

Abstract
Immune‐enhancing adjuvants usually targets antigen (Ag)‐presenting cells to tune up cellular and humoral immunity. CD141+ dendritic cells (DC) represent the professional Ag‐presenting cells in humans. In response to microbial pattern molecules, these DCs upgrade the maturation stage sufficient to improve cross‐presentation of exogenous Ag, and upregulation of MHC and costimulators, allowing CD4/CD8 T cells to proliferate and liberating cytokines/chemokines that support lymphocyte attraction and survival. These DCs also facilitate natural killer‐mediated cell damage. Toll‐like receptors (TLRs) and their signaling pathways in DCs play a pivotal role in DC maturation. Therefore, providing adjuvants in addition to Ag is indispensable for successful vaccine immunotherapy for cancer, which has been approved in comparison with antimicrobial vaccines. Mouse CD8α+ DCs express TLR7 and TLR9 in addition to the TLR2 family (TLR1, 2, and 6) and TLR3, whereas human CD141+ DCs exclusively express the TLR2 family and TLR3. Although human and mouse plasmacytoid DCs commonly express TLR7/9 to respond to their agonists, the results on mouse adjuvant studies using TLR7/9 agonists cannot be simply extrapolated to human adjuvant immunotherapy. In contrast, TLR2 and TLR3 are similarly expressed in both human and mouse Ag‐presenting DCs. Bacillus Calmette–Guerin peptidoglycan and polyinosinic–polycytidylic acid are representative agonists for TLR2 and TLR3, respectively, although they additionally stimulate cytoplasmic sensors: their functional specificities may not be limited to the relevant TLRs. These adjuvants have been posted up to a certain achievement in immunotherapy in some cancers. We herein summarize the history and perspectives of TLR2 and TLR3 agonists in vaccine‐adjuvant immunotherapy for cancer.
Keywords: Antigen‐presenting dendritic cell (CD8α+ DC, CD141+ DC); BCG–CWS; PolyI:C; TICAM‐1 pathway; Toll‐like receptor 2/3
Immune adjuvants represent substances that enhance immune activation in conjunction with antigens (Ag).1 Their functions include the physical effect on Ag to stay longer in tissue or to stabilize the Ag by formulation of emulsion; yet the biological functions are to act directly on either tumor cells or immune cells. The latter have reached a mechanistic elucidation, together with the recent progress of the studies on the molecular mechanism on pattern‐sensing in the innate immune system,1, 2 where adjuvant receptors and multiple signaling pathways are involved in the maturation of dendritic cells (DCs). The properties of the adjuvant closely link the promotion of activation stages in DCs. Innate immunologists calls microbial component pattern molecules (pathogen‐associated molecular patterns [PAMPs]). Pathogen‐associated molecular patterns in microbes are recognized by pattern recognition receptors (PRRs) on the cell membrane (including organelle membrane) or in the cytoplasm.1, 2 Actually, TLRs and cytoplasmic pattern sensors have been molecularly identified for the past two decades. Thus, we found adjuvant receptors corresponding to PRRs.
Infection is usually terminated with a severe inflammation and immune response, which are rooted in the PRR response of immune cells. Many cell types, even including tumor cells, possess PRRs in a cell type‐specific manner, and myeloid cells (i.e. macrophages and DCs) play a major role in pattern sensing in the tumor microenvironment.3 Cancer cells lack the pattern molecules because they are derived from autologous cells. In vaccines against infectious agents, administration with purified antigen alone (accompanied with no pattern molecule) has led to insufficient prophylactic or therapeutic effects.4 Peptide vaccines only with single killer epitopes are ineffective as in purified infectious vaccines. Failure of clinical trials using current tumor vaccines with monovalent Ag and the lack of PAMP would have been predicted only if the executors had had sufficient knowledge on the innate immune system.5 Generally, PRR activation must be accompanied with effective Ag for successful vaccine immunotherapy.
It has been accepted that microbial pattern molecules, PAMP, are agonists for PRRs. Furthermore, PRR activation molecules are released from autologous cells and named damage‐associated molecular patterns (DAMPs). Damage‐associated molecular patterns are also released from macrophages and cancer cells to modulate the tumor microenvironment, and DAMP sometimes causes cell death (necroptosis).6 The function of DAMPs in antitumor immunity may be diverged depending on the situation. Damage‐associated molecular pattern response makes it complicated to analyze adjuvant‐specific innate immune response against cancer. These are risk factors of lifestyle‐related diseases associated with chronic inflammation. The most prominent expression of DAMP involves cancer progression, induction of autoimmune diseases, and exacerbation of latent infections (e.g., hepatitis C virus, hepatitis B virus, tuberculosis), where the PRR‐modified microenvironment forms efficiently to modify immune response to the diseases.7
Pattern molecules are found widely in proteins, nucleic acids, and lipids, and they often possess unique structures characteristic to various microorganisms (usually absent in the host).2 Pattern recognition receptors precisely discriminate the structural differences of PAMP of each microorganism (Table 1). Pattern recognition receptors are currently classified into Toll‐like receptor (TLR), RIG‐I‐like receptor, NOD‐like receptor, C‐type lectin‐like receptor, etc.2, 8 In addition, the concept has been proposed that innate immunity consists of not only the PAMP–PRR system but also the comprehensive host‐defense system, including the coagulation, complement, Dicer‐RNAi, and nuclease–exosome systems.9 Blocking activation of retrotransposon and promoting nuclear reprogramming are also reported to be a result of PRR stimulation.10, 11 Pattern recognition receptors mostly show universal distribution, with no limiting to myeloid‐lineage cells. This does not always mean that innate immunity simply conducts a trigger for DC–lymphocyte immune response; rather, it represents the systemic orchestration of cells in a biological defense and tissue repair against infectious accidents. In particular, interferons (IFNs) and inflammatory cytokines are systemically effective for immediate eradication of microbes.12 Dicer‐RNAi and nucleases are involved in elimination of foreign RNA from host cells.13, 14 These responses lead to cell growth, tissue repair, and epigenetic alterations in affected cells.15 However, these mediators simultaneously induce inflammatory responses causing endotoxin‐like signals, with systemic side‐effects being inevitable, particularly in excess cytokinemia.16 Conventional adjuvant therapy (using biological response modifier, modulin, microbial administration) has been found to reflect a process of antimicrobial immune activation including systemic response.
Table 1.
Candidates for Toll‐like receptor (TLR) adjuvants and targeted dendritic cell (DC) subsets
| Adjuvants (PAMPs) | Receptors | Ligands | DC subsets |
|---|---|---|---|
| Pam3 lipopeptides | TLR2 and TLR1 | Lipoprotein | CD141+ DC |
| Pam2 lipopeptides | TLR2 and TLR6 | Lipoprotein | CD141+ DC |
| PGN | TLR2 | Peptidoglycan | CD141+ DC |
| OspA | TLR2 and Lectin receptor? | Lipoprotein | CD141+ DC |
| polyI:C | TLR3 and MDA5 | dsRNA | CD141+ DC |
| LPS | TLR4 | Lipopolysaccharide | |
| Flagellin | TLR5, IPAF, and NAIP5 | Flagellin | |
| Imiquimod | TLR7 and TLR8 | RNA analog | pDC |
| poly‐U | TLR7 and TLR8 | RNA analog | pDC |
| Hemozoin | TLR9 | Heme‐polymer | pDC |
| Plasmid DNA | TLR9 | Non‐methylated CpG | pDC |
IPAF, ICE protease‐activating factor (or NLRC4); LPS, lipopolysaccharide; NAIP5, neuronal apoptosis inhibitory protein 5; OspA, Outer surface protein A; PAMPs, pathogen‐associated molecular patterns; pDC, plasmacytoid dendritic cell; PGN, peptidoglycan; polyI:C, polyinosinic–polycytidylic acid; poly‐U, poly uracil.
This review focuses on the adjuvant for the immunotherapy of cancer, outlining the PRR response of myeloid cells. In addition, the review discusses the ideal adjuvant with reduced side‐effects to use in combination with a therapeutic vaccine.
