

Abstract
Transforming growth factor β (TGFβ) causes the acquisition of epithelial–mesenchymal transition (EMT). Although the tumor suppressor gene PTEN (phosphatase and tensin homologue deleted from chromosome 10) can negatively regulate many signaling pathways activated by TGFβ, hyperactivation of these signaling pathways is observed in lung cancer cells. We recently showed that PTEN might be subject to TGFβ‐induced phosphorylation of its C‐terminus, resulting in a loss of its enzyme activities; PTEN with an unphosphorylated C‐terminus (PTEN4A), but not PTEN wild, inhibits TGFβ‐induced EMT. Nevertheless, whether or not the blockade of TGFβ‐induced EMT by the PTEN phosphatase activity might be attributed to the unphosphorylated PTEN C‐terminus itself has not been fully determined. Furthermore, the lipid phosphatase activity of PTEN is well characterized, whereas the protein phosphatase activity has not been determined. By using lung cancer cells carrying PTEN domain deletions or point mutants, we investigated the role of PTEN protein phosphatase activities on TGFβ‐induced EMT in lung cancer cells. The unphosphorylated PTEN C‐terminus might not directly retain the phosphatase activities and repress TGFβ‐induced EMT; the modification that keeps the PTEN C‐terminus not phosphorylated might enable PTEN to retain the phosphatase activity. PTEN4A with G129E mutation, which lacks lipid phosphatase activity but retains protein phosphatase activity, repressed TGFβ‐induced EMT. Furthermore, the protein phosphatase activity of PTEN4A depended on an essential association between the C2 and phosphatase domains. These data suggest that the protein phosphatase activity of PTEN with an unphosphorylated C‐terminus might be a therapeutic target to negatively regulate TGFβ‐induced EMT in lung cancer cells.
Keywords: β‐Catenin, epithelial–mesenchymal transition, protein phosphatase activity, PTEN, transforming growth factor β
Lung cancer remains a leading cause of cancer‐related death worldwide. New therapeutic options available for lung cancer are likely to have a limited effect on improving survival rates in these patients, due to chemotherapy resistance and early cancer dissemination.1 Mounting evidence suggests the importance of the tumor microenvironment in which cancer cells interact with fibroblasts and consequently acquire epithelial–mesenchymal transition (EMT).2, 3 Persistent loss of epithelial markers and de novo expression of mesenchymal markers are involved during development of EMT.4 Although transforming growth factor β (TGFβ) is one of the most critical tissue‐stiffening factors derived from tumor lesions, the recent studies showed that TGFβ‐induced transcription of EMT target genes such as fibronectin and vimentin is accelerated by translocation of β‐catenin from E‐cadherin complexes at the cell membrane into the cytoplasm.5, 6
Although the tumor suppressor gene PTEN (phosphatase and tensin homologue deleted from chromosome 10) can negatively regulate many signaling pathways activated by TGFβ,7 hyperactivation of the signaling pathways induced by TGFβ is often observed in lung cancer.8 Loss of PTEN expression might accelerate the development of lung cancer in vivo.9 Recent studies suggest that TGFβ stimulation represses PTEN expression in epithelial cells in vitro.6, 10 Meanwhile, phosphorylation of the PTEN C‐terminus might induce a conformational change in PTEN structure that is closely associated with the loss of PTEN activity.11, 12 The studies also indicated that the higher phosphorylation levels of the PTEN C‐terminus in malignant leukemia cells were directly associated with loss of phosphatase activity and acquisition of aberrant cell phenotypes.13, 14 We show that TGFβ stimulation modifies phosphorylation of PTEN on its C‐terminus (p‐PTEN) in lung cancer cells, and consequently leads to the loss of PTEN activities;6 unphosphorylated PTEN with four Ala substitutions of the phosphorylation sites in the PTEN C‐terminus (PTEN4A) rescues cells from TGFβ‐induced loss of PTEN activities and the acquisition of EMT.6 Nevertheless, whether or not the PTEN phosphatase activity might be attributed to the unphosphorylated PTEN C‐terminus itself has not been fully determined. Although PTEN comprises the C‐terminus and two other major domains, that is, the C2 and phosphatase domains,15 several mutants of the PTEN phosphatase domain that modulate the PTEN phosphatase activities have been described.7, 16, 17, 18 The lipid phosphatase activity of PTEN is well characterized,19 whereas the PTEN protein phosphatase activity has not been fully determined.18
In the present study, by using a Dox‐dependent expression system to express a series of PTEN domain deletions and point mutants, we have investigated the enzyme's protein phosphatase activity, by which unphosphorylated PTEN might play a critical role in inhibiting TGFβ‐induced EMT and translocation of β‐catenin in lung cancer cells.
Materials and Methods
Materials
Monoclonal mouse anti‐PTEN antibody (clone 6H2.1) was from Cascade Bioscience (Winchester, UK). Purified rabbit anti‐phospho‐PTEN (Ser380/Thr382/Thr383) antibody, rabbit anti‐pan‐Akt antibody, rabbit anti‐phospho‐Akt (Thr308) antibody, rabbit anti‐phospho‐Akt (Ser473) antibody, rabbit anti‐FAK antibody, and rabbit anti‐phospho‐FAK (Tyr397) antibody were from Cell Signaling Technology (Boston, MA, USA). Purified anti‐fibronectin antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Purified and FITC mouse anti‐E‐cadherin antibody and anti‐β‐catenin antibody were from BD Biosciences (San Diego, CA, USA). Streptavidin (SAv)‐Alexa 594 (SAv‐594)‐conjugated anti‐mouse antibody was from Invitrogen Life Technologies (Carlsbad, CA, USA). Affinity‐isolated rabbit anti‐actin antibody was from Sigma‐Aldrich (St. Louis, MO, USA). Can Get Signal was from Toyobo (Tokyo, Japan). Doxycycline, pTet‐On Advanced, pTRE‐Tight Vector, and Adeno‐X Expression System 3 were from Clontech Laboratories (Mountain View, CA, USA). PhosSTOP was from Roche Applied Science (Mannheim, Germany). Hoechst33342 was from Dojindo (Kumamoto, Japan). 1,2,4,5‐Benzenetetramine tetrahydrochloride (FAK inhibitor 14) was from Tocris Bioscience (Bristol, UK).
Plasmids and gene transfection
The Dox‐dependent gene expression system was applied to H358 cells, a human lung cancer cell line, carrying pTet‐On Advanced (H358ON).6 H358ON cells express GFP in the pTRE‐Tight vector (GFP), or GFP fused to PTEN with four Ala substitutions (S380A, T382A, T383A, and S385A) on the C‐terminus (PTEN4A) through the pTRE‐Tight Vector (GP4A).6 All of the PTEN domain‐deletion mutants used in this study were prepared from GP4A.15 Furthermore, by using the QuikChange Site‐Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA), the following functionally selective mutants of the PTEN phosphatase domain were also established: (i) PTEN C124S, in which Cys124 is replaced with Ser, which lacks both lipid and protein phosphatase activities;17 (ii) PTEN G129E, in which Glu replaces Gly129, which lacks lipid phosphatase activity but retains protein phosphatase activity;16, 17 and (iii) PTEN Y138C, in which Tyr138 is substituted with Cys, which selectively retains lipid phosphatase activity but lacks substantial protein phosphatase activity.7, 18 Adenovirus carrying the GFP‐PTEN4A C2 domain only (adeno G4A C2 only) was also established from the GFP‐PTEN4A C2 domain only in the pTRE‐Tight Vector, by using the Adeno‐X Expression System 3 (Clontech) in accordance with the manufacturer's instruction. We defined one MOI as a total of 1 × 106 pfu of adeno G4A C2 only per 1 × 106 cells per a plate.
