

Abstract
Angiogenesis is a prerequisite of tumor growth and metastasis and, thus, anti‐angiogenesis treatment has become an important part of cancer therapy. A 15‐amino acid peptide of the fibrinogen α chain, fibrinostatin, was previously found in serum samples of gastric cancer patients. Herein we demonstrated that fibrinostatin has anti‐angiogenesis activity in several angiogenesis models and it reduces tumor growth in mouse xenografts and allografts. Increased tumor necrosis and reduced microvessel density in tumors were observed in mouse xenograft models. Fibrinostatin inhibited proliferation and induced apoptosis in HUVEC, but not in cancer cells. In addition, fibrinostatin specifically entered HUVEC. Fibrinostatin also prevented migration, adhesion and tubule formation of HUVEC in vitro. A single‐dose acute toxicity testing and a repeated‐dose chronic toxicity study in the mouse, rat and monkey indicated that fibrinostatin had a wide margin of safety. Taken together, fibrinostatin shows promise as a potential anti‐angiogenesis therapeutic agent.
Keywords: Anti‐angiogenesis, fibrinogen, fibrinostatin, peptide, tumor inhibition
Angiogenesis involves the degradation of components of the extracellular matrix and then migration, proliferation and differentiation of endothelial cells from pre‐existing blood vessels to form sprouts and eventually new capillaries,1, 2, 3, 4 whereas hemostasis maintains the blood flow by regulating platelet adherence and fibrin deposition. Both systems normally appear quiescent, but become activated in response to such injury as the leaky blood vessels in solid tumors.5
Cancer has long been recognized to be associated with hypercoagulation as a result of imbalanced coagulation (fibrin deposition) and fibrinolysis (fibrin degradation). Fibrinogen, a multimeric molecule containing two of three non‐identical chains, the α, β and γ chains, is the soluble circulating precursor of fibrin. It accumulates around leaky blood vessels in solid tumors and at the host–tumor interface, which contributes to the tumor growth and spreading.6, 7 Fibrinogen can be cleaved by plasmin and thrombin and its proteolytic products can either promote or inhibit angiogenesis. Plasmin cleavage generates the carboxyl termini of the paired α, β and γ chains of fibrinogen and the central domain of fibrinogen (fibrinogen E‐fragment [FgnE]). FgnE is a potent anti‐angiogenic factor capable of inhibiting endothelial cell migration and tubule formation.8, 9 When cleaved by thrombin, fibrinogen is converted into fibrin monomers after the removal of the amino termini of both α and β chains, yielding fibrinopeptides A (FpA) and B (FpB), respectively. The fibrin‐E fragment (FnE) of fibrin degraded by plasmin was reported to promote angiogenesis. The main difference between FnE and FgnE lies in its loss of FpA.
We previously found a 15‐amino acid (aa) fragment peptide in the sera of gastric cancer patients by surface‐enhanced laser desorption/ionization and time‐of‐flight mass spectrometry (SELDI TOF‐MS). This 15‐mer peptide (DSGEGDFLAEGGGVR) was identified as the amino terminal fragment (2‐16aa) of the fibrinogen α chain, which was named as fibrinostatin.10 Compared with FpA, fibrinostatin losses the first alanine from the amino terminus of FpA, which could be a result of the cleavage by exoprotease, because differential exoprotease activities acting on the ex vivo coagulation and complement–degradation pathways could generate cancer type‐specific serum peptides.11 We and other groups have found that fibrinostatin is differentially present in the sera of patients in a variety of cancers: higher in gastric, ovarian, hepatocellular and urothelial cancers,12, 13, 14, 15 lower in prostate, bladder and thyroid cancers,11, 16 but no difference in breast cancer patients when compared with healthy controls.11 Based on these findings, we sought to evaluate the role of fibrinostatin in tumor development. Our results support the notion that fibrinostatin is a potential antagonist of tumor angiogenesis.
Materials and Methods
Cells and animals
Human umbilical vein endothelial cells were isolated and cultured as previously described.17 Human tumor cell lines of BGC‐823 (gastric carcinoma), PG (lung carcinoma) and HT‐29 (colon carcinoma) were cultured in RPMI‐1640 with 10% FBS in 5% CO2 at 37°C. HEK‐293 cells, fibroblasts (WI‐38) cells and human microvascular endothelial cells were maintained in DMEM (10% FBS) at 37°C in 5% CO2.