History of immune adjuvant for cancer
Coley's vaccine was described in the 1890s. Although his therapeutic injection of live bacteria was too radical (sometimes life‐threatening) to follow, tumor shrinkage was significantly observed in a number of patients. Then a group of the Memorial Sloan Kettering cancer center introduced the bacillus Calmette–Guerin (BCG) to anticancer therapy before the discovery of innate immunity. Lloyd Old and fellow researchers reported a large number of case studies of patients with transitional cell bladder cancer, which was cured by administering live BCG bacteria to the bladder. Current BCG therapy leads to remission of more than 70% of bladder cancers.17 In Japan, the Maruyama vaccine18 and Yamamura and Azuma's BCG–cell wall skeleton (CWS) adjuvant therapy19 have been developed as BCG‐derived adjuvants for activation of antitumor immunity; they became early pioneers of component adjuvants (that has no infectious capacities, unlike live bacteria). Maruyama vaccine contains lipoarabinomannan, whereas BCG–CWS contains mycolic acids (trehalose dimycolate), arabinogalactan, and peptidoglycan, a ligand for TLR2.20 Recently, Yamasaki et al. discovered that trehalose dimycolate and lipoarabinomannan are ligands for C‐type lectin‐like receptors Mincle and Dectin‐2, respectively.21, 22 The accumulating evidence on innate receptors has enabled us to delineate the function of BCG reagents.
In patients with postoperative cancer, solid tumor regressed in response to BCG–CWS with <2‐year follow‐up studies in early‐staged patients with lung or gastric cancer.23, 24 Statistical intergroup difference in overall survival rates was not significant in patients of the curative group. However, the intergroup difference in overall survival rates was statistically significant in patients of the non‐curative group.24 The 5‐year survival rate was ~5% higher in the BCG‐treated group than in those with conventional therapies in stage III lung cancer patients.24 In 1994, Toyoshima, Hayashi, and Kodama et al. at the Osaka Medical Center for Cancer and Cardiovascular Diseases (Osaka, Japan) were engaged in clinical studies on s.c. injection of BCG–CWS alone (with no tumor‐associated Ag [TAA] peptide) according to protocols suitable to the modern clinical trial policy. Their results suggested that the 5‐year survival rates of postoperative patients with metastatic lung cancer were more than 40% by treatment with BCG–CWS alone, 15–20% better than conventional chemotherapy.25 Considering the low side‐effects of BCG–CWS compared to chemo‐ or radiotherapy, this was an amazing result.
The researchers, however, did not aim at completion of the bacterial adjuvants for chemical synthesis of the active component, but tried to isolate active fractions from a specific strain of bacteria by biochemical procedures.26 The immune‐enhancing component of BCG–CWS was identified as peptidoglycan containing muramyl dipeptide (MDP).27, 28 However, MDP failed to exhibit full antitumor function, although MDP was synthesized as a cytokine inducer as a NOD2 ligand.29 Chemical synthesis of the active component of BCG–CWS was not successful either. This is because the peptidoglycan was technically unable to synthesize. In addition, biologically active BCG–CWS constituents including mycolic acids (ligands for Mincle), lipoarabinomannnan (a ligand for Dectin‐2), and MDP, exert functions other than tumor regression to modulate the immune system to inflammation.30 Therefore, the structure responsible for antitumor functions cannot be strictly defined in BCG derivatives, which reflects the complexity of biological products originated from microbes. The BCG–CWS of Azuma's lot was an agonist of TLR2/4 with efficient antitumor activity.20 However, a highly purified BCG–CWS (Sumitomo lot) was reportedly TLR2‐specific, and less effective for tumor regression.31 Thus, there are lot‐to‐lot differences in BCG–CWS. The Sumitomo lot is still available for BCG–CWS therapy at Osaka Medical Center for Cancer and Cardiovascular Diseases.
Hilton Levy et al. used an analog of dsRNA, namely polyinosinic–polycytidylic acid (polyI:C), which mimicked the replication intermediate of viruses, for adjuvant immunotherapy for cancer in 1960s,32 but their polyI:C was chemically synthesized with a batch‐method, where various lengths of polyI and polyC were mixed and annealed. Therefore, the product showed a smear in agarose gel with remarkable lot‐to‐lot differences,16 causing a lack of functional uniformity. High dose therapy with polyI:C induced tumor regression in many clinical trials of patients with various types of cancers,33 but many cases were accompanied with severe endotoxin‐like shock. The toxicity was described as “intolerable” and caused interruption of the clinical trials.32, 34 More recently, polyI:C was identified as a TLR3 agonist,16 but it remained undetermined whether TLR3 was involved in cytokinemia and endotoxin‐like diseases.16, 33 Systemic activation of the MAVS (mitochondrial antiviral signaling protein) pathway by polyI:C was later found to be a cause of the toxicity.16 Side‐effects of cytokine toxicity were largely attributable to the cytoplasmic polyI:C response, which would have been the cause of “intolerable” toxicity. PolyI:C induces elegant DC maturation through Toll‐IL‐1R homology domain‐containing adaptor molecule‐1 (TICAM‐1), but not myeloid differentiation primary response gene 88 (MyD88),35 and it is still in use as an adjuvant in lower doses (to avoid toxicity) to patients in combination with Ag.36 However, low doses of polyI:C can activate IFN‐α/β receptor (IFNAR) but not the TICAM‐1 pathway (see below). Efficient cross‐presentation is evoked in DCs through combinational activation of IFNAR and TLR3–TICAM‐1–IFN regulatory factor 3 (IRF3).35 As low‐dose polyI:C only mediates IFNAR‐dependent DC maturation, this appears similar to IFN therapy and provides less advantage for vaccine adjuvants. Interferon therapy using commercial type I IFN frequently brings patients adverse events.
Hydroxide aluminium and some oils (montanide, squalene etc.) have been approved as adjuvants (Table 2), but they are strong inflammation‐inducing agents. They have been used in immunotherapy as a peptide vaccine, but no satisfactory results were obtained. Hydroxide aluminium stimulates the prostaglandin system, and additionally induces production of interleukin‐1β (IL‐1β) and IL‐18 through activation of the NALP3–inflammasome system.37 Although they enhance antibody production by Th2‐skewing, their ability to activate cellular immunity (CTL and natural killer [NK] cells) appears weak.37 They have far less ability to induce cross‐presentation in DCs than TLR adjuvants. Other biologicals such as OK‐432 (Picibanil) have been used in cancer patients as an adjuvant. In addition, there are many oral intake adjuvants, α‐glucans, β‐glucans, liposaccharides, and lipopeptides, some of which may contribute to improving the quality of life in cancer patients. Although a large number of adjuvants other than those introduced here have been attempted for antitumor immunity, none of them has been formally approved for antitumor immunotherapy (Table 2).
Table 2.
Adjuvants in development for human vaccines
| Adjuvant | Formulation | In preclinical or clinical trials |
|---|---|---|
| Montanides | Water‐in‐oil emulsions | Malaria (phase I), HIV, cancer (phase I/II) |
| Saponins (QS‐21) | Aqueous | Cancer (phase II), herpes (phase I), HIV (phase I) |
| SAF | Oil‐in‐water emulsion containing squalene, Tween‐80, Pluronic™ L121 | HIV (phase I, Chiron) |
| AS03 | Oil‐in‐water emulsion containing α‐tocopherol, squalene, Tween‐80 | Pandemic flu (GSK) |
| MTP‐PtdEtn | Oil‐in‐water emulsion | HSV |
| Exotoxins | Pseudomonas aeruginosa | P. aeruginosa, cystic fibrosis (AERUGEN – Crucell/Berna) |
| Escherichia coli heat‐labile enterotoxin LT | ETEC (phase II – Iomai Corp.) | |
| ISCOMs | Phospholipids, cholesterol, QS‐21 | Influenza, HSV, HIV, HBV, malaria, cancer |
| TLR ligands | ||
| MPL®‐SE | Oil‐in‐water emulsion | Leishmania (phase I/II – IDRI) |
| Synthetic lipid A | Oil‐in‐water emulsion | Various indications (Avanti/IDRI) |
| MPL®‐AF | Aqueous | Allergy (ATL), cancer (Biomira) |
| AS01 | Liposomal | HIV (phase I), malaria (ASO1, phase III, GSK) |
| Cancer (phase II/III, Biomira/MerckKGaA) | ||
| AS02 | Oil‐in‐water emulsion containing MPL® and QS‐21 | HPV (Cervarix), HIV, tuberculosis, malaria (phase III), herpes (GSK) |
| AS04 | Alum + aqueous MPL® | HPV, HAV (GSK) |
| AS15 | AS01+ CpG | Cancer therapy (GSK) |
| RC529 | Aqueous | HBV, pneumovax |
| Cancer (ProMune – Coley/Pfizer) | ||
| Lipopeptide MALP‐2 (TLR2) | n/a | Pancreatic cancer |
| CpG‐ODN (TLR9) | n/a | HIV, HBV, HSV, anthrax (VaxImmune Coley/GSK/Chiron) |
| HBV (HEPLISAV, phase III, Dynavax) | ||
| Cancer (phase II, Dynavax) | ||
| Poly(I:C)LC (TLR3) | n/a | Cancer (IMOxine, phase I, Hybridon Inc.) |
| (YpG, CpR motif) | Cancer (IMO‐2055, phase II, Idera Pharm.) | |
| HIV (Remune, phase I, Idera/IMNR) | ||
| TLR‐9 agonist (MIDGE®) | n/a | Cancer (phase I, Mologen AG) |
| TLR‐7/8 (Imiquimod) | n/a | Melanoma (3M Pharmaceuticals) |
| HIV (preclinical), leishmaniasis | ||
| TLR‐7/8 (Resiquimod) | n/a | HSV, HCV (phase II, 3M Pharmaceuticals) |
CpG‐ODN, CpG oligodeoxy nucleotide; ETEC, enterotoxigenic Escherichia coli; GSK, GlaxoSmithKline; HAV, hepatitis A virus; HBV, hepatitis B virus; HCV, hepatitis C virus; HPV, human papillomavirus; HSV, herpes simplex virus; IDRI, Infectious Disease Research Institute; LT, lipoteichoic acid; n/a, not applicable; polyI:C, polyinosinic–polycytidylic acid; TLR, Toll‐like receptor.
