that the variability in AHR signaling following exposure to endogenous and exogenous cues presents real difficulties in distinguishing toxic responses from the adaptive physiological responses.
The most toxic H/PAH AHR ligands (most notably TCDD), exhibit high affinity for the AHR and are poor substrates for the biotransformation enzymes up-regulated following receptor activation125,126. This endows them with the ability to persistently activate the AHR119,126. This fact bolsters the rationale for the hypothesis that the endogenous AHR ligand(s) are readily metabolized. Indeed, given the numerous well-known cell-type specific functions of the AHR and the myriad AHR ligands (endogenous and exogenous)121,127,128 it is tempting to speculate that there may be multiple endogenous ligands for the AHR, perhaps specific to cell or tissue type. This notion is supported by data showing AHR target gene responses vary with different agonists.129. For example, the induction of PON1 and Bax gene expression by PAHs (e.g. 3-MC) and not TCDD highlights the agonist specific responses130,131. We recently identified a receptor target gene, stanniocalcin 2 (STC2)132, that is readily induced by the novel endogenous AHR ligand, cinnabarinnic acid133, yet is not induced by TCDD or BaP (Joshi et al. submitted)(Figure 2).
It is clear that there are numerous mechanisms (AHRR, CYP1A1-mediated ligand depletion, proteolytic degradation, and TiPARP) in place to tightly regulate receptor activity (Figure 2). The presence of myriad regulatory pathways suggests that aberrant receptor signaling is deleterious. In fact, there is plenty evidence to support this notion, including the plethora of toxic effects observed with persistent receptor activation120,134 and the developmental defects seen with receptor loss 135-137 or aberrant activation137-139. The advent of such tight coordination is consistent with the existence of AHR functions beyond the transcriptional control of a few phase I and II enzymes. While the precise nature of the AHR-regulated events that warrant such tight control remain largely unexplored, some indications have come to light in studies examining the AHR in cell cycle control and proliferation; in the next section we will review some of these paradigms
The AHR-XRE in the cell cycle
As previously discussed, the early work in the receptor field focused on CYP1A1 induction by exogenous AHR agonists36. This research gave way to studies with TCDD responsive and non-responsive mice42 and cell lines62. While studying these cell lines, Wiebel et al. reported that TCDD treatment of 5L rat hepatoma cells slowed their cell division concomitant with decreased DNA synthesis, suggesting cell cycle arrest at the G1/S phase checkpoint140 in an AHR-dependent manner141,142. A similar TCDD-induced cell cycle arrest was also observed in AHR-null cells upon re-expression of the full length AHR116. Intriguingly, it was found that loss of AHR expression in various cell lines also resulted in a prolonged cell doubling time142. The prolonged doubling time associated with loss of AHR expression appears to be a general phenomenon, having been observed in several cell types including hepa1 cells142, epithelial cells143, and breast cancer cell lines144. Therefore, paradoxically both TCDD-induced AHR activation and loss of the receptor, delay cell cycle progression through G1-phase. In AHR null embryonic fibroblasts145 this was attributed to a mechanism thought to involve retinoblastoma tumor suppressor protein (pRb) and the transcriptional cofactor p300146. Restoration of AHR expression in AHR-null cells restored the normal doubling time146, while over-expression of the AHR is known to accelerate cell cycle progression and decrease doubling time142,143. This suggests that AHR signaling in response to endogenous cues hastens cell cycle progression. The implication is that the AHR plays a central role in the G1-phase program where perturbations affecting normal receptor function—due to sustained AHR activation by TCDD or loss of the protein—delay cell cycle progression by blocking entry into S phase.
With respect to the aforementioned studies, it is important to bear in mind that many of the in vitro systems that have been used to study the role of the AHR in cell cycle progression are in fact cancer cell lines, which by their very nature have circumvented the normal checkpoint machinery in place to regulate cell division. Consequently, it is possible these studies have inadvertently overlooked important pieces to the puzzle. It is also possible that there are differences in the AHR signaling pathway between these cell lines. Taken together, while the TCDD induced AHR-mediated G1-phase cell cycle arrest phenomenon has been well-characterized, it remains altogether unclear why loss of the AHR prolongs doubling time. Presumably, the AHR is necessary to activate a gene(s) necessary for proper G1-phase progression, the absence of which prolongs the G1-phase. Nevertheless, it is clear that the AHR has an important role in the G1/S checkpoint146,147.
We have previously reported a direct interaction between the AHR and the hypophosphorylated “active” form of pRb147. We have also demonstrated that this interaction is critical for maximal AHR-mediated G1-phase cell cycle arrest148. Additionally, hyperphosphorylated pRb (ppRb) is predominant in dividing cells while hypophosphorylated pRb (pRb) is restricted to the G0 and G1 phases, imputing a cell cycle dependency to the functional consequences of the AHR/pRb interaction148. pRb inhibits cell proliferation either by binding to E2F or other transcription factors to indirectly block gene transactivation149, or by binding with E2F at target gene promoters to actively repress gene expression150-152. Puga et al. 2000 expanded on the interaction between the AHR and pRb, reporting that the proteins interacted in the absence of Arnt and suggesting a co-repressor mechanism by which the AHR/pRb complex might inhibit cell cycle progression through suppression of E2F activity153-155 (see Figure 3). This suppression of E2F activity depends on an intact LXCXE (pRb binding) motif the AHR148. Although the AHR/pRb interaction was also documented to involve a region in the receptor's transactivation domain, it has been reported that E2F transcriptional suppression occurs in the absence of functional AHR transactivation and DNA-binding domains153,155. In contrast, we have shown the AHR/pRb interaction to be necessary for maximal G1-phase cell cycle arrest and maximal AHR-mediated induction of CYP1A1 following TCDD treatment148. Indeed, in this scenario the two proteins interact, with pRb functioning as a transcriptional co-activator to induce expression of CYP1A1 and the cyclin-dependent kinase inhibitor p27kip1, to promote cell cycle arrest148,156,157 (see Figure 3). It is noteworthy that studies in rat hepatoma 5L cells and mouse oval cells have shown the AHR to induce p27kip1 gene expression following TCDD treatment156,157. However, in vivo studies in mice indicated that the cyclin-dependent kinase inhibitor, p21cip1, rather than p27kip1 is TCDD responsive158. It is noteworthy that in characterizing the importance of the LXCXE motif in G1-arrest, studies revealed that a mutant form of the AHR unable to interact with pRb still induced a modest, albeit significantly reduced G1 arrest following TCDD exposure148. This is consistent with a role for the cyclin-dependent kinase inhibitor p27kip1 (or p21cip1, as we will discuss later) in the process. Since the pRb interaction with the AHR is limited to the active hypophosphorylated form, the interaction is restricted to the G0 and G1 phases of the cell cycle. However, the AHR/Arnt complex can interact with several cofactors146,147,153, thus AHR activity can conceivably be modified throughout the cell cycle reflecting the nature of the cofactor interactions. Specifically, the AHR/Arnt complex has been shown to recruit the cofactors p300, CREB binding protein (CBP), and steroid receptor coactivator (SRC) 1 and 2146. The involvement of these cofactors attests to the complexity of AHR mediated transcriptional control. Indeed, cofactors involved in AHR signaling via the XRE can vary depending on, among other things, the presence or absence of ligand, type of ligand, amount of ligand, species of organism, and cell type (Figure 3). The involvement of these cofactors suggests that both co-activation and co-repression contribute to AHR mediated cell cycle arrest in G1-phase7,159.
Cofactor recruitment has a direct bearing on epigenetic changes associated with AHR-mediated gene expression. Several studies have reported on AHR-dependent epigenetic modifications. Indeed, evidence exists for both histone and DNA modifications in AHR signaling160-164. This includes early studies describing methylation of a CpG sequence within the core XRE motif that disrupts AHR DNA-binding165. Epigenetic modifications targeting histones H3 and H4 have been shown to play a role in cell-specific differential expression of the classic target genes, cyp1a1 and cyp1b1164,166-168. Furthermore, Lo and Matthews 2012 using ChIP-seq, identified AHR binding to DNA as far as +/− 100kb from the closest TSS169, imputing a role for epigenetic changes in gene expression. Although these studies clearly illustrate the involvement of epigenetic control in AHR-mediated transcriptional regulation, the specific modifications are likely to be gene and context dependent, and published accounts are few. For a more in-depth review of AHR binding to the cyp1a1 promoter, as well as several epigenetic changes involved, the reader is referred to Hankinson 2005 and Solaimani et al. 2013170,171.
AHR XRE cofactors and binding partners
Apart from the canonical signaling pathway involving the AHR/Arnt heterodimer, it has also been reported that the AHR interacts with the NF-κB subunits RelA172 and RelB173,174. In 2007, Vogel et al. observed that following TCDD treatment, the AHR directly interacts with RelB and binds to the IL-8 promoter. Computational analysis failed to locate an XRE motif in the IL-8 promoter sequence173. Further study revealed the presence of an eight nucleotide DNA-binding site on the IL-8 promoter, through which the AHR is capable of inducing IL-8 gene expression following dioxin exposure173. These and other reports of the AHR interacting with new protein partners to transactivate genes through novel DNA motifs highlight a shift of focus in the field toward characterizing AHR target genes regulated by non-XRE receptor-mediated mechanisms.
For a number of the aforementioned phase II enzymes induced by the AHR, it is now known (see Figure 3) that there is an AHR interaction with NRF2114,175, where the XRE-bound AHR/Arnt complex cooperates with the NRF2/Maf complex bound to proximal antioxidant response elements (AREs) located in the target gene promoter115. The prototypical target for this complex is NQO1. Additionally, a number of other UGT and GST enzymes have also been suggested as AHR targets175,176. These enzymes are responsible for the biotransformation of several carcinogenic PAHs177 including, the ultimate genotoxin benzo[a]pyrene (BaP)-7,8-dihydrodiol epoxide, formed during CYP1A1 mediated hydroxylation of the parent compound, BaP178. For a more in-depth review of the phase II targets for the AHR/NRF2 interaction see Bock 2012177.
The AHR up-regulates certain targets via an interaction with the NF-κB protein partners RelA172,174 or RelB173,179. Two such targets for the AHR/RelA interaction are c-myc172 and the inflammatory cytokine interleukin 6 (IL-6)180. Although the precise mechanism remains unclear, the data suggest the AHR binds through either ‘XRE-like’ sites in the IL-6 promoter181 or via interactions with NF-κB elements through its RelA interaction182. AHR-mediated induction of IL-6 has been linked to a number of different physiologically relevant processes including inflammation180, exogenous ligand-independent AHR activity183, and tumorigenesis181. In the case of the AHR/RelB interaction, the two proteins form a complex at a ‘XRE-like’ motif (5'-GGGTGCAT-3’) dubbed the RelBAHRE in the IL-8 promoter173, following exposure to either the diterpene plant derivative, forskolin, or TCDD179. While additional work is needed to fully understand the functional role of the interaction with the Rel proteins, these observations highlight the intricacies of AHR biology, which are far richer and more complex than suggested by the classic AHR/Arnt signaling pathway.
