

Abstract
Anti‐epidermal growth factor receptor (EGFR) treatment is an effective option for metastatic colorectal cancer (CRC) treatment. However, there are few reliable biomarkers to predict the clinical response to anti‐EGFR treatment. We investigated the genome‐wide DNA methylation status in metastatic colorectal cancer to identify associations between the methylation status and clinical response to anti‐EGFR antibody. We retrospectively reviewed the medical records of 97 patients (45 patients for the first cohort and 52 patients for the second cohort) who received anti‐EGFR treatment for KRAS wild‐type metastatic CRC. Then we analyzed the associations between genome‐wide DNA methylation status and clinical response to anti‐EGFR treatment, and evaluated the predictive power and value of the methylation status statistically. As a result, each cohort was classified into highly methylated CRC and low methylated CRC subgroups by unsupervised clustering analyses. In the first cohort, clinical outcomes were significantly better in the low methylated CRC subgroup than in the highly methylated CRC subgroup (response rate, 35.7% vs 6.3%, P = 0.03; disease control rate, 75% vs 31.3%, P = 0.005; hazard ratio for progression‐free survival, 0.27; 95% confidence interval, 0.13–0.57, P < 0.001; overall survival, 0.19; 95% confidence interval, 0.06–0.54, P < 0.001). These results were reproducible in the second cohort. The genome‐wide methylation status was a predictive factor of progression‐free survival and overall survival independently of RAS mutation status. In conclusion, we found that the genome‐wide DNA methylation status is a powerful epigenetic predictor of anti‐EGFR treatment in patients with KRAS wild‐type metastatic colorectal cancer (UMIN000005490).
Keywords: Anti‐EGFR treatment, biomarker, colorectal cancer, DNA methylation, molecular targeted therapy
Colorectal cancer (CRC) is the third most common cancer and a major cause of cancer mortality worldwide.1 Metastatic disease develops in approximately 40% of newly diagnosed patients.2 Current standard treatment for metastatic disease is chemotherapy including molecular‐targeting drugs. Standard first‐ and second‐line treatments include fluorouracil with leucovorin and irinotecan3, 4 or oxaliplatin,5, 6 alone or in combination with bevacizumab.7, 8
Anti‐epidermal growth factor receptor (EGFR) treatment using mAb (cetuximab, panitumumab) against EGFR is effective in combination with irinotecan or as a single agent for metastatic CRC that progresses despite standard first‐ and second‐line treatments.9, 10 Recent studies have also shown that anti‐EGFR treatment is an effective option when added to irinotecan‐ or oxaliplatin‐based therapy as the first‐line treatment.11
Retrospective studies have shown that mutations in genes encoding key molecules within intracellular EGFR signaling pathways are associated with the inefficacy of anti‐EGFR treatment.12, 13 Particularly, mutations in the KRAS gene at codons 12 and 13 are the most common mutations in these pathways; thus, examination of KRAS mutations is clinically useful to exclude patients who would be unresponsive to anti‐EGFR treatment.14, 15 However, there is no other reliable biomarker to predict the clinical response to anti‐EGFR treatment.
The CpG island methylator phenotype (CIMP) is a major molecular mechanism of carcinogenesis in CRC16 and is associated with disease development in approximately 20% of cases.17 CpG island methylator phenotype‐positive CRC is reportedly associated with a higher ratio of tumors with the BRAF mutation and disease onset in women, the elderly, and patients with a history of smoking.18, 19, 20 In addition to epidemiological characteristics, CIMP‐positive CRC is associated with hyperplastic polyps and sessile‐serrated polyps as precursor lesions.21 Moreover, some other classification methods about the methylation status have been reported.22 Based on these findings, prognosis and sensitivity to chemotherapy may vary according to the methylation status. Although the role of the methylation status as a prognostic factor in CRC has been reported,22, 23 no study has examined the predictive value of the genome‐wide methylation status regarding the response to CRC treatment, particularly anti‐EGFR treatment.
In this retrospective study, we investigated the genome‐wide DNA methylation status in metastatic CRC without KRAS codon 12 or 13 mutations to identify associations between methylation status and clinical response to anti‐EGFR antibody.
Materials and Methods
Patients
We retrospectively reviewed the medical records of 97 patients with histologically confirmed adenocarcinoma of metastatic CRC without KRAS codon 12 or 13 mutation who received standard chemotherapy, including anti‐EGFR antibody, at Tohoku University Hospital (TUH) (Sendai, Japan) or National Cancer Center Hospital (NCCH) (Tokyo, Japan) from 2005 to 2013. The protocol was approved by independent ethics committees of Tohoku University School of Medicine and NCCH, and all patients provided written and oral informed consent.
Tissue samples and macrodissection
Formalin‐fixed paraffin‐embedded (FFPE) archival tissue blocks of 97 primary tumors were obtained from patients. Unstained 10‐μm‐thick slices were dissected to enrich either cancer cells or normal colon mucosa under the guidance of an HE‐stained tissue slide.
Extraction of genomic DNA and quality control
Genomic DNA was extracted from macrodissected samples using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany). Quality control of extracted genomic DNA was carried out by quantitative real‐time PCR using the Infinium HD FFPE QC Kit (Illumina, San Diego, CA, USA) and calculated delta Cq value.
Mutation analyses
Mutations in KRAS exon 2 (codons 12 and 13) and BRAF V600E were analyzed by direct DNA sequencing (Tables S1 and S2). Infrequent‐RAS mutations including KRAS (codons 61 and 146) and NRAS (codons 12, 13, and 61) were analyzed by Luminex Assay (GENOSEARCH Mu‐PACK, MBL, Nagoya, Japan).