BALB/c nude mice were obtained from the Animal Center of the Chinese Academy of Medical Science (Beijing, China). Fertilized eggs of white Leghorn chickens were supplied by the Chicken Center of the Chinese Agriculture University (Beijing, China). Wistar rats and SD rats were obtained from Vital River Laboratories (Beijing, China).
Synthetic peptides and reagents
Fibrinostatin and truncated peptides, FITC‐conjugated fibrinostatin peptide and FITC‐conjugated TAT peptide were synthesized by ChinaTech Peptide (Suzhou, China). The amino acid sequences are: DSGEGDFLAEGGGVR (fibrinostatin) and GRKKRRQRRRGY (TAT).18
Xenograft and H22 allograft models
For the present study, 2 × 106 tumor cells resuspended in PBS were injected subcutaneously into the backs of mice. When tumors reached 0.1 cm3, the mice were intravenously injected with PBS control or 1, 2.5 or 5 mg/kg fibrinostatin every day for 16 days. H22 allograft model was generated as previously described.19 The mice were randomly divided into 10 groups and administered with different treatments for 7 days. Mice were sacrificed under sodium pentobarbital anesthesia, and tumor weights were measured. Tumor volume was calculated according to the equation: tumor volume = width2 × length/2.
Immunohistochemistry and microvascular density analysis
Paraffin‐embedded sections were dewaxed using standard histological procedures. To stain the microvasculature, the sections were incubated with an antibody against CD31 (ab59251; Abcam, Cambridge, UK) overnight at 4°C in a humidified chamber and bound antibody was detected using the EnVision Kit (DAKO, Carpinteria, CA, USA). In each group, differences in vascularity and number of CD31‐positive structures per microscopic field were determined for each section at ×200 magnification in a blinded fashion.20 Mean ± SD MVD/field was calculated based on four fields per section from six xenografts.
Chick chorioallantoic membrane assay
Fertilized eggs of white Leghorn chickens were incubated at 37°C in 65–70% humidity. The 6‐day‐old embryos with intact yolks were placed in a bowl and incubated at 37°C.21 A filter disk of 6‐mm diameter containing 10 μL PBS or fibrinostatin (2, 5 or 10 μg/μL) was placed onto the chick chorioallantoic membrane (CAM) of the embryos. After 48 h of incubation, vessel formation was observed under a stereomicroscope.
Zebrafish embryos assay
Transgenic zebrafish assessment was performed by Hunter Biotechnology Corporation (Hangzhou, China). Drugs were injected into the yolk sac of zebrafish 2 days after fertilization by a microinjection method. After being incubated for another 72 h, 10 zebrafish of each group were randomly selected, and fluorescence of the subintestinal vein vessels (SIV) was detected and photographed using a multi‐purpose zoom microscope system (AZ 100, Nikon). Image analysis was performed by calculating the SIV area using Nikon NIS‐Elements D 3.10 Advanced image processing software.
Cell proliferation assay
The MTT (3‐[4,5‐Dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) assay kit from Promega (Madison, WI, USA) was used to assess proliferation of HUVEC and cancer cells, as described previously.22
Flow cytometry analysis
Cells were treated with PBS or 25 μg/mL fibrinostatin for 48 h, then washed twice and fixed in 0.04% methanol at −20°C for 16 h. After staining with the propidium iodide (PI) solution, samples were then analyzed using a FACSAria flow cytometry system (BD Biosciences, San Jose, CA, USA).
DNA ladder assay
Cells were treated with PBS or fibrinostatin (25 μg/mL) for 72 h, then washed twice with ice‐cold TBS (pH 7.6) and lysed with lysis buffer (10 mM Tris‐HCl pH 8.0, 5 mM EDTA, 1% Triton‐100, 10 mM NaCl and 100 μg/mL proteinase K) for 10 min. Then 300 μL of saturated hydroxybenzene and chloroform was added for genomic DNA extraction by centrifugation of 12 000 g for 10 min, and the supernatant was collected and two volumes of ethanol with 10% 3 mol/L NaAc were added to precipitate DNA. 20 μL H2O with 1 mg/mL RNase A was used to dissolve DNA, and the DNA Ladder were analyzed in 1.2% agarose gel.
TUNEL assay
Cells were seeded on glass coverslips in 24‐well plates. After treatment with fibrinistatin (25 μg/mL) for 72 h, TUNEL assay was performed with a kit from Roche according to the manufacturer's instructions and observed under a TCS‐SP2 confocal microscopy (Leica Microsystems, Heidelberg, Germany).