A number of studies have reported on the anti-estrogenic effects of numerous exogenous AHR ligands184,185. This anti-estrogenic activity is primarily attributed to AHR-mediated transactivaton of CYP1A1 and CYP1B1, which metabolize the estrogen receptor (ER) ligand 17β-estradiol186, and AHR/Arnt DNA-binding of so-called inhibitory XREs juxtaposing ER elements in ER target genes187-189. One other example of the AHR signaling via the XRE with novel protein partners is the interaction between the AHR/Arnt complex and the estrogen receptor (ER)188 (Figure 3). AHR binding attenuates ER transactivation185,190 of a number of different ER target genes, including several cell cycle regulatory proteins188,191,192. In addition to the ‘classical’ AHR functions in inhibiting ER activity through agonist metabolism, the AHR has been shown to decrease ER activity by acting as an E3 ubiquitin ligase targeting nuclear ER degradation through the 26S proteosome pathway193.
Section III: The non-canonical pathway (NC-XRE)
In an effort to better understand differential expression of TCDD dependent and independent AHR gene batteries, several groups performed microarray analyses on RNA from tissues or cells treated with TCDD or other AHR ligands. The Pohjanvirta laboratory extensively studied transcriptomic changes in wild type (AHR+/+) and AHR null (AHR−/−) mouse livers following TCDD treatment194,195. They noted significant differences in the expression of 456 ProbeSets in wild-type mice (AHR-dependent) following TCDD treatment. The changes included a number of proteinase inhibitors, including the plasminogen activator inhibitor-1 (serpine1, PAI-1)194. To evaluate physiological target genes for the AHR (in the absence of exogenous ligand), Tijet et al. assessed the transcriptome in both the presence and absence of the AHR. In the absence of an exogenous ligand, AHR status alone accounted for changes in the expression of 392 ProbeSets194. This finding is consistent with a normal physiological function for the AHR distinct from the xenobiotic response. Compared to the 456 ProbeSets altered in wild-type mice, only 32 ProbeSets showed significantly different expression in AHR−/− mice at 19 hours post TCDD treatment194. These results underscore the essential role of the AHR in nearly all dioxin mediated transcriptional changes.
The striking difference in gene induction between the wild-type and the AHR −/− mice is not surprising given the many reports documenting the complete lack of TCDD toxicity in AHR −/− mice 196-198. Boverhof et al. studied temporal and TCDD dose-dependent changes in gene expression patterns in the mouse liver. In these experiments, custom cDNA microarrays found 349 significant (p < 0.05) gene changes at one or more doses and 255 significant gene changes at one or more time points following TCDD treatment199. Microarray analyses to determine TCDD induced changes in the human liver have been performed using the human HepG2 hepatoma cell line as well200,201. After 10 nM TCDD treatment for 18 hr, a total of 112 significant genes were identified with at least two-fold change200. Quantitative RT-PCR confirmed significant changes in human enhancer of filamentation 1(HEF1), cot, XMP (involved in cellular proliferation and met), HM74, and PAI-1, among others200. Puga et al. treated HepG cells for 8 hr with 10 nM TCDD and performed microarray analyses; among the 202 genes significantly different in expression between the treatments, they reported that PAI-1 is significantly up-regulated following dioxin exposure. Furthermore, they showed this up-regulation persisted in the presence of pretreatment with 20 μM cyclohexamide, suggesting PAI-1 is a direct AHR target gene153.
Complementary chromatin immunoprecipitation (ChIP) DNA microarray (ChIP-chip) studies examining AHR bound genomic sequences revealed that 57.8% and 48.5% of AHR enriched regions, at 2 hours and 24 hours post-TCDD treatment respectively, did not contain an XRE core sequence (5'-GCGTG-3’)202. This is consistent with several other reports using high throughput analyses203, DNA microarray129, ChIP-chip204, and high-resolution ChIP next-generation sequencing169 (ChIP-Seq). It has been suggested that genes up-regulated following TCDD treatment, which lack the known AHR DNA-binding motifs may represent secondary targets, activated by one or more of the genes the AHR directly up-regulates. Several studies have suggested that while these indirect events likely account for some portion of the changes in genes lacking an AHR motif, there is in fact a large subset of TCDD responsive genes controlled in an AHR-dependent manner that lack a classical AHR XRE. This includes the microarray studies by Puga et al. involving cyclohexamide, the ChIP-chip report by Dere et al. assessing multiple time points, and the ChIP-seq studies by Lo and Matthews in MCF-7 cells169,201,202. Given that the AHR functions as a transcription factor, and given the numerous (previously discussed) examples of the AHR interacting with multifarious protein partners to bind DNA motifs other than the XRE, it stands to reason that the AHR regulates a subset of target genes through a mechanism independent of the XRE. PAI-1 was identified as one such gene194,200,201,205. PAI-1 is a member of the serine protease inhibitor (serpine) family. It functions as the inhibitor of urokinase plasminogen activator and tissue plasminogen activator 206,207, which convert plasminogen to plasmin to facilitate fibrinolysis. PAI-1 is ubiquitously expressed but most highly so in endothelial cells, adipocytes, and hepatocytes. We were specifically interested in PAI-1 because it is known to form a complex with urokinase plasminogen activator to inhibit liver regeneration by hampering activation of Hepatocyte Growth Factor (HGF), an important liver regeneration protein208-211.
Mitchell et al. previously reported that TCDD exposure suppresses liver regeneration119, in part by increasing PAI-1 levels to inhibit urokinase plasminogen activator activity necessary for HGF activation. Huang and Elferink further investigated PAI-1 expression using a luciferase reporter assay, containing a 116 bp region of the PAI-1 promoter, in hepatoma cells and observed an AHR-dependent induction of PAI-1 expression following TCDD treatment212. Electrophoretic mobility shift assays (EMSA) performed using mouse liver nuclear extracts identified a region spanning nucleotides −116 to −76 of the PAI-1 promoter that supported TCDD inducible protein-DNA binding by the AHR212. The term Non-Consensus Xenobiotic Response Element (NC-XRE) was coined to distinguish this motif from the canonical XRE212. The hallmark of this sequence is the 5'-GGGA-3’ tetranucleotide repeat. Ensuing sequential mutation analysis of a 40 bp region revealed that the second 5'-GGGA-3’ motif substantially contributes to protein-DNA binding (AHR-NC-XRE interaction) 212. Another striking feature associated with the AHR interaction at the PAI-1 NC-XRE was the lack of Arnt binding. EMSA experiments showed that AHR binding to the NC-XRE sequence could not be competed for with a 10-fold molar excess of cold XRE oligonucleotide212. The hypothesis that the AHR/NC-XRE interaction was Arnt-independent was tested by ChIP experiments, which confirmed direct binding of the AHR to the PAI-1 promoter NC-XRE following TCDD treatment. In contrast, ChIP experiments targeting the Arnt protein failed to yield a product, suggesting that AHR binding to the NC-XRE was not dependent on Arnt. Subsequent functional studies confirmed that AHR-mediated PAI-1 transcriptional activation through the NC-XRE is in fact Arnt independent212.
It is well established that the TCDD activated AHR suppresses liver regeneration through inhibition of cell proliferation119,158,213. This role for the AHR is consistent with previous reports showing the AHR to induce G1-phase arrest in cells exposed to TCDD120,156. Indeed, the TCDD-induced AHR-mediated suppression of liver regeneration has been ascribed to attenuation of G1-phase cyclin dependent kinase 2 (CDK2) activity119. Inhibition of CDK2 has previously been linked with increased expression of the cyclin-dependent kinase inhibitors p21cip1 and p27kip1 and binding to CDK2/cyclin E complexes. p21cip1 and p27kip1 are members of the Cip/Kip family of cyclin dependent kinase (CDK) inhibitors that regulate cell cycle progression by controlling CDK2, CDK4, and CDK6 kinase activity214. p21cip1 activity is tightly regulated, principally at the transcriptional level, through both p53-dependent and p53-independent mechanisms215. Given the link between CDK2 and p21cip1/p27kip1, our lab recently set out to assess their involvement in AHR-mediated TCDD-induced suppression of liver regeneration. Liver regeneration experiments using the 70% partial hepatectomy (PH) model in p21cip1 and p27kip1 knock out mice unequivocally demonstrated that p21cip1 is required to confer the TCDD-induced suppression of liver regeneration seen in wild-type mice158. In contrast, p27kip1 was not found to play a role in the TCDD growth arrest following PH. We observed via in silico analysis that both the p21cip1 transcript 1 and transcript 2 promoters contain NC-XRE motifs. This led us to test whether p21cip1 is an AHR target gene. Subsequent ChIP experiments confirmed that at 2 hours following TCDD treatment the AHR specifically occupies the p21cip1 transcript 2 promoter158. Further, we showed the AHR is bound to the transcript 2 promoter NC-XRE at 24 hours post-TCDD treatment158. Interestingly, ChIP experiments on livers following PH in the absence of TCDD, showed that the AHR is transiently bound to the p21cip1 transcript 2 promoter158. AHR binding to the NC-XRE in the absence of TCDD suggests that PH triggers AHR activation in the absence of an exogenous ligand. Furthermore, it suggests a physiological role for the AHR in regulating p21cip1 expression during liver regeneration158. Indeed, PH-induced AHR activation in the absence of an exogenous ligand has previously been shown using CYP1A1 up-regulation as a read-out119. It has also previously been reported that PAI-1−/− mice exhibit hastened liver regeneration211, consistent with PAI-1's role in HGF activation216,217. Interestingly, we have shown that liver regeneration in PAI-1−/− mice is inhibited by TCDD to the same extent as in WT mice (Wilson et al. unpublished data). This suggests that p21cip1 plays a dominant role in TCDD-induced suppression of liver regeneration in mice158. Mechanistically, AHR-mediated increases in p21cip1 expression not only inhibit G1-phase CDK activity and cell cycle progression by suppressing pRb inactivation153,155,158, the delay in pRb inactivation is envisioned to enhance AHR activity by prolonging the AHR/pRb interaction noted above.