Genome‐wide DNA methylation analysis
Genome‐wide DNA methylation analysis was carried out using the Infinium HumanMethylation450 BeadChip (Illumina) with probes that target 485 577 CpG sites and cover 99% of the RefSeq gene. The BeadChip was scanned using the iScan, and the methylation level was calculated as (β value: calculated as intensity of methylated probe/[intensity of methylated probe + intensity of unmethylated probe]). After excluding probes specific for the X and Y chromosomes, those with ≥0.25 SD of the DNA methylation β‐value across all CRC samples were selected for further analyses.
Evaluation of methylation status using previously defined markers
The methylation status was also evaluated using two distinct sets of methylation markers, as reported previously. Then we evaluated whether the methylation status by each method was associated with efficacy of anti‐EGFR treatment. These sets of markers were mapped on the promoter regions of: (i) seven genes (CACANA1G, LOX, SLC30A10, ELMO1, HAND1, THBD, and FBN2) reported by Yagi et al.22; and (ii) five genes (CACNA1G, IGF2, NEUROG1, RUNX3, and SOCS1) reported by Weisenberger et al.18 According to the original judging systems, samples were classified into the following categories: high‐methylation epigenotype (HME), intermediate‐methylation epigenotype (IME), and low‐methylation epigenotype (LME) by Yagi's method, and CIMP‐positive and CIMP‐negative by Weisenberger's method.
Tumor response
The tumor reduction rate from treatment initiation to disease progress was evaluated in TUH and NCCH based on Response Evaluation Criteria in Solid Tumors (version 1.0) by medical oncologists. The percentage of the overall number of cases achieving a complete response or partial response or the sum of complete response, partial response, and stable disease cases was defined as the observed response rate (RR) or disease control rate (DCR), respectively.
Progression‐free survival and overall survival
Progression‐free survival (PFS) and overall survival (OS) were defined as the time from anti‐EGFR antibody treatment to disease progression or death, respectively. Disease progression was evaluated by medical oncologists at TUH or NCCH.
Statistical analyses
Statistical analyses of patient demographic factors were carried out using the χ2‐test. Analyses of patient background factors and results of anti‐EGFR treatment were carried out using JMP Pro11 software (SAS Institute Japan Co., Ltd., Tokyo, Japan). Survival curves were constructed using the Kaplan–Meier method, and differences between curves of the two subgroups were compared using the log–rank test. Associations between patient background factors, including the methylation status and PFS and OS periods after anti‐EGFR antibody treatment, were used for univariate and multivariate analyses with a Cox proportional hazards model.
Probe selection in each cohort and mutual validation
We used the random forest algorithm for building the model and the sample classification. When we built the model by using one cohort dataset, the other cohort data were used as a validation test set and vice versa. First, we built two models on each cohort dataset with the common probes between the cohorts, and we selected the efficient probes. Thereafter, we constructed the models based on the fewer efficient probes, and classified the samples of the other cohort. Finally, we selected common probes from two models made in each cohort above, and classified the cohort by the model with another cohort using these common probes.
More precise methods are described in Data S1.
Results
Patients
A total of 97 primary tumor samples without KRAS codon 12 and 13 mutations were divided into the first cohort (n = 45) and second cohort (n = 52). The two cohorts were almost well balanced in terms of baseline characteristics (Table S3). The number of previous treatment regimens in the first cohort was significantly less than that in the second cohort (P = 0.01).
Genome‐wide DNA methylation status
From the entire 485 577 probes, 3163 and 2577 probes within ≥0.25 SD of the DNA methylation β‐value across all CRC samples were selected for the first and second cohorts, respectively. Data of the selected probes were subjected to unsupervised clustering analyses. The first cohort could be clearly classified into two subgroups, namely highly methylated colorectal cancer (HMCC, n = 17) and low methylated colorectal cancer (LMCC, n = 28) (Fig. 1a). Similarly, the second cohort was also classified into HMCC (n = 17) and LMCC (n = 35) subgroups (Fig. 1b).
Figure 1.
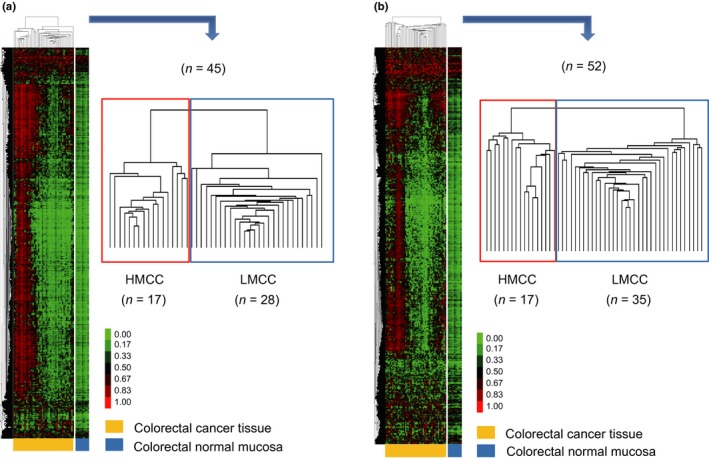
Unsupervised non‐hierarchical clustering of results of genome‐wide DNA methylation analysis for colorectal cancer. Results of genome‐wide DNA methylation analyses in the first (a) and second (b) cohorts. The first and second cohorts (n = 45 and 52, respectively) were classified into highly methylated colorectal cancer (HMCC) (n = 17 and 17, respectively) and low methylated colorectal cancer (LMCC) (n = 28 and 35, respectively) subgroups. The heat map represents the β‐value (0–1.0). Each column and row represents a case and a probe, respectively.
