

Abstract
Several studies have shown that patients with succinate dehydrogenase subunit B (SDHB) mutations have a very high risk for developing malignant paragangliomas. However, there is no consensus of what age screening for paragangliomas should start. We report a case of an 8-year-old white girl with a 3-year history of catecholamine excess-related complaints who was diagnosed with a malignant SDHB-associated mediastinal paraganglioma. The patient presented with intermittent sweating, headache, nausea, vomiting, fatigue and weight loss that had been present since she was 5 years of age. A large posterior mediastinal mass measuring 6.4 cm×3.1 cm×4.6 cm was discovered on computed tomography (CT) and magnetic resonance imaging (MRI). Laboratory data included an elevated level of urine normetanephrine of 45,400 μg/g creatinine (upper reference limit 718 μg/g) and elevated level of plasma normetanephrine of 62.4 nmol/l (upper reference limit <0.90 nmol/l). She was diagnosed with a thoracic paraganglioma and subsequently underwent surgical removal of the tumor and two lymph nodes. Histopathologic examination confirmed metastatic paraganglioma. Postoperatively, her blood pressure normalized and plasma normetanephrine levels remained normal. Our patient first presented with paraganglioma-associated signs and symptoms at the young age of 5 years. This case clearly illustrates the need for increased vigilance and screening for paragangliomas in families with SDHB at a younger age than previously thought.
Keywords: Succinate dehydrogenase subunit B (SDHB) mutation, Paraganglioma, Genetic screening, Pheochromocytoma, Malignant
Introduction
Paragangliomas are catecholamine-producing tumors that arise from chromaffin cells of the sympatho-adrenal system. Sympathetic paragangliomas may be located inside the adrenals (pheochromocytomas) or may have an extra-adrenal location [1]. Although rare, these tumors may develop in children, especially in those with underlying genetic mutations [2, 3].
In recent years several studies have been performed that describe genotype–phenotype correlations in patients with paragangliomas associated with mutations in the succinate dehydrogenase (SDH) gene family (SDH subunits B, C, D) [4–6]. Patients with disease associated with succinate dehydrogenase subunit B (SDHB) mutation appear to have an increased risk for developing multiple more aggressive and malignant tumors, and they should have careful periodic surveillance, even after successful surgical removal [6, 7]. Subsequently, family members should be offered genetic testing, and, if the findings are positive, regular screening with biochemical and imaging tests should be initiated.
Several studies have reported the development of SDHB-associated paragangliomas in childhood [2, 4]. However, the exact age at which the screening of children in these families should be commenced is still debated. It has been suggested that screening start 10 years before the earliest age of diagnosis in the family, which could be reflected by screening being started at 5–10 years of age [8, 9]. Owing to the frequently aggressive nature of the disease associated with SDHB mutations, we believe clinicians should be aware of the possible presence of pheochromocytomas or paragangliomas in very young children.
In this case report we describe an 8-year-old patient with an SDHB-associated malignant paraganglioma and provide clinicians with suggested age guidelines for screening.
Case Report
An 8-year-old white girl presented to an outside hospital in November 2007 with a prolonged history of intermittent sweating, headache, nausea, vomiting, fatigue and weight loss since the age of 5 years. In November 2007 she developed significant sweating spells and weight loss. Her grandparents, who had been her guardians since birth, were advised by her school teacher to take her to a physician. Admission to the hospital followed, and she was found to be hypertensive, with a systolic blood pressure (BP) of 140 mmHg (normal range for systolic BP in 8-year-old girls is 95–101 mmHg). Computed tomography (CT) and magnetic resonance imaging (MRI) revealed a large, posterior, mediastinal mass measuring 6.4 cm×3.1 cm× 4.6 cm that extended inferiorly through one neural foramen between T8 and T9 with spinal cord compression (Fig. 1). There were no apparent clinical sequelae to the spinal cord compression. Biochemical evaluation revealed elevated levels of urine normetanephrine of 45,400 μg/g creatinine (upper reference limit 718 μg/g creatinine) and elevated levels of plasma normetanephrine of 62.4 nmol/l (upper reference limit <0.90 nmol/l).
Fig. 1.