Fluorescence visualization for entry of peptides
To assay the entries of FITC‐labeled peptides, HUVEC, BGC‐823, PG and HT‐29 cells were incubated with FITC‐conjugated fibrinostatin (25 μg/mL) or TAT (25 μg/mL) for 3 h. The cells were then fixed with 4% paraformldehyde and permeabilized with 0.1% NP‐40 in PBS. The dye 4′,6‐diamidino‐2‐phenylindole (DAPI, 5 ng/mL) was added to stain nuclei, washed three times with PBS, and observed under a TCS‐SP2 confocal microscopy (Leica Microsystems, Heidelberg, Germany).
Cell migration assay
HUVEC (5 × 104 cells) pretreated with PBS control or 25 μg/mL fibrinostatin for 24 h were plated in the upper chamber of Transwell inserts with 8‐μm pores, and the lower chamber contained RPMI‐1640, 10% FBS (to control the HUVEC growth) with PBS or fibrinostatin (25 μg/mL). The cells were allowed to migrate for 24 h at 37°C. The mean number of cells per field that migrated across the filter was calculated based on the cell number counted in four fields at ×200 magnification per insert. Each condition was performed in triplicate.
Cell adhesion assay
Six‐well plates were coated with 200 μL/well of Matrigel (1 mg/mL; BD Biosciences) overnight. The plates were blocked with 500 μL/well of 100 μg/mL BSA for 2 h. Then 1 mL of the HUVEC suspension (2 × 105 cells/mL) pretreated with PBS control or 25 μg/mL fibrinostatin for 24 h, was added to each well and incubated for 2 h at 37°C. After washing twice with PBS, the number of attached cells was counted after methylene blue staining at ×200 magnification. Mean ± SD attached cells was calculated based on four fields per insert. Each condition was performed in triplicate.
Tubule formation assay
A total of 200 μL Matrigel (1 mg/mL) was added to each well of a 24‐well plate and allowed to polymerized for 20 min at 37°C. A suspension of HUVEC (5 × 104 cells) was plated on the Matrigel‐coated wells and then treated with PBS or fibrinostatin (10, 20 and 50 μg/mL) in RPMI‐1640 with 10% FBS and 10 ng/mL VEGF for 18 h in 5% CO2 at 37°C. The network structure of tubules was observed using a CK2 Olympus Microscope (Olympus Corporation, Tokyo, Japan). The assays were performed in triplicate.
Rat aortic ring assay
The rat aortic ring assay was performed as described previously, with slight modifications.23 Six week‐old Sprague–Dawley rats were sacrificed by CO2 inhalation and their thoracic aortas were dissected and sliced into 1‐mm thick rings. The rings were placed on a 24‐well plate and embedded in 100 μL Matrigel followed by 30 min incubation at 37°C. The wells were then overlaid with 300 μL of RPMI‐1640 containing 10% FBS with PBS control or fibrinostatin of 10, 20 and 50 μg/mL. The rings were incubated for 5 days at 37°C and observed using a CK2 Olympus microscope (×100 magnification).
Western blot
Cells lysed in RIPA buffer were subjected to western blot analysis following the standard procedure. Primary antibodies anti‐Bax mAb (sc‐7480; Santa Cruz, CA, USA), anti‐p53 mAb (sc‐126; Santa Cruz), anti‐VEGFR1 mAb (#1303‐1; Epitomics, Burlingame, CA, USA), anti‐phospho‐VEGFR1 (Tyr1213) rabbit antibody (#07‐758; Millipore, Billerica, MA, USA), anti‐VEGFR2 mAb (#2479; Cell Signaling Technology), anti‐phospho‐VEGFR2 (#2478; Cell Signaling technology, Danvers, MA, USA), anti‐phospho‐ERK1/2 mAb (sc‐7383; Santa Cruz) and anti‐Phospho‐Akt (Ser473) (#4060; Cell Signaling Technology) were used here.
Statistical analysis
Statistical analysis software package SPSS 12.0 (SPSS, Chicago, IL) was used to perform an anova test and the unpaired Student's t‐test. P‐values < 0.05 were considered statistically significant.