Subgroup analysis of baseline characteristics according to DNA methylation status
Baseline characteristics of HMCC and LMCC subgroups were analyzed (Table 1). The primary site of the HMCC subgroup tended to be the proximal colon in both cohorts (P = 0.02 and 0.04, respectively). The number of subsequent regimens was significantly less in the HMCC subgroup than in the LMCC subgroup in the first cohort (P < 0.01) but not in the second cohort (P = 0.57). The use of the combination of irinotecan and anti‐EGFR treatment was significantly less in the HMCC subgroup than in the LMCC subgroup in the first cohort (P = 0.02) but not in the second cohort (P = 0.7). There was no significant difference in other baseline characteristics between subgroups in the two cohorts.
Table 1.
Baseline characteristics of study patients of highly methylated colorectal cancer (HMCC) and low methylated colorectal cancer (LMCC) subgroups in two independent cohorts
| Variable | First cohort | P‐value | Second cohort | P‐value | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All samples | HMCC | LMCC | All samples | HMCC | LMCC | |||||||||
| n | % | n | % | n | % | n | % | n | % | n | % | |||
| Total | 45 | 100 | 17 | 37.8 | 28 | 62.2 | 52 | 100 | 17 | 43.8 | 35 | 56.3 | ||
| Gender | 0.79† | 0.29† | ||||||||||||
| Male | 28 | 66.2 | 11 | 64.7 | 17 | 60.7 | 38 | 73.1 | 14 | 82.4 | 24 | 68.6 | ||
| Female | 17 | 37.8 | 6 | 35.3 | 11 | 39.3 | 14 | 26.9 | 3 | 17.6 | 11 | 31.4 | ||
| Median age, years (range) | 0.70‡ | 0.15‡ | ||||||||||||
| 61 (32–77) | 61 (33–73) | 61.5 (32–77) | 61 (29–83) | 63 (39–83) | 60 (29–79) | |||||||||
| Primary site | 0.02† | 0.04† | ||||||||||||
| Proximal | 12 | 26.7 | 8 | 47.1 | 4 | 14.3 | 15 | 33.3 | 8 | 47.1 | 7 | 20.0 | ||
| Cecum | 2 | 4.4 | 1 | 5.9 | 1 | 3.6 | 2 | 4.4 | 1 | 5.9 | 1 | 2.9 | ||
| Ascending | 7 | 15.6 | 5 | 29.4 | 2 | 7.1 | 8 | 17.8 | 6 | 35.3 | 2 | 5.7 | ||
| Transverse | 3 | 6.7 | 2 | 11.8 | 1 | 3.6 | 5 | 11.1 | 1 | 5.9 | 4 | 11.4 | ||
| Distal | 33 | 73.3 | 9 | 52.9 | 24 | 85.7 | 37 | 82.2 | 9 | 52.9 | 28 | 80.0 | ||
| Descending | 1 | 2.2 | 0 | 0.0 | 1 | 3.6 | 1 | 2.2 | 1 | 5.9 | 0 | 0.0 | ||
| Sigmoid | 6 | 13.3 | 1 | 5.9 | 5 | 17.9 | 13 | 28.9 | 2 | 11.8 | 11 | 31.4 | ||
| Rectum | 26 | 57.8 | 8 | 47.1 | 18 | 64.3 | 23 | 51.1 | 6 | 35.3 | 17 | 48.6 | ||
| Stage at diagnosis | 0.68† | 0.94† | ||||||||||||
| I | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | ||
| II | 2 | 4.4 | 1 | 5.9 | 1 | 3.6 | 3 | 6.7 | 1 | 5.9 | 2 | 5.7 | ||
| III | 13 | 28.9 | 6 | 35.3 | 7 | 25.0 | 17 | 37.8 | 5 | 29.4 | 12 | 34.3 | ||
| IV | 30 | 66.7 | 10 | 58.8 | 20 | 71.4 | 32 | 71.1 | 11 | 64.7 | 21 | 60 | ||
| Number of organs with metastasis | 0.53‡ | 0.84‡ | ||||||||||||
| 0 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | 2 | 4.4 | 0 | 0 | 2 | 5.7 | ||
| 1 | 22 | 48.9 | 9 | 20 | 13 | 28.9 | 22 | 48.9 | 8 | 47.1 | 14 | 40.0 | ||
| 2 | 21 | 46.7 | 8 | 17.8 | 13 | 28.9 | 23 | 51.1 | 7 | 41.2 | 16 | 45.7 | ||
| 3 | 2 | 4.4 | 0 | 0.0 | 2 | 4.4 | 5 | 11.1 | 2 | 11.8 | 3 | 8.6 | ||
| BRAF mutation | 0.38† | 0.17† | ||||||||||||
| + | 3 | 7.5 | 2 | 11.8 | 1 | 4.3 | 3 | 5.9 | 2 | 12.5 | 1 | 2.9 | ||
| − | 37 | 92.5 | 15 | 88.2 | 22 | 95.7 | 48 | 94.1 | 14 | 87.5 | 34 | 97.1 | ||
| NA | 5 | 0 | 5 | 1 | 1 | 0 | ||||||||
| No. of previous regimens | 0.33‡ | 0.57‡ | ||||||||||||
| 0 | 5 | 11.1 | 0 | 0.0 | 5 | 17.9 | 0 | 0.0 | 0 | 0.0 | 0 | 0.0 | ||
| 1 | 4 | 8.9 | 2 | 11.8 | 2 | 7.1 | 5 | 8.2 | 2 | 11.8 | 3 | 8.6 | ||
| 2 | 33 | 73.3 | 14 | 82.4 | 19 | 67.9 | 33 | 65.3 | 11 | 64.7 | 22 | 62.9 | ||
| ≥3 | 3 | 6.7 | 1 | 5.9 | 2 | 7.1 | 14 | 26.5 | 4 | 23.5 | 10 | 28.6 | ||
| No. of following regimens | <0.01‡ | 0.57‡ | ||||||||||||
| 0 | 29 | 64.4 | 17 | 100.0 | 12 | 42.9 | 33 | 61.2 | 12 | 70.6 | 21 | 60 | ||
| 1 | 10 | 22.2 | 0 | 0.0 | 10 | 35.7 | 14 | 26.5 | 3 | 17.6 | 11 | 31.4 | ||
| ≥2 | 6 | 13.3 | 0 | 0.0 | 6 | 21.4 | 5 | 10.2 | 2 | 11.8 | 3 | 8.6 | ||
| Type of anti‐EGFR therapy | 0.02† | 0.70† | ||||||||||||
| Monotherapy | 12 | 26.7 | 8 | 47.1 | 4 | 14.3 | 14 | 26.5 | 4 | 23.5 | 10 | 28.6 | ||
| Combination with irinotecan | 33 | 73.3 | 9 | 52.9 | 24 | 85.7 | 38 | 73.5 | 13 | 76.5 | 25 | 71.4 | ||
†χ2‐test. ‡Wilcoxon test. NA, Not Available.
