

Abstract
Radiotherapy is widely applied for treatment of esophageal squamous cell carcinoma (ESCC). The Rad51‐related protein XRCC3 plays roles in the recombinational repair of DNA double‐strand breaks to maintain chromosome stability and repair DNA damage. The present study aimed to investigate the effect of XRCC3 on the radiotherapy response of ESCC and the underlying mechanisms of the roles of XRCC3 in ESCC radiosensitivity. XRCC3 expression in ESCC cells and tissues was higher than that in normal esophageal epithelial cells and corresponding adjacent noncancerous esophageal tissue. High XRCC3 expression was positively correlated with resistance to chemoradiotherapy in ESCC and an independent predictor for short disease‐specific survival of ESCC patients. Furthermore, the therapeutic efficacy of radiotherapy in vitro and in vivo was substantially increased by knockdown of XRCC3 in ESCC cells. Ectopic overexpression of XRCC3 in both XRCC3‐silenced ESCC cells dramatically enhanced ESCC cells' resistance to radiotherapy. Moreover, radiation resistance conferred by XRCC3 was attributed to enhancement of homologous recombination, maintenance of telomere stability, and a reduction of ESCC cell death by radiation‐induced apoptosis and mitotic catastrophe. Our data suggest that XRCC3 protects ESCC cells from ionizing radiation‐induced death by promoting DNA damage repair and/or enhancing telomere stability. XRCC3 may be a novel radiosensitivity predictor and promising therapeutic target for ESCC.
Keywords: Esophageal squamous cell carcinoma, homologous recombination, radiotherapy, telomere stability, XRCC3
Esophageal cancer is one of the most common cancers and the sixth leading cause of cancer‐related deaths worldwide.1 Esophageal squamous cell carcinoma (ESCC) is the predominant histological type of esophageal cancer in East Asian, with 5‐year survival of <30%. Clinically, most ESCC patients present with locally advanced disease for which chemoradiotherapy (CRT) is considered as the standard treatment. Even with recent advancements in diagnostics and therapies, the survival rate of ESCC patients is still poor.2, 3, 4 Currently, only a primary complete response (CR) to CRT and the tumor‐node‐metastasis (TNM) stage of ESCC are widely accepted as prognostic factors.5 Thus, there is still an urgent need to identify novel markers that can predict CRT responses and/or outcomes of ESCC patients.
Radiotherapy damages DNA directly and causes double‐strand breaks (DSB) that are considered as the main lethal form of DNA damage caused by ionizing radiation (IR). Error prone, nonhomologous end joining (NHEJ) and high fidelity, error‐free homologous recombination (HR) are the two main repair pathways for DSB.6, 7, 8 Previous studies have shown that NHEJ acts during any phase of the cell cycle and is the primary mechanism for the repair of DSB induced by IR.9 However, high expression of HR‐associated proteins is associated with increased resistance to CRT in several types of human cancer cells.10, 11, 12 Furthermore, many inhibitors targeting the HR pathway can delay DSB repair and increase the sensitivity of cancer cells to IR.13 These findings suggest that HR plays a critical role in the repair of DSB induced by IR and may be an effective target for radiotherapy of cancer.
In mammals, the core of the HR pathway includes MRN (MRE11, RAD50 and NSB1), which form a complex, RAD51 and RAD51 paralogs (Rad51B, Rad51C, Rad51D, XRCC2, and XRCC3, BRCA1, BRCA2, RAD52, RAD54, and RAD54B).14 The X‐ray repair complementing defective repair in Chinese hamster cell (XRCC3) protein, which shares some homology and interacts with RAD51, has roles in both the initiation and late stages of HR and controls the fidelity of HR.15, 16 It has been reported that polymorphisms of the XRCC3 gene are positively associated with an increased risk for numerous cancers and may be effective biomarkers for genetic susceptibility to these diseases. In addition, XRCC3‐deficient cells are more sensitive to cisplatin by suppression of HR and increases in chromosomal instability.17, 18, 19 These data indicate that XRCC3 has roles in tumorigenesis and cancer therapy. However, the details of XRCC3 expression and its clinical significance in the prognosis and therapeutic responses of cancers, including ESCC, have not been elucidated.
Here, we report for the first time that upregulation of XRCC3 in ESCC tissue is closely associated with CRT resistance in ESCC patients and predicts poor patient survival. Furthermore, we investigated the possible effects and molecular mechanism of XRCC3 in the radiosensitivity of ESCC in vitro and in vivo.
Materials and Methods
Patients and tissue specimens
A total of 60 paraffin‐embedded tissue samples were obtained from 60 patients with ESCC at Tianjin Medical University Cancer Institute and Hospital. We also collected 20 specimens of normal esophageal epithelial tissue. All tissues were obtained by esophagoscopy as diagnostic biopsies. Patients who received definitive CRT at the Department of Radiotherapy between January 2008 and January 2010 were enrolled in this study. All 60 patients had received CRT with cisplatin‐based chemotherapy and the same concomitant radiotherapy (daily dose of 1.8–2.0 Gy to a total of 60–70 Gy over 6–7 weeks). Tumor staging was carried out according to the 6th edition of the TNM classification of the International Union Against Cancer (UICC, 2002). The clinicopathological characteristics of the patients are summarized in Table 1. The study was approved by the medical ethics committee of the institute.
Table 1.
Clinico‐pathological correlation of XRCC3 expression in ESCC
| Variables | Cases | High expression (%) | P † |
|---|---|---|---|
| Age (years) | |||
| ≤55‡ | 33 | 21 (63.6) | 0.662 |
| >55 | 27 | 18 (66.7) | |
| Gender | |||
| Male | 46 | 30 (65.2) | 0.513 |
| Female | 14 | 9 (64.2) | |
| WHO grade | |||
| G1 | 15 | 10 (66.7) | 0.631 |
| G2 | 27 | 17 (63.0) | |
| G3/4 | 18 | 12 (66.7) | |
| Tumor size (cm) | |||
| ≤6§ | 40 | 25 (62.5) | 0.140 |
| >6 | 20 | 14 (70.0) | |
| T status | |||
| T2 | 17 | 11 (64.7) | 0.541 |
| T3 | 24 | 15 (62.5) | |
| T4 | 19 | 13 (68.4) | |
| N status | |||
| N0 | 23 | 15 (65.2) | 0.715 |
| N1 | 37 | 24 (64.9) | |
| M status | |||
| M0 | 33 | 20 (60.6) | 0.116 |
| M1‐lym | 27 | 19 (70.4) | |
| CRT response | |||
| CR | 15 | 5 (33.3) | 0.002 |
| Not CR | 45 | 34 (75.6) | |
CR,complete response; CRT, chemoradiotherapy; DSS, disease‐specific survival; ESCC, esophageal squamous cell carcinoma; M, metastases; M1‐lym, distant lymph node metastasis; N, node; T, tumor. †χ2‐test. ‡Mean age. §Mean tumor size.