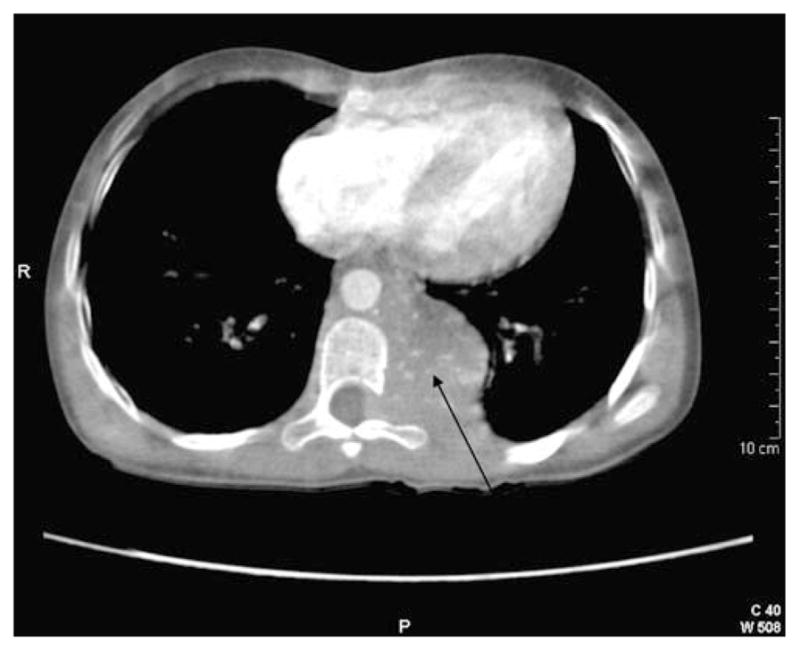
CT reveals a large, posterior, mediastinal paraganglioma (arrow) that extends inferiorly through one neural foramen
She was started on α-adrenergic blockade with phenoxybenzamine and underwent surgical resection of the tumor. At surgery, the mediastinal tumor and the intraspinal component were removed. Pathological review of the specimen confirmed diagnosis of the paraganglioma and identified metastatic spread in one of two adjacent lymph nodes. After surgery [18F]-fluorodeoxyglucose positron emission tomography and [123I]-meta-iodobenzylguanidine (MIBG) scintigraphy suggested either no evidence of a tumor or the presence of tumor that did not take up any of the two compounds.
The patient was referred to the National Institutes of Health (NIH) for further clinical and genetic evaluation of her metastatic paraganglioma. Her plasma catecholamine and metanephrine levels were within normal limits. Genetic testing revealed a mutation in the SDHB gene (G change to T at cDNA nucleotide 418 in exon 4) (c.418G.T), known to be associated with familial paragangliomas. After genetic testing had been performed on members of her family, it was found that her father and paternal grandfather had findings positive for the SDHB mutation as well but had not developed any pheochromocytoma or paraganglioma so far.
Discussion
We describe an 8-year-old patient with a malignant thoracic paraganglioma associated with a mutation in the SDHB gene. Since her symptoms had been present for more than 3 years prior to presentation, we believe that she is one of the youngest patients with paraganglioma reported to date. This case suggests that the screening of SDHB mutation carriers for paragangliomas and pheochromocytomas should commence at a younger age than previously suggested.
Signs and symptoms of pheochromocytoma or sympathetic paraganglioma may be highly variable, and most, but not all, are related to catecholamine excess. In children pheochromocytomas are more frequently familial, extra-adrenal, bilateral and multifocal than in adults [10–13]. In addition to the well-known symptoms and signs associated with catecholamine excess, such as hypertension, headache and palpitations, children more often present with excess sweating, nausea, vomiting, weight loss, polyuria, and visual disturbances than adults [11, 14]. Although our patient had presented with signs and symptoms of catecholamine excess at the age of 5 years, her diagnosis had been delayed for several years, because complaints were found to be nonspecific. Recently, Corcoran et al. reported an atypical presentation of a 6-year-old girl with catecholamine excess based on a pheochromocytoma associated with von Hippel–Lindau syndrome. In that report the need for routine measurement of blood pressure was exemplified [15]. The highly variable presentation of disease in children, in combination with a low prevalence, may lead to a missed diagnosis with considerable consequences.