Association between genome‐wide DNA methylation status and treatment response
The association between genome‐wide DNA methylation status and the clinical outcome of anti‐EGFR treatment with regard to RR, DCR, PFS, and OS was analyzed. Overall RR was 25% in all cases. In the first cohort, RR and DCR were significantly higher in the LMCC subgroup than in the HMCC subgroup (35.7% vs 6.3%, P = 0.03 for RR; 75% vs 31.3%, P = 0.005 for DCR) (Table S4), and differences were validated in the second cohort (34.3% vs 5.9%, P = 0.03 for RR; 77.1% vs 47.1%, P = 0.03 for DCR). In the first cohort, hazard ratios (HRs) for PFS and OS among patients in the LMCC subgroup in comparison with the HMCC subgroup were 0.27 (95% confidence interval [CI], 0.13–0.57; P < 0.001) and 0.19 (95% CI, 0.06–0.54; P < 0.001), respectively (Fig. 2). The median periods among patients in LMCC and HMCC subgroups were 197 and 72 days for PFS, and 24.9 and 5.6 months for OS, respectively. These results were validated in the second cohort. Hazard ratios for PFS and OS among patients in the LMCC subgroup in comparison with the HMCC subgroup were 0.22 (95% CI, 0.11–0.45; P < 0.001) and 0.35 (95% CI, 0.13–0.98; P = 0.03), respectively (Fig. 2). The median periods among patients in LMCC and HMCC subgroups were 191 and 70 days for PFS, and 14.1 and 9.3 months for OS, respectively. Furthermore, we analyzed RR, DCR, PFS, and OS in patients who used anti‐EGFR as a third line of treatment. As the sample size of patients who had been administered anti‐EGFR antibody for third‐line treatment was small, we combined the cohorts for analysis. As a result, these findings were also confirmed only in third‐line patients (Fig. S1, Table S5).
Figure 2.
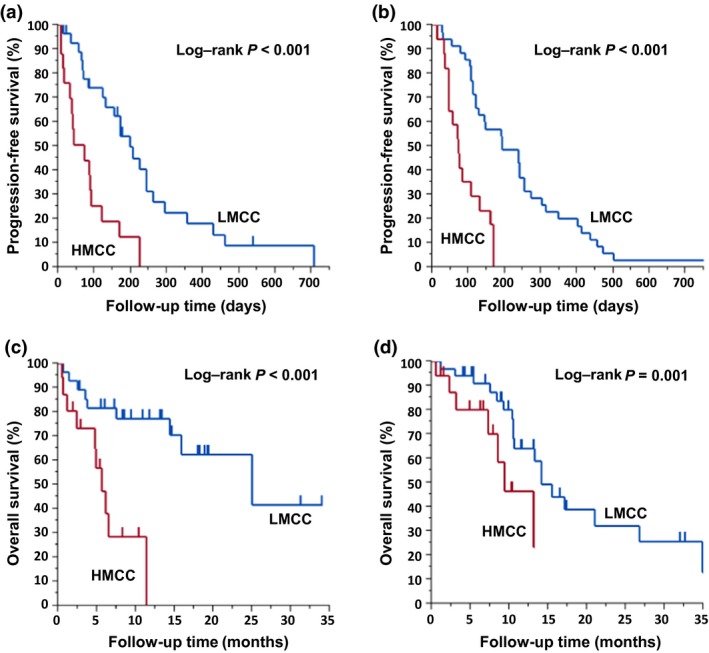
Kaplan–Meier curves for progression‐free survival and overall survival after anti‐epidermal growth factor receptor (EGFR) treatment in the first and second cohorts of colorectal cancer patients according to genome‐wide DNA methylation status. Panels (a,b) Progression‐free survival after anti‐EGFR treatment for the highly methylated colorectal cancer (HMCC) subgroup (red line) and the low methylated colorectal cancer (LMCC) subgroup (blue line) in the first and second cohorts, respectively. (c,d) Overall survival after anti‐EGFR treatment for the HMCC subgroup (red line) and the LMCC subgroup (blue line) in the first and second cohorts, respectively.
Comparison with known predictors for unresponsiveness to anti‐EGFR treatment
To examine whether the differences in clinical outcome of anti‐EGFR treatment between HMCC and LMCC subgroups was affected by cases with the infrequent‐RAS mutation, 97 cases were classified into three subgroups, RAS wild‐type HMCC subgroup (28 cases), RAS wild‐type LMCC subgroup (58 cases), and infrequent‐RAS mutation subgroup (11 cases), based on the results of the infrequent‐RAS mutation analyses. The anti‐EGFR treatment outcomes were then compared among these subgroups. The RR and DCR of the RAS wild‐type HMCC subgroup were found to be significantly lower than those of the RAS wild‐type LMCC subgroup (3.7% vs 37.9%, P < 0.01 for RR; 40.7% vs 79.3%, P < 0.001 for DCR) (Table S6). The PFS and OS of the RAS wild‐type HMCC subgroup were significantly shorter than that of the RAS wild‐type LMCC subgroup (HR = 0.22; 95% CI, 0.13–0.37; P < 0.001; and HR = 0.24; 95% CI, 0.11–0.53; P < 0.001, respectively) (Fig. 3). The treatment outcome of the infrequent‐RAS mutation subgroup was significantly inferior to that of the RAS wild‐type LMCC subgroup in DCR, PFS, and OS except for RR. However, there was no significant difference in the treatment outcome of the infrequent‐RAS mutation subgroup with the RAS wild‐type HMCC subgroup.