Evaluation and follow up
When patients completed the treatments, the CRT effect was evaluated clinically for primary lesions based on esophagography and CT 4 weeks after CRT according to the World Health Organization criteria. Complete response (CR), partial response (PR), no change (NC) and progressive disease (PD) were achieved in 15, 31, 13 and 1 patient, respectively. Of the 45 patients who did not achieve CR, 12 cases received adjuvant chemotherapy, and 2 cases received radical esophagectomy. The other patients did not receive any anti‐tumor treatments until tumor progression. The patients were followed every 3 months for the first year and then every 6 months for the next 2 years, and thereafter annually. The diagnostic examinations consisted of esophagography, CT, chest X‐ray, abdominal ultrasonography and bone scanning when necessary to detect recurrence and/or metastasis.
Cell lines and culture
Primary cultures of normal esophageal epithelial cells were established from fresh specimens of adjacent noncancerous esophageal tissue according to a previous study.20 ESCC cell lines KYSE30, KYSE150, KYSE410 and KYSE510 were cultured in RPMI 1640 and the TE‐1 cell line was maintained in DMEM at 37°C in a humidified atmosphere with 5% CO2. Media were supplemented with 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin.
Reverse transcription PCR, RT‐PCR
The expression of XRCC3, XRCC2 and Rad51C mRNA was analyzed by a RT‐PCR assay. The total RNA, which was extracted using TRIZOL (Invitrogen, Carlsbad, CA, USA), was used for cDNA synthesis with Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega, Madison, WI, USA). RT‐PCR determined the expression pattern of XRCC3 mRNA in each of the ESCC cells. Expression data were normalized to GAPDH. cDNA was subjected to PCR with primers for XRCC3 (Forward, 5′‐AACCCGCGGGAGAGTCCCCA‐3′ and Reverse, 5′‐AAAGCCTGTGGGAGGCCCGA‐3′), GAPDH (Forward, 5′‐GTTCGACAGTCAGCCGCATCT‐3′ and Reverse, 5′‐CCTGCAAATGAGCCCCAGCCT‐3′), Rad51C (Forward, 5′‐TTCGCTGTCGTGACTACACA‐3′ and Reverse, 5′‐TGCCAACCTTTGCTTTCGGT‐3′) and XRCC2 (Forward, 5′‐TGGATAGACCGCGTCAATGG‐3′ and Reverse, 5′‐GCTGCCATGCCTTACAGAGA‐3′). Amplification consisted of 30 cycles of 30 s at 94°C, 30 s at 56°C and 60 s at 72°C.
Western blot assay
Cells were lysed in lysis buffer. Protein concentration was determined using a BCA kit. SDS‐PAGE and western blotting were done according to standard procedures. Proteins were detected with antibodies recognizing XRCC3, Rad51, Rad51b, Rad51c, Rad51d (Abcam, Cambridge, MA, USA) and PARP, cleaved‐PARP, Caspase‐3, cleaved‐Caspase‐3 (Cell Signaling Technology, Danvers, MA, USA). GAPDH (Santa Cruz Biotechnology, California, CA, USA) was used as loading control.
Immunohistochemistry
Paraffin‐embedded tissues were cut into 4‐μm sections and rehydrated through a graded alcohol series, subjected to heating for antigen retrieval, allowed to cool to room temperature, immersed in 3% hydrogen peroxide for 5–10 min, and then rinsed with PBS. The sections were incubated with normal goat serum to block nonspecific binding, followed by incubation overnight with primary antibodies at 4°C, and then rinsed with PBS. Subsequently, the sections were incubated with a secondary antibody for 15 min at 37°C, washed with PBS and then stained with diaminobenzidine. Finally, the sections were counterstained with hematoxylin, dehydrated and mounted.
Immunohistochemistry evaluation
The expression of XRCC3 was assessed by three independent pathologists (D. Xie, J. Cheng and D. Qian), who were blinded to clinical follow‐up data. Their conclusions were in complete agreement for 85% of cases, indicating that this scoring method was highly reproducible. If two or all three agreed with the scoring results, the value was selected. If the results were completely different, then the pathologists worked collaboratively to confirm the score.
For evaluation of XRCC3 staining, a semi‐quantitative scoring criterion was used, in which both staining intensity and positive areas were recorded. A staining index (values 0–12) obtained as the intensity of positive staining (weak, 1; moderate, 2; strong, 3) and the proportion of immune‐positive cells of interest (0%, 0; <10%, 1; 10–50%, 2; 51–80%, 3; >80%, 4) were calculated. Finally, cases were classified into two different groups: low expression cases (score 0–6) and cases with high expression (score 8–12).
Construction of the recombinant lentivrial vector
Plasmid containing the validated shRNA targeted XRCC3 was cloned into the vectors. pLLU2G (kindly provided by Professor Dan Xie, Cancer Center, Sun Yet‐Sen University) used in this study are derived from pLL3.7, contain separate GFP and short hairpin RNA (shRNA) expression elements and are required for lentiviral packaging. The target sequences of XRCC3 for constructing lentiviral shRNA are 5′‐TTCATCGAGCACGTGGCCGAT‐3′ (shXRCC3). For rescue experiments, a XRCC3 construct resistant to the shRNA used (shXRCC3) (mutations underlined: 5′‐TTTATAGAACATGTCGCAGAT‐3′; the mutations do not affect XRCC3 protein sequence) was cloned into a pCDH cDNA expression lentivector (System Biosciences, Mountain View, CA, USA). Then the lentiviral expression construct and packaging plasmid mix was co‐transfected into 293 cells to generate the recombinant lentivirus (according to the manual).