Cells
H358ON cells was maintained in RPMI supplemented with 2 mmol/L l‐glutamine, Antibiotic‐Antimycotic (Thermo Fisher Scientific, Inc., Waltham, MA), and 10% FCS.20 To evaluate the effect of TGFβ, the cells were treated with TGFβ at 2 ng/mL and analyzed for mRNA levels and protein levels at the indicated time points (see below). To evaluate the effect of PTEN transduction on TGFβ stimulation, H358ON cells expressing Dox‐dependent PTEN constructs were incubated with 1 μg/mL Dox at for 24 h at 37°C before TGFβ treatment.6 In the adeno reconstitution model, H358ON cells expressing Dox‐dependent G4A G129E lacking the PTEN C2 domain (G4A G129E Δ C2) were pretreated with 10 MOI of adeno G4A C2 only 24 h before Dox treatment.
Western blot analysis
For whole‐cell extracts, cells were harvested in ice‐cold lysis buffer and cleared by centrifugation.21, 22 The samples were then subjected to SDS‐PAGE and analyzed by immunoblotting. To detect phosphorylation levels of the targeted proteins, Phosphostop was added to the lysis buffer and Can Get Signal was added to the dilution solution for primary and secondary antibodies. β‐actin was evaluated as a loading control.
Immunofluorescence and confocal laser scanning microscopy
Immunocytochemistry was carried out as previously described.6, 23 To evaluate the effect of PTEN4A or PTEN mutants on the TGFβ‐induced translocation of β‐catenin, H358ON cells expressing Dox‐dependent PTEN mutants were incubated with anti‐β‐catenin antibody, followed by SAv‐594‐conjugated anti‐mouse antibody. Nuclear staining was carried out using Hoechst 33342. The distribution of β‐catenin was determined by confocal laser scanning microscopy (LSM 5 PASCAL; Carl Zeiss, Jena, Germany). The fluorescence intensities of β‐catenin and the nucleus were evaluated by using imaging software (LSM Software ZEN 2008; Carl Zeiss), which plotted fluorescence intensities over a random cross‐section of the cells.24, 25 The subcellular distribution of PTEN was determined by confocal laser scanning microscopy. A minimum of five randomly selected high‐power fields were examined per sample to measure fluorescence intensity in the nucleus and the cytoplasm.26 To determine localization of β‐catenin in the cell membrane, double immunostaining was carried out for β‐catenin and E‐cadherin in the cells.27
Statistical analysis
The results were analyzed using the Mann–Whitney U‐test for comparison between any two groups, and by non‐parametric equivalents of anova or multiple comparisons. A value of P < 0.05was considered to indicate statistical significance.
Results
Unphosphorylated PTEN C‐terminus might not directly inhibit TGFβ‐induced EMT in H358 cells
To determine whether the PTEN C‐terminus with four Ala substitutions might directly modulate the acquisition of TGFβ‐induced EMT, we established H358ON cells Dox‐dependently expressing either GFP‐PTEN4A with deletion of the tail (G4A Δ tail) or GFP‐PTEN4A tail only (G4A tail only), which lacks both C2 and phosphatase domains (Fig. 1a). First, the expression of these mutants was verified by Western blotting of cells grown in the absence or presence of Dox. Although G4A tail expression induced by Dox was detected by an anti‐PTEN antibody that recognizes the PTEN C‐terminus, expression of G4A Δ tail expression was not detected (Fig. 1b,c). In contrast, Western blotting with anti‐GFP antibody showed the expression of GFP‐tagged proteins with the sizes expected for the G4A Δ tail and G4A tail, respectively (Fig. 1b,c). Next, we evaluated the effect of G4A Δ tail and G4A tail on TGFβ‐induced signaling pathways. We recently showed that TGFβ‐induced phosphorylation of Akt at Thr308 and Ser473 (Akt308 and Akt473) and FAK was repressed in H358 ON cells with G4A (Fig. S1a,b).6 We evaluated the effects of G4A tail protein and GFP4A Δ tail protein on these phosphorylation signals (Fig. 1d–g, S1c,d). De novo expression of G4A tail protein did not inhibit TGFβ‐induced phosphorylation of Akt308, Akt473, or FAK (Figs 1e,g, S1d). In contrast, these phosphorylation signals were inhibited by de novo GFP4A Δ tail protein in H358ON cells (Figs 1d,f, S1c). To evaluate the effect of the PTEN mutants on TGFβ‐induced EMT, Western blotting analysis for fibronectin5, 28 and E‐cadherin5, 29 was carried out after treatment with vehicle or TGFβ for 48 h in the absence or presence of Dox. A previous study showed that compensatory induction of PTEN4A repressed TGFβ‐induced EMT through complete blockade of β‐catenin translocation to the cytoplasm and the nucleus.6 Furthermore, double immunostaining showed colocalization of β‐catenin and E‐cadherin on the cell membrane in the cells (Fig. S1e). There was no reduction in the increasing fibronectin/E‐cadherin ratio (F/E ratio) in TGFβ‐treated cells expressing G4A tail (Fig. 1j); however, expression of G4A Δ tail yielded a significant decrease in the F/E ratio (Fig. 1i), similar to those in H358ON cells with G4A (Fig. 1h). Localization of β‐catenin was evaluated in TGFβ‐treated H358ON cells expressing Dox‐dependent G4A tail and G4A Δ tail protein by immunofluorescence coupled with confocal microscopy. β‐catenin appeared localized on the cell membrane in H358ON cells expressing Dox‐dependent G4A tail or G4A Δ tail when no TGFβ was added (Fig. 1m–p). Translocation of β‐catenin into the cytoplasm and the nucleus was observed after TGFβ stimulation in H358ON cells expressing G4A tail protein (Fig. 1o,p). In contrast, β‐catenin was completely retained on the cell membrane in H358ON cells after TGFβ stimulation in H358ON cells expressing de novo GFP4A Δ tail protein (Fig. 1m,n), similar to those in H358ON cells with G4A (Fig. 1k,l). Furthermore, TGFβ‐induced EMT and β‐catenin translocation into the cytoplasm and the nucleus was also not blocked in H358ON cells expressing GFP‐PTEN wild tail only (Fig. S2a–d). Although we recently showed that TGFβ‐induced phosphorylation of FAK was repressed in H358ON cells with G4A, treatment by a FAK inhibitor targeting Tyr397 did not block TGFβ‐induced EMT or translocation in H358ON cells expressing GFP.6 To demonstrate that inhibition of TGFβ‐induced EMT and β‐catenin translocation in H358ON cells with unphosphorylated PTEN might be independent of repression of phosphorylation of TGFβ‐induced FAK, H358ON cells expressing G4A tail with TGFβ stimulation were treated with a FAK inhibitor targeting Tyr397. Although phosphorylation of FAK was completely inhibited by a FAK inhibitor 14, TGFβ‐induced EMT and β‐catenin translocation into the cytoplasm and the nucleus remained persistent in H358ON cells expressing G4A tail (Fig. S2e–h). Taken together, these data suggested that the unphosphorylated PTEN C‐terminus itself might not directly retain the phosphatase activities and repress TGFβ‐induced EMT; the modification that keeps the PTEN C‐terminus not phosphorylated might enable PTEN to retain the phosphatase activity and inhibit TGFβ‐induced EMT.
Figure 1.