Figure 3.
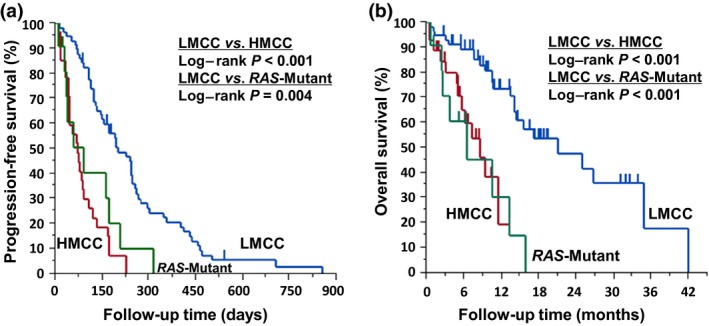
Kaplan–Meier curves for progression‐free survival and overall survival after anti‐epidermal growth factor receptor treatment in the unified cohort (first and second cohorts) of colorectal cancer patients according to genome‐wide DNA methylation status and infrequent‐RAS mutation status. Progression‐free survival (a) and overall survival (b) in the unified cohort according to RAS‐wild highly methylated colorectal cancer (HMCC) subgroup (red line, n = 28), RAS‐wild low methylated colorectal cancer (LMCC) subgroup (blue line, n = 58), and RAS‐mutant subgroup (green line, n = 11) by Luminex Assay. (a) ; . (b) ; .
A waterfall plot showing the maximal reduction rate of each case is shown in Figure 4. Among 80 RAS wild‐type cases, whose reduction rates were able to be determined, 73.7% (42/57) of the RAS wild‐type LMCC cases showed reduction of tumor size, whereas 78.3% (18/23) of the RAS wild‐type HMCC cases showed increases in size.
Figure 4.
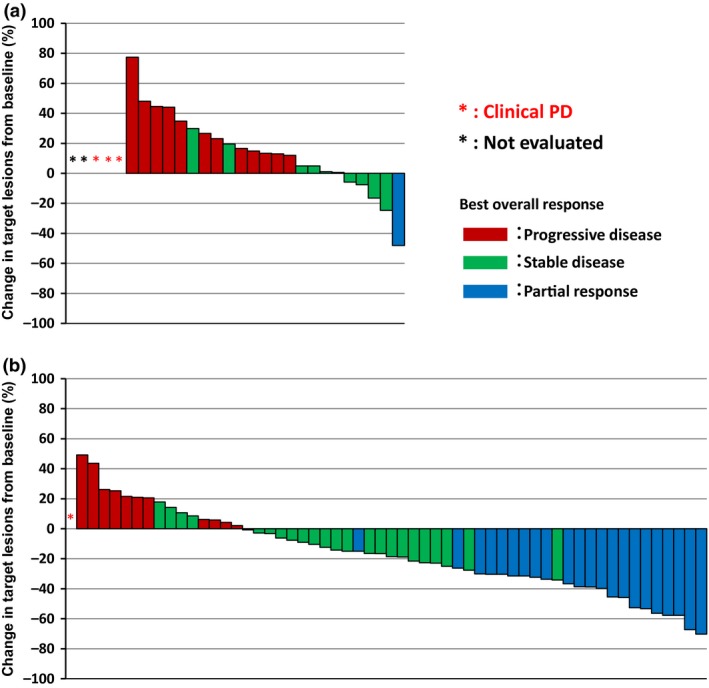
Waterfall plot of the maximal reduction rate in colorectal cancer target lesions for each case. Waterfall plots include 28 highly methylated colorectal cancer (HMCC) cases without RAS mutations (a) and 58 low methylated colorectal cancer (LMCC) cases without RAS mutations (b). Each column represents a case, and the color indicates best overall response for anti‐epidermal growth factor receptor treatment. Blue, partial response; green, stable disease; red, progressive disease (PD).
Predictive power of DNA methylation status for PFS and OS
To identify factors associated with differences in PFS and OS, six parameters (methylation status, infrequent‐RAS mutation status, primary site, number of previous regimens, type of anti‐EGFR treatment, and BRAF mutation status) were evaluated using Cox regression analyses. In both univariate and multivariate analyses, the methylation status was the strongest predictive factor of PFS (HR = 0.23; 95% CI, 0.14–0.38; P < 0.001, HR = 0.23; 95% CI, 0.13–0.41; P < 0.001, respectively, Table 2) and OS (HR = 0.25; 95% CI, 0.12–0.50; P < 0.001, HR = 0.35; 95% CI, 0.16–0.74; P = 0.006, respectively, Table 2).
Table 2.