Toll‐like receptor expression in DC subsets
For mice, bone marrow‐derived DC (BMDC), a representative of myeloid DC (mDC) and plasmacytoid DC (pDC), are prepared from bone marrow cells using granulocyte/macrophage colony‐stimulating factor (GM‐CSF) or Flt3 ligand, respectively.1 Langerhans cells are prepared by treatment of bone marrow cells with GM‐CSF, IL‐4, and transforming growth factor‐β.38 Additional DC subsets are separated from the spleen and intestine using FACS. A submucosal DC subset for Ag presentation is CD103+ DC, which is developed from the same origin as CD8α+ DC.39 For humans, monocyte‐derived DC is used as mDC,40 and they show significantly different properties from the CD141+ (BDCA3) DC subset, a representative antigen‐presenting cell (APC), in the peripheral blood.41, 42, 43 On the other hand, peripheral blood pDC can be separated using BDCA4,40 and have a similar TLR7/9‐expressing profile to mouse pDC.1, 41 BDCA3 represents a thrombomodulin epitope whereas BDCA4 represents a neuropilin‐1 epitope.
Distribution of TLRs in human DC subsets and blood cells is shown in Figure 1, where the TLR proteins were determined using anti‐human TLR antibody. Mouse TLRs in terms of protein expression have not been addressed with mouse BMDC or pDC, as no suitable mAb was available for their assessment.1 However, PCR analyses suggested that mouse BMDC express TLR7 and TLR9 as in pDC, though a report of protein analysis suggested that mouse CD8a+ DC express TLR9 but not TLR7 (41). Anyhow, their properties entirely differ from those of human monocyte‐derived DC or CD141+ DC (Table 3). A representative Ag‐presenting DC subset is CD8α+ DC in mouse spleen.
Figure 1.
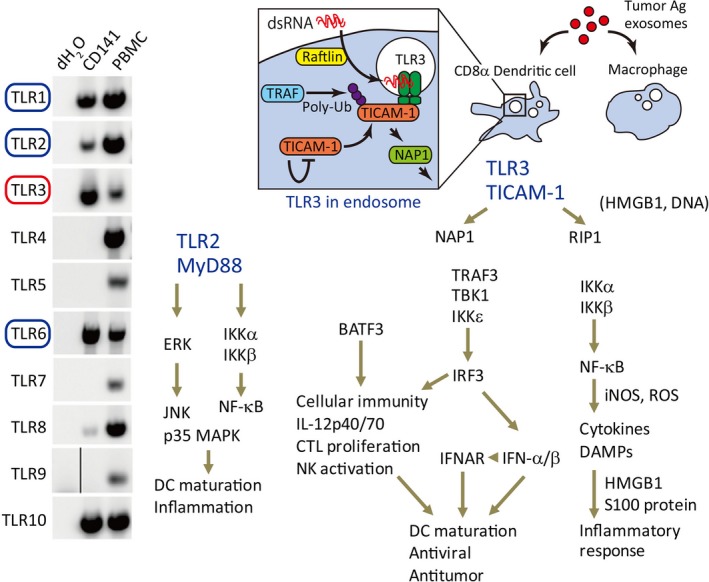
Human dendritic cell (DC) response to Toll‐like receptor (TLR)2/3 adjuvants. Human CD141+ DC corresponds to mouse CD8α+ DC, and functions as a main antigen (Ag)‐presenting cell. CD141+ DC express the TLR2 family (TLR1, 2, and 6) and TLR3 but does not express other TLRs. Hence, this type of antigen‐presenting DC cannot respond to LPS, lipopolysaccharide; flagellin, imiquimod, or CpG‐ODN, oligodeoxy nucleotide. TLR2 is surface‐expressed and captures its agonists on the membrane whereas TLR3 is expressed in endosomes, where TLR3 encounters dsRNA. The TLR3–TLR adaptor molecule‐1 (TICAM‐1) pathway is unique in the induction of interleukin (IL)‐12p70 and interferon (IFN)‐independent cross‐presentation. Both pathways also accompany inflammation. The possible pathways for DC maturation by TLR2 and TLR3 are depicted. DAMPs, damage‐associated molecular patterns; HMGB1, high mobility group box protein1; IFNAR, IFN‐α/β receptor; IKK, IκB kinase; iNOS, inducible nitric oxide synthase; IRF3, IFN regulatory factor 3; MyD88, myeloid differentiation primary response gene 88; NAP1, NF‐κB‐activating kinase‐associated protein 1; NF‐κB, nuclear factor‐κB; NK, natural killer; RIP1, receptor‐interacting protein kinase 1; ROS, reactive oxygen species; TBK1, TANK‐binfing kinase 1; TRAF, TNF receptor‐associated factor. Panel A was quoted from Ref.39.
Table 3.
Expression of Toll–like receptors (TLR) in human and murine dendritic cell (DC) subsets
| TLR1 | TLR2 | TLR3 | TLR4 | TLR5 | TLR6 | TLR7 | TLR8 | TLR9 | TLR10 | References | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Human | |||||||||||
| Myeloid DCs (CD11c+) | + | + | + | + | + | + | + | +/− | − | + | 1 |
| CD11c+/CD141+ DC | + | ++ | +++ | − | − | + | − | +/− | − | + | 39 |
| Monocyte‐derived DCs | + | + | + | + | + | +/− | +/− | + | − | + | 1,40 |
| Plasmacytoid DCs (CD11c− BDCA2+ BDCA4+) | +/− | − | − | − | − | − | + | − | + | + | 42 |
| Mouse | |||||||||||
| Conventional DCs (CD11chigh B220−) | |||||||||||
| CD4+ | + | + | − | + | + | + | + | − | + | − | 1 |
| CD4−CD8a− | + | + | +/− | + | + | + | +/− | − | + | − | 1 |
| CD8α+ | + | + | + | + | − | + | − | − | + | − | 1 |
| Plasmacytoid DCs (CD11clow B220+ PDCA‐1+) | + | + | − | + | +/− | + | + | − | + | − | 1 |
Human CD141+ DC of APC do not express TLR7 or TLR9.42 Although mouse CD8α+ DC (a counterpart of human CD141+ DC) express moderate TLR4 and TLR5, human CD141+ DC do not express them.39, 40, 41 Human and mouse APC commonly express high levels of the TLR2 family (TLR1, 2, and 6) and TLR3 proteins (Table 3). In intestine, however, CD103+ DC reside in the submucosal region, and express TLR3, TLR7, and TLR9 and function as an Ag‐presenting cell similar to CD141+ DC.41 Plasmacytoid DC generally express TLR7 and TLR9, but not other TLRs.42 Human but not mouse mDC express TLR8.3 CpG‐ODN (oligodeoxy nucleotide) has low immune‐enhancing function because human APC DCs exert limited TLR9 compared to mouse equivalents. The in vivo immune‐enhancing function of CpG may be supported by pDC and CD103+ DC with TLR9.42, 43
In summary, Ag‐presenting DC must be activated by adjuvant in evoking antitumor response. The subsets of DCs are CD141+ DC in human and CD8α+ DC in mouse.39, 41, 44 Their TLR repertoires differ from the conventional DCs, MoDC, or BMDC, prepared from the reported methods.3 Mouse CD8α+ DC recognizes DNA/RNA by TLR7/9 as in pDC and matures for Ag‐presentation,44 but human CD141+ DC expresses only TLR2/3.39 To induce efficient TAA presentation, TLR2/3 agonist is essential to complement the lack of PAMP in antitumor immunotherapy in addition to TAA in humans.