Clonogenic survival assay
Survival following radiation exposure was defined as the ability of the cells to maintain their clonogenic capacity and to form colonies. KYSE 30 and TE‐1 ESCC cells were counted and seeded for colony formation in six‐well plates with 50–5000 cells per well. After incubation intervals of 14–21 days, colonies were stained with crystal violet and manually counted. Colonies consisting of 50 cells or more were scored, and five replicate wells containing 10–150 colonies per well were counted for each treatment.
In vivo tumor growth assay
Female 4‐week‐old athymic nude mice were used for in vivo experiments. TE‐1‐ shXRCC3 cells and the corresponding control TE‐1 cells were injected subcutaneously on the lateral aspect of the rear leg. When tumors of the control group grew to a volume of approximately 500 mm3, a total radiation dose of 6 Gy (2 Gy/fraction every other day for 3 days) was delivered locally using a Pantak X‐ray source to animals restrained in custom lead jigs. Tumor diameters were measured with calipers every 3 days, and tumor volumes were calculated using the formula (width2 × length/2). All the procedures are in accordance with the guidelines of the laboratory animal ethics committee of Tianjin Medical University.
Annexin‐V‐FITC/propidium iodide flow cytometry apoptosis assay
Annexin V‐FITC and propidium iodide stains were used to determine the percentage of cells undergoing apoptosis. Briefly, after exposure to radiation, cells were stained in the dark for 15 min at room temperature. Each sample was then subjected to analyses by flow cytometry (BD FACSCanto II Flow Cytometer; BD Biosciences, Bedford, MA, USA).
Immunofluorescence
Cells grown in coverslips were washed twice in PBS and incubated in 3.7% formaldehyde for 10 min at room temperature. After permeabilization and blocking with 2% normal goat serum and 0.5% Triton X‐100 for 30 min, the cells were double immunostained with the mouse anti‐TRF1 antibody (GeneTex, Alton Pkwy Irvine, CA, USA) and rabbit anti‐γH2AX antibody (Cell Signaling Technology) in a humidified chamber for 1 h. Primary antibodies were visualized by DyLight 549 conjugated Goat anti‐Mouse IgG (Thermo Scientific, Waltham, MA, USA) and Alexa Fluor 488 conjugated Goat anti‐Rabbit IgG (Invitrogen). DNA were stained with DAPI. Images were collected on the Olympus FluoView confocal microscopes and analyzed with FV10‐ASW viewer software (Olympus, Tokyo, Japan). The telomere dysfunction induced foci (TIF) was evidenced by the colocalizations of the DNA damage factor γH2AX with the constitutive telomere protein component TRF1.21 Cells with at least five telomeric γH2AX foci were scored as TIF positive. Data reported are averages of three independent experiments.
Immunoprecipitation
Cells (2 × 107) were lysated and sonicated in a cell lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X‐100, 2.5 mM sodium pyrophosphate, 10 mg/mL protease inhibitor cocktail) on ice. Equal amounts of cell lysates precleared with protein A/G agarose (Calbiochem, Darmstadt, Germany) were incubated with the primary antibody with gentle rocking overnight at 4°C. Immune complexes were then precipitated by incubating them with proteinA/G agarose for 2 h at 4°C. Immunoprecipitates were washed five times with a cell lysis buffer. After boiling in 20 mL 2 × SDS sample buffer, the samples were analyzed by western blotting. Western blot analysis followed the standard procedures and was repeated at least three times for each protein tested.
Statistical analysis
Statistical analyses were performed using the SPSS 17.0 statistical software package. Data from in vitro experiments are presented as the mean ± standard error (SE) and were assessed by the two‐tailed Student's t‐test. Associations between XRCC3 expression and the clinicopathological features of ESCC patients were assessed by the χ2‐test. Disease‐specific survival (DSS) was defined as the time from diagnosis to cancer‐related death, which was analyzed using the Kaplan–Meier method and compared using the log‐rank test. Multivariate survival analysis was performed on all parameters that were found to be significant in univariate analysis using the Cox‐regression model. A value of P < 0.05 was considered statistically significant.
Results
High expression of XRCC3 in esophageal squamous cell carcinoma cell lines and tissues
In this study, the mRNA and protein levels of XRCC3 were examined by RT‐PCR (Fig. 1a) and western blotting (Fig. 1b), respectively. All five ESCC cell lines expressed higher levels of XRCC3 than the normal cell line. Next, the expression of XRCC3 was detected by immunohistochemistry (IHC) in 60 ESCC samples and 20 normal control specimens of esophageal mucosa. According to the scoring criteria detailed above, high expression of XRCC3 was observed in 65% (39/60) of the ESCC samples. However, only 10% (2/20) of the normal esophageal epithelial mucosa specimens showed high expression of XRCC3 (Fig. 1c–e, P < 0.01). Consistent with the in vitro data, XRCC3 was strongly upregulated in these ESCC tissues.
Figure 1.
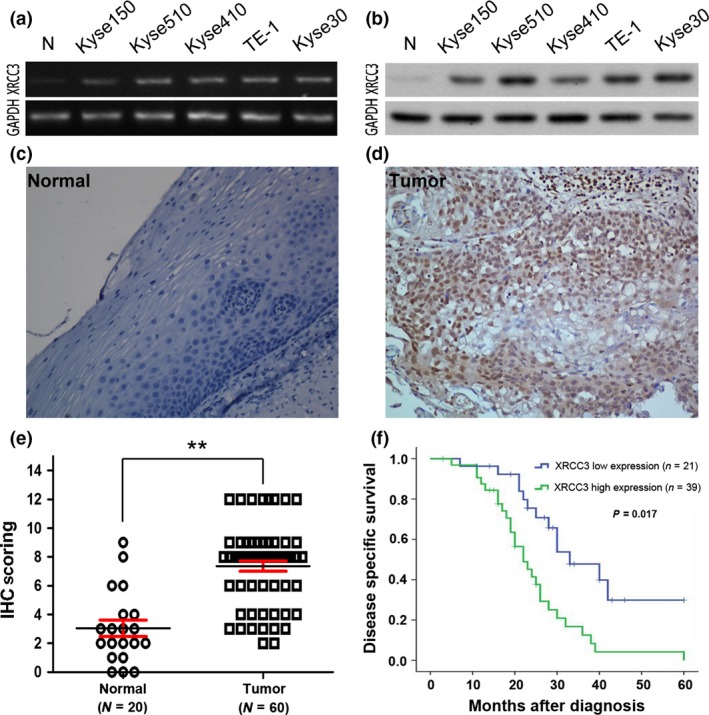
XRCC3 expression in esophageal squamous cell carcinoma (ESCC) cell lines and tissues and its prognostic significance in ESCC patients. (a,b) RT‐PCR (a) and western blotting (b) showed that the levels of XRCC3 in five ESCC cell lines (Kyse150, Kyse510, Kyse410, TE‐1 and Kyse30) were higher than that in a normal esophageal epithelial cell line (N). (c,d) Immunohistochemistry staining of XRCC3 in human esophageal tissues. A specimen of normal esophageal mucosa (case 7) was negative for XRCC3 (c) and an ESCC sample (case 32) exhibited high expression of XRCC3, in which more than 90% of carcinoma cells demonstrated positive staining for XRCC3 (d). (e) Statistical analysis revealed significantly higher expression of XRCC3 in ESCC (**P < 0.01, Student's t‐test). (f) High expression of XRCC3 was associated with a poor prognosis in 60 ESCC patients. Kaplan–Meier plots show disease‐specific survival (DSS) curves according to XRCC3 expression levels in the primary tumor (P = 0.017, log‐rank test).