Cox regression analysis for clinical outcome of anti‐epidermal growth factor receptor (EGFR) therapy for colorectal cancer
| Variable | Univariate | Multivariate | ||||||
|---|---|---|---|---|---|---|---|---|
| PFS | OS | PFS | OS | |||||
| HR (95% CI) | P‐value† | HR (95% CI) | P‐value† | HR (95% CI) | P‐value† | HR (95% CI) | P‐value† | |
|
Methylation status (LMCC versus HMCC) |
0.23 | <0.001 | 0.25 | <0.001 | 0.23 | <0.001 | 0.35 | 0.006 |
| (0.14–0.38) | (0.12–0.50) | (0.13–0.41) | (0.16–0.74) | |||||
|
Infrequent‐RAS mutation status (wild versus mutant) |
0.54 | 0.100 | 0.32 | 0.010 | 0.84 | 0.64 | 0.37 | 0.030 |
| (0.29–1.13) | (0.15–0.76) | (0.44–1.80) | (0.17–0.91) | |||||
|
Primary site (distal versus proximal) |
0.83 | 0.430 | 0.55 | 0.080 | 1.08 | 0.78 | 0.64 | 0.210 |
| (0.53–1.34) | (0.30–1.08) | (0.65–1.83) | (0.33–1.31) | |||||
|
No. of previous regimens (one or less versus two or more) |
0.73 | 0.290 | 0.44 | 0.080 | 0.85 | 0.61 | 0.48 | 0.150 |
| (0.38–1.29) | (0.13–1.10) | (0.43–1.54) | (0.14–1.26) | |||||
|
Type of anti‐EGFR treatment (combination with irinotecan versus monotherapy) |
0.53 | 0.010 | 0.64 | 0.270 | 0.57 | 0.03 | 0.59 | 0.220 |
| (0.34–0.87) | (0.31–1.45) | (0.35–0.96) | (0.28–1.39) | |||||
|
BRAF mutation status (wild versus mutant) |
0.96 | 0.940 | 0.94 | 0.930 | 0.97 | 0.95 | 0.88 | 0.870 |
| (0.42–2.82) | (0.28–5.80) | (0.41–2.89) | (0.23–5.51) | |||||
†χ2 test. CI, confidence interval; HMCC, highly methylated colorectal cancer; HR, hazard ratio; LMCC, low methylated colorectal cancer; OS, overall survival; PFS, progression‐free survival.
Predictive value of DNA methylation status
We compared results of classification by our method with those of classification by methylation markers used in two previous studies. First, 97 cases were divided into three subgroups according to the markers proposed by Yagi et al. The number of HME, IME, and LME cases was 7, 16, and 74, respectively. All HME and IME cases were classified as HMCC by our method. We then classified 97 cases by the markers reported by Weisenberger et al. As a result, 18 cases were considered CIMP‐positive and 79 CIMP‐negative. Except for one, all CIMP‐positive cases were included in the HMCC subgroup (Fig. S2).
To clarify the correlation between classification by these markers and the clinical outcome of anti‐EFGR treatment, we compared clinical outcomes between subgroups classified by the reported methylation markers. As a result, there was clear correlation between methylation status and the clinical outcome of anti‐EGFR treatment (Fig. S3), and our method showed the strongest correlation with the clinical outcome.
Selection of most reliable markers
To clarify the reproducibility of the classification of each cohort, we examined the prediction accuracy of the model made in each cohort (see Materials and Methods) when we classified the cohort by a model made in the other cohort. We obtained 1744 common probes out of 3163 and 2577 probes, which were used in each cohort. Then we extracted the efficient 140 and 128 probes in the first cohort and second cohort, respectively, and classified the second cohort and the first cohort using either of these probes. The correct prediction rates were 94.2% (three HMCC cases were judged incorrectly) and 97.8% (one LMCC case was judged incorrectly). To further select markers that were highly reliable for prediction of anti‐EGFR treatment sensitivity, we used the common 24 probes among the 140 and 128 probes (Table S7). By the model made in the first and second cohorts using the 24 probes, the correct answer rates were 98.1% (only one HMCC case was judged incorrectly) and 100%, respectively.
Discussion
We found that metastatic CRC without KRAS codon 12 or 13 mutations could be clearly classified into two subgroups by genome‐wide DNA methylation analysis: one‐third to the HMCC subgroup and two‐thirds to the LMCC subgroup. The percentage of HMCC cases was higher than that reported in previous studies (~25%), as the genome‐wide analysis adopted in this study was able to detect highly methylated cases maximally (Fig. S2). We also found that genome‐wide DNA methylation status was clearly correlated with the clinical outcome of anti‐EGFR treatment and was an independent predictive factor of PFS and OS after anti‐EGFR treatment. According to any of the distinct sets of methylation markers, the highly methylated subgroups showed inferior clinical outcomes. Importantly, as the method that is high in detectability of the DNA methylation status, predictability of the effect of treatment of the anti‐EGFR therapy is high, strongly supporting this idea. In previous studies, RRs of these tumors with KRAS codon 12 or 13 mutation following monotherapy and combination therapy were 0–1.2% and 0–13%, respectively.24, 25, 26, 27 The median PFS of anti‐EGFR monotherapy and combination therapy with irinotecan for cases with KRAS codon 12 or 13 mutations was 52–54 and 48‐150 days, respectively.24, 25, 26, 27 Because 73.2% of cases in our cohort received anti‐EGFR antibody with irinotecan (Table S3), it is rational that the clinical outcome of anti‐EGFR treatment for the HMCC subgroup (RR, 6.1%; PFS, 70 days) was similar to that for CRC cases with a KRAS codon 12 or 13 mutation.
In addition to KRAS codon 12 and 13 mutations, mutations in several other codons of KRAS and NRAS (called the infrequent‐RAS mutation) were recently established as unresponsiveness predictors of anti‐EGFR treatment.28, 29 When the analysis was limited to RAS wild‐type cases, clinical outcome of anti‐EGFR treatment for the HMCC subgroup was significantly inferior to that for the LMCC subgroup and was similar to that for CRC cases with a KRAS codon 12 or 13 mutation (RR, 3.7%; PFS, 70 days). These results strongly support that genome‐wide DNA methylation status is a predictive marker for anti‐EGFR treatment outcome and could eliminate non‐responders from cases without the KRAS codon 12 or 13 mutation, and also cases without the infrequent‐RAS mutation.
