

Abstract
Mammalian target of rapamycin (mTOR) is a serine–threonine kinase that acts downstream of the phosphatidylinositol 3‐kinase signaling pathway and regulates a wide range of cellular functions including transcription, translation, proliferation, apoptosis, and autophagy. Whereas genetic alterations that result in mTOR activation are frequently present in human cancers, whether the mTOR gene itself becomes an oncogene through somatic mutation has remained unclear. We have now identified a somatic non‐synonymous mutation of mTOR that results in a leucine‐to‐valine substitution at amino acid position 2209 in a specimen of large cell neuroendocrine carcinoma. The mTOR(L2209V) mutant manifested marked transforming potential in a focus formation assay with mouse 3T3 fibroblasts, and it induced the phosphorylation of p70 S6 kinase, S6 ribosomal protein, and eukaryotic translation initiation factor 4E–binding protein 1 in these cells. Examination of additional tumor specimens as well as public and in‐house databases of cancer genome mutations identified another 28 independent non‐synonymous mutations of mTOR in various cancer types, with 12 of these mutations also showing transforming ability. Most of these oncogenic mutations cluster at the interface between the kinase domain and the FAT (FRAP, ATM, TRRAP) domain in the 3‐D structure of mTOR. Transforming mTOR mutants were also found to promote 3T3 cell survival, and their oncogenic activity was sensitive to rapamycin. Our data thus show that mTOR acquires transforming activity through genetic changes in cancer, and they suggest that such tumors may be candidates for molecularly targeted therapy with mTOR inhibitors.
Keywords: Cancer genomics, molecularly targeted therapy, mammalian target of rapamycin, oncogene, somatic mutation
Mammalian target of rapamycin (mTOR) is a serine–threonine kinase and a component, together with the rapamycin‐sensitive regulatory associated protein of mTOR or the rapamycin‐insensitive companion of mTOR, of mTOR complex 1 (mTORC1) or mTORC2, respectively.1 From its amino‐terminus, mTOR contains HEAT (huntingtin, elongation factor 3, protein phosphatase 2A, TORI) repeat, FAT (FRAP, ATM, TRRAP), FRB (FK506‐rapamycin binding), amino‐terminal lobe of the kinase, LBE (LST8 binding element), carboxyl‐terminal lobe of the kinase, and FATC (FRAPP, ATM, TOR at C‐terminus) domains. Whereas the molecular mechanism by which the kinase activity of mTOR is regulated remains unclear, mTOR acts downstream of the phosphatidylinositol 3‐kinase (PI3K) signaling pathway. One cascade leading to mTOR activation, for instance, involves activation of the serine–threonine kinase AKT in a manner dependent on PI3K, phosphorylation of tuberous sclerosis complex (TSC) 1/2 by AKT, consequent loading of Rheb with GTP, and activation of mTORC1. Ligand binding to many receptor‐type tyrosine kinases thus results in the activation of mTOR through PI3K.
Activated mTOR regulates a plethora of cellular activities including gene transcription, translation, cell growth and proliferation, apoptosis, and autophagy.1, 2, 3 For example, mTOR phosphorylates and thereby activates p70 S6 kinase (p70S6K), which is a positive regulator of cell size and mRNA translation.4 Also, eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) becomes directly phosphorylated by mTOR, and dissociated from eukaryotic translation initiation factors, allowing ribosome entry into 5′‐cap site of mRNAs.
Consistent with the importance of mTOR in the regulation of cell growth and proliferation as well as of protein synthesis, multiple genetic alterations that lead to mTOR activation have been identified in human cancers.5, 6 Activating somatic mutations in the catalytic subunit of PI3K, for instance, are among the most frequent oncogenic changes in tumors. Loss‐of‐function mutations of phosphatase and tensin homolog, which counteracts PI3K activity, are also frequently detected. Activating mutations of AKT1 and AKT3 have been associated with breast cancer and melanoma, respectively, and genetic loss of TSC1 has been observed. Such genetic changes commonly lead to the activation of mTOR, and blockade of such signaling with mTOR‐specific inhibitors has proven to be of clinical benefit.