High expression of XRCC3 in esophageal squamous cell carcinoma is associated with chemoradiotherapy resistance and predicts poor patient survival
Table 1 summarizes the rate of high expression of XRCC3 with respect to several standard clinicopathological features in the ESCC patients. A relationship was not found between high XRCC3 expression and most patient characteristics, including age, gender, TNM stage, tumor grade and tumor size. However, further analysis showed that high expression of XRCC3 was significantly associated with CRT resistance in ESCC patients. Among the 60 ESCC patients, CR, PR, NC and PD were achieved in 15, 31, 13 and 1 patient, respectively, at the time of evaluation. In these cases, high expression of XRCC3 was observed more frequently in the non‐CR group than in the CR group (P = 0.002, Table 1). Moreover, in univariate analysis, high expression of XRCC3 was correlated closely with poor DSS (P = 0.017, Fig. 1f). Further multivariate analysis showed that XRCC3 expression as well as a CRT response and M stage were independent predictors of DSS (P = 0.011, 0.007 and 0.041, respectively; Table 2).
Table 2.
Multivariate Cox regression analysis for DSS in ESCC patients
| Factors | HR | 95%CI | P |
|---|---|---|---|
| XRCC3 Expression | 2.296 | 1.557–4.520 | 0.011 |
| CRT response | 2.973 | 1.429–6.985 | 0.007 |
| N stage | 1.421 | 0.493–3.247 | 0.173 |
| M stage | 1.310 | 0.652–3.017 | 0.041 |
CI, confidence interval; DSS, disease‐specific survival; ESCC, esophageal squamous cell carcinoma; HR, hazard ratio.
Depletion of XRCC3 enhances esophageal squamous cell carcinoma cell radiosensitivity in vitro and in vivo
Radiotherapy is widely applied to ESCC patients and has a central role in the therapeutic strategy against ESCC. To explore the roles of XRCC3 in the radiotherapy response of ESCC cells, we infected KYSE30 and TE‐1 cells with a lentivirus carrying XRCC3 short hairpin RNA (shXRCC3) and the corresponding control scramble shRNA. The knockdown efficiency of XRCC3 was confirmed by western blotting (Fig. 2a). The results showed that inhibition of XRCC3 had no significant effect on the colony formation capacity of untreated cells (Fig. 2b). However, after IR treatment, clonogenic survival was obviously reduced in both Kyse30‐shXRCC3 and TE‐1‐shXRCC3 cell lines compared with control cells (Fig. 2c). To ensure that the levels of XRCC3 modulate the radiosensitivity of ESCC cells, we replenished the levels of XRCC3 in XRCC3‐silenced Kyse30 and TE‐1 cells by infection with recombinant lentivirus encoding a XRCC3 construct resistant to the used shRNA (shXRCC3 + XRCC3) (Fig. 2a). We observed that after ectopic overexpression of XRCC3 in both XRCC3‐silenced ESCC cells, the survival capacity of the cells under IR treatment was substantially enhanced (Fig. 2c). Next, we investigated whether XRCC3 knockdown affected ESCC cell responses to IR in vivo. We found that depletion of XRCC3 did not affect tumor growth under normal conditions but xenograft tumor volumes were decreased significantly compared with the control group after the same IR treatment (Fig. 2d). These results suggest a radiosensitizing effect by suppression of XRCC3.
Figure 2.