A subsequent study of AHR binding to the NC-XRE identified a novel AHR DNA-binding partner 14. Wilson et al. focused on the Kruppel-like factor (KLF) protein family due to sequence homology between the NC-XRE and the KLF family DNA-binding motif as well as a previous study showing KLF4 to regulate CYP1A1 gene expression218. The KLF family of transcription factors is related to the specificity protein factor family due to similarities in the C-terminal DNA binding domains219. Due to high C-terminal sequence homology shared by KLF family members, most KLF transcription factors bind to a similar DNA-motif known as a ‘GC box’ on a number of functionally diverse target genes220,221. Apart from the homology in the zinc-finger C-terminal regions, the KLF family proteins have significantly less sequence homology in the N-terminal portions, which disposes them to unique individual regulatory pathways222. Structural and functional studies showed that the C-terminus of the AHR and N-terminus of KLF6 are necessary for both the mouse and human proteins to interact14. Interestingly, KLF6 binding to the NC-XRE requires three arginine residues juxtaposing the KLF6 zinc-finger DNA binding domain, rather than the zinc-finger domain itself. In fact, the entire zinc-finger region is expendable for NC-XRE binding. It should be noted that loss of the zinc-finger domain did not preclude NC-XRE binding in the EMSA experiments14, this region contains the NLS (Figure 1) required for KLF6 nuclear localization and thus the zinc-finger domain is likely necessary in cells and in vivo. Furthermore, these experiments also showed that key AHR residues critical for XRE DNA-binding were not required for NC-XRE DNA-binding14.
KLF6 target genes regulate a number of different processes including cellular differentiation, proliferation, and apoptosis220,222. KLF6 mutations, loss of heterozygosity, and over-expression are linked to a number of cancers, including hepatocellular carcinoma223, gastric cancer224, pancreatic cancer225, colorectal cancer226, prostate cancer227,228, and astrocytic glioma229. In this light, KLF6 is considered to be a tumor suppressor according to Knudson's two hit model230. It has been demonstrated to up-regulate E-cadherin231, transforming growth factor β1232, and insulin-like growth factor 1 receptor233. KLF6 is known to activate expression of p21cip1 in a p53-independent manner222, which leads to the inhibition of cell cycle progression and decreased hepatocyte proliferation223,227,234,235. A recent study has shown that p21cip1 knockdown in dividing cells impairs this KLF6 induced G1-phase cell cycle arrest236. Certainly, in the case of E-cadherin, we obtained evidence that the promoter harbors a NC-XRE and recruits the AHR/KLF6 complex in a TCDD-dependent manner (Figure 4), suggesting that the AHR and KLF6 are partners in anti-tumor signaling pathways.
Fig. 4. E-cadherin is an AHR/KLF6 target gene.

E-cadherin is a tumor suppressor protein involved in cell-cell adhesion. Loss of E-cadherin expression (or function) correlates positively with cancer progression and metastasis. Mice were treated by oral gavage with vehicle (−) or 20 μg/kg TCDD (+) for 2 hours. Chromatin immunoprecipitations (ChIP) were performed on livers using antibodies against the indicated proteins and the E-cadherin promoter (−220 to +37) encompassing a NC-XRE, amplified by PCR. The data show TCDD-dependent recruitment of the AHR and KLF6 to the E-cadherin promoter. This sequence was independently shown to harbor a KLF6 binding site in 3 separate human cancer cell lines261.
Collectively, the studies by Huang et al., Wilson et al., and Jackson et al. have contributed to a characterization of what we have dubbed the non-canonical pathway, which parallels AHR/Arnt signaling (Figure 5). Nuclear localization of the AHR, stimulated by an exogenous or endogenous ligand drives the KLF6/AHR interaction, NC-XRE binding, and subsequent transcriptional events. In this context, the AHR is expected to modulate expression of both XRE- and NC-XRE-driven target genes14,158,212, and we envision that both pathways are activated concomitantly, but function to alter expression of distinct subsets of target genes.
Fig. 5. The NC-XRE pathway.
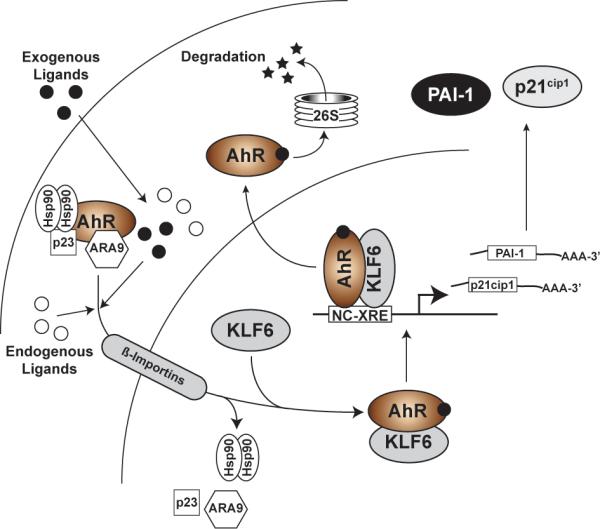
In the NC-XRE pathway, as in the canonical pathway, the AHR is activated by endogenous or exogenous ligands, tostimulate nuclear translocation. Unlike the canonical pathway, the NC-XRE pathway relies on an AHR association with KLF6 and recruitment to NC-XRE binding sites flanking target genes, including PAI-1 and p21cip1.
Section IV: Cross-talk between the XRE and the NC-XRE pathways
As noted above, the AHR is known to function in regulating cell cycle progression involving both XRE- and NC-XRE-driven mechanisms 140-142,158. The evidence showing that the AHR and Arnt are necessary for proper AHR mediated cell cycle progression143,145, coupled with our finding that AHR-mediated p21cip1 expression involves the NC-XRE158, suggests that the NC-XRE and XRE pathways conspire in their control of G1-phase cell cycle progression. p21cip1 expression decreases CDK2 activity214, leading to decreased CDK2-mediated pRb hyperphosphorylation and inactivation237. One downstream consequence of NC-XRE mediated p21cip1 induction is the prolonged presence of hypophosphorylated ‘active’ pRb (Figure 6). Since this form of pRb interacts with the AHR to facilitate XRE-mediated target gene expression, it is tempting to speculate that the NC-XRE might synergize XRE-driven target gene expression, most notably of CYP1A1238(Figure 3). Such a scenario predicts that maximal induction of CYP1A1 would lead to enhanced metabolic clearance of AHR ligands and provide a counter balance to prevent sustained AHR signaling and subsequent deleterious consequences.
Fig. 6. Cross-talk between the XRE- and NC-XRE-driven pathways during cell cycle progression.

AHR activation and nuclear translocation results in induction of XRE- and NC-XRE-driven target genes. In cycling cells, prolonged activation of the AHR results in enhanced p21cip1 expression and inhibition of cyclin-dependent kinase 2 (CDK2) activity, thus preventing pRb inactivation resulting in cell cycle arrest. Induction of CYP1A1—which is enhanced by the AHR interaction with pRb—results in metabolic clearance of receptor ligands and subsequent inactivation of the AHR. This in turn, silences p21cip1 expression allowing CDK2 to inactivate pRb by hyperphosphorylation, thus facilitating G1 phase cell cycle progression. The equilibrium between the XRE and NC-XRE pathways and consequent effects on AHR activity thus influences passage through G1 phase.
In the context of G1-phase cell cycle progression, the evidence clearly shows that sustained AHR activity in response to TCDD induction results in cell cycle arrest. This occurs because TCDD resists metabolic degradation and thus constitutes a persistent agonist that sustains AHR activity. Why loss of the AHR also leads to prolonged cell doubling time is less clear145,146. One possibility is that the AHR contributes to growth promoting processes during G1-phase that are lost when the receptor is absent. Indeed there is evidence to support this notion, including recent studies showing AHR induced cell proliferation in cancer cell lines239,240 possibly through a mechanism involving the Akt pathway241,242. There is also evidence to suggest that AHR activation up-regulates the proliferative transcription factor c-Myc through an NF-κB element via the AHR/RelA interaction172. Finally, it is known that AHR activation leads to increased expression of the early response proto-oncogenes Fos and Jun 243,244, possibly through activation of the p38 MAPK pathway245. The implication is that the AHR regulates both positive and negative growth promoting signals that provide balance to an orchestrated program regulating passage through G1-phase of the cell cycle while ensuring that entry into S phase does not occur prematurely. The evidence for endogenous AHR activation during liver regeneration following PH—and possibly during cellular proliferation in general—is entirely consistent with that viewpoint. We previously observed in the p21cip1 knock out mice, that the absence of p21cip1 expression in the regenerating liver also prevented the transient AHR-mediated induction of CYP1A1158. This suggests that the absence of p21cip1 hastens G1-phase pRb inactivation due to enhanced CDK-mediated hyperphosphorylation, which in turn curtails CYP1A1 induction. This interplay between p21cip1 and CYP1A1 induction further highlights the cross-talk between NC-XRE and XRE driven transcriptional events. Although our findings with the p27kip1 knock out mouse model indicated that this CDK inhibitor plays no role in the TCDD-induced growth inhibition during liver regeneration158, p27kip1 is a known AHR target gene in cell lines suggesting that it may yet play a role in G1-phase passage in vivo under certain conditions156,157,238. Our results with the PH model and the proposed signaling pathwaypredict that liver regeneration would be delayed in liver specific conditional AHR knock out mice subjected to PH. However, these experiments have not been performed to date.
The recent discovery of the NC-XRE and the research showing its role in regulating novel AHR target genes has shed new light on the landscape of AHR-mediated transcriptional regulation. While our picture of AHR signaling likely remains incomplete, it is tempting to speculate that NC-XRE mediated genes represent a functionally distinct and hitherto uncharacterized class of AHR target genes. This is especially intriguing given transcriptomic studies noted above showing large percentages of TCDD induced AHR target genes lacking known AHR DNA-binding motifs169,201-204. While none of these studies have directly investigated the connection between the AHR and KLF6, a 2011 study by Dere et al. found via RegionMiner analysis that KLF6 and ZF9 (another name for KLF6) transcription factor matrices were over-represented in AHR enriched regions at both 2 and 24 hours post TCDD treatment (1.41 and 1.37 fold for KLF6 matrices, 1.78 and 1.74 for ZF9 matrices, respectively)202. Interestingly, they also noted an over-representation of KLF6 binding-sites within 50 base pairs of a XRE at 2 and 24 hours following TCDD treatment (2.36 and 2.87 fold respectively)202. Further, it was noted in a 2012 study using ChIP-Seq that KLF family binding sites are over-represented in AHR/Arnt target genes in MCF-7 cells treated with TCDD, though these results do not specify which KLF family member binding-site is over-represented (supplemental information)169. These are not the only striking commonalities between the KLF6 and AHR fields. Studies assessing the role of KLF6 in cell-cycle progression suggest that KLF6 controls G1 phase cell cycle progression through a mechanism which affects pRb hyperphosphorylation234,246. Specifically, it has been reported that akin to the AHR, loss of KLF6 prolongs cell doubling time247. These studies suggest that while KLF6 up-regulates p21cip1 expression in a number of experimental paradigms, like the AHR, KLF6 also plays an important role in driving the G1/S-phase transition in dividing cells234,246,248,249. Interestingly, studies have also reported that loss of KLF6 leads to tumor promotion following chronic diethylnitrosoamine (DEN) exposure, with increases in hepatocellular carcinoma and these tumors mirroring the gene signatures of highly aggressive human hepatocellular carcinoma250,251. Tumor promotion following DEN treatment has also been shown for the AHR in combination with various exogenous ligands252-254. Further, KLF6 has recently been suggested to function in T-cell activation255 and in NF-κB signaling via p65-dependent transcription256; the AHR has also been implicated in T-cell activation257 and in NF-κB signaling with p65258. Moreover, while total knock-out of the AHR in mice is not lethal, it does result in significantly decreased litter sizes. whereas total KLF6 knock-out mice are embryonic lethals. Furthermore, the livers in AHR−/− and KLF6+/− mice exhibit phenotypic commonalities135,223,247. These examples serve to highlight the parallels that exist between AHR and KLF6 action, consistent with their role as a transcriptional complex.