Dendritic cell subsets and effector induction
Effector cells can be evaluated by the Ag‐dependent proliferation of T cells, CTL, Th1, Th2, Th17, and regulatory T cells (Treg), and Ag‐independent NK activation (Fig. 2). Natural killer cells are activated through cytokines/mediators and by cell–cell contact, where an NK‐activating ligand on mDCs stimulates NK receptors on NK cells.45 Natural killer‐activating cytokines such as IL‐15, IL‐18, IFN‐α/β, and IL‐12 are released from mDCs to act on NK cells.46 Cytotoxic T lymphocytes are a result of activation of CD8α+ T cells; this process is promoted by cross‐presentation through class I upregulation by mDCs. Interleukin‐2 from lymphocytes is additionally required for T cell proliferation and long‐term survival. Other effectors are a result of the activation of CD4+ T cells by mDC class II presentation. The CD4+ T cells are classified into subsets, including Th1, Th2, Th17, and Treg. The master transcription factors to Th1, Th2, Th17, and Treg are T‐bet, GATA‐3, RORgT, and Foxp3, respectively.47 Additional CD4 subsets may exist under differential regulations. T cell proliferation and activation are closely associated with DC maturation stage in the priming phase, which is regulated under epigenetic control.
Figure 2.
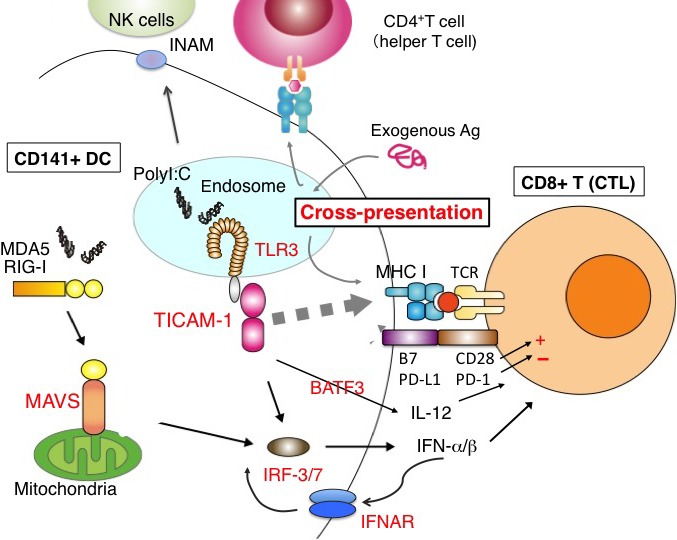
Immune response to tumor‐associated antigen (TAA) + RNA adjuvant in CD141+ dendritic cells (DC). When a soluble antigen (Ag) and dsRNA are taken up into DC, the DC upregulates MHC class II and activates CD4 T cells. MHC class I is upregulated by dsRNA response. The Ag falls in a cytoplasm, and enters the TAP1‐dependent processing to degrade peptides in the proteasome. This process is called cross‐presentation. Toll‐like receptor 3 (TLR3) recognizes dsRNA and promotes cross‐presentation of the TAA peptides. CD80/86 upregulation and interleukin (IL)‐12/type I interferon (IFN) production is simultaneously induced in TLR3 activation. Natural killer (NK) cells are also activated by the IFN‐inducible NK activating ligands. All these responses are through the TLR adaptor molecule‐1 (TICAM‐1)–IFN regulatory factor 3 (IRF3) axis. BATF3, basic leucine zipper transcription factor; IFNAR, IFN‐α/β receptor; INAM, IRF3‐dependent NK activation molecule; MAVS, mitochondrial antiviral signaling protein; MDA5, melanoma differentiation associated gene 5; polyI:C, polyinosinic–polycytidylic acid; RIG‐1, retinoic acid‐inducible gene I.
The mechanism by which DCs selectively induce various effectors remains molecularly unclear. The mechanism as to what molecules are associated with the effector‐inducing event is largely unknown. Also, the mechanism by which cross‐presentation is induced for exogenous tumor Ag remains unknown. Our reports indicate that TICAM‐1‐inducible genes do not directly link the trigger of cross‐presentation48 (Takeda Y, Azuma M, and Seya T, unpublished data). Although IFNAR‐inducible genes participate in cross‐presentation, they are independent of TICAM‐1‐mediated cross‐presentation.49 Recently, Sec61 translocon was identified as a cross‐presentation promoting factor downstream of TICAM‐1.50 The Sec61 protein movement from the endoplasmic reticulum to the endosome membrane without any gene induction would explain the TICAM‐1‐mediated promotion of cross‐priming involved in Ag‐presenting DCs. There appear several modes of cross‐presentation that distinctly involve TICAM‐1 (IRF3) and IFNAR (STAT1/2).
Certain DC subsets seem to associate with preferential induction of a particular effector. If the root that imparts directionality to the immune system is a DC, Ag per se does not have the ability to command the strategy for immune activation but dictates the specificity for the activated immune cells. The strategy is reflected in the induced effector, such as antibody, NK, CTL, Th17, and Treg. In tumor, an increase of PD‐1 on T lymphocytes is often found to link exhaustion.51 The effector switch appears to be regulated by the stage of DC maturation, and therefore by adjuvant. In fact, murine splenic CD8α+ DC are likely to induce Treg52 and NK cells,53 depending on the adjuvant tested. Lamina propria pDC in response to mouse intestinal flora promote IgA production.54 CD70+/CD11c+ DC induce Th17 cells by adenosine triphosphate (ATP) of intestinal bacteria.55 Bone marrow‐derived DC can activate NK cells through the TICAM‐1 pathway in polyI:C‐stimulated DC.56
Dendritic cell‐mediated NK cell activation in tumor immunity by adjuvant
Bone marrow‐derived DC drive antitumor NK cytotoxicity, depending on the TICAM‐1 pathway.56 This NK activation is attributable to cell–cell contact between BMDC and NK cells rather than humoral mediators such as cytokines, induced by DCs.57 Therefore, the key for the mechanisms of induction of antitumor NK cells would be a membrane molecule on DC that promotes the surface expression by the TICAM‐1 pathway (Fig. 2). This DC–NK contact and cytokines including IL‐2, IL‐12, IL15, IL‐18, and IFN are involved in total NK cell activation, IFN‐gamma production and cytotoxicity.45, 46 Dendritic cell‐mediated NK activation is attributable to IRF3 but not to IRF7, as NK activation is not affected in IRF7 KO BMDC but is severely hampered in IRF3 BMDC.57 Thus, the NK induction pathway in mDC uses the transcription factor IRF3 in TICAM‐1 downstream. With screening methods of the candidate cDNA to express lentiviral vectors in IRF3‐deficient BMDC, it is possible to identify NK activation molecules.57 We have identified the IRF3‐dependent NK activation molecule (INAM, Fam26F) as an NK‐activating molecule of DCs. The INAM specifically connects BMDCs with NK cells (Fig. 3). This molecule strongly promotes NK activation in DC but does not induce NK activation in response to other cell types expressing INAM. Notably, CD8a+ DC barely induce NK tumoricidal activity but induce IFN‐g production in response to dsRNA, unlike BMDC( 57 , 58 ). As a membrane protein similar to tetraspanin, INAM has a molecular weight of 45 kDa and a sugar chain with post‐translational modification. It is mainly distributed to the spleen and lymph node cells. The INAM protein is expected to make a loop‐like structure in two locations on the cell surface from the predicted sequence.57
Figure 3.
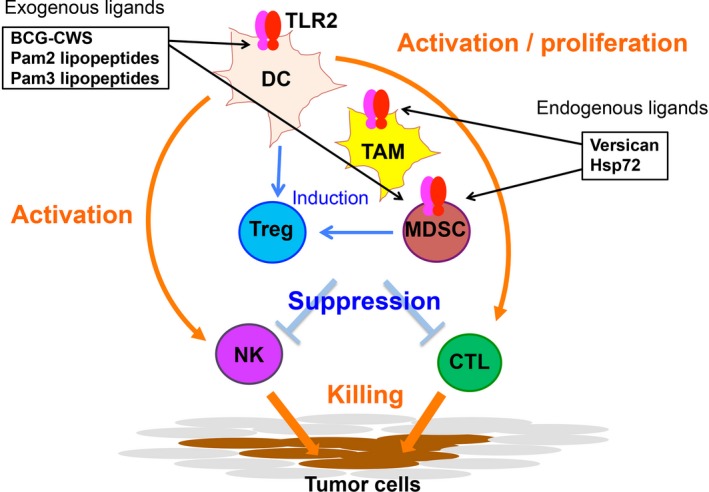
Myeloid cells responding to adjuvants. Toll‐like receptor 2 (TLR2) response is illustrated. Tumor‐associated macrophages (TAM), myeloid‐derived suppressor cells (MDSC), and dendritic cells (DC) express TLR2, which senses exogenous and endogenous ligands of TLR2. These cells are originated from bone marrow. They are activated in response to the ligands through the myeloid differentiation primary response gene 88 (MyD88) pathway. MDSC and TAM suppress natural killer (NK) activation and CTL proliferation by inducing regulatory T cells (Treg). DCs in tumor turn active by recognition of TLR2 ligands to activate NK cells and CTL. Tumor microenvironment controls the balance of activation/suppression of immune cells. Tumor cells express TLR2, which may support tumor progression in response to TLR2 ligands. BCG, bacillus Calmette–Guerin; CWS, cell wall skeleton; Hsp77, heat shock protein 77.