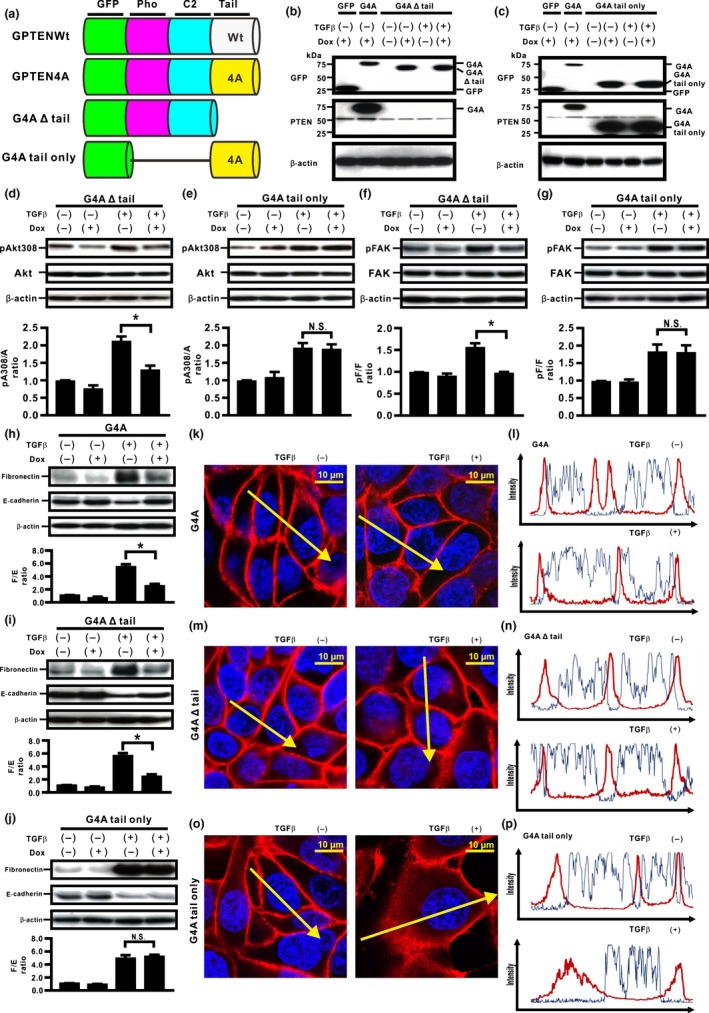
Unphosphorylated PTEN C‐terminus itself might not directly inhibit transforming growth factor β (TGFβ)‐induced epithelial–mesenchymal transition in H358 lung cancer cells. (a) Schematic diagram of PTEN deletion mutants. We established H358ON cells Dox‐dependently expressing either GFP‐PTEN4A with deletion of the tail (G4A Δ tail) or GFP‐PTEN4A tail only (G4A tail only), which lacks both C2 and phosphatase (Pho) domains. A representative blot from three independent experiments is shown as (b) G4A Δ tail and (c) G4A tail only. (d–g) Total and phosphorylated Akt308 (d,e) and FAK (f,g) at the indicated time points after treatment with vehicle or TGFβ (20 min for Akt308 (a) and 12 h for FAK (f), respectively) were analyzed by Western blotting. The ratio of phosphorylated protein to total protein (pA308/A and pF/F) is presented as the intensity level relative to that in H358ON cells treated with vehicle in the absence of Dox. *P < 0.05. NS, not significant. (h–j) Fibronectin (F) and E‐cadherin (E) were analyzed by Western blotting ((h) G4A; (i) G4A Δ tail; (j) G4A tail only). The F/E ratio is shown compared to that in cells treated with vehicle in the absence of Dox. (k–p) Fluorescence intensity of β‐catenin was evaluated ((k,l) G4A; (m,n) G4A Δ tail; (o,p) G4A tail only). Left and right images in (k,m,o) show cells without and with TGFβ stimulation, respectively. Upper and lower panels in (l,n,p) plot the fluorescence intensity of β‐catenin (red) and nucleus (blue) over a cross‐section of the cells without and with TGFβ stimulation along the selected yellow arrows in (k, m, o), respectively. GPTENWt, GFP‐PTENWild.
Both C2 and phosphatase domains of unphosphorylated PTEN might be essential for inhibiting TGFβ‐induced EMT in H358 cells
To determine the biological roles of the C2 and phosphatase domains of PTEN4A in the inhibition of TGFβ‐induced EMT, we established H358ON cells expressing Dox‐dependent GFP‐PTEN4A lacking the phosphatase domain (G4A Δ phosphatase), GFP‐PTEN4A lacking the C2 domain (G4A Δ C2), and GFP‐PTEN4A containing the C2 domain only (G4A C2 only) (Fig. 2a). The expression of these mutants was verified by Western blotting of cells grown in the absence or presence of Dox. Western blotting with anti‐GFP antibody showed the expression of GFP‐tagged proteins of the expected size of G4A Δ phosphatase, G4A Δ C2, and G4A C2 only, in Dox‐treated cells (Fig. 2b–d). First, we evaluated PTEN phosphatase activities in cells expressing these mutants. Whereas Dox‐treated H358ON cells expressing PTEN4A or G4A Δ tail showed repression of TGFβ‐induced FAK and Akt activation through PTEN phosphatase activities (Fig. 1d,f and data not shown), expression of G4A Δ phosphatase, G4A Δ C2, or G4A C2 only did not appear to inhibit TGFβ‐induced FAK and Akt activation in H358On cells (Fig. 2e–j), indicating that the phosphatase activities in PTEN4A might be orchestrated by both the C2 and the phosphatase domains. Western blot analysis for EMT markers showed that there was no reduction in the increasing F/E ratio in TGFβ‐treated cells expressing G4A Δ phosphatase, G4A Δ C2, or G4A C2 only (Fig. 3a–c); thus, these mutants had no effect on TGFβ‐induced EMT. To elucidate the underlying mechanism, immunofluorescence was carried out in cells expressing these deletion mutants. Treatment with TGFβ induced β‐catenin translocation from the cell membrane to the cytoplasm and the nucleus in H358ON cells expressing each of the mutants (Fig. 3d–i). Taken together, these results indicate that the close association of both the C2 and phosphatase domains might be essential to inhibit the acquisition of TGFβ‐induced EMT and to block the underlying mechanisms in lung cancer cells.
Figure 2.
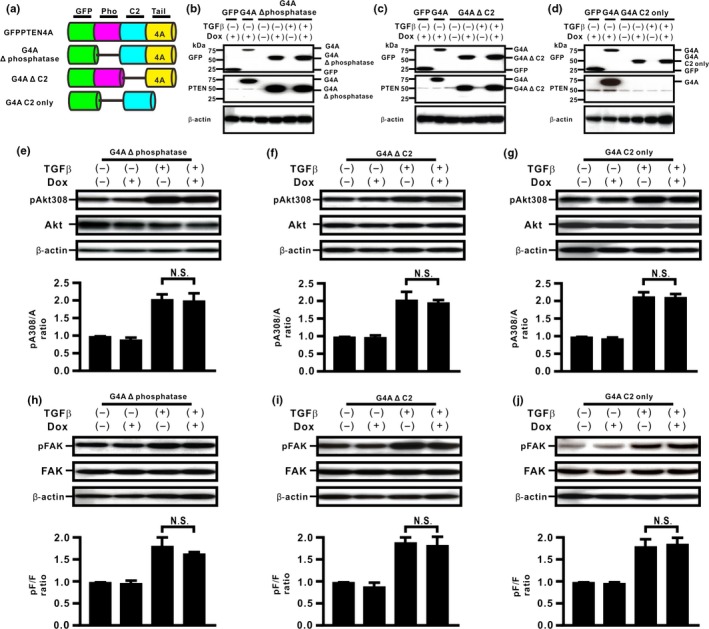
Phosphatase and C2 domains of unphosphorylated PTEN are essential for inhibition of transforming growth factor β (TGFβ)‐induced signaling pathways in H358 lung cancer cells. (a) Schematic diagram of PTEN deletion mutants. We established H358ON cells expressing Dox‐dependent GFP‐PTEN4A lacking the phosphatase (Pho) domain (G4A Δ phosphatase), GFP‐PTEN4A lacking the C2 domain (G4A Δ C2), and GFP‐PTEN4A containing the C2 domain only (G4A C2 only). A representative blot from three independent experiments is shown as G4A Δ phosphatase (b), G4A Δ C2 (c), and G4A C2 only (d). (e–j) Total and phosphorylated Akt308 (e–g) and FAK (h–j) after treatment with vehicle or TGFβ were analyzed by Western blotting. The ratio of phosphorylated protein to total protein (pA308/A and pF/F) is presented as the intensity level relative to that in H358ON cells treated with vehicle in the absence of Dox. Data shown represent the means ± SE. The experiment was repeated three times with similar results. *P < 0.05. NS, not significant.