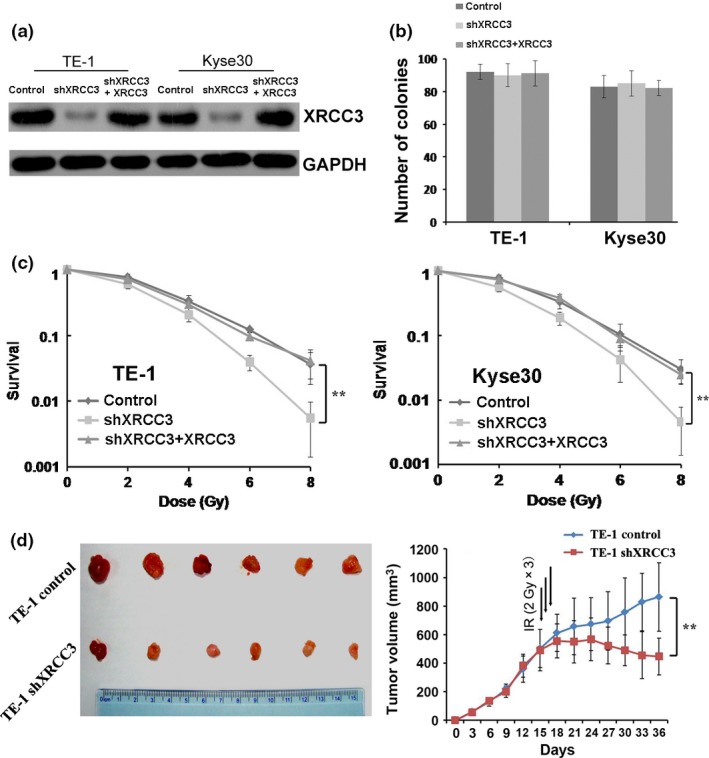
Depletion of XRCC3 enhances esophageal squamous cell carcinoma (ESCC) cell radiosensitivity in vitro and in vivo. (a) A shRNA targeting XRCC3 mRNA (shXRCC3) was introduced into two ESCC cell lines (TE‐1 and Kyse30) for stable knockdown of XRCC3 through recombinant lentiviral infection. Then, pCDH‐XRCC3 lentiviral particles were transduced into the above XRCC3‐silenced ESCC cells (shXRCC3 + XRCC3) to replenish XRCC3 expression. The levels of XRCC3 were examined by western blotting. (b) XRCC3 levels had no effect on colony formation of Kyse30 and TE‐1 cells. Surviving colonies (>50 cells/colony) were counted and are shown in the bar chart. (c) Silencing of XRCC3 enhanced radiosensitivity in both TE‐1 and Kyse30 cells. The responses of ESCC cells to ionizing radiation (IR) were examined by clonogenic survival assays. After ectopic overexpression of XRCC3 in both XRCC3‐silenced ESCC cells, the survival capacity of the cells under IR treatment was substantially enhanced. (Data represent the mean ± SE derived from three individual experiments with triplicate wells. Error bars, SE.) (d) Inhibition of XRCC3 enhanced the therapeutic effect of IR on TE‐1 cell xenografts. XRCC3 knockdown did not affect the growth of tumors before IR treatment. The mean tumor volumes in shXRCC3 and control groups were 502.6 ± 134.72 mm3 and 490.7 ± 144.28 mm3, respectively, at day 15 after tumor cell transplantation (n = 6, P = 0.73, Student's t‐test). After 6 Gy of IR treatment, the mean tumor volume in the shXRCC3 group was 447.2 ± 129.66 mm3, which was significantly smaller than the 863.9 ± 239.53 mm3 in the control group at day 36 after tumor cell transplantation (n = 6, P = 0.01, Student's t‐test). Values represent the mean tumor volume ± SE.
Inhibition of XRCC3 promotes ionizing radiation‐induced apoptosis and mitotic catastrophe in esophageal squamous cell carcinoma cells
Apoptosis and mitotic catastrophe (MC) are considered as the main forms of cell death induced by IR. First, we examined the effect of XRCC3 depletion on apoptosis by flow cytometry. Compared with control cells, inhibition of XRCC3 did not appear to induce KYSE30 or TE‐1 cell apoptosis. However, a significant increase in the proportion of apoptotic cells was found when the cells were treated with 4 or 6 Gy IR (Fig. 3a). Next, we analyzed the effect of XRCC3 depletion on IR‐induced MC. KYSE30 and TE‐1 cells were stained with DAPI after 6 Gy IR treatment and then cultured for 48 h. Compared with control cells, the incidence of the micronuclear phenotype, a hallmark of MC, was remarkably increased in XRCC3‐silenced cells treated with IR (Fig. 3b). Consistently, suppression of XRCC3 in KYSE30 and TE‐1 cells induced significant increases in cleaved PARP and cleaved caspase‐3 in cells treated with 6 Gy IR (Fig. 3c). Moreover, western blot analysis indicated significant downregulation of XRCC2 and RAD51C in both KYSE30‐shXRCC3 and TE‐1‐shXRCC3 cells (Fig. 3d). In contrast, after restoration of XRCC3, the altered cell apoptosis, MC, protein expression of XRCC2 and RAD51C were all recovered (Fig. 3a–d). However, as measured by RT‐PCR, the mRNA levels of Rad51C and XRCC2 were not altered by XRCC3 knockdown in ESCC cells (Fig. S1a). In addition, an immunoprecipitate assay was performed with the XRCC3 antibody, and we detected Rad51C and XRCC2 in the XRCC3 immunoprecipitates (Fig. S1b).
Figure 3.
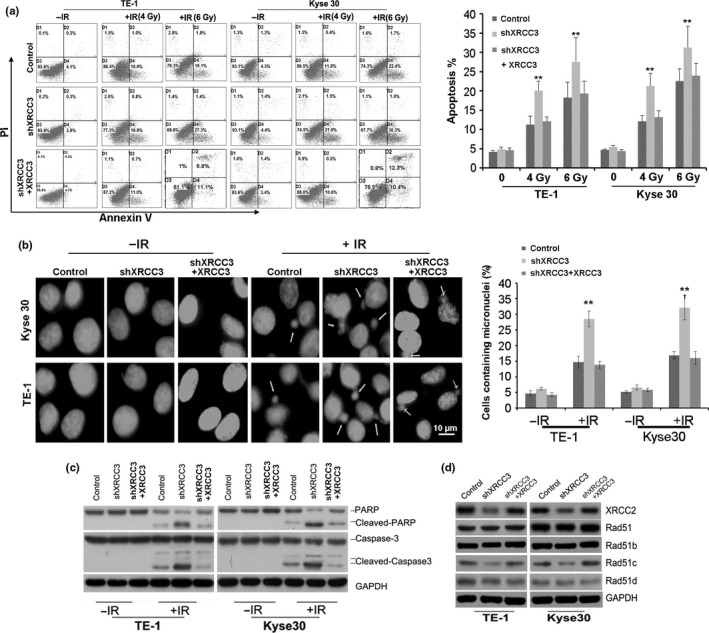
Silencing of XRCC3 promotes ionizing radiation (IR)‐induced apoptosis and mitotic catastrophe (MC) in esophageal squamous cell carcinoma (ESCC) cells. (a) Silencing of XRCC3 promoted IR‐induced apoptosis in both Kyse30 and TE‐1 cells. Annexin V and propidium iodide staining was used to determine the percentage of cells undergoing apoptosis. The percentage of cells with apoptosis is also shown (right). Data represent mean values and SE (*P < 0.05; **P < 0.01, Student's t‐test). (b) Inhibition of XRCC3 increased micronuclei in both Kyse30 and TE‐1 cells exposed to 6 Gy IR. Cells were stained with DAPI and examined for nuclear morphology. Arrows indicate micronuclei in interphase (left). The percentage of cells with micronuclei is also shown (right). Data represent mean values and SE. (Bar equals 10 um. *P < 0.05; **P < 0.01, Student's t‐test.) (c) Silencing of XRCC3 increased the levels of cleaved PARP and cleaved caspase‐3 in both TE‐1 and Kyse30 cells exposed to 6 Gy IR. (d) Inhibiton of XRCC3 in ESCC cells downregulated the protein expression of XRCC2 and Rad51c. However, XRCC3 knockdown did not alter the expression of Rad51, Rad51b or Rad51c. GAPDH was used as a loading control. After restoration of XRCC3, the altered cell apoptosis, mitotic catastrophe (MC), protein expression of XRCC2 and RAD51C were all recovered. All data are derived from three individual experiments.