Section V: Conclusions
Given that almost four decades of research in the AHR field has yet to identify the precise role of the AHR in TCDD toxicity, it is our contention that a greater emphasis on the AHR/KLF6 complex bound to NC-XRE sites will fill some of the existing gaps in understanding as it relates to TCDD toxicity and AHR action in general. Accordingly, the identification of the AHR/KLF6 complex and NC-XRE recognition site constitute a paradigm shift in AHR biology. Future studies focused on this novel complex should be considered a research priority, and hold promise to reward the field with unanticipated discoveries that have eluded the field to date. While we have clearly demonstrated the role of the NC-XRE in the regulation of several genes using ChIP, future studies should leverage the considerable power of the ‘omics’ approaches to characterize the NC-XRE pathway at the genomic, transcriptomic, and proteomic levels. Such an approach can determine the extent to which novel gene regulation through the NC-XRE—and conceivably hitherto unknown AHR DNA-binding motifs—is involved in the well-known but poorly understood functions of the AHR. These technologies can be employed to address old questions about AHR function, like the proliferative role of the AHR in the G1/S transition or the pathophysiologies of TCDD induced wasting syndrome. The recent observations in the studies that examined the NC-XRE also highlighted the cross-talk that exists between NC-XRE- and XRE-driven AHR target genes to regulate complex signaling pathways. This interplay is clearly exemplified by the cell cycle control studies, but it is likely that similar observations will be made for the repertoire of genes involved in other signaling cascades regulated by the AHR.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors would like to thank Dr. Michael Patterson for helpful discussion during the preparation of this manuscript.
This work was funded by grants from the National Institute of Environmental Health Sciences T32ES007254 and F31ES023305 (to DPJ) and P30ES006676 (to CJE). The taxpayers are gratefully acknowledged as well.
Literature Cited
- 1.Poland A, Glover E, Kende AS. Journal of Biological Chemistry. 1976;251:4936–4946. [PubMed] [Google Scholar]
- 2.Denison MS, Soshilov AA, He G, DeGroot DE, Zhao B. Toxicol. Sci. 2011;124:1–22. doi: 10.1093/toxsci/kfr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stevens EA, Mezrich JD, Bradfield CA. Immunology. 2009;127:299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Veldhoen M, Duarte JH. Curr. Opin. Immunol. 2010;22:747–752. doi: 10.1016/j.coi.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Abdelrahim M, Ariazi E, Kim K, Khan S, Barhoumi R, Burghardt R, Liu S, Hill D, Finnell R, Wlodarczyk B, Jordan VC, Safe S. Cancer Research. 2006;66:2459–2467. doi: 10.1158/0008-5472.CAN-05-3132. [DOI] [PubMed] [Google Scholar]
- 6.Lindén J, Lensu S, Tuomisto J, Pohjanvirta R. Front Neuroendocrinol. 2010;31:452–478. doi: 10.1016/j.yfrne.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Marlowe JL, Puga A. J. Cell. Biochem. 2005;96:1174–1184. doi: 10.1002/jcb.20656. [DOI] [PubMed] [Google Scholar]
- 8.Chopra M, Schrenk D. Crit. Rev. Toxicol. 2011;41:292–320. doi: 10.3109/10408444.2010.524635. [DOI] [PubMed] [Google Scholar]
- 9.Wells PG, Lee CJJ, McCallum GP, Perstin J, Harper PA. Handbook of experimental pharmacology. 2010;196:131–162. doi: 10.1007/978-3-642-00663-0_6. [DOI] [PubMed] [Google Scholar]
- 10.Dietrich C, Kaina B. Carcinogenesis. 2010;31:1319–1328. doi: 10.1093/carcin/bgq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White SS, Birnbaum LS. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2009;27:197–211. doi: 10.1080/10590500903310047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korashy HM, El-Kadi AOS. Drug metabolism reviews. 2006;38:411–450. doi: 10.1080/03602530600632063. [DOI] [PubMed] [Google Scholar]
- 13.Bock KW, Köhle C. Biochemical Pharmacology. 2006;72:393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
- 14.Wilson SR, Joshi AD, Elferink CJ. J. Pharmacol. Exp. Ther. 2013;345:419–429. doi: 10.1124/jpet.113.203786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denison MS, Nagy SR. Annual Review of Pharmacology and Toxicology. 2003:309–334. doi: 10.1146/annurev.pharmtox.43.100901.135828. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen LP, Bradfield CA. Chem. Res. Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alfred LJ, Gelboin HV. Science. 1967;157:75–76. doi: 10.1126/science.157.3784.75. [DOI] [PubMed] [Google Scholar]
- 18.Nebert DW, Gelboin HV. Journal of Biological Chemistry. 1968;243:6242–6249. [PubMed] [Google Scholar]
- 19.Nebert DW, Gelboin HV. Journal of Biological Chemistry. 1968;243:6250–6261. [PubMed] [Google Scholar]
- 20.Yamagiwa K, Ichikawa K. The Journal of Cancer Research. 1918;3:1–29. [Google Scholar]
- 21.Yamagiwa K, Murayama K. The Journal of Cancer Research. 1924;8:119–136. [Google Scholar]
- 22.Narat JK. The Journal of Cancer Research. 1925;9:135–147. [Google Scholar]
- 23.Cook JW, Hewett CL, Hieger I. J. Chem. Soc. 1933:395–405. [Google Scholar]
- 24.Barry G, Cook JW, Hewett CL, Haslewood GAD, Hieger I, Kennaway FRS. Proceedings of the Royal Society. 1935;117:313–351. [Google Scholar]
- 25.Conney AH, Miller EC, Miller JA. Cancer Research. 1956;16:450–459. [PubMed] [Google Scholar]
- 26.Conney AH, Miller EC, Miller JA. Journal of Biological Chemistry. 1957;228:753–766. [PubMed] [Google Scholar]
- 27.Conney AH, Gillette JR, Inscoe ER, Trams ER, Posner HS. Science. 1959;130:1478–1479. doi: 10.1126/science.130.3387.1478. [DOI] [PubMed] [Google Scholar]
- 28.White J, White A. Journal of Biological Chemistry. 1939;131:149–161. [Google Scholar]
- 29.Friedewald WF, Rous P. Journal of Experimental Medicine. 1944;80:101–126. doi: 10.1084/jem.80.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poland A, Glover E. Molecular Pharmacology. 1973;9:736–747. [PubMed] [Google Scholar]
- 31.Poland A, Glover E. Environmental Health Perspectives. 1973:245–251. doi: 10.1289/ehp.7305245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kimbrough RD, Carter CD, Liddle JA, Cline RE. Arch. Environ. Health. 1977;32:77–86. doi: 10.1080/00039896.1977.10667259. [DOI] [PubMed] [Google Scholar]
- 33.Baccarelli A, Pesatori AC, Masten SA, Patterson DG., Jr Toxicology Letters. 2004;149:287–293. doi: 10.1016/j.toxlet.2003.12.062. [DOI] [PubMed] [Google Scholar]
- 34.Sorg O, Zennegg M, Schmid P, Fedosyuk R, Valikhnovskyi R, Gaide O, Kniazevych V, Saurat J-H. The Lancet. 2009;374:1179–1185. doi: 10.1016/S0140-6736(09)60912-0. [DOI] [PubMed] [Google Scholar]
- 35.Kimmig J, Schulz KH. Dermatologica. 1957;115:540–546. [PubMed] [Google Scholar]
- 36.Gupta BN, Vos JG, Moore JA, Zinkl JG, Bullock BC. Environmental Health Perspectives. 1973:125–150. doi: 10.1289/ehp.7305125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gasiewicz TA, Holscher MA, Neal RA. Toxicology and applied Pharmacology. 1980;54:469–488. doi: 10.1016/0041-008x(80)90174-x. [DOI] [PubMed] [Google Scholar]
- 38.Thomas PE, Kouri RE, Hutton JJ. Biochem Genet. 1972:157–168. doi: 10.1007/BF00486400. [DOI] [PubMed] [Google Scholar]
- 39.Schwetz BA, Norris JM, Sparschu GL, Rowe VK, Gehring PJ, E. J. L, G. C. G Environmental Health Perspectives. 1973;5:87–99. doi: 10.1289/ehp.730587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McConnell EE, Moore JA, Dalgard DW. Toxicology and Applied Pharmacology. 1978 doi: 10.1016/s0041-008x(78)80042-8. [DOI] [PubMed] [Google Scholar]
- 41.Henck JM, New MA, Kociba RJ, Rao KS. Toxicology and applied Pharmacology. 1981;59:405–407. doi: 10.1016/0041-008x(81)90212-x. [DOI] [PubMed] [Google Scholar]
- 42.Gielen JE, Goujon FM, Nebert DW. Journal of Biological Chemistry. 1972;247:1125–1137. [PubMed] [Google Scholar]
- 43.Poland AP, Glover E, Robinson JR, Nebert DW. Journal of Biological Chemistry. 1974;249:5599–5606. [PubMed] [Google Scholar]
- 44.Guenthner TM, Nebert DW. Journal of Biological Chemistry. 1977;252:8981–8989. [PubMed] [Google Scholar]
- 45.Okey AB, Bondy GP, Mason ME, Kahl GF, Eisen HJ, Guenthner TM, Nebert DW. Journal of Biological Chemistry. 1979;254:11636–11648. [PubMed] [Google Scholar]
- 46.Chang C-Y, Smith DR, Prasad VS, Sidman CL, Nebert DW, Puga A. Pharmacogenetics and Genomics. 1993;3:312. doi: 10.1097/00008571-199312000-00005. [DOI] [PubMed] [Google Scholar]
- 47.Okey AB, Bondy GP, Mason ME, Nebert DW, Forster-Gibson CJ, Muncan J, Dufresne MJ. Journal of Biological Chemistry. 1980;255:11415–11422. [PubMed] [Google Scholar]