Figure 3.
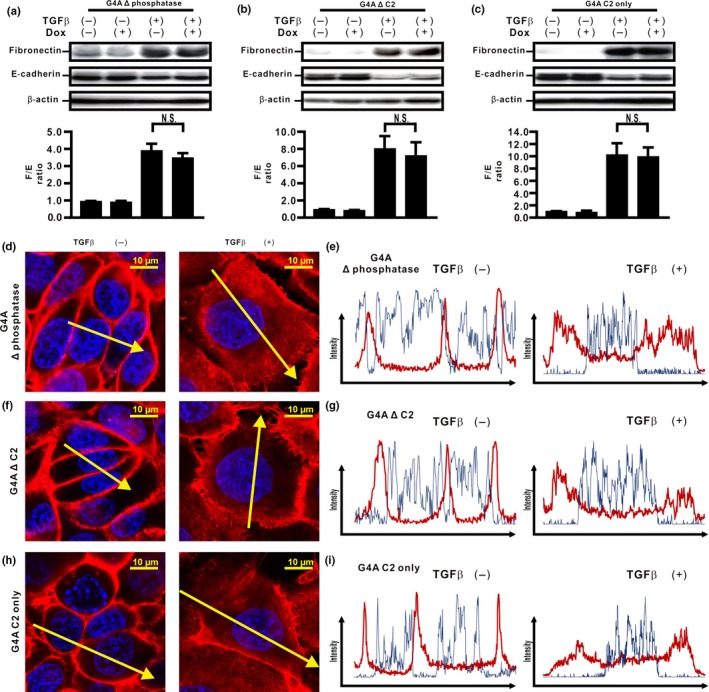
Phosphatase and C2 domains of unphosphorylated PTEN are essential for inhibition of transforming growth factor β (TGFβ)‐induced epithelial–mesenchymal transition in H358 cells. (a–c) Fibronectin (F) and E‐cadherin (E) were analyzed by Western blotting. The F/E ratio is shown in comparison to that in cells treated with vehicle in the absence of Dox. A representative blot from three independent experiments is shown. Data shown represent the means ± SE. The experiment was repeated three times with similar results. (d–i) The fluorescence intensity of β‐catenin was evaluated. Left and right images in (d,f,h) show cells without and with TGFβ stimulation, respectively. Upper and lower panels in (e,g,i) plot the fluorescence intensity of β‐catenin (red) and nucleus (blue) over a cross‐section of cells without and with TGFβ stimulation along the selected yellow arrows in (d, f, h), respectively. Data shown are representative of at least three independent experiments.
Unphosphorylated PTEN might inhibit TGFβ‐induced EMT through protein phosphatase activity in H358 cells
Compensatory expression of PTENWild (PTENWt) inhibits TGFβ‐induced PI3K/Akt signaling in lung cancer cells, whereas it only partially inhibits TGFβ‐induced EMT.6 Therefore, we hypothesized that the protein phosphatase activity of PTEN might be involved in the inhibition of TGFβ‐induced EMT caused by PTEN4A in H358ON cells. To evaluate the role of PTEN phosphatase activity, therefore, we established H358ON cells expressing Dox‐dependent GFP‐PTEN4A with C124S (G4A C124S), GFP‐PTEN4A with G129E (G4A G129E), and GFP‐PTEN4A with Y138C (G4A Y138C) (Fig. 4a–d), three point mutants that show distinct characteristics of phosphatase activity. First, we determined whether TGFβ‐induced FAK and Akt activation could be modulated by these point mutants. De novo G4A C124S expression did not inhibit TGFβ‐induced FAK and Akt activation (Fig. 4e–h), indicating that H358ON cells expressing G4A C124S retain neither the lipid phosphatase activity nor the protein phosphatase activity of PTEN. De novo G4A G129E expression inhibited TGFβ‐induced FAK activation, but not Akt activation (Fig. 4f,i), indicating that H358ON cells expressing G4A G129E retain the protein phosphatase activity but lack the lipid activity of PTEN. In contrast, de novo G4A Y138C expression inhibited TGFβ‐induced Akt activation, but not FAK activation (Fig. 4g,j), indicating that H358ON cells expressing G4A Y138C retain the lipid phosphatase but lack the protein phosphatase activity of PTEN. Next, we used H358ON cells expressing these mutants to evaluate the effect of PTEN phosphatase activities on EMT. There was no reduction in the increasing F/E ratio in TGFβ‐treated cells expressing G4A C124S even when Dox was added (Fig. 5a). In addition, expression of G4A G129E repressed the TGFβ‐induced EMT phenotype (Fig. 5b), whereas de novo expression of G4A Y138C did not inhibit TGFβ‐induced acquisition of the EMT phenotype (Fig. 5c). Furthermore, localization of β‐catenin was evaluated in these cells (Fig 5d‐i). Although TGFβ‐induced translocation of β‐catenin from the cell membrane to the cytoplasm and the nucleus was not blocked in H358ON cells expressing Dox‐dependent G4A C124S or G4A Y138C (Fig. 5d,e,h,i), de novo expression of G4A G129E completely retained localization of β‐catenin on the cell membrane (Fig. 5f,g). These findings indicate that the protein phosphatase activity of PTEN, but not the lipid phosphatase activity, might be involved in inhibiting TGFβ‐induced EMT and in blocking the associated β‐catenin translocation.
Figure 4.