In the literature several children with paragangliomas have been described who were carriers of the SDHB mutation. Ludwig et al. estimated that an SDHB mutation is present in 20% of the childhood pheochromocytoma cases [2]. Recently, Bockenhauer et al. reported on a young boy with an SDHB mutation-associated pheochromocytoma at the age of 7 years who presented with a 2-month history of typical complaints [16]. In our case report we describe a patient who, although already presenting with signs and symptoms at the age of 5 years, was finally diagnosed with metastatic paraganglioma after a delay of 3 years. Metastases might have been prevented, had the disease been suspected in an earlier phase. Unfortunately, SDHB mutations were not known to be present in the family, and she was only later found to carry an SDHB mutation, so initial suspicion was low.
In contrast to individuals with other sporadic or familial pheochromocytomas, SDHB mutation carriers may already have malignant disease at the time of initial diagnosis. In a cohort of patients that were SDHB mutation-positive, metastases were identified in 22% during their first surgery, with sites of distant metastases including lung, liver, bone and lymph nodes [4]. The elevated levels of norepinephrine and its metabolite normetanephrine that were found in our patient are consistent with the notion that extra-adrenal paragangliomas rarely secrete epinephrine [17]. Most SDHB-related paragangliomas have a noradrenergic (and rarely dopaminergic) biochemical phenotype. This probably reflects a reduced expression of phenylethanolamine-N-methyltransferase, the enzyme that converts norepinephrine to epinephrine [6, 18].
Although, in recent years, there has been the suggestion that screening in families with a known SDHB mutation should be started at a younger age, no consensus, regarding the age, has been reached so far. Experts working in this field have recommended starting such surveillance 10 years before the earliest age of diagnosis in the family, or, more specifically at 5–10 years of age for SDHB mutation carriers [8, 9]. In this case report we describe a young child with a malignant thoracic paraganglioma associated with SDHB, which further exemplifies the aggressive nature of the disease. Our patient most likely first presented with paraganglioma-associated signs and symptoms at the very young age of 5 years, if not earlier. In general, blood pressure should be routinely measured in children presenting with atypical complaints. This case clearly illustrates the importance of appropriate and early screening for catecholamine excess (by measurements of either plasma or urine metanephrines) in children who are known to carry an SDHB gene mutation. However, some children with SDHB mutation may have biochemically silent pheochromocytomas or paragangliomas [19]. In such situations we recommend that the first imaging screening test (CT or MRI) be performed when the child is around the age of 5 years. Based on this report and the data from the literature, we would recommend that children in families with SDHB be screened for the mutation at the age of 5 years or earlier, and, if the findings for such children are found to be positive, biochemical and imaging screening (preferably MRI) should be started as early as 5 years of age.
Follow-up biochemical screening should be performed every 1–2 years. If the results are positive, such screening should be followed by whole-body imaging, preferably by MRI. Although very rare, biochemically silent pheochromocytomas could be missed when only biochemical follow up is used; therefore, imaging studies would be recommended for those aged 5–10 years and, thereafter, every 2–3 years.
Footnotes
The authors have nothing to disclose.
Contributor Information
Tamara Prodanov, Reproductive and Adult Endocrinology Program, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA.
Bas Havekes, Reproductive and Adult Endocrinology Program, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA. Department of Endocrinology and Metabolism, Leiden University Medical Center, Leiden, The Netherlands.
Katherine L. Nathanson, Division of Medical Genetics, Department of Medicine, University of Pennsylvania School of Medicine, Philadelphia, PA, USA
Karen T. Adams, Reproductive and Adult Endocrinology Program, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA
Karel Pacak, Email: karel@mail.nih.gov, Reproductive and Adult Endocrinology Program, National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, USA. Section on Medical Neuroendocrinology, Reproductive and Adult, Endocrinology Program, NICHD, NIH, Building 10, CRC, 1-East, Room 1-3140, 10 Center Drive, MSC-1109, Bethesda, MD 20892-1109, USA.