- 48.Hannah RR, Nebert DW, Eisen HJ. Journal of Biological Chemistry. 1981;256:4584–4590. [PubMed] [Google Scholar]
- 49.Greenlee WF, Poland A. Journal of Biological Chemistry. 1979;254:9814–9821. [PubMed] [Google Scholar]
- 50.Tierney B, Weaver D, Heintz NH, Schaeffer WI, Bresnick E. Archives of biochemistry and biophysics. 1980;200:513–523. doi: 10.1016/0003-9861(80)90383-5. [DOI] [PubMed] [Google Scholar]
- 51.Tukey RH, Hannah RR, Negishi M, Nebert DW. Cell. 1982;31:275–284. doi: 10.1016/0092-8674(82)90427-5. [DOI] [PubMed] [Google Scholar]
- 52.Israel DI, Whitlock JP., Jr. Journal of Biological Chemistry. 1984;259:5400–5402. [PubMed] [Google Scholar]
- 53.Jones P, Galeazzi D, Fisher J, Whitlock J. Science. 1985;227:1499–1502. doi: 10.1126/science.3856321. [DOI] [PubMed] [Google Scholar]
- 54.Jones PB, Durrin LK, Galeazzi DR, Whitlock JP., Jr. Proceedings of the National Academy of Sciences. 1986;83:2802–2806. doi: 10.1073/pnas.83.9.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Denison MS, Fisher JM, Whitlock JP., Jr. Proc. Natl. Acad. Sci. U.S.A. 1988;85:2528–2532. doi: 10.1073/pnas.85.8.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Denison MS, Fisher JM, Whitlock JP., Jr. Journal of Biological Chemistry. 1988;263:17221–17224. [PubMed] [Google Scholar]
- 57.Shen ES, Whitlock JP., Jr. Journal of Biological Chemistry. 1992;267:6815–6819. [PubMed] [Google Scholar]
- 58.Watson AJ, Hankinson O. Journal of Biological Chemistry. 1992;266:6874–6878. [PubMed] [Google Scholar]
- 59.Lusska A, Shen E, Whitlock JP., Jr. Journal of Biological Chemistry. 1993;268:6575–6580. [PubMed] [Google Scholar]
- 60.Prokipcak RD, Okey AB. Archives of biochemistry and biophysics. 1988;267:811–828. doi: 10.1016/0003-9861(88)90091-4. [DOI] [PubMed] [Google Scholar]
- 61.Perdew GH. Biochemical and Biophysical Research Communications. 1992;182:55–62. doi: 10.1016/s0006-291x(05)80111-1. [DOI] [PubMed] [Google Scholar]
- 62.Miller AG, Israel D, Whitlock JP., Jr. Journal of Biological Chemistry. 1983;258:3523–3527. [PubMed] [Google Scholar]
- 63.Hankinson O. Somatic Cell Genet. 1983;9:497–514. doi: 10.1007/BF01543050. [DOI] [PubMed] [Google Scholar]
- 64.Hankinson O, Andersen RD, Birren BW, Sander F, Negishi M, Nebert DW. J. Biol. Chem. 1985;260:1790–1795. [PubMed] [Google Scholar]
- 65.Hankinson O. Proc. Natl. Acad. Sci. U.S.A. 1979;76:373–376. doi: 10.1073/pnas.76.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hankinson O. Somatic Cell Genet. 1981;7:373–388. doi: 10.1007/BF01542983. [DOI] [PubMed] [Google Scholar]
- 67.Legraverend C, Hannah RR, Eisen HJ, Owens IS, Nebert DW, Hankinson O. J. Biol. Chem. 1982;257:6402–6407. [PubMed] [Google Scholar]
- 68.Henry EC, Rucci G, Gasiewicz TA. Biochemistry. 1989;28:6430–6440. doi: 10.1021/bi00441a041. [DOI] [PubMed] [Google Scholar]
- 69.Elferink CJ, Gasiewicz TA, Whitlock JP., Jr. Journal of Biological Chemistry. 1990;265:20708–20712. [PubMed] [Google Scholar]
- 70.Gasiewicz TA, Elferink CJ, Henry EC. Biochemistry. 1991;30:2909–2916. doi: 10.1021/bi00225a026. [DOI] [PubMed] [Google Scholar]
- 71.Hoffman EC, Reyes H, Chu FF, Sander F, Conley LH, Brooks BA, Hankinson O. Science. 1991;252:954–958. doi: 10.1126/science.1852076. [DOI] [PubMed] [Google Scholar]
- 72.Reyes H, Reisz-Porszasz S, Hankinson O. Science. 1992;256:1193–1195. doi: 10.1126/science.256.5060.1193. [DOI] [PubMed] [Google Scholar]
- 73.Bradfield CA, Glover E, Poland A. Molecular Pharmacology. 1991 [PubMed] [Google Scholar]
- 74.Burbach KM, Poland A, Bradfield CA. Proc. Natl. Acad. Sci. U.S.A. 1992;89:8185–8189. doi: 10.1073/pnas.89.17.8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dolwick KM, Schmidt JV, Carver LA, Swanson HI, Bradfield CA. Molecular Pharmacology. 1993;4:911–917. [PubMed] [Google Scholar]
- 76.Denis M, Wikström AC, Gustafsson JA. Journal of Biological Chemistry. 1987 [Google Scholar]
- 77.Perdew GH. J. Biol. Chem. 1988;263:13802–13805. [PubMed] [Google Scholar]
- 78.Pongratz I, Mason GG, Poellinger L. Journal of Biological Chemistry. 1992;267:13728–13734. [PubMed] [Google Scholar]
- 79.McGuire J, Whitelaw ML, Pongratz I, Gustafsson JA, Poellinger L. Mol. Cell. Biol. 1994;14:2438–2446. doi: 10.1128/mcb.14.4.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Probst MR, Reisz-Porszasz S, Agbunag RV, Ong MS, Hankinson O. Molecular Pharmacology. 1993;44:511–518. [PubMed] [Google Scholar]
- 81.Pollenz RS, Sattler CA, Poland A. Molecular Pharmacology. 1994;45:428–438. [PubMed] [Google Scholar]
- 82.Reisz-Porszasz S, Probst MR, Fukunaga BN, Hankinson O. Mol. Cell. Biol. 1994;14:6075–6086. doi: 10.1128/mcb.14.9.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fukunaga BN, Probst MR, Reisz-Porszasz S, Hankinson O. Journal of Biological Chemistry. 1995;270:29270–29278. doi: 10.1074/jbc.270.49.29270. [DOI] [PubMed] [Google Scholar]
- 84.Ikuta T, Eguchi H, Tachibana T, Yoneda Y, Kawajiri K. Journal of Biological Chemistry. 1998;273:2895–2904. doi: 10.1074/jbc.273.5.2895. [DOI] [PubMed] [Google Scholar]
- 85.Kewley RJ, Whitelaw ML, Chapman-Smith A. Int. J. Biochem. Cell Biol. 2004;36:189–204. doi: 10.1016/s1357-2725(03)00211-5. [DOI] [PubMed] [Google Scholar]
- 86.Evans MR, Card PB, Gardner KH. Proceedings of the National Academy of Sciences. 2009;106:2617–2622. doi: 10.1073/pnas.0808270106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rezvani HR, Ali N, Nissen LJ, Harfouche G, de Verneuil H, Taïeb A, Mazurier F. Journal of Investigative Dermatology. 2011;131:1793–1805. doi: 10.1038/jid.2011.141. [DOI] [PubMed] [Google Scholar]
- 88.Chen HS, Perdew GH. Journal of Biological Chemistry. 1994;269:27554–27558. [PubMed] [Google Scholar]
- 89.Perdew G, Bradfield C. IUBMB Life. 1996;39:589–593. doi: 10.1080/15216549600201651. [DOI] [PubMed] [Google Scholar]
- 90.Meyer BK, Pray-Grant MG, Heuvel JPV, Perdew GH. Mol. Cell. Biol. 1998;18:978–988. doi: 10.1128/mcb.18.2.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ma Q, Whitlock JP., Jr. Journal of Biological Chemistry. 1997;272:8878–8884. [PubMed] [Google Scholar]
- 92.Carver LA, Bradfield CA. Journal of Biological Chemistry. 1997;272:11452–11456. doi: 10.1074/jbc.272.17.11452. [DOI] [PubMed] [Google Scholar]
- 93.Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. Cell Stress Chaperones. 1996;1:237–250. doi: 10.1379/1466-1268(1996)001<0237:apomci>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kazlauskas A, Poellinger L, Pongratz I. J. Biol. Chem. 1999;274:13519–13524. doi: 10.1074/jbc.274.19.13519. [DOI] [PubMed] [Google Scholar]
- 95.Soshilov A, Denison MS. J. Biol. Chem. 2008;283:32995–33005. doi: 10.1074/jbc.M802414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Henry EC, Gasiewicz TA. Molecular Pharmacology. 2003;63:392–400. doi: 10.1124/mol.63.2.392. [DOI] [PubMed] [Google Scholar]
- 97.Ikuta T, Tachibana T, Watanabe J, Yoshida M, Yoneda Y, Kawajiri K. J. Biochem. 2000;127:503–509. doi: 10.1093/oxfordjournals.jbchem.a022633. [DOI] [PubMed] [Google Scholar]
- 98.Backlund M, Ingelman-Sundberg M. Cellular Signalling. 2005;17:39–48. doi: 10.1016/j.cellsig.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 99.Pongratz I, Strömstedt PE, Mason GG, Poellinger L. J. Biol. Chem. 1991;266:16813–16817. [PubMed] [Google Scholar]
- 100.Carrier F, Owens RA, Nebert DW, Puga A. Mol. Cell. Biol. 1992;12:1856–1863. doi: 10.1128/mcb.12.4.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mahon MJ, Gasiewicz TA. Archives of biochemistry and biophysics. 1995;318:166–174. doi: 10.1006/abbi.1995.1217. [DOI] [PubMed] [Google Scholar]
- 102.Elferink CJ, Whitlock JP., Jr. Journal of Biological Chemistry. 1990;265:5718–5721. [PubMed] [Google Scholar]
- 103.Jones PB, Durrin LK, Fisher JM, Whitlock JP., Jr. Mol. Cell. Biol. 1986 [PubMed] [Google Scholar]
- 104.Swanson HI, Chan WK, Bradfield CA. J. Biol. Chem. 1995;270:26292–26302. doi: 10.1074/jbc.270.44.26292. [DOI] [PubMed] [Google Scholar]
- 105.Mimura J, Ema M, Sogawa K, Fujii-Kuriyama Y. Genes & Development. 1999;13:20–25. doi: 10.1101/gad.13.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ma Q, Baldwin KT. Journal of Biological Chemistry. 2000;275:8432–8438. doi: 10.1074/jbc.275.12.8432. [DOI] [PubMed] [Google Scholar]