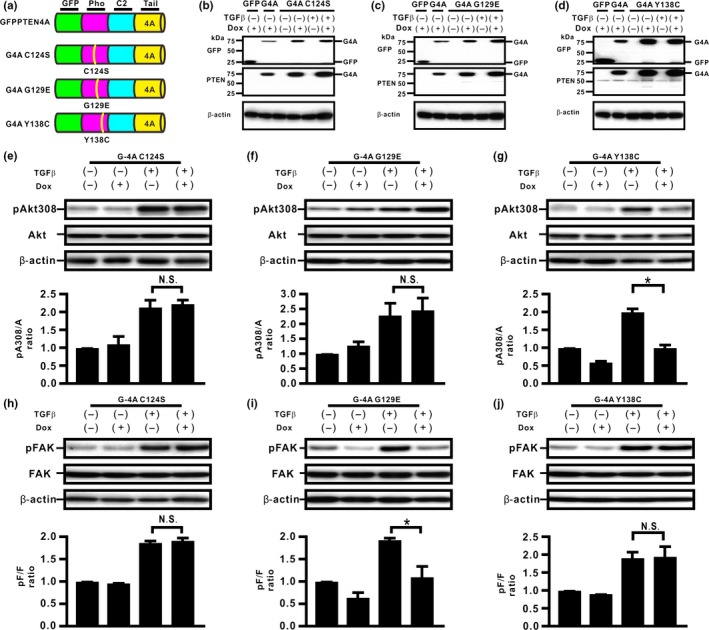
Unphosphorylated PTEN with point mutants in the phosphatase (Pho) domain might differentially modulate transforming growth factor β (TGFβ)‐induced signaling pathways in H358 lung cancer cells. (a) Schematic diagram of some of the PTEN deletion mutants. We established H358ON cells expressing Dox‐dependent GFP‐PTEN4A with C124S (G4A C124S), GFP‐PTEN4A with G129E (G4A G129E), and GFP‐PTEN4A with Y138C (G4A Y138C). A representative blot from three independent experiments is shown as G4A C124S (b), G4A G129E (c), and G4A Y138C (d). (e–j) Total and phosphorylated Akt308 (e–g) and FAK (h–j) after treatment with vehicle or TGFβ were analyzed by Western blotting. The ratio of phosphorylated protein to total protein (pA308/A and pF/F) is presented as the intensity level relative to that in H358ON cells treated with vehicle in the absence of Dox. Data shown represent the means ± SE. The experiment was repeated three times with similar results. *P < 0.05. NS, not significant.
Figure 5.
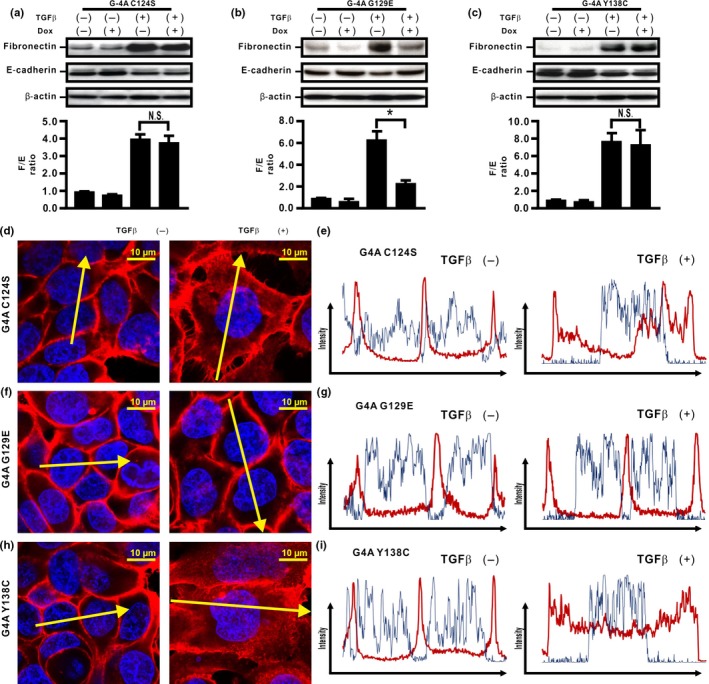
Unphosphorylated PTEN might inhibit transforming growth factor β (TGFβ)‐induced epithelial–mesenchymal transition through protein phosphatase (Pho) activity in H358 lung cancer cells. (a–c) Fibronectin (F) and E‐cadherin (E) were analyzed by Western blotting. The F/E ratio is shown in comparison to that in cells treated with vehicle in the absence of Dox. A representative blot from three independent experiments is shown. Data shown represent the means ± SE. The experiment was repeated three times with similar results. (d–i) The fluorescence intensity of β‐catenin was evaluated. Left and right images in (d,f,h) show cells without and with TGFβ stimulation, respectively. Upper and lower panels in (e,g,i) plot the fluorescence intensity of β‐catenin (red) and nucleus (blue) over a cross‐section of cells without and with TGFβ stimulation along the selected yellow arrows in (d, f, h), respectively. Data shown are representative of at least three independent experiments.
C2 domain might be essential for protein phosphatase activity of unphosphorylated PTEN in H358 cells
To determine whether the association of both the C2 and phosphatase domains might be essential for PTEN4A to show protein phosphatase activity, we established H358ON cells expressing G4A G129E lacking the PTEN C2 domain (G4A G129E Δ C2) (Fig. 6a). Western blotting verified that the cells expressed protein of the expected sized (Fig. 6b). De novo expression of G4A G129E Δ C2 did not inhibit TGFβ‐induced FAK activation (Fig. 6c), indicating the close association of both the C2 and phosphatase domains might be essential for the protein phosphatase activity of PTEN4A. In TGFβ‐treated cells expressing the G4A G129E Δ C2 domain, there was no reduction in the increasing F/E ratio (Fig. 6d) and translocation of β‐catenin from the cell membrane into the cytoplasm and the nucleus (Fig. 6e,f) was observed. Finally, to determine whether the PTEN C2 domain is essential for the inhibition of TGFβ‐induced EMT by protein phosphatase activity, a model of PTEN C2 domain reconstitution was established in H358ON cells expressing Dox‐dependent G4A G129E Δ C2 using adeno G4A C2 only (Fig. 6g,h). Reconstitution of the PTEN C2 domain by adeno G4A C2 only led to the inhibition of both TGFβ‐induced FAK activation in H358ON cells expressing G4A G129E Δ C2 (Fig. 6i), TGFβ‐induced EMT (Fig. 6j), in addition to blockade of β‐catenin translocation from the cell membrane into the cytoplasm and nucleus (Fig. 6k,l). Taken together, these data suggest that compensatory induction of PTEN4A might negatively regulate TGFβ‐induced EMT and the associated translocation of β‐catenin through its protein phosphatase activity, for which the C2 and the phosphatase domains might both be essential.
Figure 6.