Using a method able to evaluate the genome‐wide DNA methylation status, patients who can expect benefits from anti‐EGFR treatment can be narrowed down further. Obviously, we need a method that is both simple and highly reliable to assess the genome‐wide DNA methylation status for clinical use. The reproducibility of the classification in the two cohorts was thought to be actually high because the other cohort could be separated exactly when a model made in one cohort was used. Also, the probes that were important to classify both cohorts were thought to be comprised of probes with a similar methylation profile. From these results, the finally selected 24 probes can classify colorectal cancer into HMCC or LMCC subgroups with very high precision, and the transition to the detection system that is simpler and easier for clinical application is possible. We are now planning a prospective study and prospective–retrospective analysis to confirm our findings.
We did not investigate why susceptibility to the anti‐EGFR antibody varied according to the DNA methylation status. ALX4 had been identified as a chemosensitive methylation candidates to cetuximab regimens by in vitro chemosensitivity assay,30 and six probes corresponding to ALX4 were included in 1744 common probes out of 3163 and 2577 probes, which were used in each cohort. According to the epidemiological and pathological findings, biological features differ depending on the DNA methylation status.31 Therefore, more precise mechanisms should be elucidated by further molecular analyses including whole exome sequencing and comprehensive expression analyses.
Recently, Garrido‐Laguna et al.32 reported that decitabine, a hypomethylating agent, in combination with panitumumab was effective in some patients previously treated with cetuximab. The susceptibility for the anti‐EGFR antibody of HMCC patients may also improve by adding these agents. The methylation status and hypomethylating agents could change the treatment strategy of anti‐EGFR therapy for metastatic CRC without RAS mutation.
This study is the first to indicate that genome‐wide DNA methylation status is an epigenetic marker and independent predictive factor of anti‐EGFR treatment outcomes in patients with metastatic CRC without the KRAS codon 12 or 13 mutation.
Disclosure Statement
Dr. Ishioka received grant support from Chugai, Taiho, Bristol‐Myers Squibb, Daiichi‐Sankyo, Merck Serono, Yakult, Ono, and Novartis. All remaining authors have no conflicts of interest.
Supporting information
Data S1. Detailed methods.
Data S2. Supplementary references.
Fig. S1. Kaplan–Meier curves for progression‐free survival and overall survival after anti‐epidermal growth factor receptor treatment in third‐line patients in the unified cohort according to genome‐wide DNA methylation status.
Fig. S2. Mutual relations of the cases classified in the highly methylated group by each method.
Fig. S3. Kaplan–Meier curves for progression‐free survival after anti‐epidermal growth factor receptor treatment in the unified cohort according to DNA methylation status.
Table S1. Polymerase chain reaction and sequencing primers for KRAS and BRAF mutation analyses.
Table S2. Polymerase chain reaction conditions for KRAS and BRAF mutation analyses.
Table S3. Baseline characteristics of the study patients in the overall population and two independent cohorts.
Table S4. Tumor response of highly methylated colorectal cancer (HMCC) and low methylated colorectal cancer (LMCC) subgroups in two independent cohorts.
Table S5. Tumor response of highly methylated colorectal cancer (HMCC) and low methylated colorectal cancer (LMCC) subgroups in third‐line patients.
Table S6. Tumor response of highly methylated colorectal cancer (HMCC), low methylated colorectal cancer (LMCC), and RAS‐mutant subgroups.
Table S7. Annotation list of selected 24 probes.
Acknowledgments
This study was supported by a grant from the Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT) by the Ministry of Education, Culture, Sports, Science and Technology of Japan. Dr. Ouchi was a postgraduate student of the Medical Oncologist Course, Tohoku Cancer Professional Training Promotion Plan. We thank our patients and medical and technical staff, especially Ms. Hiromi Nakano and Ms. Hiroko Meguro.
Cancer Sci 106 (2015) 1722–1729
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61: 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Van Cutsem E, Oliveira J, Group EGW. Advanced colorectal cancer: ESMO clinical recommendations for diagnosis, treatment and follow‐up. Ann Oncol 2009; 20(Suppl 4): 61–3. [DOI] [PubMed] [Google Scholar]
- 3. Douillard JY, Cunningham D, Roth AD et al Irinotecan combined with fluorouracil compared with fluorouracil alone as first‐line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet 2000; 355: 1041–7. [DOI] [PubMed] [Google Scholar]
- 4. Tournigand C, André T, Achille E et al FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol 2004; 22: 229–37. [DOI] [PubMed] [Google Scholar]
- 5. de Gramont A, Figer A, Seymour M et al Leucovorin and fluorouracil with or without oxaliplatin as first‐line treatment in advanced colorectal cancer. J Clin Oncol 2000; 18: 2938–47. [DOI] [PubMed] [Google Scholar]
- 6. Goldberg RM, Sargent DJ, Morton RF et al A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J Clin Oncol 2004; 22: 23–30. [DOI] [PubMed] [Google Scholar]
- 7. Hurwitz H, Fehrenbacher L, Novotny W et al Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004; 350: 2335–42. [DOI] [PubMed] [Google Scholar]
- 8. Saltz LB, Clarke S, Díaz‐Rubio E et al Bevacizumab in combination with oxaliplatin‐based chemotherapy as first‐line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 2008; 26: 2013–9. [DOI] [PubMed] [Google Scholar]
- 9. Cunningham D, Humblet Y, Siena S et al Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan‐refractory metastatic colorectal cancer. N Engl J Med 2004; 351: 337–45. [DOI] [PubMed] [Google Scholar]