- 107.MacPherson L, Tamblyn L, Rajendra S, Bralha F, McPherson JP, Matthews J. Nucleic Acids Research. 2012;41:1604–1621. doi: 10.1093/nar/gks1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.MacPherson L, Ahmed S, Tamblyn L, Krutmann J, Förster I, Weighardt H, Matthews J. Int J Mol Sci. 2014;15:7939–7957. doi: 10.3390/ijms15057939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reiners JJ, Jones CL, Hong N, Clift RE, Elferink C. Mol. Carcinog. 1997;19:91–100. doi: 10.1002/(sici)1098-2744(199707)19:2<91::aid-mc4>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 110.Arpiainen S, Raffalli-Mathieu F, Lang M, Polkonen O, Hakkola J. Molecular Pharmacology. 2005;67:1325–1333. doi: 10.1124/mol.104.008078. [DOI] [PubMed] [Google Scholar]
- 111.Sutter TR, Tang YM, Hayes CL, Wo YY, Jabs EW, Li X, Yin H, Cody CW, Greenlee WF. J. Biol. Chem. 1994;269:13092–13099. [PubMed] [Google Scholar]
- 112.Friling RS, Bensimon A, Tichauer Y, Daniel V. Proceedings of the National Academy of Sciences. 1990;87:6258–6262. doi: 10.1073/pnas.87.16.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Dunn TJ, Lindahl R, Pitot HC. Journal of Biological Chemistry. 1988;263:10878–10886. [PubMed] [Google Scholar]
- 114.Ma Q, Kinneer K, Bi Y, Chan AM, Kan YW. Biochem. J. 2004;377:205. doi: 10.1042/BJ20031123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang L, He X, Szklarz GD, Bi Y, Rojanasakul Y, Ma Q. Archives of biochemistry and biophysics. 2013;537:31–38. doi: 10.1016/j.abb.2013.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weiss C, Kolluri SK, Kiefer F, Gottlicher M. Experimental Cell Research. 1996;226:154–163. doi: 10.1006/excr.1996.0214. [DOI] [PubMed] [Google Scholar]
- 117.Gu YZ, Hogenesch JB, Bradfield CA. Annual Review of Pharmacology and Toxicology. 2000;40:519–561. doi: 10.1146/annurev.pharmtox.40.1.519. [DOI] [PubMed] [Google Scholar]
- 118.Tsuji G, Takahara M, Uchi H, Matsuda T, Chiba T, Takeuchi S, Yasukawa F, Moroi Y, Furue M. J. Invest. Dermatol. 2012;132:59–68. doi: 10.1038/jid.2011.194. [DOI] [PubMed] [Google Scholar]
- 119.Mitchell KA, Lockhart CA, Huang G, Elferink CJ. Molecular Pharmacology. 2006;70:163–170. doi: 10.1124/mol.106.023465. [DOI] [PubMed] [Google Scholar]
- 120.Mitchell KA, Elferink CJ. Biochemical Pharmacology. 2009;77:947–956. doi: 10.1016/j.bcp.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McMillan BJ, Bradfield CA. Molecular Pharmacology. 2007;72:487–498. doi: 10.1124/mol.107.037259. [DOI] [PubMed] [Google Scholar]
- 122.Hahn ME. Chemico-Biological Interactions. 2002;141:131–160. doi: 10.1016/s0009-2797(02)00070-4. [DOI] [PubMed] [Google Scholar]
- 123.Powell-Coffman JA, Bradfield CA, Wood WB. Proceedings of the National Academy of Sciences. 1998;95:2844–2849. doi: 10.1073/pnas.95.6.2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Uno S, Dalton TP, Derkenne S, Curran CP, Miller ML, Shertzer HG, Nebert DW. Molecular Pharmacology. 2004;65:1225–1237. doi: 10.1124/mol.65.5.1225. [DOI] [PubMed] [Google Scholar]
- 125.Shimada T, Fujii-Kuriyama Y. Cancer Science. 2004;95:1–6. doi: 10.1111/j.1349-7006.2004.tb03162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ma Q, Lu AYH. Drug Metabolism and Disposition. 2007;35:1009–1016. doi: 10.1124/dmd.107.015826. [DOI] [PubMed] [Google Scholar]
- 127.McMillan BJ, Bradfield CA. Proc. Natl. Acad. Sci. U.S.A. 2007;104:1412–1417. doi: 10.1073/pnas.0607296104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hao N, Lee KL, Furness SGB, Bosdotter C, Poellinger L, Whitelaw ML. Molecular Pharmacology. 2012;82:1082–1093. doi: 10.1124/mol.112.078873. [DOI] [PubMed] [Google Scholar]
- 129.Pansoy A, Ahmed S, Valen E, Sandelin A, Matthews J. Toxicol. Sci. 2010;117:90–100. doi: 10.1093/toxsci/kfq096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gouédard C, Barouki R, Morel Y. Mol. Cell. Biol. 2004;24:5209–5222. doi: 10.1128/MCB.24.12.5209-5222.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matikainen T, Perez GI, Jurisicova A, Pru JK, Schlezinger JJ, Ryu HY, Laine J, Sakai T, Korsmeyer SJ, Casper RF, Sherr DH, Tilly JL. Nat. Genet. 2001;28:355–360. doi: 10.1038/ng575. [DOI] [PubMed] [Google Scholar]
- 132.Harper TA, Joshi AD, Elferink CJ. J. Pharmacol. Exp. Ther. 2013;344:579–588. doi: 10.1124/jpet.112.201111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lowe MM, Mold JE, Kanwar B, Huang Y, Louie A, Pollastri MP, Wang C, Patel G, Franks DG, Schlezinger J, Sherr DH, Silverstone AE, Hahn ME, McCune JM. PLoS ONE. 2014;9:e87877. doi: 10.1371/journal.pone.0087877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Budinsky RA, Schrenk D, Simon T, Van den Berg M, Reichard JF, Silkworth JB, Aylward LL, Brix A, Gasiewicz T, Kaminski N, Perdew G, Starr TB, Walker NJ, Rowlands JC. Crit. Rev. Toxicol. 2014;44:83–119. doi: 10.3109/10408444.2013.835787. [DOI] [PubMed] [Google Scholar]
- 135.Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6731–6736. doi: 10.1073/pnas.93.13.6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lund AK, Goens MB, Kanagy NL, Walker MK. Toxicology and applied Pharmacology. 2003;193:177–187. doi: 10.1016/j.taap.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 137.Vasquez A, Atallah-Yunes N, Smith FC, You X, Chase SE, Silverstone AE, Vikstrom KL. Cardiovasc Toxicol. 2003;3:153–163. doi: 10.1385/ct:3:2:153. [DOI] [PubMed] [Google Scholar]
- 138.Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Toxicological sciences. 2002;67:63–74. doi: 10.1093/toxsci/67.1.63. [DOI] [PubMed] [Google Scholar]
- 139.Burns KA, Zorrilla LM, Hamilton KJ, Reed CE, Birnbaum LS, Korach KS. Toxicological sciences. 2013;136:514–526. doi: 10.1093/toxsci/kft208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gottlicher M, Wiebel FJ. Toxicology and applied Pharmacology. 1991;111:496–503. doi: 10.1016/0041-008x(91)90253-b. [DOI] [PubMed] [Google Scholar]
- 141.Wiebel FJ, Klose U, Kiefer F. Toxicology Letters. 1991 doi: 10.1016/0378-4274(91)90130-x. [DOI] [PubMed] [Google Scholar]
- 142.Ma Q, Whitlock JP., Jr. Mol. Cell. Biol. 1996;16:2144–2150. doi: 10.1128/mcb.16.5.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shimba S, Komiyama K, Moro I, Tezuka M. J. Biochem. 2002;132:795–802. doi: 10.1093/oxfordjournals.jbchem.a003289. [DOI] [PubMed] [Google Scholar]
- 144.Barhoover MA, Hall JM, Greenlee WF, Thomas RS. Molecular Pharmacology. 2010;77:195–201. doi: 10.1124/mol.109.059675. [DOI] [PubMed] [Google Scholar]
- 145.Elizondo G, Fernandez-Salguero P, Sheikh MS, Kim G-Y, Fornace AJ, Lee KS, Gonzalez FJ. Molecular. 2000;57:1056–1063. [PubMed] [Google Scholar]
- 146.Tohkin M, Fukuhara M, Elizondo G, Tomita S, Gonzalez FJ. Molecular Pharmacology. 2000;58:845–851. doi: 10.1124/mol.58.4.845. [DOI] [PubMed] [Google Scholar]
- 147.Ge N-L, Elferink CJ. J. Biol. Chem. 1998;273:22708–22713. doi: 10.1074/jbc.273.35.22708. [DOI] [PubMed] [Google Scholar]
- 148.Elferink CJ, Ge NL, Levine A. Molecular Pharmacology. 2001;59:664–673. doi: 10.1124/mol.59.4.664. [DOI] [PubMed] [Google Scholar]
- 149.Helin K, Harlow E, Fattaey A. Mol. Cell. Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Weintraub SJ, Prater CA, Dean DC. Nature. 1992;358:259–261. doi: 10.1038/358259a0. [DOI] [PubMed] [Google Scholar]
- 151.Weintraub SJ, Chow KN, Luo RX, Zhang SH, He S, Dean DC. Nature. 1995;375:812–815. doi: 10.1038/375812a0. [DOI] [PubMed] [Google Scholar]
- 152.Chang C-C, Sue Y-M, Yang N-J, Lee Y-H, Juan S-H. PLoS ONE. 2014;9:e92793–e92793. doi: 10.1371/journal.pone.0092793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Puga A, Barnes SJ, Dalton TP, Chang CY, Knudsen ES, Maier MA. J. Biol. Chem. 2000;275:2943–2950. doi: 10.1074/jbc.275.4.2943. [DOI] [PubMed] [Google Scholar]
- 154.Elferink CJ. Prog Cell Cycle Res. 2003;5:261–267. [PubMed] [Google Scholar]
- 155.Marlowe JL, Knudsen ES, Schwemberger S, Puga A. J. Biol. Chem. 2004;279:29013–29022. doi: 10.1074/jbc.M404315200. [DOI] [PubMed] [Google Scholar]
- 156.Kolluri S, Weiss C, Koff A, Gottlicher M. Genes & Development. 1999;13:1742–1753. doi: 10.1101/gad.13.13.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Faust D, Kletting S, Ueberham E, Dietrich C. Toxicology Letters. 2013;223:73–80. doi: 10.1016/j.toxlet.2013.08.022. [DOI] [PubMed] [Google Scholar]
- 158.Jackson DP, Li H, Mitchell KA, Joshi AD, Elferink CJ. Molecular Pharmacology. 2014:533–541. doi: 10.1124/mol.113.089730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Huang G, Elferink CJ. Molecular Pharmacology. 2005;67:88–96. doi: 10.1124/mol.104.002410. [DOI] [PubMed] [Google Scholar]