Whether mTOR itself becomes a bona fide oncoprotein as a result of somatic genomic alterations in cancer has remained elusive. Forced introduction of amino acid substitutions into mTOR has been shown to result in activation of its kinase function or to confer transforming activity,7, 8, 9 whereas deletion of the inhibitory domain also leads to mTOR activation,10 revealing an oncogenic potential for mTOR. Sato et al.11 searched non‐synonymous mTOR mutations in the COSMIC cancer mutation database (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/), and found seven independent mutations. Then they revealed that E2419K, S2215Y, and R2505P substitutions elevate enzymatic activity of mTOR, but failed to demonstrate transforming ability. Thus, it is not settled yet if mTOR mutations found in human cancer confer direct transforming potential.
We have now identified somatic single nucleotide variations in a specimen of large cell neuroendocrine carcinoma (LCNEC), one of which results in a Leu2209‐to‐Val substitution in mTOR. The mTOR(L2209V) mutant was found not only to mediate marked phosphorylation of p70S6K but also to possess transforming ability when expressed in mouse 3T3 fibroblasts. Moreover, approximately half of the amino acid substitutions in mTOR listed in the cancer database similarly confer oncogenic activity.
Materials and Methods
Clinical specimens and cell lines
Surgically resected tumor and paired normal specimens from LCNEC patients were analyzed with written informed consent. This study was approved by the institutional ethics committees of The University of Tokyo (Tokyo, Japan) and Chiba University (Chiba, Japan). Mouse 3T3 and HEK293T cells were obtained from ATCC (Manassas, VA, USA), and were maintained in DMEM‐F12 supplemented with 10% FBS and 2 mM l‐glutamine (all from Invitrogen, Carlsbad, CA, USA).
Exome analysis with next generation sequencer
Exon fragments were isolated from genomic DNA of an LCNEC tumor (#G4T) and its matched normal control specimen with the use of a SureSelect Human All Exon kit (Agilent Technologies, Santa Clara, CA, USA). Nucleotide sequencing of these fragments was carried out with the HiSeq2500 platform (Illumina, San Diego, CA, USA) with the paired‐end option. We selected only sequence reads with a Q value of ≥20 at each base, and further extracted unique reads that were subsequently mapped to the reference human genome sequence (hg19) with the use of the Bowtie 2 algorithm (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml). Mismatches were discarded if: (i) a given read contained ≥3 independent mismatches; (ii) they were already present in the “1000 genomes” database (http://www.1000genomes.org) or included in the normal human genome variations of our in‐house database; or (iii) they were supported by only one strand of the genome. Somatic mutations were called with the MuTect algorithm (http://www.broadinstitute.org/cancer/cga/mutect) and annotated with SnpEff (http://snpeff.sourceforge.net). A genomic region corresponding to Leu2209 of mTOR was amplified by PCR with the primers 5′‐CCTCCTACCTTGGCATTACA‐3′ and 5′‐CGCTCCCACTGTTCCTTACA‐3′ (forward and reverse, respectively), and the PCR products were sequenced by the dye‐termination method.
Functional analyses
Wild‐type human mTOR cDNA was obtained by PCR, and its mutant forms were generated with the use of a QuickChange site‐directed mutagenesis kit (Agilent Technologies). All cDNA sequences were verified by the dye‐termination method and were then ligated into the pMXS retroviral vector (Cell Biolabs, San Diego, CA, USA). The recombinant plasmids were introduced together with packaging plasmids (Takara Bio, Shiga, Japan) into HEK293T cells in order to obtain recombinant infectious virus particles.
For a focus formation assay, 3T3 cells were infected with the ecotropic recombinant retroviruses with the use of 4 μg/mL polybrene (Sigma‐Aldrich, St. Louis, MO, USA) for 24 h, and further cultured in DMEM‐F12 supplemented with 5% calf serum (Invitrogen) for up to 2 weeks. Cell transformation was assessed either by phase‐contrast microscopy or by staining with the Giemsa solution.
For immunoblot analyses, 3T3 cells were lysed by NP‐40 lysis buffer containing 1% NP‐40, 50 mM Tris‐HCl, 150 mM NaCl, 1 mM sodium fluoride, 1 mM sodium vanadate, and 1 mM PMSF (all from Sigma‐Aldrich). Cell lysates were separated by 7.5% SDS‐PAGE, and subjected to immunoblot analysis. Antibodies used were #2972 for mTOR, #9202 for p70S6K, #9234 for phosphorylated p70S6K, #2217 for S6RP, #4857 for phosphorylated S6RP, #9644 for 4EBP1, #2855 for phosphorylated 4EBP1, #4691 for AKT, #4060 for phosphorylated AKT, and #4970 for ACTB (all from Cell Signaling Technology, Danvers, MA, USA). Rapamycin was obtained from Merck Millipore (Billerica, MA, USA).