- 10. Van Cutsem E, Peeters M, Siena S et al Open‐label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy‐refractory metastatic colorectal cancer. J Clin Oncol 2007; 25: 1658–64. [DOI] [PubMed] [Google Scholar]
- 11. Heinemann V, von Weikersthal LF, Decker T et al FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first‐line treatment for patients with metastatic colorectal cancer (FIRE‐3): a randomised, open‐label, phase 3 trial. Lancet Oncol 2014; 15: 1065–75. [DOI] [PubMed] [Google Scholar]
- 12. Di Nicolantonio F, Martini M, Molinari F et al Wild‐type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008; 26: 5705–12. [DOI] [PubMed] [Google Scholar]
- 13. De Roock W, Claes B, Bernasconi D et al Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy‐refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010; 11: 753–62. [DOI] [PubMed] [Google Scholar]
- 14. Lièvre A, Bachet JB, Boige V et al KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 2008; 26: 374–9. [DOI] [PubMed] [Google Scholar]
- 15. Van Cutsem E, Köhne CH, Hitre E et al Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 2009; 360: 1408–17. [DOI] [PubMed] [Google Scholar]
- 16. Toyota M, Ahuja N, Ohe‐Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999; 96: 8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ogino S, Cantor M, Kawasaki T et al CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 2006; 55: 1000–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weisenberger DJ, Siegmund KD, Campan M et al CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–93. [DOI] [PubMed] [Google Scholar]
- 19. Weisenberger DJ, Trinh BN, Campan M et al DNA methylation analysis by digital bisulfite genomic sequencing and digital MethyLight. Nucleic Acids Res 2008; 36: 4689–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Samowitz WS, Albertsen H, Sweeney C et al Association of smoking, CpG island methylator phenotype, and V600E BRAF mutations in colon cancer. J Natl Cancer Inst 2006; 98: 1731–8. [DOI] [PubMed] [Google Scholar]
- 21. Torlakovic EE, Gomez JD, Driman DK et al Sessile serrated adenoma (SSA) vs. traditional serrated adenoma (TSA). Am J Surg Pathol 2008; 32: 21–9. [DOI] [PubMed] [Google Scholar]
- 22. Yagi K, Akagi K, Hayashi H et al Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res 2010; 16: 21–33. [DOI] [PubMed] [Google Scholar]
- 23. Dahlin AM, Palmqvist R, Henriksson ML et al The role of the CpG island methylator phenotype in colorectal cancer prognosis depends on microsatellite instability screening status. Clin Cancer Res 2010; 16: 1845–55. [DOI] [PubMed] [Google Scholar]
- 24. Karapetis CS, Khambata‐Ford S, Jonker DJ et al K‐ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008; 359: 1757–65. [DOI] [PubMed] [Google Scholar]
- 25. Amado RG, Wolf M, Peeters M et al Wild‐type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 2008; 26: 1626–34. [DOI] [PubMed] [Google Scholar]
- 26. Peeters M, Price TJ, Cervantes A et al Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second‐line treatment in patients with metastatic colorectal cancer. J Clin Oncol 2010; 28: 4706–13. [DOI] [PubMed] [Google Scholar]
- 27. Soeda H, Shimodaira H, Gamoh M et al Phase II trial of cetuximab plus irinotecan for oxaliplatin‐ and irinotecan‐based chemotherapy‐refractory patients with advanced and/or metastatic colorectal cancer: evaluation of efficacy and safety based on KRAS mutation status (T‐CORE0801). Oncology 2014; 87: 7–20. [DOI] [PubMed] [Google Scholar]
- 28. Douillard JY, Oliner KS, Siena S et al Panitumumab‐FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013; 369: 1023–34. [DOI] [PubMed] [Google Scholar]
- 29. Schwartzberg LS, Rivera F, Karthaus M et al PEAK: a randomized, multicenter phase II study of panitumumab plus modified fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) or bevacizumab plus mFOLFOX6 in patients with previously untreated, unresectable, wild‐type KRAS Exon 2 metastatic colorectal cancer. J Clin Oncol 2014; 32: 2240–7. [DOI] [PubMed] [Google Scholar]
- 30. Kim JC, Lee HC, Cho DH et al Genome‐wide identification of possible methylation markers chemosensitive to targeted regimens in colorectal cancers. J Cancer Res Clin Oncol 2011; 137: 1571–80. [DOI] [PubMed] [Google Scholar]
- 31. Hinoue T, Weisenberger DJ, Lange CP et al Genome‐scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012; 22: 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Garrido‐Laguna I, McGregor KA, Wade M et al A phase I/II study of decitabine in combination with panitumumab in patients with wild‐type (wt) KRAS metastatic colorectal cancer. Invest New Drugs 2013; 31: 1257–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Detailed methods.
Data S2. Supplementary references.
Fig. S1. Kaplan–Meier curves for progression‐free survival and overall survival after anti‐epidermal growth factor receptor treatment in third‐line patients in the unified cohort according to genome‐wide DNA methylation status.
Fig. S2. Mutual relations of the cases classified in the highly methylated group by each method.
Fig. S3. Kaplan–Meier curves for progression‐free survival after anti‐epidermal growth factor receptor treatment in the unified cohort according to DNA methylation status.
Table S1. Polymerase chain reaction and sequencing primers for KRAS and BRAF mutation analyses.
Table S2. Polymerase chain reaction conditions for KRAS and BRAF mutation analyses.
Table S3. Baseline characteristics of the study patients in the overall population and two independent cohorts.
Table S4. Tumor response of highly methylated colorectal cancer (HMCC) and low methylated colorectal cancer (LMCC) subgroups in two independent cohorts.
Table S5. Tumor response of highly methylated colorectal cancer (HMCC) and low methylated colorectal cancer (LMCC) subgroups in third‐line patients.
Table S6. Tumor response of highly methylated colorectal cancer (HMCC), low methylated colorectal cancer (LMCC), and RAS‐mutant subgroups.
Table S7. Annotation list of selected 24 probes.