Results
Fibrinostatin inhibits in vivo tumor growth but does no harm to the hosts
Fibrinostatin was first found in the serum of gastric cancer patients in our previous study by SELDI TOF‐MS analysis.10 To investigate the function(s) of fibrinostatin in vivo, we synthesized the 15‐amino acid peptide (fibrinostatin) and utilized three tumor xenograft models (gastric cancer, lung cancer and colorectal cancer). Chemotherapeutic agent cyclophosphamide (CTX) was used as a positive control. We found that fibrinostatin significantly inhibited xenograft tumor growth in mice (Fig. 1a–c). For the gastric cancer BGC‐823 in mice, the mean tumor volume and weight were 0.515 ± 0.228 cm3 and 0.61 ± 0.24 g in the fibrinostatin‐treated group, compared with 1.932 ± 0.652 cm3 and 1.92 ± 0.40 g in the control group (P < 0.05). The maximal inhibition rates of tumor growth and tumor volume by fibrinostatin were 68.3% and 36%, respectively. The inhibition of fibrinostatin acts in a dose‐dependent manner (Fig. 1a). Similar inhibition was observed in the colon (HT‐29) and lung (PG) cancer in mice (Fig. 1b,c). We also conducted a toxicology study in pre‐clinical animal models according to Good Laboratory Practice (GLP) standards and found no observable side effects (data not shown).
Figure 1.

Fibrinostatin inhibits tumor growth in vivo in mouse xenograft models. Fibrinostatin inhibited tumor growth in BGC‐823 gastric cancer cells (a), HT‐29 colon cancer cells (b) and PG lung cancer cells (c) bearing BALB/c nude mice. Cyclophosphamide (CTX), a chemotherapuetic agent, was used as a positive control. Bars represent mean ± SD. *P < 0.05; **P < 0.01; *** P < 0.001.
Fibrinostatin induces necrosis and decreases microvascular density in tumors
To explore the effects of fibrinostatin on tumor growth and tumor angiogenesis, we dissected and analyzed all the xenografts from the mice by histological H&E staining. We observed necrotic areas in the fibrinostatin‐treated tumors, in which more lymphocyte and erythrocyte exudation and some necrotic tumor cells were identified, with significantly less areas of necrosis in the control tumors (Fig. 2a). We then assessed the microvascular density (MVD) by immunostaining endothelial cells with an anti‐CD31 antibody and calculated MVD as a percentage of tumor section area using the Chalkley grid counting method.24 We found that the MVD in the fibrinostatin‐treated group was significantly decreased compared with the control group (P < 0.05) (Fig. 2b). These results suggest that the fibrinostatin's anti‐tumor activity is associated with inhibition of tumor angiogenesis.
Figure 2.
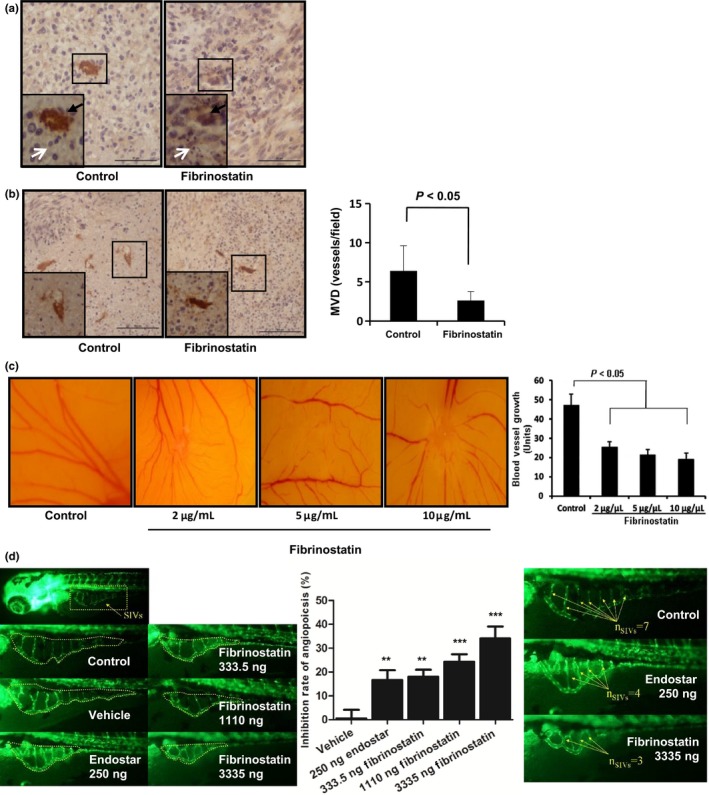
Fibrinostatin inhibits angiogenesis in vivo. (a) Tumor sections from BGC‐823 xenografts were immunostained with anti‐CD31 antibody. Black arrowheads point to the stained vessels. White arrowheads point to tumor cells (left) and necrotic tumor cells (right). Scale bar = 50 μm (b) Tumor sections from BGC‐823 xenografts immunostained with anti‐CD31 antibody were used to examine the vessel number. (×200 magnification, left). Boxed regions show CD31‐positive structures magnified at ×400 in the insets. Mean ± SD microvascular density (MVD) per field was calculated (right). P < 0.05 (unpaired Student's t‐test). Scale bar = 100 μm. (c) Fibrinostatin inhibited angiogenesis in chick chorioallantoic membrane (CAM). (d) Fibrinostatin inhibited angiogenesis in transgenic zebrafish. *** P < 0.001; **P < 0.01; (Student's t‐test).