For the cell number analysis, 1 × 104 of 3T3 cells at 48 h after the infection with recombinant retrovirus were seeded into each well of 96‐well plates. The next day, the medium was changed to DMEM‐F12 with various concentrations of calf serum, and further cultured for 36 h. Cell number was assessed with a luminometer by using CellTiter‐Glo Luminescent Cell Viability assay (Promega, Madison, WI, USA). At the same time, cells were subjected to measuring the caspase 3/caspase 7 activity by using the Caspase‐Glo 3/7 assay system (Promega).
Results
mTOR(L2209V) is a transforming mutant
Full‐exome sequencing for an LCNEC specimen (#G4T) and its paired normal cells (PBMCs) obtained from a 70‐year‐old man identified a total of 167 somatic missense single nucleotide variations (Table S1) including one for a Leu2209‐to‐Val substitution in mTOR (Fig. 1a, Table S2). Sanger sequencing of the #G4T genome confirmed a heterozygous C‐to‐G mutation in codon 2209 (Fig. 1b). Given that it has been unclear whether mTOR can function as an oncoprotein as a result of its own genomic changes in cancer, we examined the transforming ability of mTOR(L2209V) in a focus formation assay. Expression of mTOR(L2209V) induced marked transformation of mouse 3T3 cells, whereas that of the wild‐type protein did not (Fig. 1c). As a positive control, the v‐Ras oncoprotein also readily transformed 3T3 cells, although the morphology of the transformed cells was distinct from that of those expressing mTOR(L2209V). These results indicated that mTOR can become a bona fide oncoprotein as a result of a somatic mutation found in human cancer.
Figure 1.
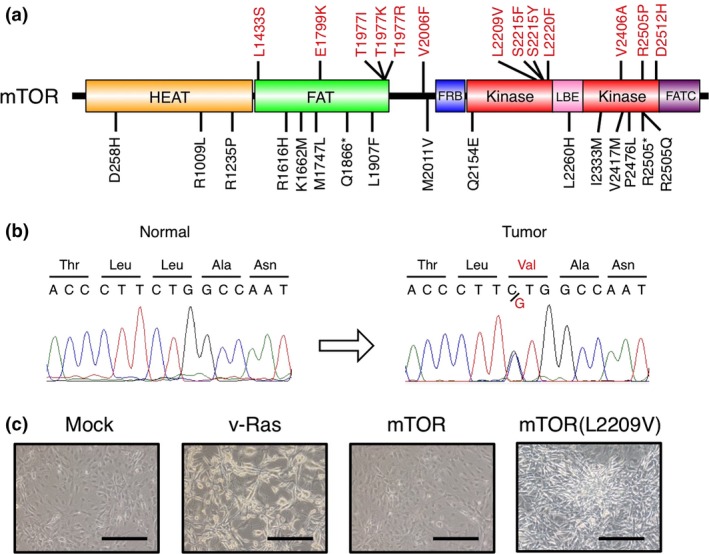
Transforming activity of the mammalian target of rapamycin (mTOR)(L2209V) mutant. (a) Domain organization of human mTOR showing the positions of non‐synonymous mutations (trans‐forming ones shown in red) detected in human cancers. (b) Sequencing electrophoretograms for PCR products corresponding to the region of the mTOR gene containing codon 2209 that were obtained from large cell neuroendocrine carcinoma tumor #G4T or its paired normal sample. (c) Mouse 3T3 fibroblasts infected with recombinant retroviruses encoding v‐Ras, mTOR, or mTOR(L2209V), or with the corresponding empty virus (Mock), were cultured in the presence of 5% calf serum for 12 days and then examined by phase‐contrast microscopy. Scale bar = 100 μm. FAT, FRAP, ATM, TRRAP; FATC, FRAPP, ATM, TOR at C‐terminus; FRB, FK506‐rapamycin binding; HEAT, huntingtin, elongation factor 3, protein phosphatase 2A, TORI; LBE, LST8 binding element.
Various amino acid substitutions confer transforming ability on mTOR
Our exome sequencing of other LCNEC samples did not reveal additional tumors positive for mTOR(L2209V), but identified two independent somatic non‐synonymous mutations (p.D258H and p.R1235P) of mTOR (data not shown). Searches of the COSMIC database as well as our in‐house sequencing data for mTOR mutations identified an additional 26 amino acid substitutions in various cancer specimens and the OCUM‐2M gastric cancer cell line (Fig. 1a, Table S2).12 Of note, recurrent sites for missense mutations, including Thr1977 and Ser2215, were apparent.