It is presumed that INAM is involved in the configuration of the immune synapse of the BMDC–NK intersurface. When the BMDCs overexpressing INAM are adoptively transferred to tumor‐bearing mice, regression of NK‐sensitive tumor occurs rapidly.58 If NK cells are removed from the mice by NK1.1 Ab, tumor (B16 melanoma) regression no longer occurs.56 This suggests that INAM is an essential factor that drives the induction of antitumor NK cells. However, INAM is a tetraspanin‐like molecule that is unlikely to mediate direct NK–DC interaction. Other partner molecules associated with INAM in the membrane synapse may act as an NK‐activating molecule in this context.
Pattern recognition receptors in antigen‐presenting DCs and cross‐presentation
Adjuvants usually target DC for immune enhancement. Human CD141+ DC specifically express TLR2 and 3, but do not express TLR4, 5, 7, or 9 (Fig. 1, Table 3). Toll‐like receptor 2 recognizes bacterial lipopeptides and peptidoglycan, and activates the MyD88 pathway (Table 1).2 Toll‐like receptor 3 recognizes stem‐structured RNA, and activates the TICAM‐1 pathway.59 Therefore, we explain the differences in cross‐presentation response of these two pathways in DCs. Some adjuvants primarily promote antibody production, whereas others evoke cellular immunity in the antitumor environment. The latter adjuvants are preferable when tumor antigens are taken up in DCs. Proteins and long‐chain peptides are appropriate as TAAs, as they are endocytozed and provide multivalent epitopes involving CD4 activation. It is TLR2 and TLR3 that directly promote the antigen presentation in human APC.39
Toll‐like receptor 3 adjuvant
The immunostimulatory function of TLR3 adjuvant is to induce inflammatory cytokines and chemokines, high expression of MHC, upregulation of costimulatory molecules, promotion of cross‐presentation, production of type I IFN, and the production of IL‐12 (Fig. 1). Type I IFN induces T cell proliferation and releases the exhaustion of CD8 T cells to confer long live on T cells. However, IFN‐α is relatively weak in the induction of long live of T cells due to the upregulation of PD‐1 compared to IL‐12p70 in the OT‐1 adoptive transfer system.51 Type I IFN further activates CD4 helper and NK cells.60 These lymphocytes generally maintain their antitumor activity by type I IFN or IL‐12 supplied by DCs, and IL‐12 is important for the exertion of T cell cytotoxicity in tumor.61 Th is is in part due to the fact that PD‐1 is upregulated on T cells by IFN but not IL‐12. However, the levels of PD‐1 in lymphocytes are variable depending on the conditions of the tumor microenvironment, which critically affects cytolytic activity of tumor‐infiltrated lymphocytes.
A good activation marker of lymphocytes is IFN‐γ.62 The levels of IFN‐γ reflect the active behaviors of multiple lymphocytes, and are influenced by the type of adjuvant. Interleukin‐12 is produced depending on Batf3–TLR3 signaling (TICAM‐1 pathway),48, 63 but not on the MyD88 pathway, which is used in most TLR signaling. The TLR3 ligand usually promotes T cell infiltration into tumor, which may be partly due to the release of CXCL10 and 11 around the tumor.48 Other factors, including CCL5, reported to participate in T cell tumor infiltration,64 are also upregulated by the TICAM‐1 signal. Hence, these chemokines are all induced by the TLR3–TICAM‐1 pathway.48
Polyinosinic–polycytidylic acid has been used as a TLR3 agonist, but is now approved as a broad agonist for cytoplasmic RNA sensors, such as MDA5 RIG‐I, DDX1, DDX3, and DDX21 in addition to TLR3.16 Most of these, with the exception of TLR3 and DDX1, are cytoplasmic, mitochondrial antiviral signaling protein (MAVS) activators with ubiquitous distribution. Type I IFN induced by the MAVS pathway is an effector in cytoplasmic RNA sensing and to some extent improves DC maturation. As polyI:C gains access to TLR3 in endosomes as well as these RNA sensors in the cytoplasm, it causes endotoxin‐like cytokine toxicity.16 Without RIG‐I/MDA5 activation, no cytokine storm is observed in mice having polyI:C treatment, while TLR3‐mediated DC maturation is kept intact.63
We chemically synthesized a TLR3‐specific agonist, ARNAX, and found that exclusive stimulation of TLR3 without activation of the MAVS pathway by ARNAX attained robust antitumor cellular immunity with no increase of serum cytokines.63 The results support the finding that the TLR3–TICAM‐1 pathway upregulates only a few genes without the participation of type I IFN, (Takeda et al., unpublished data). It is expected that ARNAX downregulates PD‐1 in DC‐primed lymphocytes. We defined ARNAX as a non‐inflammatory, DC‐priming adjuvant. ARNAX consists of 5′‐cap of GpC DNA and ~140 bp dsRNA, which contains no part of the human genome sequences and thus is independent of RNAi response.63 ARNAX will be the function‐defined non‐inflammatory adjuvant with high safety. In combination with various peptide vaccines, preclinical tests of ARNAX are in progress.
Toll‐like receptor 2 adjuvant
Toll‐like receptor 2 evokes cross‐presentation secondary to activation of the MyD88 pathway.65 MyD88 conforms a fundamental pathway inducing inflammation. In this context, TLR2 ligand induces tumor‐associated inflammation, including inflammatory cytokines with activation of DC, as well as tumor‐infiltrating macrophages (Figs 1, 3). A typical ligand of TLR2 is Pam2 lipopeptide including MALP‐2 or MALP‐2s.66 This adjuvant barely induces IL‐12 or type I IFN, but induces high levels of inflammatory cytokines including tumor necrosis factor‐α (TNF‐α),66 as well as activation of IL‐1 receptor‐associated kinase (IRAK).2 In some cell types, TLR2 can link to TICAM‐1 and induce small amounts of IFN‐β and IL‐12.67 We found TLR2 expressed on tumor‐infiltrated myeloid‐derived suppressor cells (MDSC) and tumor‐associated macrophages (TAM).68 These tumor‐supporting myeloid cells are then activated to promote tumor expansion, invasion, and metastasis. Hence, TLR2 signal makes tumor progress, although it acts on DCs to mature. A successful example of clinical trials was reported on MALP‐2 adjuvant therapy in patients with pancreatic cancer.69 MALP‐2 may be effective for some types of cancer with minimal macrophages.
PD‐1 is expressed in CD8+ T cells. Effective cases of PD‐1/PD‐L1 are <30% in solid tumors,70, 71 although severe side‐effects appear induced by PD‐1 Ab therapy in some cases. In cases with tumor regression by anti‐PD‐1 Ab therapy, tumor cells express high PD‐L1 expression, as observed in Hodgkin's lymphoma.72 Recruiting lymphocytes with low PD‐1 expressions to tumor foci thus makes tumors shrink.73 The TLR2 agonists may downregulate PD‐1 on CTL in the tumor microenvironment.74 A question is whether the combination of Ags and TLR2 adjuvants resolve the ineffective properties of PD‐1‐expressing lymphocytes in the tumor microenvironment in cancer patients.
Pattern recognition receptors in tumor‐infiltrated macrophages
Myeloid cells are essential for the organogenic process in the life. Native organs contain resident macrophages originated from the yolk sac or fetal liver. The tissue‐resident macrophages adapt to the organ environment to protect the organ from infections. Macrophages consist of a variety of subsets, most of which are highly sensitive to microbial patterns and release DAMPs (such as HMGB1 and dsDNA) in response to TLR stimulation.7 Because tumor is a kind of organ, it includes a variety of myeloid cells around vessels.75 Tumors are developed as aging along somatic or epigenetic gene‐modification process, myeloid cells are supplied from the bone marrow rather than the yolk sac for tumorigenesis. Tumor‐infiltrating myeloid cells endow a unique microenvironment to tumors (Fig. 3). Tumor‐associated macrophages are F4/80+ and Gr‐1−, whereas MDSC come to the fraction of Gr‐1+ cells.75 Individual tumors have a distinct distribution profile of different TAM:MDSC ratios.