Fibrinostatin inhibits vessel formation in vivo
To test the direct effect of fibrinostatin on angiogenesis, we applied the chick chorioallantoic membrane assay for vessel formation. We found that the 4‐day treatment of fibrinostatin dramatically inhibited microvessel formation in a dose‐dependent manner, whereas the control group had normal vascularization (Fig. 2c).
Effects of fibrinostatin on angiogenesis in zebrafish
The anti‐angiogenic effect of fibrinostatin was further confirmed using a transgenic zebrafish model; zebrafish express enhanced green fluorescent protein (EGFP) in their endothelial cells. Observations demonstrated that both fibrinostatin and Endostar (as a positive control) inhibited the development of subintestinal vein vessels and had no tocixity to zebrafish. Fibrinostatin was administrated at 333.5, 1110 and 3335 ng and the inhibition rates of angiogenesis were 17.6 ± 3.0%, 23.9 ± 3.1% and 33.7 ± 5.0%, respectively (Fig. 2d left and middle panel). When micro‐injected with 3335 ng of fibrinostatin, the number of subintestinal vascular vessels was decreased to 3–4, which was less than that of the control group (7–8; Fig. 2d right panel).
Fibrinostatin inhibits endothelial cell migration, adhesion and tubule formation
The above results suggest that fibrinostatin may act on endothelial cells. To investigate this possibility, we isolated HUVEC from the vein of human umbilical cords and tested the effect of fibrinostatin on HUVEC. Migration and adhesion of vascular endothelial cells are critical steps for angiogenesis. We first determined the effect of fibrinostatin on the migration of HUVEC using a transwell chamber assay. We found that the migration of HUVEC treated with fibrinostatin was significantly reduced compared with that of the control cells (mean 89 ± 36 cells/chamber vs mean 265 ± 82 cells/chamber, P < 0.05; Fig. 3a).
Figure 3.
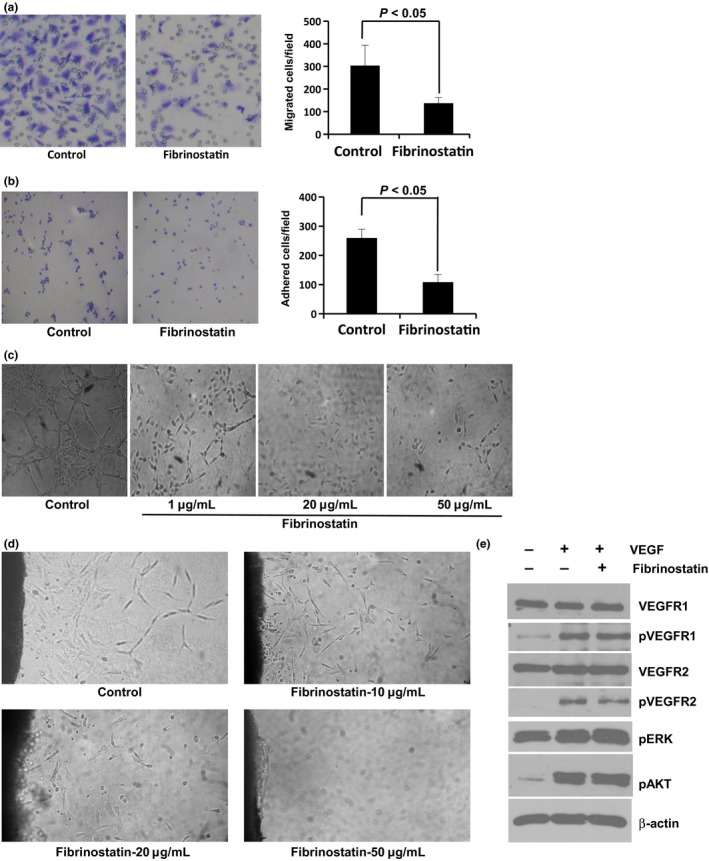
Fibrinostatin inhibits migration, adhesion and tubule formation of endothelial cells. The inhibitory effects of fibrinostatin on HUVEC migration (a), adhesion (b) and tubule formation (c). (d) Fibrinostatin inhibits the outgrowth of tubules from the rat aortic rings embedded in Matrigel. (e) Fibrinostatin does not interfere with VEGF signal pathway.