We constructed recombinant retroviruses encoding each of the identified mTOR mutants and used them for focus formation assays with mouse 3T3 cells. Unexpectedly, approximately half of these mutants manifested transforming activity (Fig. 2, Fig. S1). Whereas the morphology of the transformed cells appeared similar (Fig. S1), the extent of the malignant change (the number of piled‐up foci) varied substantially among the mutants. For example, mTOR(T1977K), mTOR(T1977R), mTOR(S2215F), and mTOR(S2215Y) showed marked transforming potential, whereas that of mTOR(L2220F) and mTOR(D2512H) appeared less pronounced.
Figure 2.
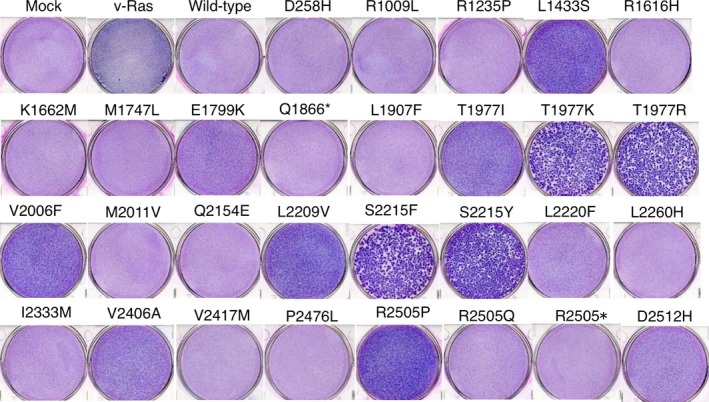
Transforming amino acid changes in mammalian target of rapamycin (mTOR). Mouse 3T3 cells were infected with recombinant retroviruses encoding each mTOR mutant, wild‐type mTOR, or v‐Ras or with the corresponding empty virus (Mock). The cells were cultured in the presence of 5% calf serum for 10 days and then stained with Giemsa solution.
Intracellular signaling evoked by oncogenic mTOR
The various transformed 3T3 cell lines were then examined for the activation status of signaling molecules downstream to mTOR (Fig. 3a). First, immunoblot analysis with antibodies to mTOR revealed that each mTOR mutant was expressed at a similar level, although some mutants (such as R2505P) were more abundant than the others. Phosphorylation of p70S6K at Thr389 was induced by most of the transforming mutants, although such induction was minimal or non‐existent for mTOR(E1799K), mTOR(L2220F), and mTOR(D2512H). Of note, the ability of the mutants to phosphorylate p70S6K appeared to correlate approximately with their transforming potential (Fig. 2, Fig. S1). Activated p70S6K phosphorylates S6 ribosomal protein (S6RP), which in turn promotes mRNA translation. Marked phosphorylation of S6RP at Ser235/236 was evident in the 3T3 cell lines that manifested increased activation of p70S6K (Fig. 3a). Similarly, 4EBP1, another substrate of mTOR, was phosphorylated at Thr37/46 in 3T3 cells expressing the transforming mTOR mutants, although to a lesser extent compared with S6RP phosphorylation.
Figure 3.