Toll‐like receptor 3 adjuvant
Most subsets of macrophages express TLR3. Necroptosis occurs in tissue‐resident macrophages in response to polyI:C, which acts on TLR3.76 In contrast, tumor‐infiltrating macrophages adapt to the tumor environment and show a different response. Tumor‐associated macrophages are regarded as M2 macrophages in the M1/M2 classification, and promote tumor development.77 They induce hemorrhagic necrosis of tumor in response to TLR3 stimulation.75 This is attributable to the rapid onset of TNF‐α. The TLR3 signal converts TAM to macrophages with tumoricidal activity. Myeloid‐derived suppressor cells also support immune suppression and tumor progression.78 The mechanism of myeloid‐mediated tumor progression is based on the oxidative reaction of reactive oxygen species caused by the expression of inducible nitric oxide synthase. Induction of inducible nitric oxide synthase occurs in response to polyI:C not only in macrophages but also in tumor cells, stromal cells, or lymphocytes.75
Toll‐like receptor 2 adjuvant
Myeloid‐derived suppressor cells are known to systemically increase in quantity by TLR2 stimulation and enhance immunosuppressive activity.79 Toll‐like receptor 2 is expressed in tumor cells as well as macrophages, and endogenous TLR2 ligands, such as versican,80 are released from the tumor. Toll‐like receptor 2‐dependent tumor growth is defined by the overall response of these complex reactions, and MDSC play a central part in promotion of tumorigenesis. The TAM response to TLR2 ligands should be shown together with a TLR response to MDSC (Fig. 3).
Tumor cells responding to adjuvants
Tumor cells express TLR2 and TLR3 in many cases, depending on their origin. In pancreatic cancer, TLR2 agonist therapy leads to tumor regression. However, TLR2 adjuvant directs growth of tumor cells in many cases because it activates the MyD88 pathway.80 The MyD88 responses in both tumor and macrophages lead the tumor toward malignancy, that is, promoting growth, invasion, and metastasis of tumors. In contrast, TLR3 shows a limited expression in homeostatic myeloid and epithelial cells, and often induces upregulation in cancerous tissue in other cells.81 Hence, TLR3 is expressed in many tumor cells, but mostly does not induce tumor growth in response to RNA stimuli. Activation of the TLR3–TICAM‐1 pathway in tumor cells has been reported to trigger cell death, in some cases apoptosis or necrosis.82 Either type of cell death is induced through the receptor‐interacting protein kinase 3 pathway of TLR3 signaling in tumor cells (Fig. 4). Polyinosinic–polycytidylic acid particularly triggers necroptosis in tumor cells, which would rarely occur in the absence of caspase 8 in tumor cells.82 Thus, TLR3 adjuvant acts on both tumor and immune cells for tumor regression. In either case, inflammation profoundly associates with the TLR3 response, which includes an alteration of epigenetic status in cells that leads to innate immune activation as well as tumor regression. The interaction between tumor and immune cells may be modified by cell debris or exosomes, which is produced in response to virus or dsRNA stimulation.83, 84 Viral RNA and polyI:C affect the promotion of tissue recovery by stem cell activation. In fact, nuclear reprogramming happens in response to TLR3 stimulation in human fibroblasts.11
Figure 4.
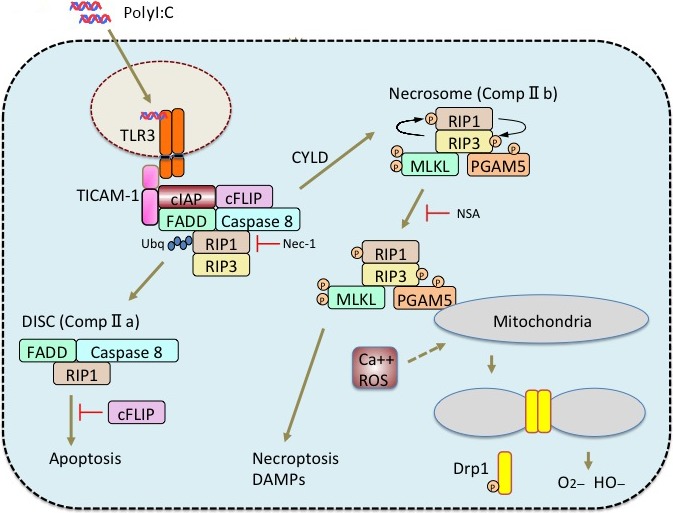
Cell death pathway in tumor cells. If RNAs possess an RNase‐resistant stem structure, they are stably incorporated into endosomes of tumor cells. They are captured by Toll‐like receptor 3 (TLR3) in the endosome and evoke activation of the receptor‐interacting protein kinase (RIP)1/3 cell death pathways. In some tumor cells, two cell death pathways are activated as in macrophages: caspase 8‐dependent and ‐independent pathways. The two pathways are schematically illustrated. Apoptosis or necroptosis occurs through the presence or absence of the function of caspase 8. The latter involves the scission of mitochondria and oxidative stress. cFLIP, cellular FLICE (FADD‐like IL‐1β‐converting enzyme)‐inhibitory protein; cIAP, cellular inhibitor of apoptosis protein 1; CYLD, tumor suppressor cylindromatosis; DISC, death‐inducing signaling complex; DAMPs, damage‐associated molecular patterns; FADD, Fas‐associated death domain protein; MLKL, Mixed lineage kinase domain‐like; NSA, necrosulfonamide; PGAM5, phosphoglycerate mutase family member 5; polyI:C, polyinosinic–polycytidylic acid; ROS, reactive oxygen species; TICAM‐1, TLR adaptor molecule‐1.
Conclusion
Adjuvants induce the activation of the immune system as well as modulate tumor cells in conjunction with macrophages.85 These comprehensive responses are converged into tumor regression. Adjuvants activate the cellular immunity in addition to humoral immunity by acting on DCs. An Ag‐presenting DC is CD141+ DC in humans, which exclusively expresses TLR2 and TLR3; in mouse, it is CD8α+ DC, which additionally express TLR7/9. In human studies, adjuvants must be TLR2 or TLR3 agonists to expect an Ag‐presenting response for cancer immunotherapy. Stimulation with TLR3 makes tumor vaccines effective, and there is no induction of tumor invasion, proliferation, cytokinemia, or toxic diseases. However, many TLRs (including TLR2) strongly activate the MyD88 pathway, which shows an example of not only immune activation but also tumor growth or progression secondary to inflammation. Our point is that TLR3‐specific agonists are the best adjuvants for vaccine immunotherapy in cancer therapeutics, given that they do not induce cytokinemia. Dendritic cells, tumor cells, and tumor‐infiltrating myeloid cells have their unique adjuvant responses, which is an issue to be considered individually.
Disclosure Statement
The authors have no conflict of interest.
Acknowledgments
We are grateful to Drs. H. Oshiumi (Kumamoto University, Kumamoto, Japan), T. Nakatsura (National Institute of Cancer, Tokyo, Japan), T. Ebihara (Washington University, MO, USA) and N. Sato (Sapporo Medical University, Sapporo, Japan) for their fruitful discussions. Ms. H. Sato is gratefully acknowledged for her secretarial management. This work was supported in part by Grants‐in‐Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (Specified Project for “Carcinogenic Spiral”) and the Ministry of Health, Labor, and Welfare of Japan, and by the Program of Founding Research Centers for Emerging and Reemerging Infectious Diseases, MEXT. Financial support by the Takeda Science Foundation, the Yasuda Cancer Research Foundation, Kato memorial bioscience foundation (HS) and the Iskra Foundation is gratefully acknowledged.
Cancer Sci 106 (2015) 1659–1668
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan; Ministry of Health, Labor and Welfare of Japan; Takeda Science Foundation; Yasuda Cancer Research Foundation; Kato memorial bioscience foundation (HS); Iskra Foundation.