We then coated the 24‐well plate with Matrigel and examined the adhesion of HUVEC with or without treatment of fibrinostatin by counting the number of cells attached on the Matrigel membrane. Our data showed an approximate 50% reduction of HUVEC adhesion by fibrinostatin treatment compared with that of the control cells (mean 96 ± 39 vs mean 186 ± 72, P < 0.05; Fig. 3b).
Matrigel tubule formation assay was used as an in vitro model to study the effect of fibrinostatin. HUVEC were pretreated with fibrinostatin and angiogenesis was assessed after an 18‐h incubation period on Matrigel. Untreated cells formed a branching and anastomosing network of capillary‐like tubules with multi‐centric junctions, whereas the fibrinostatin‐treated cells had reduced branching points, tubule number and length. HUVEC treated with 50 μg/mL of fibrinostatin grew into complete single cells and no capillary‐like tubule formed (Fig. 3c).
The fibrinostatin‐induced inhibition of tubule formation was further confirmed by the rat aortic ring assay. After culturing the aortic rings for 5 days, the tubules grown from the rings were observed in the control group, but the tubule formation was inhibited in the fibrinostatin‐treated group in a dose‐dependent manner. At the dosage of 50 μg/mL fibrinostatin, complete abrogation of capillary growth was observed, whereas the tubule structure was formed even far from the ring in the control (Fig. 3d).
Fibrinostatin has no effect on vascular endothelial growth factor signal pathway
Because VEGF and its receptors play key roles in angiogenesis, we further tested whether fibrinostatin interferes with VEGF signal pathway. The phosphorylations of VEGFR1, VEGFR2, AKT and ERK were increased when treated with VEGF (10 ng/mL) for 15 min. However, pretreatment with fibrinostatin had no inhibitory effect (Fig. 3e). These results reflected that fibrinostatin's effect is independent of VEGF signal pathway.
Fibrinostatin inhibits the proliferation of endothelial cells but not of cancer cells, possibly by induction of apoptosis
To clarify the possible mechanisms of fibrinostatin on angiogenesis, we tested the effect of fibrinostatin on HUVEC proliferation by MTT assay. We found that fibrinostatin significantly inhibited HUVEC proliferation in a concentration‐dependent manner (10–50 μg/mL). At 20 μg/mL concentration, fibrinostatin almost reached the plateau of inhibition (P < 0.05) (Fig. 4a). In contrast, fibrinostatin had no distinguishable effects on proliferations of BGC‐823, PG and HT‐29 cancer cells. We also tested the inhibitory effect of fibrinostatin on HMEC, fibroblasts WI‐38 cells and HEK‐293 cells. We found that fibrinostatin did not inhibit the proliferation of fibroblasts WI‐38 cells or HEK‐293 cells, but could inhibit the growth of HMEC (Fig. S1). These results demonstrated that fibrinostatin has a specific inhibitory effect on endothelial cells, but not on cancerous or other untransformed cells. By flow cytometry analysis, we further found that in comparison with the control cells, fibrinostatin treatment resulted in almost 10‐fold increase in the percentage of sub‐G1 cell, but had no obvious effect on those of cancer cells (Fig. 4b), indicating that fibrinostatin inhibits HUVEC proliferation by inducing apoptosis. In addition, we found that significantly more DNA fragmentation was generated in fibrinostatin‐treated HUVEC than in controls by the DNA Ladder assay (Fig. 4c).
Figure 4.