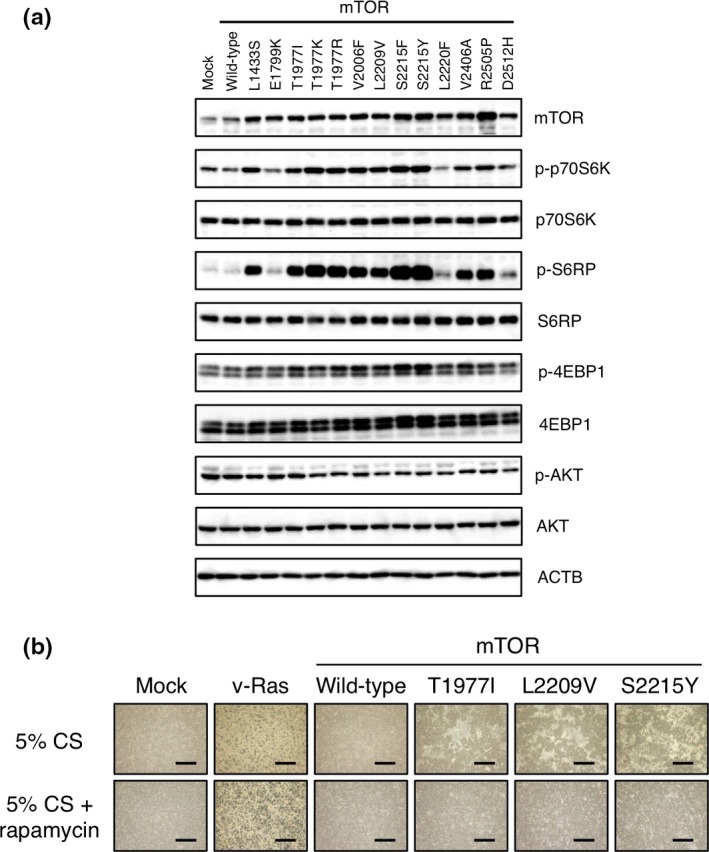
Intracellular signaling evoked by transforming mammalian target of rapamycin (mTOR) mutants and rapamycin sensitivity of transforming activity. (a) Mouse 3T3 cells were lysed 3 days after infection with recombinant retroviruses encoding wild‐type or transforming mutants of mTOR. The cell lysates were subjected to immunoblot analysis with antibodies to total or phosphorylated (p‐) forms of the indicated proteins. ACTB served as a loading control. (b) Mouse 3T3 cells infected with recombinant retroviruses encoding v‐Ras, wild‐type mTOR, mTOR(T1977I), mTOR(L2209V), or mTOR(S2215Y) were cultured in the presence of 5% calf serum (CS) and with or without 100 pM rapamycin for 12 days and then examined by phase‐contrast microscopy. Scale bar = 1 mm.
Whereas mTORC2 directly phosphorylates AKT, activated p70S6K phosphorylates and negatively regulates insulin receptor substrate 1.13 As insulin receptor substrate 1 is an important upstream activator of PI3K–AKT, phosphorylation of p70S6K, thus, evokes a physiological negative feedback loop toward AKT.5 Phosphorylation of AKT at Ser473 was slightly attenuated in 3T3 cells expressing active mTOR mutants compared with that in those expressing the wild‐type protein (Fig. 3a), consistent with a previous observation.8
Transforming activity of mTOR mutants is rapamycin sensitive
Mammalian target of rapamycin acts as a catalytic subunit of both mTORC1 and mTORC2. Given that the macrolide rapamycin inhibits the activity of mTORC1, but not that of mTORC2 in vitro,14 we tested whether the transforming activity of mTOR mutants is sensitive to rapamycin. Mouse 3T3 cells expressing T1977I, L2209V, or S2215Y mutants of mTOR (which are among those with the greatest transforming activity) were subjected to a focus formation assay in the absence or presence of 100 pM rapamycin. Whereas all three mutants (but not the wild‐type protein) generated malignant foci in the absence of rapamycin, this effect was not apparent in the presence of the drug (Fig. 3b, Fig. S2). The transforming ability of these mTOR mutants is thus sensitive to rapamycin.
Cell survival signaling by oncogenic mTOR mutants
Active mTORC1 and mTORC2 induce cell survival signals.15 We therefore examined whether transforming mutation of mTOR promotes cell survival. The 3T3 cells infected with recombinant retroviruses encoding wild‐type mTOR, mTOR(T1971I), mTOR(L2209V), or mTOR(S2215Y) were cultured in the presence of 5%, 0.3%, or 0% calf serum for 36 h (Fig. 4a,b). Whereas cells infected with the empty virus proliferated in the presence of 5% serum, they ceased to do so in the presence of 0.3% or 0% serum. Similarly, 3T3 cells infected with the virus for wild‐type mTOR decreased in number as the serum concentration decreased. In contrast, cells expressing the transforming mutants of mTOR continued to proliferate even in the absence of serum. Cells expressing v‐Ras also generated abnormal foci under all three serum conditions. The serum deprivation‐induced activation of caspase 3/caspase 7 was partially suppressed when 3T3 cells expressed v‐Ras or active mTOR mutants (Fig. 4c). Therefore, oncogenic mutations of mTOR confer anti‐apoptotic functions as well.
Figure 4.