Inhibition of XRCC3 enhances ionizing radiation‐induced DNA damage and telomere dysfunction
Homologous recombination plays important roles in DSB repair and telomere maintenance. Therefore, we investigated whether downregulation of XRCC3 influences DNA damage repair and telomere stability. After 48 h of IR treatment, the unrepaired DNA damage detected by r‐H2AX using immunofluorescence was significantly increased in XRCC3‐silenced ESCC cells compared with the control cells (Fig. 4a,b). Furthermore, the incidence of telomere dysfunction as judged by telomere dysfunction induced foci (TIF) assays was increased remarkably in XRCC3‐knockdown KYSE30 and TE‐1 cells compared with control cells after IR exposure (Fig. 4a,c). After replenishment of XRCC3 in both XRCC3‐silenced KYSE30 and TE‐1 cells, the altered levels of IR‐induced DNA damage and telomere dysfunction were all recovered (Fig. 4a–c). Collectively, these results suggest that elevated XRCC3 levels in ESCC may confer resistance to IR‐induced apoptosis and MC by enhancing DSB damage repair and avoiding telomere dysfunction under IR conditions.
Figure 4.
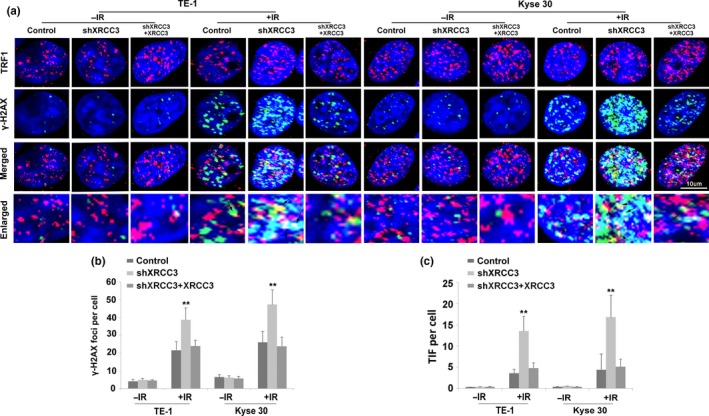
Silencing of XRCC3 promotes ionizing radiation (IR)‐induced DNA damage and telomere dysfunction in both Kyse30 and TE‐1 cells. (a) Silencing of XRCC3 by shXRCC3 increased the percentage of telomere dysfunction induced foci (TIF)‐positive cells among both TE‐1 and Kyse30 cells. Cells were fixed and double stained with an anti‐TRF1 mouse monoclonal antibody to mark telomeres (red) and an anti‐phosphorylated histone H2A.X (Ser‐139) rabbit monoclonal antibody to indicate DNA damage‐activated γH2AX foci (green). Nuclei were counterstained with DAPI (blue). Merged images show DNA damage at telomeres. Arrows indicate double‐stained areas (yellow). (b) Quantification of the average numbers of IR‐induced γH2AX foci per cell. (c) Quantification of the average numbers of IR‐induced TIF per cell. After replenishment of XRCC3 in both XRCC3‐silenced KYSE30 and TE‐1 cells, the altered levels of IR‐induced DNA damage and telomere dysfunction were recovered. 48 h after irradiation, cells were fixed to perform an immunofluorescence assay. All data are derived from three individual experiments. Data represent mean values and SE. (Bar equals 10 um. *P < 0.05; **P < 0.01, Student's t‐test.)
Discussion
XRCC3 is located in the 14q32.3 chromosome region and plays a key role during HR that is essential for DSB repair and maintaining chromosomal stability.17, 22 To date, many reports have shown that polymorphisms in the XRCC3 gene are associated with an increased risk for various solid tumors, including breast, bladder and lung cancers.23, 24, 25 However, the expression profile and clinical significance of XRCC3 protein in cancer remain unknown. To investigate the potential role of XRCC3 in ESCC, we first examined the expression level of XRCC3 protein by IHC in 20 normal esophageal mucosa tissues and 60 ESCC cases with complete follow‐up information. For the first time, our results clearly show that XRCC3 expression is frequently higher in ESCC tissues than in normal esophageal mucosal tissues. Further correlation analyses revealed that high expression of XRCC3 was closely correlated with ESCC resistance to CRT. Moreover, high XRCC3 expression was a strong and independent predictor for poor survival of this disease. Taken together, these results underscore a potentially important role of XRCC3 in the development and therapeutic responses of ESCC.
Clinically, radiotherapy is widely applied to ESCC patients who present with locally advanced disease.26 In this study, all ESCC patients had received cisplatin‐based chemotherapy and radiotherapy. A previous study has shown that XRCC3 induces cisplatin resistance in MCF‐7 human breast cancer cells by stimulation of Rad51‐related recombinational repair.19 Similarly, in the present study, we observed that inhibition of XRCC3 increased the sensitivity of ESCC cells to IR in vitro and in vivo. To the best of our knowledge, apoptosis and MC are the two main mechanisms by which IR induces cell death.27, 28, 29 MC is a delayed type of cell death executed through necrosis, delayed apoptosis or induced senescence days after radiotherapy initiation, which can explain why clinical regression of solid tumors is slow.27 Thus, we examined the effect of XRCC3 inhibition on IR‐induced apoptosis and MC using annexin V‐FITC/propidium iodide (PI) staining together with flow cytometry and detection of micronuclei, respectively, which are a hallmark of MC.30 Our data revealed that XRCC3 knockdown enhanced apoptotic cell death and MC induced by IR. Furthermore, silencing XRCC3 resulted in a significant increase of proteolytic cleavage of caspase‐3 and PARP in cells treated with IR. Therefore, upregulation of XRCC3 might enable ESCC cells to overcome apoptosis and MC‐associated cell death and, in turn, promote the development of therapeutic resistance. A two‐hybrid analysis shows that XRCC3 interacts with the human Rad51C protein, which shows apparent 1:1 stoichiometry.31 The DNA‐binding activity of XRCC3‐Rad51c and Rad51C protein expression is drastically decreased in the absence of XRCC3,32, 33 which was supported by our results. Moreover, we found that expression of XRCC2, another Rad51 paralog involved in HR, was decreased in XRCC3‐deficient ESCC cells. However, as measured by RT‐PCR, the mRNA levels of Rad51C and XRCC2 were not altered by XRCC3 knockdown in TE‐1 cells (Fig. S1a). Next, we performed immunoprecipitation with XRCC3 antibody, and detected Rad51C and XRCC2 in the XRCC3 immunoprecipitates (Fig. S1b). These results indicated that XRCC3 protein could interact with XRCC2 and Rad51C in ESCC cells, directly or indirectly. Therefore, inhibition of XRCC3 might prevent the interaction of XRCC3, Rad51C and XRCC2, and could cause instability of these proteins. In sum, these findings strongly suggest that inhibition of XRCC3 in ESCC disrupts the HR pathway, which plays important roles in chromosome stability and in controlling the cell response to DNA‐damaging agents and IR.