References
- 1. Iwasaki A, Medzhitov R. Toll‐like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987–95. [DOI] [PubMed] [Google Scholar]
- 2. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140: 805–20. [DOI] [PubMed] [Google Scholar]
- 3. Goutagny N, Estornes Y, Hasan U, Lebecque S, Caux C. Targeting pattern recognition receptors in cancer immunotherapy. Target Oncol 2012; 7: 29–54. [DOI] [PubMed] [Google Scholar]
- 4. Matsumoto M, Azuma M, Seya T. Adjuvant Immunotherapy for cancer: basic research to clinical bench In: Seya T, ed. Inflammation and Immunity in Cancer. Japan: Springer, 2015; 229–42. [Google Scholar]
- 5. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10: 909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kono H, Rock KL. How dying cells alert the immune system to danger. Nat Rev Immunol 2008; 8: 279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ikushima H, Yanai H, Taniguchi T. Innate immune receptor signaling and IRF family transcription factors; good deeds and misdeeds in oncogenesis In: Seya T, ed. Inflammation and Immunity in Cancer. Japan: Springer, 2015; 85–101. [Google Scholar]
- 8. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 2015; 33: 257–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Seya T, Shime H, Matsumoto M. Functional alteration of tumor‐infiltrating myeloid cells in RNA adjuvant therapy In: Yamaguchi Y, ed. Cancer Immunotherapy. Springer, 2015; (in press). [PubMed] [Google Scholar]
- 10. Yu P, Lübben W, Slomka H, Gebler J et al Nucleic acid‐sensing Toll‐like receptors are essential for the control of endogenous retrovirus viremia and ERV‐induced tumors. Immunity 2012; 37: 867–79. [DOI] [PubMed] [Google Scholar]
- 11. Lee J, Sayed N, Hunter A et al Activation of innate immunity is required for efficient nuclear reprogramming. Cell 2012; 151: 547–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Honda K, Taniguchi T. IRFs: master regulators of signalling by Toll‐like receptors and cytosolic pattern‐recognition receptors. Nat Rev Immunol 2006; 6: 644–58. [DOI] [PubMed] [Google Scholar]
- 13. Aliyari R, Ding SW. RNA‐based viral immunity initiated by the Dicer family of host immune receptors. Immunol Rev 2009; 227 (1): 176–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leong CR, Matsumoto M, Suzuki T, Oshiumi H, Seya T. Nucleic acid sensors involved in the recognition of hepatitis B virus (HBV) in the liver‐specific in vivo transfection mouse models‐Pattern recognition receptors and sensors for HBV. Med Sci. 2015; 3: 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Oshiumi H. Links between recognition and degradation of cytoplasmic viral RNA in innate immune response. Rev Med Virol 2015; (in press). [DOI] [PubMed] [Google Scholar]
- 16. Seya T, Azuma M, Matsumoto M. Targeting TLR3 with no RIG‐I/MDA5 activation is effective in immunotherapy for cancer. Expert Opin Ther Targets 2013; 17: 533–44. [DOI] [PubMed] [Google Scholar]
- 17. Lamm DL, Blumenstein BA, Crawford ED et al A randomized trial of intravesical doxorubicin and immunotherapy with bacille Calmette‐Guérin for transitional‐cell carcinoma of the bladder. N Engl J Med 1991; 325: 1205–9. [DOI] [PubMed] [Google Scholar]
- 18. Maruyama C. Treatment of malignant tumors by an extract from tubercle bacilli (tuberculosis vaccine). Nihon Ika Daigaku Zasshi 1971; 38: 267–76. [PubMed] [Google Scholar]
- 19. Azuma I, Ribi EE, Meyer TJ, Zbar B. Biologically active components from mycobacterial cell walls. I. Isolation and composition of cell wall skeleton and component P3. J Natl Cancer Inst 1974; 52: 95–101. [DOI] [PubMed] [Google Scholar]
- 20. Uehori J, Matsumoto M, Tsuji S et al Simultaneous blocking of human Toll‐like receptors 2 and 4 suppresses myeloid dendritic cell activation induced by Mycobacterium bovis bacillus Calmette‐Guérin peptidoglycan. Infect Immun 2003; 71: 4238–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ishikawa E, Ishikawa T, Morita YS et al Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C‐type lectin Mincle. J Exp Med 2009; 206: 2879–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yonekawa A, Saijo S, Hoshino Y et al Dectin‐2 is a direct receptor for mannose‐capped lipoarabinomannan of mycobacteria. Immunity 2014; 41: 402–13. [DOI] [PubMed] [Google Scholar]
- 23. Ochiai T, Sato H, Sato H et al Randomly controlled study of chemotherapy versus chemoimmunotherapy in postoperative gastric cancer patients. Cancer Res 1983; 43: 3001–7. [PubMed] [Google Scholar]
- 24. Yasumoto K, Manabe H, Yanagawa E et al Nonspecific adjuvant immunotherapy of lung cancer with cell wall skeleton of Mycobacterium bovis Bacillus Calmette‐Guérin. Cancer Res 1979; 39: 3262–7. [PubMed] [Google Scholar]
- 25. Kodama K, Higashiyama M, Takami K et al Innate immune therapy with a BCG cell wall skeleton after radical surgery for non‐small cell lung cancer: a case control study. Surg Today 2009; 39: 194–200. [DOI] [PubMed] [Google Scholar]
- 26. Azuma I, Seya T. Development of immunoadjuvants for immunotherapy of cancer. Int Immunopharmacol 2001; 1: 1249–59. [DOI] [PubMed] [Google Scholar]
- 27. Tsuji S, Matsumoto M, Takeuchi O et al Maturation of human dendritic cells by cell wall skeleton of Mycobacterium bovis bacillus Calmette‐Guérin: involvement of toll‐like receptors. Infect Immun 2000; 68: 6883–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uehori J, Fukase K, Akazawa T et al Dendritic cell maturation induced by muramyl dipeptide (MDP) derivatives: monoacylated MDP confers TLR2/TLR4 activation. J Immunol 2005; 174: 7096–103. [DOI] [PubMed] [Google Scholar]
- 29. Girardin SE, Boneca IG, Viala J et al Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003; 278: 8869–72. [DOI] [PubMed] [Google Scholar]
- 30. Toyonaga K, Miyake Y, Yamasaki S. Characterization of the receptors for mycobacterial cord factor in Guinea pig. PLoS ONE 2014; 9: e88747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Murata M. Activation of Toll‐like receptor 2 by a novel preparation of cell wall skeleton from Mycobacterium bovis BCG Tokyo (SMP‐105) sufficiently enhances immune responses against tumors. Cancer Sci 2008; 99: 1435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Levine AS, Sivulich M, Wiernik PH, Levy HB. Initial clinical trials in cancer patients of polyriboinosinic‐polyribocytidic acid stabilized with poly‐l‐Lysine, in carboxy methylcellulose [polyI:CLC], a highly effective interferon inducer. Cancer Res 1979; 39: 1645–50. [PubMed] [Google Scholar]
- 33. Galluzzi L, Vacchelli E, Eggermont A et al Trial Watch: experimental Toll‐like receptor agonists for cancer therapy. Oncoimmunology 2012; 1: 699–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Absher M, Stinebring WR. Endotoxin‐like properties of poly I. poly C, an interferon stimulator. Nature 1969; 223: 715. [DOI] [PubMed] [Google Scholar]
- 35. Azuma M, Ebihara T, Oshiumi H, Matsumoto M, Seya T. Cross‐priming for antitumor CTL induced by soluble Ag + polyI: C depends on the TICAM‐1 pathway in mouse CD11c(+)/CD8α(+) dendritic cells. Oncoimmunology 2012; 1: 581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sabbatini P, Tsuji T, Ferran L et al Phase I trial of overlapping long peptides from a tumor self‐antigen and poly‐ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin Cancer Res 2012; 18: 6497–508. [DOI] [PubMed] [Google Scholar]
- 37. Marichal T, Ohata K, Bedoret D et al DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med 2011; 17: 996–1002. [DOI] [PubMed] [Google Scholar]
- 38. Geissmann F, Prost C, Monnet JP, Dy M, Brousse N, Hermine O. Transforming growth factor β1, in the presence of granulocyte/macrophage colony‐stimulating factor and interleukin 4, induces differentiation of human peripheral blood monocytes into dendritic Langerhans cells. J Exp Med 1998; 187: 961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jongbloed SL, Kassianos AJ, McDonald KJ et al Human CD141+ (BDCA‐3)+ dendritic cells (DC) represent a unique myeloid DC subset that cross‐presents necrotic cell antigens. J Exp Med 2010; 207: 1247–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shortman K, Nail SH. Steady‐state and inflammatory dendritic cell development. Nat Rev Immunol 2007; 7: 19–30. [DOI] [PubMed] [Google Scholar]
- 41. Poulin LF, Salio M, Griessinger E et al Characterization of human DNGR‐1+ BDCA3+ leukocytes as putative equivalents of mouse CD8alpha+ dendritic cells. J Exp Med 2010; 207: 1261–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kadowaki N, Ho S, Antonenko S et al Subsets of human dendritic cell precursors express different toll‐like receptors and respond to different microbial antigens. J Exp Med 2001; 194: 863–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edwards AD, Diebold SS, Slack EM et al Toll‐like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol 2003; 33: 827–33. [DOI] [PubMed] [Google Scholar]