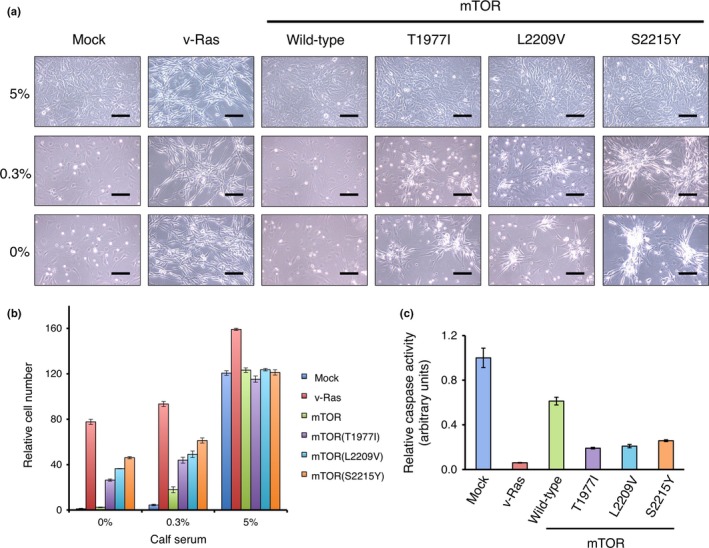
Promotion of cell survival by oncogenic mammalian target of rapamycin (mTOR) mutants. (a) Mouse 3T3 cells infected with recombinant retroviruses encoding v‐Ras, wild‐type mTOR, mTOR(T1977I), mTOR(L2209V) or mTOR(S2215Y), or empty virus (Mock) were cultured in the presence of 5%, 0.3%, or 0% calf serum for 36 h and then examined by phase‐contrast microscopy. Scale bar = 100 μm. (b) The same sets of 3T3 cells after incubation for 36 h with various concentrations of calf serum were subjected to cell number assessment with a luciferase‐based assay. The luciferase activity of each fraction is normalized by that of the mock‐infected 3T3 cells cultured without calf serum, and is shown as mean ± SD of three independent experiments. (c) Caspase 3/caspase 7 activity was measured in 3T3 by a luciferase‐based assay after culturing without calf serum for 36 h. Luciferase activity in each fraction is normalized by the corresponding cell number, and further normalized by that of the mock‐infected 3T3 cells. Data are means ± SD in triplicate experiments.
Discussion
We have here shown that mTOR becomes activated and acquires direct transforming ability as a result of amino acid substitutions found in human cancer cells. The mTOR gene thus becomes an oncogene through genetic alteration. However, we failed to observe tumor formation in a nude mouse tumorigenicity assay for 3T3 cells expressing mTOR mutants, whereas those expressing v‐Ras did generate s.c. tumors in the same observation period (data not shown). The transforming ability of mTOR mutants may, therefore, be less potent than that of v‐Ras.
As expected, oncogenic mTOR mutants activated downstream signaling molecules such as p70S6K, S6RP, and 4EBP1 in 3T3 cells. Furthermore, cells expressing such mTOR mutants were more resistant to nutrient deprivation than were those expressing the wild‐type protein. Our observations that the proliferation of cells expressing these mTOR mutants was sensitive to rapamycin and that AKT was not activated in these cells suggest that such mutants exert their transforming action through mTORC1. Artificially generated active mutants of mTOR similarly confer resistance to nutrient deprivation and induce phosphorylation of S6RP.7 However, given that long‐time treatment with rapamycin can also affect mTORC2 activity in some cell types,16 whether mTORC1 is the only complex that mediates the starvation resistance of cells expressing transforming mTOR mutants requires further examination.
Yang et al.17 reported that artificially generated transforming mutations are widely distributed in the 3‐D structure of mTOR. They are enriched in several “limiting active‐site access” regions including the catalytic cleft and the amino‐terminal portion of FAT. In contrast, most of the naturally occurring transforming mutations (E1799K, L2209V, S2215F/Y, L2220F, V2406A, R2505P, and D2512H) cluster in an unexpectedly narrow region of the structure (Fig. 5). They are thus localized at an interacting surface between the carboxyl‐terminal lobe of the kinase domain and the FAT domain, the latter of which forms a “C”‐shaped clamp for the former. Whether these mutations loosen the binding between the FAT and kinase domains and thereby render the kinase domain more open and active remains to be determined. Our findings suggest that the FAT domain plays a key role in the regulation of mTOR activity.
Figure 5.