It is interesting that XRCC3 knockdown without IR treatment had no impact on the colony formation of ESCC cells in the present study. Our results were consistent with some previous studies that showed that XRCC3 is not required for the survival of some cancer cells under normal conditions.34, 35 However, another study reports that depletion of XRCC3 leads to induction of p53‐dependent cell death and inhibits the proliferation of MCF‐7 breast cancer cells.36 Together these studies suggest that the functions of XRCC3 in tumorigenesis are complicated and may be tumor‐type‐specific.
The HR pathway plays a key role in telomere replication and capping, and is important for maintenance of telomere integrity.37 Telomeres that can no longer exert end‐protective functions are dysfunctional. Telomere dysfunction results in inappropriate chromosomal end‐to‐end fusions leading to MC and subsequent breakages that require further repair.38, 39 Maintenance of telomere stability is required for the growth of almost all cancer cells. Some studies have demonstrated that disruption of telomere maintenance enhances the sensitivity of cancer cells to chemotherapeutic agents and IR.40, 41 In the present study, downregulation of XRCC3 in ESCC cells resulted in disruption of telomere stability and an increase in radiosensitivity. These results suggested that the XRCC3‐mediated HR pathway may be critical for telomere stability in ESCC cells under DNA‐damaging conditions.
In summary, we describe the expression pattern of XRCC3 in human ESCC for the first time. High expression of XRCC3, as detected by IHC, may be a novel predictor of aggressive ESCC with CRT resistance and an independent prognostic factor for ESCC patients treated with CRT. Further analyses revealed that the XRCC3‐dependent HR pathway might be critical for telomere stability in ESCC cells and accounts for decreases in cell death induced by radiation‐induced apoptosis and MC. Thus, targeting the XRCC3‐mediated HR pathway may be a promising therapeutic strategy to improve therapeutic effects in ESCC patients.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1 Interactions of XRCC3, XRCC2 and Rad51C in ESCC (TE‐1) cells.
Acknowledgments
This study was supported by the Doctor Supporting Foundation of Tianjin Medical University Cancer Institute and Hospital (Nos. B1302 and B1305) and the National Nature Science Foundation of China (Nos. 81401948, 81472182 and 81472797), and the Tianjin Municipal Science and Technology Commission (Nos. 15JCQNJC11800 and 15JCYBJC25500).
Cancer Sci 106 (2015) 1678–1686
Funding Information
This study was supported by the Doctor Supporting Foundation of Tianjin Medical University Cancer Institute and Hospital (Nos. B1302 and B1305) and the National Nature Science Foundation of China (Nos. 81401948, 81472182 and 81472797) and Tianjin Municipal Science and Technology Commission (Nos. 15JCQNJC11800 and 15JCYBJC25500).
Reference
- 1. Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet 2013; 381: 400–12. [DOI] [PubMed] [Google Scholar]
- 2. Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol 2013; 19: 5598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin Y, Totsuka Y, He Y et al Epidemiology of esophageal cancer in Japan and China. J Epidemiol 2013; 23: 233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nakajima M, Kato H. Treatment options for esophageal squamous cell carcinoma. Expert Opin Pharmacother 2013; 14: 1345–54. [DOI] [PubMed] [Google Scholar]
- 5. Tahara M, Ohtsu A, Hironaka S et al Clinical impact of criteria for complete response (CR) of primary site to treatment of esophageal cancer. Jpn J Clin Oncol 2005; 35: 316–23. [DOI] [PubMed] [Google Scholar]
- 6. Jackson SP, Bartek J. The DNA‐damage response in human biology and disease. Nature 2009; 461: 1071–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Helleday T, Lo J, van Gent DC, Engelward BP. DNA double‐strand break repair: from mechanistic understanding to cancer treatment. DNA Repair 2007; 6: 923–35. [DOI] [PubMed] [Google Scholar]
- 8. Aparicio T, Baer R, Gautier J. DNA double‐strand break repair pathway choice and cancer. DNA Repair 2014; 19: 169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang C, Lees‐Miller SP. Detection and repair of ionizing radiation‐induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Rad Oncol 2013; 86: 440–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beucher A, Birraux J, Tchouandong L et al ATM and Artemis promote homologous recombination of radiation‐induced DNA double‐strand breaks in G2. EMBO J 2009; 28: 3413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Thompson LH. Recognition, signaling, and repair of DNA double‐strand breaks produced by ionizing radiation in mammalian cells: the molecular choreography. Mutat Res 2012; 751: 158–246. [DOI] [PubMed] [Google Scholar]
- 12. Ward A, Khanna KK, Wiegmans AP. Targeting homologous recombination, new pre‐clinical and clinical therapeutic combinations inhibiting RAD51. Cancer Treat Rev 2015; 41: 35–45. [DOI] [PubMed] [Google Scholar]
- 13. Huang F, Mazin AV. Targeting the homologous recombination pathway by small molecule modulators. Bioorg Med Chem Lett 2014; 24: 3006–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Carvalho JF, Kanaar R. Targeting homologous recombination‐mediated DNA repair in cancer. Expert Opin Ther Targets 2014; 18: 427–58. [DOI] [PubMed] [Google Scholar]
- 15. Brenneman MA, Wagener BM, Miller CA, Allen C, Nickoloff JA. XRCC3 controls the fidelity of homologous recombination: roles for XRCC3 in late stages of recombination. Mol Cell 2002; 10: 387–95. [DOI] [PubMed] [Google Scholar]