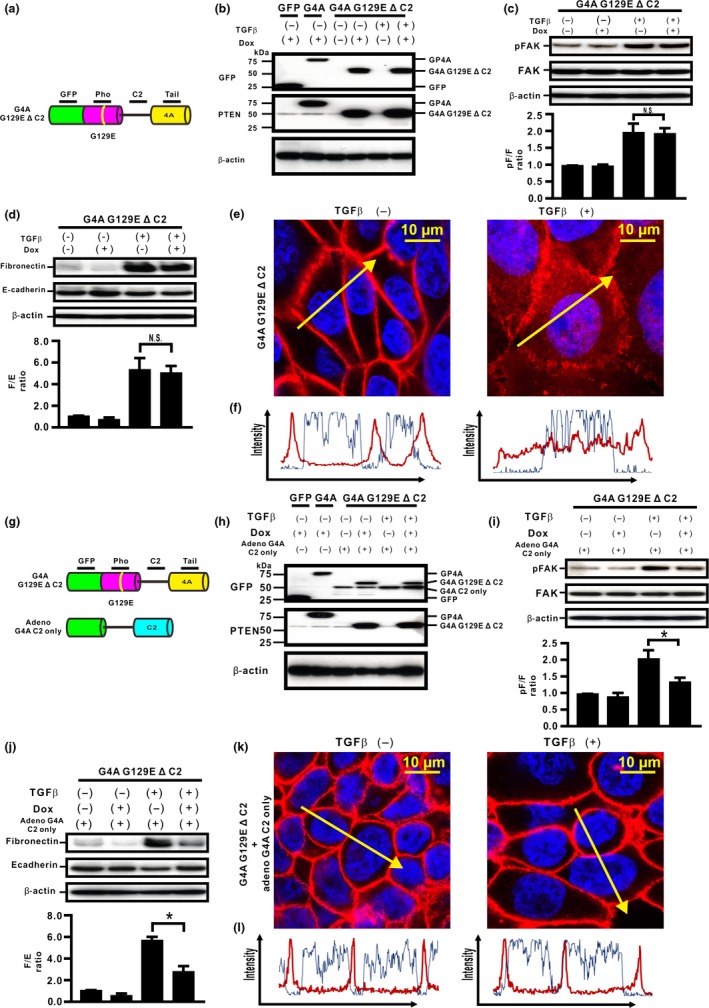
C2 domain is essential for unphosphorylated PTEN to inhibit transforming growth factor β (TGFβ)‐induced epithelial–mesenchymal transition through the protein phosphatase (Pho) activity in H358 lung cancer cells. (a) Schematic diagram of G4A G129EΔ C2. We established H358ON cells expressing G4A G129E lacking the PTEN C2 domain (G4A G129E Δ C2). A representative blot from three independent experiments is shown as G4A G129E Δ C2 (b). (c) Total and phosphorylated FAK after treatment with vehicle or TGFβ were analyzed by Western blotting. A representative blot from three independent experiments is shown (top). The ratio of phosphorylated protein to total protein (pF/F) is presented as the intensity level relative to that in H358ON cells treated with vehicle in the absence of Dox (bottom). (D) Fibronectin (F) and E‐cadherin (E) were analyzed by Western blotting. The F/E ratio is shown in comparison to that in cells treated with vehicle in the absence of Dox. (e,f) The fluorescence intensity of β‐catenin was evaluated. Left and right images in (e) show cells without and with TGFβ stimulation, respectively. Left and right panels in (f) plot the fluorescence intensity of β‐catenin (red) and nucleus (blue) over a cross‐section of cells without and with TGFβ stimulation along the selected yellow arrows in (e), respectively. Data shown are representative of at least three independent experiments. (g) A model of PTEN C2 domain reconstitution was established in H358ON cells expressing Dox‐dependent G4A G129E Δ C2 by using adenovirus carrying G4A C2 only. A representative blot from three independent experiments is shown as G4A G129E Δ C2 and G4A C2 only (h). (i) Total and phosphorylated FAK in adeno‐transfected H358ON cells expressing Dox‐dependent G4A G129E Δ C2 in the absence or presence of Dox and/or TGFβ stimulation were analyzed by Western blotting. (j) Fibronectin and E‐cadherin were analyzed by Western blotting. (k,l) The fluorescence intensity of β‐catenin in adeno‐transfected H358ON cells expressing Dox‐dependent G4A G129E Δ C2 was evaluated. (k) Left and right images show cells without and with TGFβ stimulation along the selected yellow arrows in (k), respectively. (l) Upper and lower panels plot the fluorescence intensity of β‐catenin (red) and nucleus (blue) over a cross‐section of cells without and with TGFβ stimulation, respectively. Data shown are representative of at least three independent experiments.
Discussion
There is an increasing awareness of the tumor microenvironment, in which intercellular communication between cancer‐associated fibroblasts30, 31 and cancer cells might influence the behavior of lung cancer cells.2 Aberrant activation of the inducers of EMT, such as TGFβ stimulation and persistent hypoxia, is often observed in tumor microenvironment; as a consequence, lung cancer cells acquire the malignant phenotypes.20, 32, 33 Therefore, prevention of the EMT process, in which excessive ECM production and loss of alveolar epithelial cell integrity are involved,4 is one of the most critical approaches for treating lung cancer cells.34 Although intercellular transfer of the newly proposed PTEN variants such as alternative transcriptional PTEN (PTEN‐Long) and monoubiquitinated PTEN (exosomal PTEN)35, 36 might be proposed as a biopharmaceutical approach to treat lung cancer cells that lose functional PTEN,9 intercellular transfer of PTENWt into cancer cells in the tumor microenvironment might not be enough to exert a therapeutic effect because PTENWt remains subject to TGFβ‐induced phosphorylation of its C‐terminus, resulting in a loss of its enzyme activities.6, 37, 38 Furthermore, to successfully achieve exogenous transfer of the targeted molecule, research on therapeutically important proteins has focused on miniaturization of protein‐specific functions because of size constraints in exogenous administration.39 The PTEN C‐terminus with four Ala substitutions cannot be phosphorylated on its C‐terminus, even after TGFβ stimulation.6 Indeed, our data suggest that compensatory induction of PTEN4A successfully represses the TGFβ‐induced acquisition of EMT in lung cancer cells, whereas de novo PTENWt expression shows limited repression of TGFβ‐induced EMT, probably due to TGFβ‐induced phosphorylation of its C‐terminus.6 Nevertheless, whether or not the unphosphorylated PTEN C‐terminus itself might directly modulate the TGFβ‐induced acquisition of EMT in lung cancer cells has not been fully determined. The present data showed that the PTEN C‐terminus itself (G4A tail) might not be involved in the lipid and protein phosphatase activities of PTEN, as evaluated by the levels of TGFβ‐induced Akt and FAK phosphorylation, respectively. Furthermore, the PTEN C‐terminus did not appear to directly modulate TGFβ‐induced EMT in lung cancer cells, or to inhibit translocation of β‐catenin from the cell membrane into the cytoplasm and the nucleus in cells treated with TGFβ. Meanwhile, PTEN lacking the C‐terminus but including the catalytic core of the C2 and phosphatase domains (G4A Δ tail) repressed TGFβ‐induced phosphorylation of FAK and Akt to basal levels through PTEN phosphatase activities, consistent with previous data.40, 41 The cells expressing G4A Δ tail did not undergo TGFβ‐induced EMT, and β‐catenin remained localized on the cell membrane in cells treated with TGFβ, compatible with the cell expressing PTEN4A (GP4A). The unphosphorylated PTEN C‐terminus might be critical for stable intramolecular interactions between the C2 and phosphatase domains of PTEN.40, 42 Taken together, these data suggested that the approach that keeps the PTEN C‐terminus not phosphorylated might be crucial for the unphosphorylated PTEN to inhibit TGFβ‐induced EMT.
Although the C2 and phosphatase domains, that is, the minimal catalytic core of PTEN, are required to interact with the plasma membrane to inhibit Akt activity through the lipid phosphatase activity,18, 40 whether or not the close association between the C2 and phosphatase domains might be essential to exert protein phosphatase activity and negatively regulate TGFβ‐induced EMT in lung cancer cells remains unclear. To determine the contribution of the C2 and phosphatase domains, we evaluated the effect of several PTEN domain deletion mutants in TGFβ‐stimulated lung cancer cells. Although PTEN4A negatively regulates TGFβ‐induced Akt and FAK phosphorylation, we considered that a PTEN mutant lacking either the phosphatase domain (G4A Δ phosphatase) or the C2 domain (G4A Δ C2) might lose both its lipid and protein phosphatase activities. Compensatory induction of G4A Δ phosphatase could not retain the protein activity and blunt TGFβ‐induced EMT. These findings strengthened the hypothesis that the phosphatase domain involves the essential regions required to exert protein phosphatase activity.7, 16, 17, 18 Meanwhile, when G4A Δ C2, in which the phosphatase domain is involved, was induced, protein phosphatase activity could not be restored and TGFβ‐induced EMT remained persistent. PTEN lacking the C2 domain or the phosphatase domain could not inhibit TGFβ‐induced translocation of β‐catenin from the cell membrane into the cytoplasm and the nucleus. A previous study indicates that PTEN lipid phosphatase activity might involve the association of both the C2 and phosphatase domains.40 Taken together, these data indicate that both the C2 and phosphatase domains might be essential to exert phosphatase activity and negatively regulate TGFβ‐induced EMT.