- 44. Fujimoto K, Karuppuchamy T, Takemura N et al A new subset of CD103+CD8alpha+ dendritic cells in the small intestine expresses TLR3, TLR7, and TLR9 and induces Th1 response and CTL activity. J Immunol 2011; 186: 6287–95. [DOI] [PubMed] [Google Scholar]
- 45. Andoniou CE, van Dommelen SL, Voigt V et al Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol 2005; 6: 1011–9. [DOI] [PubMed] [Google Scholar]
- 46. Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans‐presenting interleukin 15. Immunity 2007; 26: 503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Finotto S, Glimcher L. T cell directives for transcriptional regulation in asthma. Springer Semin Immunopathol 2004; 25: 281–94. [DOI] [PubMed] [Google Scholar]
- 48. Azuma M, Takeda Y, Nakajima H et al BATF3 fundamentally supports TLR3‐derived IL‐12 induction in CD8α+ dendritic cells, which promotes antitumor T cell responses by Poly(I:C). Cancer Res 2015; (in press). [Google Scholar]
- 49. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol 2014; 12: e1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zehner M, Marschall AL, Bos E et al The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross‐presentation to CD8(+) T cells. Immunity 2015; 42: 850–63. [DOI] [PubMed] [Google Scholar]
- 51. Gerner MY, Heltemes‐Harris LM, Fife BT, Mescher MF. Cutting edge: IL‐12 and type I IFN differentially program CD8 T cells for programmed death 1 re‐expression levels and tumor control. J Immunol 2013; 191: 1011–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yamazaki S, Iyoda T, Tarbell K et al Direct expansion of functional CD25+ CD4+ regulatory T cells by antigen‐processing dendritic cells. J Exp Med 2003; 198: 235–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miyake T, Kumagai Y, Kato H et al Poly I:C‐induced activation of NK cells by CD8alpha+ dendritic cells via the IPS‐1 and TRIF‐dependent pathways. J Immunol 2009; 183: 2522–8. [DOI] [PubMed] [Google Scholar]
- 54. Tezuka H, Abe Y, Iwata M et al Regulation of IgA production by naturally occurring TNF/iNOS‐producing dendritic cells. Nature 2007; 448: 929–33. [DOI] [PubMed] [Google Scholar]
- 55. Atarashi K, Nishimura J, Shima T et al ATP drives lamina propria T(H)17 cell differentiation. Nature 2008; 455: 808–12. [DOI] [PubMed] [Google Scholar]
- 56. Akazawa T, Ebihara T, Okuno M et al Antitumor NK activation induced by the Toll‐like receptor 3‐TICAM‐1 (TRIF) pathway in myeloid dendritic cells. Proc Natl Acad Sci USA 2007; 104: 252–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ebihara T, Azuma M, Oshiumi H et al Identification of a polyI:C‐inducible membrane protein that participates in dendritic cell‐mediated natural killer cell activation. J Exp Med 2010; 207: 2675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kasamatsu J, Azuma M, Oshiumi H et al INAM plays a critical role in IFN‐γ production by NK cells interacting with polyinosinic‐polycytidylic acid‐stimulated accessory cells. J Immunol 2014; 193: 5199–207. [DOI] [PubMed] [Google Scholar]
- 59. Tatematsu M, Nishikawa F, Seya T, Matsumoto M. Toll‐like receptor 3 recognizes incomplete stem structures in single‐stranded viral RNA. Nat Commun 2013; 4: 1833. [DOI] [PubMed] [Google Scholar]
- 60. Hervas‐Stubbs S, Perez‐Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I. Direct effects of type I interferons on cells of the immune system. Clin Cancer Res 2011; 17: 2619–27. [DOI] [PubMed] [Google Scholar]
- 61. Colombo MP, Trinchieri G. Interleukin‐12 in anti‐tumor immunity and immunotherapy. Cytokine Growth Factor Rev 2002; 13: 155–68. [DOI] [PubMed] [Google Scholar]
- 62. Matsumoto M, Seya T, Kikkawa S et al Interferon gamma‐producing ability in blood lymphocytes of patients with lung cancer through activation of the innate immune system by BCG cell wall skeleton. Int Immunopharmacol 2001; 1: 1559–69. [DOI] [PubMed] [Google Scholar]
- 63. Matsumoto M, Tatematsu M, Nishikawa F et al Defined TLR3‐specific adjuvant that induces NK and CTL activation without significant cytokine production in vivo. Nat Commun 2015; 6: 6280. [DOI] [PubMed] [Google Scholar]
- 64. Conforti R, Ma Y, Morel Y et al Opposing effects of toll‐like receptor (TLR3) signaling in tumors can be therapeutically uncoupled to optimize the anticancer efficacy of TLR3 ligands. Cancer Res 2010; 70: 490–500. [DOI] [PubMed] [Google Scholar]
- 65. Shen KY, Song YC, Chen IH et al Molecular mechanisms of TLR2‐mediated antigen cross‐presentation in dendritic cells. J Immunol 2014; 192: 4233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Azuma M, Sawahata R, Akao Y et al The peptide sequence of diacyl lipopeptides determines dendritic cell TLR2‐mediated NK activation. PLoS ONE 2010; 5: e12550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Stack J, Doyle SL, Connolly DJ et al TRAM is required for TLR2 endosomal signaling to type I IFN induction. J Immunol 2014; 193: 6090–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shime H, Matsumoto M, Seya T. The role of innate immune signaling in regulation of tumor‐associated myeloid cells In: Seya T, ed. Inflammation and Immunity in Cancer. Japan: Springer; 2015; 25–48. [Google Scholar]
- 69. Schmidt J, Welsch T, Jäger D, Mühlradt PF, Büchler MW, Märten A. Intratumoural injection of the toll‐like receptor‐2/6 agonist ‘macrophage‐activating lipopeptide‐2’ in patients with pancreatic carcinoma: a phase I/II trial. Br J Cancer 2007; 97: 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Brahmer JR, Tykodi SS, Chow LQ et al Safety and activity of anti‐PD‐L1 antibody in patients with advanced cancer. N Engl J Med 2012; 366: 2455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Topalian SL, Hodi FS, Brahmer JR et al Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012; 366: 2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ansell SM, Lesokhin AM, Borrello I et al PD‐1 blockade with Nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med 2015; 372: 311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tumeh PC, Harview CL, Yearley JH et al PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515: 568–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hernández‐Ruiz J, Salaiza‐Suazo N, Carrada G et al CD8 cells of patients with diffuse cutaneous leishmaniasis display functional exhaustion: the latter is reversed, in vitro, by TLR2 agonists. PLoS Negl Trop Dis 2010; 4: e871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Shime H, Matsumoto M, Oshium i H et al Toll‐like receptor 3 signaling converts tumor‐supporting myeloid cells to tumoricidal effectors. Proc Natl Acad Sci USA 2012; 109: 2066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. de Bouteiller O, Merck E, Hasan UA et al Recognition of double‐stranded RNA by human toll‐like receptor 3 and downstream receptor signaling requires multimerization and an acidic pH. J Biol Chem 2005; 280: 38133–45. [DOI] [PubMed] [Google Scholar]
- 77. Sica A, Larghi P, Mancino A et al Macrophage polarization in tumour progression. Semin Cancer Biol 2008; 18: 349–55. [DOI] [PubMed] [Google Scholar]
- 78. Shime H, Kojima A, Maruyama A et al Myeloid‐derived suppressor cells confer tumor‐suppressive functions on natural killer cells via polyinosinic:polycytidylic acid treatment in mouse tumor models. J Innate Immun 2014; 6: 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Maruyama A, Shime H, Takeda Y, Azuma M, Matsumoto M, Seya T. Pam2 lipopeptides systemically increase myeloid‐derived suppressor cells through TLR2 signaling. Biochem Biophys Res Commun 2015; 457: 445–50. [DOI] [PubMed] [Google Scholar]
- 80. Kim S, Takahashi H, Lin WW et al Carcinoma‐produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature 2009; 457: 102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Salaun B, Zitvogel L, Asselin‐Paturel C et al TLR3 as a biomarker for the therapeutic efficacy of double‐stranded RNA in breast cancer. Cancer Res 2011; 71: 1607–14. [DOI] [PubMed] [Google Scholar]
- 82. Takemura R, Takaki H, Okada S et al PolyI:C‐induced, TLR3/RIP3‐dependent necroptosis backs up immune effector‐mediated tumor elimination in vivo . Cancer Immunol Res 2015; 3: 902–14. [DOI] [PubMed] [Google Scholar]
- 83. Atay S, Godwin AK. Tumor‐derived exosomes: a message delivery system for tumor progression. Commun Integr Biol 2014; 7: e28231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ebihara T, Shingai M, Matsumoto M, Wakita T, Seya T. Hepatitis C virus‐infected hepatocytes extrinsically modulate dendritic cell maturation to activate T cells and natural killer cells. Hepatology 2008; 48: 48–58. [DOI] [PubMed] [Google Scholar]
- 85. Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annu Rev Immunol 2015; 33: 445–74. [DOI] [PubMed] [Google Scholar]