- 160.Schnekenburger M, Talaska G, Puga A. Mol. Cell. Biol. 2007;27:7089–7101. doi: 10.1128/MCB.00838-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schnekenburger M, Peng L, Puga A. Biochim. Biophys. Acta. 2007;1769:569–578. doi: 10.1016/j.bbaexp.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gomez-Duran A, Ballestar E, Carvajal-Gonzalez JM, Marlowe JL, Puga A, Esteller M, Fernandez-Salguero PM. J. Mol. Biol. 2008;380:1–16. doi: 10.1016/j.jmb.2008.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Englert NA, Turesky RJ, Han W, Bessette EE, Spivack SD, Caggana M, Spink DC, Spink BC. Biochemical Pharmacology. 2012;84:722–735. doi: 10.1016/j.bcp.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kurita H, Schnekenburger M, Ovesen JL, Xia Y, Puga A. Toxicologial Sciences. 2014;139:121–132. doi: 10.1093/toxsci/kfu027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shen ES, J P Whitlock J. Journal of Biological Chemistry. 1989;264:17754–17758. [PubMed] [Google Scholar]
- 166.Taylor RT, Wang F, Hsu EL, Hankinson O. Toxicological sciences. 2009;107:1–8. doi: 10.1093/toxsci/kfn217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Beedanagari SR, Taylor RT, Bui P, Wang F, Nickerson DW, Hankinson O. Molecular Pharmacology. 2010;78:608–616. doi: 10.1124/mol.110.064899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ovesen JL, Schnekenburger M, Puga A. Toxico. Sci. 2011;121:123–131. doi: 10.1093/toxsci/kfr032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lo R, Matthews J. Toxicol. Sci. 2012;130:349–361. doi: 10.1093/toxsci/kfs253. [DOI] [PubMed] [Google Scholar]
- 170.Hankinson O. Archives of biochemistry and biophysics. 2005;433:379–386. doi: 10.1016/j.abb.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 171.Solaimani P, Damoiseaux R, Hankinson O. Toxicol. Sci. 2013;136:107–119. doi: 10.1093/toxsci/kft191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim DW, Gazourian L, Quadri SA, Romieu-Mourez R, Sherr DH, Sonenshein GE. Oncogene. 2000;19:5498–5506. doi: 10.1038/sj.onc.1203945. [DOI] [PubMed] [Google Scholar]
- 173.Vogel CFA, Sciullo E, Matsumura F. Biochemical and Biophysical Research Communications. 2007;363:722–726. doi: 10.1016/j.bbrc.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tian Y, Ke S, Denison MS, Rabson AB, Gallo MA. J. Biol. Chem. 1999;274:510–515. doi: 10.1074/jbc.274.1.510. [DOI] [PubMed] [Google Scholar]
- 175.Auyeung DJ, Kessler FK, Ritter JK. Molecular Pharmacology. 2003;63:119–127. doi: 10.1124/mol.63.1.119. [DOI] [PubMed] [Google Scholar]
- 176.Yeager RL, Reisman SA, Aleksunes LM, Klaassen CD. Toxicological sciences. 2009;111:238–246. doi: 10.1093/toxsci/kfp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bock KW. Biochemical Pharmacology. 2012;83:833–838. doi: 10.1016/j.bcp.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 178.Eaton DL, Bammler TK. Toxico. Sci. 1999;49:156–164. doi: 10.1093/toxsci/49.2.156. [DOI] [PubMed] [Google Scholar]
- 179.Vogel CFA, Sciullo E, Li W, Wong P, Lazennec G, Matsumura F. Mol. Endocrinol. 2007;21:2941–2955. doi: 10.1210/me.2007-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hollingshead BD, Beischlag TV, DiNatale BC, Ramadoss P, Perdew GH. Cancer Research. 2008;68:3609–3617. doi: 10.1158/0008-5472.CAN-07-6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.DiNatale BC, Schroeder JC, Francey LJ, Kusnadi A, Perdew GH. Journal of Biological Chemistry. 2010;285:24388–24397. doi: 10.1074/jbc.M110.118570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen P-H, Chang H, Chang JT, Lin P. Oncogene. 2012;31:2555–2565. doi: 10.1038/onc.2011.438. [DOI] [PubMed] [Google Scholar]
- 183.DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS, Laurenzana EM, Omiecinski CJ, Perdew GH. Toxicological sciences. 2010;115:89–97. doi: 10.1093/toxsci/kfq024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Zacharewski TR, Bondy KL, McDonell P, Wu ZF. Cancer Research. 1994 [PubMed] [Google Scholar]
- 185.Safe S, Wormke M, Samudio I. Journal of Mammary Gland Biology and Neoplasia. 2000;5:295–306. doi: 10.1023/a:1009550912337. [DOI] [PubMed] [Google Scholar]
- 186.Safe S, Wang F, Porter W, Duan R, McDougal A. Toxicology Letters. 1998;102:343–347. doi: 10.1016/s0378-4274(98)00331-2. [DOI] [PubMed] [Google Scholar]
- 187.Gillesby BE, Stanostefano M, Porter W, Safe S, Wu ZF, Zacharewski TR. Biochemistry. 1997;36:6080–6089. doi: 10.1021/bi962131b. [DOI] [PubMed] [Google Scholar]
- 188.Wang W, Smith R, Safe S. Archives of biochemistry and biophysics. 1998;356:239–248. doi: 10.1006/abbi.1998.0782. [DOI] [PubMed] [Google Scholar]
- 189.Ohtake F, Takeyama K-I, Matsumoto T, Kitagawa H, Yamamoto Y, Nohara K, Tohyama C, Krust A, Mimura J, Chambon P, Yanagisawa J, Fujii-Kuriyama Y, Kato S. Nature. 2003;423:545–550. doi: 10.1038/nature01606. [DOI] [PubMed] [Google Scholar]
- 190.Safe S, Wormke M. Chem. Res. Toxicol. 2003;16:807–816. doi: 10.1021/tx034036r. [DOI] [PubMed] [Google Scholar]
- 191.Duan R, Porter W, Samudio I, Vyhlidal C, Kladde M, Safe S. Molecular Endocrinology. 1999;13:1511–1521. doi: 10.1210/mend.13.9.0338. [DOI] [PubMed] [Google Scholar]
- 192.Porter W, Wang F, Duan R, Qin C, Castro-Rivera E, Kim K, Safe S. J. Mol. Endocrinol. 2001;26:31–42. doi: 10.1677/jme.0.0260031. [DOI] [PubMed] [Google Scholar]
- 193.Ohtake F, Fujii-Kuriyama Y, Kato S. Biochemical Pharmacology. 2009;77:474–484. doi: 10.1016/j.bcp.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 194.Tijet N, Boutros PC, Moffat ID, Okey AB, Tuomisto J, Pohjanvirta R. Molecular Pharmacology. 2006;69:140–153. doi: 10.1124/mol.105.018705. [DOI] [PubMed] [Google Scholar]
- 195.Boutros PC, Bielefeld KA, Pohjanvirta R, Harper PA. Toxicological sciences. 2009;112:245–256. doi: 10.1093/toxsci/kfp191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Toxicology and applied Pharmacology. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 197.Mimura J, Yamashita K, Nakamura K, Morita M, Takagi TN, Nakao K, Ema M, Sogawa K, Yasuda M, Katsuki M, Fujii-Kuriyama Y. Genes to Cells. 1997;2:645–654. doi: 10.1046/j.1365-2443.1997.1490345.x. [DOI] [PubMed] [Google Scholar]
- 198.Peters JM, Narotsky MG, Elizondo G, Fernandez-Salguero PM, Gonzalez FJ, Abbott BD. Toxico. Sci. 1999;47:86–92. doi: 10.1093/toxsci/47.1.86. [DOI] [PubMed] [Google Scholar]
- 199.Boverhof DR, Burgoon LD, Tashiro C, Chittim B, Harkema JR, Jump DB, Zacharewski TR. Toxicol. Sci. 2005;85:1048–1063. doi: 10.1093/toxsci/kfi162. [DOI] [PubMed] [Google Scholar]
- 200.Frueh FW, Hayashibara KC, Brown PO, Whitlock JP., Jr. Toxicology Letters. 2001;122:189–203. doi: 10.1016/s0378-4274(01)00364-2. [DOI] [PubMed] [Google Scholar]
- 201.Puga A, Maier A, Medvedovic M. Biochemical Pharmacology. 2000;60:1129–1142. doi: 10.1016/s0006-2952(00)00403-2. [DOI] [PubMed] [Google Scholar]
- 202.Dere E, Lo R, Celius T, Matthews J, Zacharewski T. BMC Genomics. 2011:12. doi: 10.1186/1471-2164-12-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kinehara M, Fukuda I, Yoshida K-I, Ashida H. Genes Genet. Syst. 2008;83:455–468. doi: 10.1266/ggs.83.455. [DOI] [PubMed] [Google Scholar]
- 204.Lo R, Celius T, Forgacs AL, Dere E, MacPherson L, Harper P, Zacharewski T, Matthews J. Toxicology and applied Pharmacology. 2011;257:38–47. doi: 10.1016/j.taap.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 205.Son D-S, Rozman K. Arch Toxicol. 2002;76:404–413. doi: 10.1007/s00204-002-0354-6. [DOI] [PubMed] [Google Scholar]
- 206.Binder BR, Christ G, Gruber F, Grubic N, Hufnagl P, Krebs M, Mihaly J, Prager GW. Physiology. 2002;17:56–61. doi: 10.1152/nips.01369.2001. [DOI] [PubMed] [Google Scholar]
- 207.Durand M, Bodker JS, Christensen A, Hansen DDM,M, Jensen JK, Kjelgaard S, Mathiasen L, Pedersen KE, Skeldal S, Wind T, Andreasen PA. Journal of Thrombosis and Haemostasis. 2004;91:438–449. doi: 10.1160/TH03-12-0784. [DOI] [PubMed] [Google Scholar]
- 208.Michalopoulos GK, DeFrances MC. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 209.Irigoyen JP, Muñoz-Cánoves P, Montero L, Koziczak M, Nagamine Y. CMLS, Cell. Mol. Life Sci. 1999;56:104–132. doi: 10.1007/PL00000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tanaka M, Okada K, Ueshima S, Imano M, Ohyanagi H, Carmeliet P, Matsuo O. Fibrinolysis and Proteolysis. 2001;15:2–8. [Google Scholar]
- 211.Shimizu M, Hara A, Okuno M, Matsuno H, Okada K, Ueshima S, Matsuo O, Niwa M, Akita K, Yamada Y, Yoshimi N, Uematsu T, Kojima S, Friedman SL, Moriwaki H, Mori H. Hepatology. 2001;33:569–576. doi: 10.1053/jhep.2001.22650. [DOI] [PubMed] [Google Scholar]