Previous studies have shown that both LY294002, a PI3K/Akt inhibitor, and rapamycin, an mTOR‐specific inhibitor do not rescue EMT.43, 44 To determine which of the PTEN phosphatase activities, that is, lipid or protein phosphatase activity, might negatively control TGFβ‐induced EMT in lung cancer cells, we established lung cancer cells expressing previously characterized PTEN mutants with different alterations in phosphatase activities.7, 16, 17, 18 De novo expression of the unphosphorylated PTEN mutants without protein phosphatase activity (C124S and Y138C) did not repress TGFβ‐induced EMT in lung cancer cells, whereas unphosphorylated PTEN with protein phosphatase activity (G129E) did repress TGFβ‐induced EMT;41 in addition, de novo expression of G129E, but not C124S or Y138C, inhibited TGFβ‐induced β‐catenin translocation from the cell membrane into the cytoplasm and nucleus. Together, our data suggest a new role of PTEN protein phosphatase activity to negatively regulate the acquisition of TGFβ‐induced EMT in lung cancer cells.
Although the close association of both the PTEN C2 and phosphatase domains might be essential for unphosphorylated PTEN to block TGFβ‐induced EMT, whether or not the close association of both the PTEN C2 and phosphatase domains might be essential for PTEN protein phosphatase activity remains elusive. Therefore, we reconstituted PTEN in cells expressing G4A G129E Δ C2 by adeno GP4A C2 only. De novo G4A G129E Δ C2 showed no PTEN protein phosphatase activity in lung cancer cells stimulated by TGFβ, and abrogated the inhibition of TGFβ‐induced EMT in lung cancer cells. Subsequently, reconstitution of the PTEN C2 domain in these cells by adeno GP4A C2 only restored PTEN protein phosphatase activity and its associated inhibition of TGFβ‐induced EMT. Thus, although the protein phosphatase activity of PTEN has not been fully characterized,18 we have shown for the first time that a critical association between the C2 and phosphatase domains might be essential for unphosphorylated PTEN to retain its protein phosphatase activity and to inhibit TGFβ‐induced EMT in lung cancer cells. Although a recent study suggests that TGFβ‐induced transcription of EMT target genes such as fibronectin and vimentin is accelerated by translocation of β‐catenin from E‐cadherin complexes at the cell membrane into the cytoplasm,5 the exact mechanism by which PTEN with an unphosphorylated C‐terminus might block TGFβ‐induced β‐catenin translocation from the cell membrane into the cytoplasm, remains elusive. Nevertheless, our data indicate that PTEN protein phosphatase activity might be closely associated with inhibiting the translocation of β‐catenin from the cell membrane, possibly through the blockade of β‐catenin phosphorylation.45, 46, 47
In summary, we demonstrated that PTEN with an unphosphorylated C‐terminus exerted its functions through its protein phosphatase activity, which in turn might depend on an essential association between the C2 and phosphatase domains. PTEN4A might negatively regulate TGFβ‐induced EMT and the associated translocation of β‐catenin through its protein phosphatase activity. These data suggest that the protein phosphatase activity of PTEN with an unphosphorylated C‐terminus might be a therapeutic target to negatively regulate TGFβ‐induced EMT during development of lung cancers.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Evaluation of Akt activation by the mutants of PTEN and localization of β‐catenin on the cell membrane in H358 lung cancer cells.
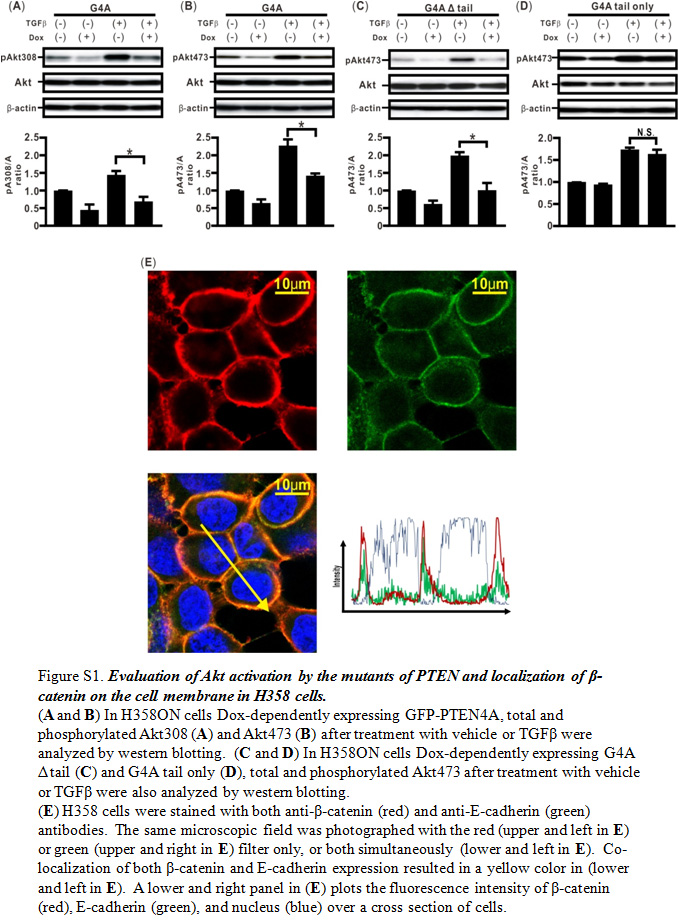{kind=link}
Fig. S2. PTEN wild C‐terminus and FAK activation might not directly inhibit transforming growth factor β (TGFβ)‐induced epithelial–mesenchymal transition in H358 lung cancer cells.
{kind=link}
Acknowledgments
This work was supported by Kowa Life Science Foundation and Grants‐in‐Aid for Scientific Research (C) (21590987 and 24591162), and also supported by a grant to the Diffuse Lung Diseases Research Group from the Ministry of Health, Labor and Welfare, Japan.
Cancer Sci 106 (2015) 1693–1704
Funding Information
Kowa Life Science Foundation; Ministry of Health, Labour and Welfare, Japan.