- 16. Liu N, Lamerdin JE, Tebbs RS et al XRCC2 and XRCC3, new human Rad51‐family members, promote chromosome stability and protect against DNA cross‐links and other damages. Mol Cell 1998; 1: 783–93. [DOI] [PubMed] [Google Scholar]
- 17. Khlifi R, Rebai A, Hamza‐Chaffai A. Polymorphisms in human DNA repair genes and head and neck squamous cell carcinoma. J Genet 2012; 91: 375–84. [DOI] [PubMed] [Google Scholar]
- 18. Osti MF, Nicosia L, Agolli L et al Potential role of single nucleotide polymorphisms of XRCC1, XRCC3, and RAD51 in predicting acute toxicity in rectal cancer patients treated with preoperative radiochemotherapy. Am J Clin Oncol 2015. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 19. Xu ZY, Loignon M, Han FY, Panasci L, Aloyz R. Xrcc3 induces cisplatin resistance by stimulation of Rad51‐related recombinational repair, S‐phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther 2005; 314: 495–505. [DOI] [PubMed] [Google Scholar]
- 20. Qian D, Zhang B, Zeng XL et al Inhibition of human positive cofactor 4 radiosensitizes human esophageal squmaous cell carcinoma cells by suppressing XLF‐mediated nonhomologous end joining. Cell Death Dis 2014; 5: e1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Qian D, Zhang B, He LR et al The telomere/telomerase binding factor PinX1 is a new target to improve the radiotherapy effect of oesophageal squamous cell carcinomas. J Pathol 2013; 229: 765–74. [DOI] [PubMed] [Google Scholar]
- 22. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 2010; 11: 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manuguerra M, Saletta F, Karagas MR et al XRCC3 and XPD/ERCC2 single nucleotide polymorphisms and the risk of cancer: a HuGE review. Am J Epidemiol 2006; 164: 297–302. [DOI] [PubMed] [Google Scholar]
- 24. He XF, Wei W, Li JL et al Association between the XRCC3 T241M polymorphism and risk of cancer: evidence from 157 case‐control studies. Gene 2013; 523: 10–19. [DOI] [PubMed] [Google Scholar]
- 25. Qureshi Z, Mahjabeen I, Baig R, Kayani M. Correlation between selected XRCC2, XRCC3 and RAD51 gene polymorphisms and primary breast cancer in women in Pakistan. Asian Pac J Cancer Prev 2014; 15: 10225–9. [DOI] [PubMed] [Google Scholar]
- 26. Berger B, Belka C. Evidence‐based radiation oncology: oesophagus. Radiother Oncol 2009; 92: 276–90. [DOI] [PubMed] [Google Scholar]
- 27. Eriksson D, Stigbrand T. Radiation‐induced cell death mechanisms. Tumour Biol 2010; 31: 363–72. [DOI] [PubMed] [Google Scholar]
- 28. Roninson IB, Broude EV, Chang BD. If not apoptosis, then what? Treatment‐induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat 2001; 4: 303–13. [DOI] [PubMed] [Google Scholar]
- 29. Ianzini F, Kosmacek EA, Nelson ES et al Activation of meiosis‐specific genes is associated with depolyploidization of human tumor cells following radiation‐induced mitotic catastrophe. Cancer Res 2009; 69: 2296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eriksson D, Lofroth PO, Johansson L, Riklund KA, Stigbrand T. Cell cycle disturbances and mitotic catastrophes in HeLa Hep2 cells following 2.5 to 10 Gy of ionizing radiation. Clin Cancer Res 2007; 13: 5501s–8s. [DOI] [PubMed] [Google Scholar]
- 31. Kurumizaka H, Ikawa S, Nakada M et al Homologous‐pairing activity of the human DNA‐repair proteins Xrcc3.Rad51C. Proc Natl Acad Sci U S A 2001; 98: 5538–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lio YC, Schild D, Brenneman MA, Redpath JL, Chen DJ. Human Rad51C deficiency destabilizes XRCC3, impairs recombination, and radiosensitizes S/G2‐phase cells. J Biol Chem 2004; 279: 42313–20. [DOI] [PubMed] [Google Scholar]
- 33. Wiese C, Collins DW, Albala JS, Thompson LH, Kronenberg A, Schild D. Interactions involving the Rad51 paralogs Rad51C and XRCC3 in human cells. Nucleic Acids Res 2002; 30: 1001–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Compton SA, Choi JH, Cesare AJ, Ozgur S, Griffith JD. Xrcc3 and Nbs1 are required for the production of extrachromosomal telomeric circles in human alternative lengthening of telomere cells. Cancer Res 2007; 67: 1513–19. [DOI] [PubMed] [Google Scholar]
- 35. Chang JF, Lin ST, Hung E et al Nuclear proteomics with XRCC3 knockdown to reveal the development of doxorubicin‐resistant uterine cancer. Toxicol Sci 2014; 139: 396–406. [DOI] [PubMed] [Google Scholar]
- 36. Loignon M, Amrein L, Dunn M, Aloyz R. XRCC3 depletion induces spontaneous DNA breaks and p53‐dependent cell death. Cell Cycle 2007; 6: 606–11. [DOI] [PubMed] [Google Scholar]
- 37. Tacconi EM, Tarsounas M. How homologous recombination maintains telomere integrity. Chromosoma 2014; 124: 119–30. [DOI] [PubMed] [Google Scholar]
- 38. Verdun RE, Karlseder J. Replication and protection of telomeres. Nature 2007; 447: 924–31. [DOI] [PubMed] [Google Scholar]
- 39. Cosme‐Blanco W, Shen MF, Lazar AJ et al Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53‐dependent cellular senescence. EMBO Rep 2007; 8: 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Uziel O, Beery E, Dronichev V et al Telomere shortening sensitizes cancer cells to selected cytotoxic agents: in vitro and in vivo studies and putative mechanisms. PLoS ONE 2010; 5: e9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pal J, Gold JS, Munshi NC, Shammas MA. Biology of telomeres: importance in etiology of esophageal cancer and as therapeutic target. Transl Res 2013; 162: 364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Interactions of XRCC3, XRCC2 and Rad51C in ESCC (TE‐1) cells.