References
- 1.Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet. 2005;366:665–675. doi: 10.1016/S0140-6736(05)67139-5. [DOI] [PubMed] [Google Scholar]
- 2.Ludwig AD, Feig DI, Brandt ML, Hicks MJ, Fitch ME, Cass DL. Recent advances in the diagnosis and treatment of pheochromocytoma in children. Am J Surg. 2007;194:792–796. doi: 10.1016/j.amjsurg.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 3.de Krijger RR, Petri BJ, van Nederveen FH, Korpershoek E, de Herder WW, De Muinck Keizer-Schrama SM, Dinjens WN. Frequent genetic changes in childhood pheochromocytomas. Ann N Y Acad Sci. 2006;1073:166–176. doi: 10.1196/annals.1353.017. [DOI] [PubMed] [Google Scholar]
- 4.Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PL, Elston M, Gimm O, Henley D, Herman P, Murday V, Niccoli-Sire P, Pasieka JL, Rohmer V, Tucker K, Jeunemaitre X, Marsh DJ, Plouin PF, Robinson BG. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 5.Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C European-American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–951. doi: 10.1001/jama.292.8.943. [DOI] [PubMed] [Google Scholar]
- 6.Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype–phenotype correlations in patients with succinate dehydrogenase subunit B-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2007;92:779–786. doi: 10.1210/jc.2006-2315. [DOI] [PubMed] [Google Scholar]
- 7.Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul JL, Strompf L, Schlumberger M, Bertagna X, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 8.Young WF, Jr, Abboud AL. Paraganglioma—all in the family (editorial) J Clin Endocrinol Metab. 2006;91:790–792. doi: 10.1210/jc.2005-2758. [DOI] [PubMed] [Google Scholar]
- 9.Benn DE, Robinson BG. Genetic basis of phaeochromocytoma and paraganglioma. Best Pract Res Clin Endocrinol Metab. 2006;20:435–450. doi: 10.1016/j.beem.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Perel Y, Schlumberger M, Marguerite G, Alos N, Revillon Y, Sommelet D, De Lumley L, Flamant F, Dyon JF, Lutz P, Heloury H, Lemerle J. Pheochromocytoma and paraganglioma in children: a report of 24 cases of the French Society of Pediatric Oncology. Pediatr Hematol Oncol. 1997;14:413–422. doi: 10.3109/08880019709028771. [DOI] [PubMed] [Google Scholar]
- 11.Ciftci AO, Tanyel FC, Senocak ME, Buyukpamukcu N. Pheochromocytoma in children. J Pediatr Surg. 2001;36:447–452. doi: 10.1053/jpsu.2001.21612. [DOI] [PubMed] [Google Scholar]
- 12.Pham TH, Moir C, Thompson GB, Zarroug AE, Hamner CE, Farley D, van Heerden J, Lteif AN, Young WF., Jr Pheochromocytoma and paraganglioma in children: a review of medical and surgical management at a tertiary care center. Pediatrics. 2006;118:1109–1117. doi: 10.1542/peds.2005-2299. [DOI] [PubMed] [Google Scholar]
- 13.Barontini M, Levin G, Sanso G. Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann N Y Acad Sci. 2006;1073:30–37. doi: 10.1196/annals.1353.003. [DOI] [PubMed] [Google Scholar]
- 14.Hack HA. The perioperative management of children with phaeochromocytoma. Paediatr Anaesth. 2000;10:463–476. doi: 10.1046/j.1460-9592.2000.00504.x. [DOI] [PubMed] [Google Scholar]
- 15.Corcoran J, Bird C, Side L, Lakhoo K, Ryan F. Diagnosis at dusk: malignant hypertension and phaeochromocytoma in a 6-year-old girl. Emerg Med Australas. 2008;20:66–69. doi: 10.1111/j.1742-6723.2007.01049.x. [DOI] [PubMed] [Google Scholar]
- 16.Bockenhauer D, Rees L, Neumann H, Foo Y. A sporadic case of paraganglioma undetected by urine metabolite screening. Pediatr Nephrol. 2008;23:1889–1891. doi: 10.1007/s00467-008-0841-y. [DOI] [PubMed] [Google Scholar]
- 17.Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, Brouwers FM, Pacak K. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem. 2005;51:735–744. doi: 10.1373/clinchem.2004.045484. [DOI] [PubMed] [Google Scholar]
- 18.Feldman JM, Blalock JA, Zern RT, Wells SA., Jr The relationship between enzyme activity and the catecholamine content and secretion of pheochromocytomas. J Clin Endocrinol Metab. 1979;49:445–451. doi: 10.1210/jcem-49-3-445. [DOI] [PubMed] [Google Scholar]
- 19.Holwitt D, Neifeld J, Massey G, Lanning D. Case report of an 11-year-old child with a nonfunctional malignant pheochromocytoma. J Pediatr Surg. 2007;42:E13–E15. doi: 10.1016/j.jpedsurg.2007.08.046. [DOI] [PubMed] [Google Scholar]