References
- 1. Rocken M. Early tumor dissemination, but late metastasis: insights into tumor dormancy. J Clin Invest 2010; 120: 1800–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010; 140: 883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kojima Y, Acar A, Eaton EN et al Autocrine TGF‐beta and stromal cell‐derived factor‐1 (SDF‐1) signaling drives the evolution of tumor‐promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A 2010; 107: 20009–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol 2006; 7: 131–42. [DOI] [PubMed] [Google Scholar]
- 5. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial–mesenchymal transition through beta‐catenin‐T‐cell factor‐4‐dependent expression of transforming growth factor‐beta3. Mol Biol Cell 2008; 19: 4875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aoyama D, Hashimoto N, Sakamoto K et al Involvement of TGFbeta‐induced phosphorylation of the PTEN C‐terminus on TGFbeta‐induced acquisition of malignant phenotypes in lung cancer cells. PLoS ONE 2013; 8: e81133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davidson L, Maccario H, Perera NM et al Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene 2010; 29: 687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gharaee‐Kermani M, Gyetko MR, Hu B et al New insights into the pathogenesis and treatment of idiopathic pulmonary fibrosis: a potential role for stem cells in the lung parenchyma and implications for therapy. Pharm Res 2007; 24: 819–41. [DOI] [PubMed] [Google Scholar]
- 9. Yanagi S, Kishimoto H, Kawahara K et al Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest 2007; 117: 2929–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li DM, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor beta. Cancer Res 1997; 57: 2124–9. [PubMed] [Google Scholar]
- 11. Odriozola L, Singh G, Hoang T et al Regulation of PTEN activity by its carboxyl‐terminal autoinhibitory domain. J Biol Chem 2007; 282: 23306–15. [DOI] [PubMed] [Google Scholar]
- 12. Rahdar M, Inoue T, Meyer T et al A phosphorylation‐dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci U S A 2009; 106: 480–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martins LR, Lucio P, Silva MC et al Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood 2010; 116: 2724–31. [DOI] [PubMed] [Google Scholar]
- 14. Shehata M, Schnabl S, Demirtas D et al Reconstitution of PTEN activity by CK2 inhibitors and interference with the PI3‐K/Akt cascade counteract the antiapoptotic effect of human stromal cells in chronic lymphocytic leukemia. Blood 2010; 116: 2513–21. [DOI] [PubMed] [Google Scholar]
- 15. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13: 283–96. [DOI] [PubMed] [Google Scholar]
- 16. Furnari FB, Huang HJ, Cavenee WK. The phosphoinositol phosphatase activity of PTEN mediates a serum‐sensitive G1 growth arrest in glioma cells. Cancer Res 1998; 58: 5002–8. [PubMed] [Google Scholar]
- 17. Myers MP, Pass I, Batty IH et al The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci U S A 1998; 95: 13513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tibarewal P, Zilidis G, Spinelli L et al PTEN protein phosphatase activity correlates with control of gene expression and invasion, a tumor‐suppressing phenotype, but not with AKT activity. Sci Signal 2012; 5: ra18. [DOI] [PubMed] [Google Scholar]
- 19. Chalhoub N, Baker SJ. PTEN and the PI3‐kinase pathway in cancer. Annu Rev Pathol 2009; 4: 127–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sakamoto K, Hashimoto N, Kondoh Y et al Differential modulation of surfactant protein D under acute and persistent hypoxia in acute lung injury. Am J Physiol Lung Cell Mol Physiol 2012; 303: L43–53. [DOI] [PubMed] [Google Scholar]
- 21. Ikenoue T, Inoki K, Zhao B et al PTEN acetylation modulates its interaction with PDZ domain. Cancer Res 2008; 68: 6908–12. [DOI] [PubMed] [Google Scholar]
- 22. Song MS, Carracedo A, Salmena L et al Nuclear PTEN regulates the APC‐CDH1 tumor‐suppressive complex in a phosphatase‐independent manner. Cell 2011; 144: 187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takagi Y, Hashimoto N, Phan SH et al Erythromycin‐induced CXCR4 expression on microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 2009; 297: L420–31. [DOI] [PubMed] [Google Scholar]
- 24. Otero JJ, Fu W, Kan L et al Beta‐catenin signaling is required for neural differentiation of embryonic stem cells. Development 2004; 131: 3545–57. [DOI] [PubMed] [Google Scholar]
- 25. Samuel MS, Lopez JI, McGhee EJ et al Actomyosin‐mediated cellular tension drives increased tissue stiffness and beta‐catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011; 19: 776–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu T, Nozaki Y, Phan SH. Regulation of telomerase activity in rat lung fibroblasts. Am J Respir Cell Mol Biol 2002; 26: 534–40. [DOI] [PubMed] [Google Scholar]
- 27. Hashimoto N, Phan SH, Imaizumi K et al Endothelial–mesenchymal transition in bleomycin‐induced pulmonary fibrosis. Am J Respir Cell Mol Biol 2010; 43: 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morel AP, Hinkal GW, Thomas C et al EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin‐low tumors in transgenic mice. PLoS Genet 2012; 8: e1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zeisberg M, Neilson EG. Biomarkers for epithelial–mesenchymal transitions. J Clin Invest 2009; 119: 1429–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Orimo A, Gupta PB, Sgroi DC et al Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF‐1/CXCL12 secretion. Cell 2005; 121: 335–48. [DOI] [PubMed] [Google Scholar]
- 31. Zeisberg EM, Potenta S, Xie L et al Discovery of endothelial to mesenchymal transition as a source for carcinoma‐associated fibroblasts. Cancer Res 2007; 67: 10123–8. [DOI] [PubMed] [Google Scholar]
- 32. Keith B, Simon MC. Hypoxia‐inducible factors, stem cells, and cancer. Cell 2007; 129: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Massague J. TGFbeta in cancer. Cell 2008; 134: 215–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Acloque H, Adams MS, Fishwick K et al Epithelial–mesenchymal transitions: the importance of changing cell state in development and disease. J Clin Invest 2009; 119: 1438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Putz U, Howitt J, Doan A et al The tumor suppressor PTEN is exported in exosomes and has phosphatase activity in recipient cells. Sci Signal 2012; 5: ra70. [DOI] [PubMed] [Google Scholar]
- 36. Hopkins BD, Fine B, Steinbach N et al A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 2013; 341: 399–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kotelevets L, van Hengel J, Bruyneel E et al Implication of the MAGI‐1b/PTEN signalosome in stabilization of adherens junctions and suppression of invasiveness. FASEB J 2005; 19: 115–7. [DOI] [PubMed] [Google Scholar]
- 38. Siesser PM, Hanks SK. The signaling and biological implications of FAK overexpression in cancer. Clin Cancer Res 2006; 12: 3233–7. [DOI] [PubMed] [Google Scholar]
- 39. Nagahara H, Vocero‐Akbani AM, Snyder EL et al Transduction of full‐length TAT fusion proteins into mammalian cells: TAT‐p27Kip1 induces cell migration. Nat Med 1998; 4: 1449–52. [DOI] [PubMed] [Google Scholar]
- 40. Lee JO, Yang H, Georgescu MM et al Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 1999; 99: 323–34. [DOI] [PubMed] [Google Scholar]
- 41. Leslie NR, Yang X, Downes CP et al PtdIns(3,4,5)P(3)‐dependent and ‐independent roles for PTEN in the control of cell migration. Curr Biol 2007; 17: 115–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Malaney P, Pathak RR, Xue B et al Intrinsic disorder in PTEN and its interactome confers structural plasticity and functional versatility. Sci Rep 2013; 3: 2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lamouille S, Derynck R. Cell size and invasion in TGF‐beta‐induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J Cell Biol 2007; 178: 437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zuo W, Chen YG. Specific activation of mitogen‐activated protein kinase by transforming growth factor‐beta receptors in lipid rafts is required for epithelial cell plasticity. Mol Biol Cell 2009; 20: 1020–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Piedra J, Martinez D, Castano J et al Regulation of beta‐catenin structure and activity by tyrosine phosphorylation. J Biol Chem 2001; 276: 20436–43. [DOI] [PubMed] [Google Scholar]
- 46. Kim Y, Kugler MC, Wei Y et al Integrin alpha3beta1‐dependent beta‐catenin phosphorylation links epithelial Smad signaling to cell contacts. J Cell Biol 2009; 184: 309–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chapman HA. Epithelial responses to lung injury: role of the extracellular matrix. Proc Am Thorac Soc 2012; 9: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Evaluation of Akt activation by the mutants of PTEN and localization of β‐catenin on the cell membrane in H358 lung cancer cells.
Fig. S2. PTEN wild C‐terminus and FAK activation might not directly inhibit transforming growth factor β (TGFβ)‐induced epithelial–mesenchymal transition in H358 lung cancer cells.