- 212.Huang G, Elferink CJ. Molecular Pharmacology. 2012;81:338–347. doi: 10.1124/mol.111.075952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Mitchell KA, Wilson SR, Elferink CJ. Toxicology. 2010;276:103–109. doi: 10.1016/j.tox.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E. Mol. Biol. Cell. 1995;6:387–400. doi: 10.1091/mbc.6.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Gartel AL, Tyner AL. Experimental Cell Research. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- 216.Mars WM, Jo M, Gonias SL. Biochem. J. 2005;390:311–315. doi: 10.1042/BJ20042028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Wang H, Zhang Y, Heuckeroth RO. FEBS Lett. 2007;581:3098–3104. doi: 10.1016/j.febslet.2007.05.049. [DOI] [PubMed] [Google Scholar]
- 218.Zhang W, Shields JM, Sogawa K, Fujii-Kuriyama Y, Yang VW. J. Biol. Chem. 1998;273:17917–17925. doi: 10.1074/jbc.273.28.17917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Suske G, Bruford E, Philipsen S. Genomics. 2005;85:551–556. doi: 10.1016/j.ygeno.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 220.Philipsen S, Suske G. Nucleic Acids Research. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Bieker JJ. Journal of Biological Chemistry. 2001;276:34355–34358. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 222.Andreoli V, Gehrau RC, Bocco JL. IUBMB Life. 2010;62:896–905. doi: 10.1002/iub.396. [DOI] [PubMed] [Google Scholar]
- 223.Narla G, Kremer-Tal S, Matsumoto N, Zhao X, Yao S, Kelley K, Tarocchi M, Friedman SL. Oncogene. 2007;26:4428–4434. doi: 10.1038/sj.onc.1210223. [DOI] [PubMed] [Google Scholar]
- 224.Sangodkar J, Shi J, Difeo A, Schwartz R, Bromberg R, Choudhri A, McClinch K, Hatami R, Scheer E, Kremer-Tal S, Martignetti JA, Hui A, Leung WK, Friedman SL, Narla G. Eur. J. Cancer. 2009;45:666–676. doi: 10.1016/j.ejca.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hartel M, Narla G, Wente MN, Giese NA, Martignoni ME, Martignetti JA, Friess H, Friedman SL. Eur. J. Cancer. 2008;44:1895–1903. doi: 10.1016/j.ejca.2008.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Reeves HL, Narla G, Ogunbiyi O, Haq AI, Katz A, Benzeno S, Hod E, Harpaz N, Goldberg S, Tal-Kremer S, Eng FJ, Arthur MJP, Martignetti JA, Friedman SL. Gastroenterology. 2004;126:1090–1103. doi: 10.1053/j.gastro.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 227.Narla G, Heath K, Reeves HL, Li D, Giono L, Kimmelman A, Glucksman M, Narla J, Eng F, Chan AM, Ferrari A, Martignetti JA, Friedman SL. Science. 2001;294:2563–2566. doi: 10.1126/science.1066326. [DOI] [PubMed] [Google Scholar]
- 228.Narla G, Difeo A, Reeves HL, Schaid DJ, Hirshfeld J, Hod E, Katz A, Isaacs WB, Hebbring S, Komiya A, McDonnell SK, Wiley KE, Jacobsen SJ, Isaacs SD, Walsh PC, Zheng SL, Chang B-L, Friedrichsen DM, Stanford JL, Ostrander EA, Chinnaiyan AM, Rubin MA, Xu J, Thibodeau SN, Friedman SL, Martignetti JA. Cancer Research. 2005;65:1213–1222. doi: 10.1158/0008-5472.CAN-04-4249. [DOI] [PubMed] [Google Scholar]
- 229.Jeng Y. International journal of cancer. 2003;105:625–629. doi: 10.1002/ijc.11123. [DOI] [PubMed] [Google Scholar]
- 230.Knudson AG. Proceedings of the National Academy of Sciences. 1971;68:820–823. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.DiFeo A, Narla G, Camacho-Vanegas O, Nishio H, Rose SL, Buller RE, Friedman SL, Walsh MJ, Martignetti JA. Oncogene. 2006;25:6026–6031. doi: 10.1038/sj.onc.1209611. [DOI] [PubMed] [Google Scholar]
- 232.Botella LM, Sanz-Rodriguez F, Komi Y, Fernandez-L A, Varela E, Garrido-Martin EM, Narla G, Friedman SL, Kojima S. Biochem. J. 2009;419:485. doi: 10.1042/BJ20081434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rubinstein M, Idelman G, Plymate SR, Narla G, Friedman SL, Werner H. Endocrinology. 2004;145:3769–3777. doi: 10.1210/en.2004-0173. [DOI] [PubMed] [Google Scholar]
- 234.Benzeno S, Narla G, Allina J, Cheng GZ, Reeves HL, Banck MS, Odin JA, Diehl JA, Germain D, Friedman SL. Cancer Research. 2004;64:3885–3891. doi: 10.1158/0008-5472.CAN-03-2818. [DOI] [PubMed] [Google Scholar]
- 235.Lang UE, Kocabayoglu P, Cheng GZ, Ghiassi-Nejad Z, Muñoz U, Vetter D, Eckstein DA, Hannivoort RA, Walsh MJ, Friedman SL. Oncogene. 2013;32:4557–4564. doi: 10.1038/onc.2012.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Trucco LD, Andreoli V, Núñez NG, Maccioni M, Bocco JL. The FASEB Journal. 2014;28:5262–5276. doi: 10.1096/fj.14-251884. [DOI] [PubMed] [Google Scholar]
- 237.Giacinti C, Giordano A. Oncogene. 2006;25:5220–5227. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 238.Levine-Fridman A, Chen L, Elferink CJ. Molecular Pharmacology. 2004;65:461–469. doi: 10.1124/mol.65.2.461. [DOI] [PubMed] [Google Scholar]
- 239.Xie G, Peng Z, Raufman J-P. AJP: Gastrointestinal and Liver Physiology. 2012;302:G1006–G1015. doi: 10.1152/ajpgi.00427.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chen YJ, Hung CM, Kay N, Chen CC, Kao YH. Cancer Letters. 2012;319:223–231. doi: 10.1016/j.canlet.2012.01.017. [DOI] [PubMed] [Google Scholar]
- 241.Goode GD, Ballard BR, Manning HC, Freeman ML, Kang Y, Eltom SE. Int. J. Cancer. 2013;133:2769–2780. doi: 10.1002/ijc.28297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Xu G, Li Y, Yoshimoto K, Wu Q, Chen G, Iwata T. Toxicology Letters. 2014;224:362–370. doi: 10.1016/j.toxlet.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 243.Puga A, Nebert DW, Carrier F. DNA Cell Biol. 1992;11:269–281. doi: 10.1089/dna.1992.11.269. [DOI] [PubMed] [Google Scholar]
- 244.Ashida H, Nagy S, Matsumura F. Biochemical Pharmacology. 2000;59:741–751. doi: 10.1016/s0006-2952(99)00387-1. [DOI] [PubMed] [Google Scholar]
- 245.Weiss C, Faust D, Dürk H, Kolluri SK, Pelzer A, Schneider S, Dietrich C, Oesch F, Göttlicher M. Oncogene. 2005;24:4975–4983. doi: 10.1038/sj.onc.1208679. [DOI] [PubMed] [Google Scholar]
- 246.Sirach E, Bureau C, Péron JM, Pradayrol L, Vinel JP, Buscail L, Cordelier P. Cell Death Differ. 2007;14:1202–1210. doi: 10.1038/sj.cdd.4402114. [DOI] [PubMed] [Google Scholar]
- 247.Matsumoto N, Kubo A, Liu H, Akita K, Laub F, Ramirez F, Keller G, Friedman SL. Blood. 2006;107:1357–1365. doi: 10.1182/blood-2005-05-1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.D'Astolfo DS, Gehrau RC, Bocco JL, Koritschoner NP. Cell Death Differ. 2008;15:613–616. doi: 10.1038/sj.cdd.4402299. [DOI] [PubMed] [Google Scholar]
- 249.Liu J, Du T, Yuan Y, He Y, Tan Z, Liu Z. Mol Cell Biochem. 2010;335:29–35. doi: 10.1007/s11010-009-0237-8. [DOI] [PubMed] [Google Scholar]
- 250.Tarocchi M, Hannivoort R, Hoshida Y, Lee UE, Vetter D, Narla G, Villanueva A, Oren M, Llovet JM, Friedman SL. Hepatology. 2011;54:522–531. doi: 10.1002/hep.24413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Vetter D, Cohen-Naftaly M, Villanueva A, Lee YA, Kocabayoglu P, Hannivoort R, Narla G, M Llovet J, Thung SN, Friedman SL. Hepatology. 2012;56:1361–1370. doi: 10.1002/hep.25810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Poland A, Palen D, Glover E. Nature. 1982;300:271–273. doi: 10.1038/300271a0. [DOI] [PubMed] [Google Scholar]
- 253.Davis BJ, McCurdy EA, Miller BD, Lucier GW, Tritscher AM. Cancer Research. 2000;60:5414–5419. [PubMed] [Google Scholar]
- 254.Kennedy GD, Nukaya M, Moran SM, Glover E, Weinberg S, Balbo S, Hecht SS, Pitot HC, Drinkwater NR, Bradfield CA. Toxicological sciences. 2014;140:135–143. doi: 10.1093/toxsci/kfu065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Palau N, Julià A, Ferrándiz C, Puig L, Fonseca E, Fernández E, López-Lasanta M, Tortosa R, Marsal S. BMC Genomics. 2013;14:825. doi: 10.1186/1471-2164-14-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Li S, Pei X, Zhang W, Xie HQ, Zhao B. Int J Mol Sci. 2014;15:6475–6487. doi: 10.3390/ijms15046475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Prigent L, Robineau M, Jouneau S, Morzadec C, Louarn L, Vernhet L, Fardel O, Sparfel L. Eur. J. Immunol. 2014;44:1330–1340. doi: 10.1002/eji.201343920. [DOI] [PubMed] [Google Scholar]
- 258.Vogel CFA, Khan EM, Leung PSC, Gershwin ME, Chang WLW, Wu D, Haarmann-Stemmann T, Hoffmann A, Denison MS. Journal of Biological Chemistry. 2014;289:1866–1875. doi: 10.1074/jbc.M113.505578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Li D, Yea S, Chen Z, Narla G, Banck M, Laborda J, Tan S, Friedman JM, Friedman SL, Walsh MJ. Journal of Biological Chemistry. 2005;280:26941–26952. doi: 10.1074/jbc.M500463200. [DOI] [PubMed] [Google Scholar]
- 260.Rodríguez E, Aburjania N, Priedigkeit NM, Difeo A, Martignetti JA. PLoS ONE. 2010;5:e12639. doi: 10.1371/journal.pone.0012639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Calderon MR, Verway M, An BS, DiFeo A, Bismar TA, Ann DK, Martignetti JA, Shalom-Barak T, White JH. Journal of Biological Chemistry. 2012;287:8662–8674. doi: 10.1074/jbc.M111.311605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.