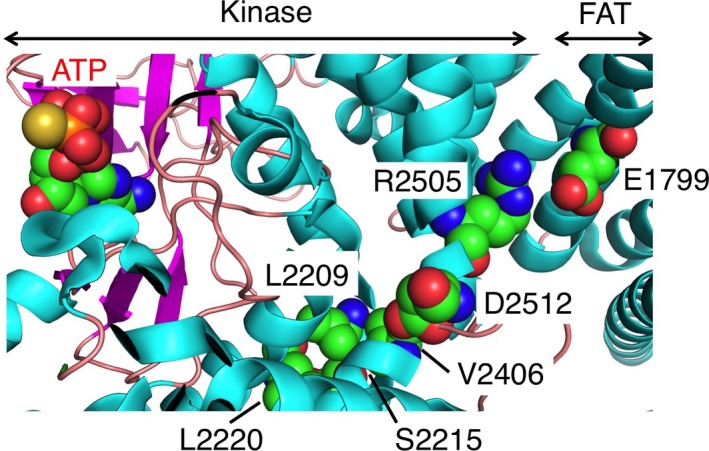
Cluster of transforming amino acid substitutions at the interaction surface between the carboxyl‐terminal lobe of the kinase domain and the FAT (FRAP, ATM, TRRAP) domain of mammalian target of rapamycin (mTOR). The structure of human mTOR (ID “4JSP” in the Protein Data Bank, http://www.pdb.org) is presented schematically, with α helices and β sheets shown in cyan and magenta, respectively. Oncogenic mutation sites (E1799, L2209, S2215, L2220, V2406, R2505, and D2512) and ATP are shown as spherical models. The positions of the kinase and FAT domains are also indicated.
The FRB domain is thought to function as a gatekeeper for the mTOR catalytic site.17 It prevents the entry of non‐substrate peptides, but allows that of genuine ones. Importantly, two activating mutation sites (Thr1977 and Val2006) are positioned close to each other in the vicinity of the FRB domain (Fig. S3). Substitution of these residues thus likely affects the structure or position of the FRB domain.
In general, transforming mutations of protein tyrosine kinases cluster in the activation loop of the catalytic domain. In contrast, groups of activating mutation sites, such as those in the vicinity of the FRB and FAT domains, are located outside of the catalytic domain in mTOR. This finding may be due to the fact that mTOR is intrinsically active and negatively regulated by the FRB and FAT domains.17
We have thus shown that mTOR becomes an oncokinase in human cancer as a result of amino acid substitutions, and that the transforming activity of this oncokinase is sensitive to rapamycin. Inhibitors of mTOR are currently in use clinically for the treatment of renal cell carcinoma, but whether such tumors harbor oncogenic mTOR mutations is not examined in the clinics. Our results have shown that transforming mTOR mutants are distributed across various cancer types (albeit at a low frequency), suggesting that clinical trials with mTOR inhibitors are warranted for individuals with such tumors positive for oncogenic mTOR mutations.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Focus formation assay for mammalian target of rapamycin (mTOR) mutants. Mouse 3T3 cells were infected with recombinant retroviruses encoding each mTOR mutant shown in Figure 1(a), wild‐type mTOR, or v‐Ras or with the corresponding empty virus (Mock). The cells were cultured for 10 days in the presence of 5% calf serum and then examined by phase‐contrast microscopy. Scale bar = 100 μm.
{kind=link}
Fig. S2. Transforming activity of mammalian target of rapamycin (mTOR) mutants is rapamycin sensitive. Mouse 3T3 cells infected with recombinant retroviruses encoding v‐Ras, wild‐type mTOR, mTOR(T1977I), mTOR(L2209V), or mTOR(S2215Y) were cultured in the presence of 5% calf serum and with or without 100 pM rapamycin for 12 days and then stained with Giemsa solution.
{kind=link}
Fig. S3. Positions of Thr1977 and Val2006 in human mammalian target of rapamycin (mTOR). The 3‐D structure of mTOR is shown schematically as in Figure 5. Thr1977, Val2006, and ATP are shown as spherical models.
{kind=link}
Table S1. Somatic missense single nucleotide variations detected in patient #G4T with large cell neuroendocrine carcinoma.
Table S2. Non‐synonymous mutations of mammalian target of rapamycin (mTOR) in human cancer.
Acknowledgments
This study was supported in part by a grant for Leading Advanced Projects for medical innovation from the Japan Agency for Medical Research and Development, by a grant for Research on Development of New Drugs from the Ministry of Health, Labor, and Welfare of Japan, and by Grants‐in‐Aid for Scientific Research (C) and for Young Scientists (B) from the Japan Society for the Promotion of Science.