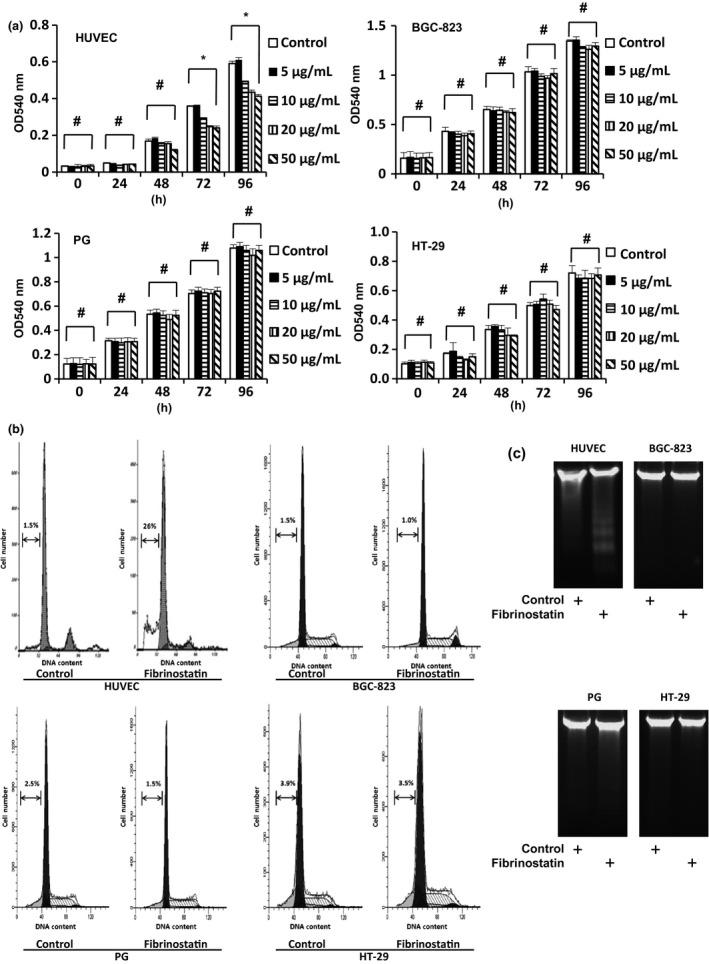
Fibrinostatin inhibits proliferation and induces apoptosis of endothelial cells specifically. (a) The effects of fibrinostatin on the proliferation of HUVEC, BGC‐823, PG and HT‐29 cells were determined with an MTT assay. *P < 0.05. #P > 0.05, anova. (b) Flow cytometry analysis of cells treated with PBS or fibrinostatin and stained with propidium iodide. The percentage of the sub‐G1 cells is presented. (c) DNA ladder analysis of cells treated with PBS or 25 μg/mL fibrinostatin.
TUNEL assay was further used to confirm these findings. Consistent with flow cytometry analysis and DNA ladder results, fibrinostatin treatment in HUVEC caused 20.51 ± 6.62% positive staining, while almost no positive staining was observed in cancer cell groups (Fig. 5a,b). By Western blot analysis, we found that fibrinostatin upregulated p53 and Bax levels in HUVEC (Fig. 5c).
Figure 5.
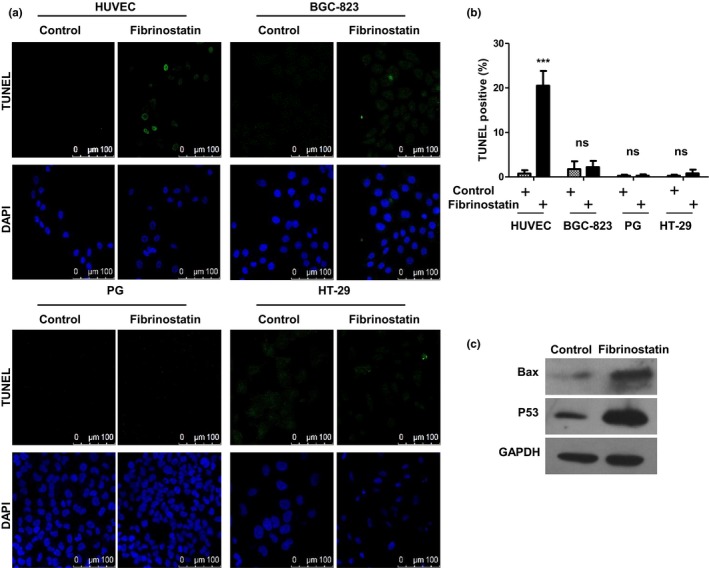
Fibrinostatin induces apoptosis of endothelial cells specifically. (a) TUNEL staining analysis of cells treated with PBS or 25 μg/mL fibrinostatin. Scale bar, 100 μm. (b) The percentage of positive TUNEL staining is presented; mean ± SD ***P < 0.001; ns, no significance (Student's t‐test). (c) Protein lysates were prepared from HUVEC treated with PBS or 25 μg/mL fibrinostatin for 48 h. The lysates were then subjected to western blot analysis of Bax, p53 and GAPDH expression.