Cancer Sci 106 (2015) 1687–1692
Funding Information
Japan Agency for Medical Research and Development; Ministry of Health, Labor, and Welfare of Japan; Japan Society for the Promotion of Science.
References
- 1. Sarbassov DD, Ali SM, Sabatini DM. Growing roles for the mTOR pathway. Curr Opin Cell Biol 2005; 17: 596–603. [DOI] [PubMed] [Google Scholar]
- 2. Bader AG, Kang S, Zhao L, Vogt PK. Oncogenic PI3K deregulates transcription and translation. Nat Rev Cancer 2005; 5: 921–9. [DOI] [PubMed] [Google Scholar]
- 3. Xu K, Liu P, Wei W. mTOR signaling in tumorigenesis. Biochim Biophys Acta 2014; 1846: 638–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev 2004; 18: 1926–45. [DOI] [PubMed] [Google Scholar]
- 5. Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer 2006; 6: 729–34. [DOI] [PubMed] [Google Scholar]
- 6. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer 2009; 9: 550–62. [DOI] [PubMed] [Google Scholar]
- 7. Urano J, Sato T, Matsuo T, Otsubo Y, Yamamoto M, Tamanoi F. Point mutations in TOR confer Rheb‐independent growth in fission yeast and nutrient‐independent mammalian TOR signaling in mammalian cells. Proc Natl Acad Sci USA 2007; 104: 3514–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ohne Y, Takahara T, Hatakeyama R et al Isolation of hyperactive mutants of mammalian target of rapamycin. J Biol Chem 2008; 283: 31861–70. [DOI] [PubMed] [Google Scholar]
- 9. Murugan AK, Alzahrani A, Xing M. Mutations in critical domains confer the human mTOR gene strong tumorigenicity. J Biol Chem 2013; 288: 6511–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sekulic A, Hudson CC, Homme JL et al A direct linkage between the phosphoinositide 3‐kinase‐AKT signaling pathway and the mammalian target of rapamycin in mitogen‐stimulated and transformed cells. Cancer Res 2000; 60: 3504–13. [PubMed] [Google Scholar]
- 11. Sato T, Nakashima A, Guo L, Coffman K, Tamanoi F. Single amino‐acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 2010; 29: 2746–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yashiro M, Chung YS, Nishimura S, Inoue T, Sowa M. Establishment of two new scirrhous gastric cancer cell lines: analysis of factors associated with disseminated metastasis. Br J Cancer 1995; 72: 1200–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci Signal 2005; 2005: pe4. [DOI] [PubMed] [Google Scholar]
- 14. Jacinto E, Loewith R, Schmidt A et al Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 2004; 6: 1122–8. [DOI] [PubMed] [Google Scholar]
- 15. Hung CM, Garcia‐Haro L, Sparks CA, Guertin DA. mTOR‐dependent cell survival mechanisms. Cold Spring Harb Perspect Biol 2012; 4: a008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sarbassov DD, Ali SM, Sengupta S et al Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22: 159–68. [DOI] [PubMed] [Google Scholar]
- 17. Yang H, Rudge DG, Koos JD, Vaidialingam B, Yang HJ, Pavletich NP. mTOR kinase structure, mechanism and regulation. Nature 2013; 497: 217–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Focus formation assay for mammalian target of rapamycin (mTOR) mutants. Mouse 3T3 cells were infected with recombinant retroviruses encoding each mTOR mutant shown in Figure 1(a), wild‐type mTOR, or v‐Ras or with the corresponding empty virus (Mock). The cells were cultured for 10 days in the presence of 5% calf serum and then examined by phase‐contrast microscopy. Scale bar = 100 μm.
Fig. S2. Transforming activity of mammalian target of rapamycin (mTOR) mutants is rapamycin sensitive. Mouse 3T3 cells infected with recombinant retroviruses encoding v‐Ras, wild‐type mTOR, mTOR(T1977I), mTOR(L2209V), or mTOR(S2215Y) were cultured in the presence of 5% calf serum and with or without 100 pM rapamycin for 12 days and then stained with Giemsa solution.
Fig. S3. Positions of Thr1977 and Val2006 in human mammalian target of rapamycin (mTOR). The 3‐D structure of mTOR is shown schematically as in Figure 5. Thr1977, Val2006, and ATP are shown as spherical models.
Table S1. Somatic missense single nucleotide variations detected in patient #G4T with large cell neuroendocrine carcinoma.
Table S2. Non‐synonymous mutations of mammalian target of rapamycin (mTOR) in human cancer.