Fibrinostatin enters endothelial cells but not cancer cells
To investigate why fibrinostatin inhibited the proliferation of endothelial cells specifically, we labeled fibrinostatin with a fluorescence dye FITC and stained the HUVEC and three cancer cell lines. To our surprise, we observed intracellular localization of FITC‐fibrinostatin in HUVEC cells, but not in cancer cells (Fig. 6). However, FITC‐labeled TAT peptide derived from the transactivator of transcription of human immunodeficiency virus, which is well known as a cell‐penetrating peptide,25 entered both HUVEC and the different cancer cells. These findings further support the endothelial cell‐specific functions of fibrinostatin, and the mechanism is under investigated.
Figure 6.
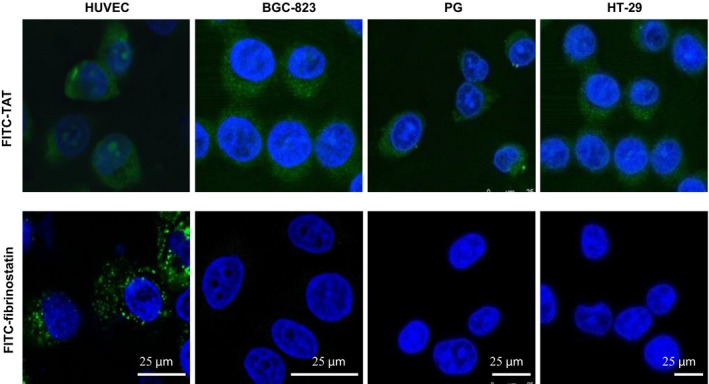
Fibrinostatin enters endothelial cell specifically. Cells were incubated with 25 μg/mL FITC‐conjugated fibrinostatin (green) or TAT (green, positive control), fixed, stained with DAPI (blue), and observed under a confocal microscope. Scale bar = 25 μm.
Identification of key inhibitory sequence of fibrinostatin
We further synthesized a series of deleted peptides of fibrinostatin and tested their inhibitory effect on tumor growth in a H22 allograft model to identify the key inhibitory motif. We found that three amino acid deletions in the amino or carboxyl terminus did not affect its anti‐tumor activity. However, more amino acid deletions in the N‐terminal or the C‐terminal strongly decreased its anti‐tumor activity (Fig. 7a). These results suggested that the six amino acids (DFLAEG) of fibrinostatin comprised the key inhibitory sequence. In addition, we performed tubule formation and migration assays to define the effect of these deleted peptides. Consistent with in vivo experiment results, peptides C3d, N3d, N6d and N5C4d also inhibit tubule formation and migration of HUVEC in vitro (Fig. 7b,c).
Figure 7.

Identification of key sequence of fibrinastatin. (a) The inhibitory effects of truncated peptides on tumor growth were assessed in murine H22 allografts. Cyclophosphamide (CTX) was used as a positive control. Inhibition rates were calculated compared with the control group. The inhibitory effects of fibrinostatin deleted peptides on HUVEC tubule formation (b) and migration (c) are presented; mean ± SD *P < 0.05; **P < 0.01; ***P < 0.001 compared with the control group (Student's t‐test).
Discussion
The hemostatic system has long been shown to regulate angiogenesis involving various hemostatic precursor proteins and cleavage of these proteins by proteolysis, which can generate novel cryptic fragments as either positive or negative factors to control the rate of angiogenesis.5, 26, 27 Some cryptic fragments can antagonize angiogenesis, such as Domain 5 of kininogen, fragment‐1 and ‐2 of prothrombin and cleaved AT‐III (serpin antithrombin).28, 29, 30 Fibrinostatin detected in the serum is the amino terminal fragment (2–16 aa) derived from the precusor fibrinogen α chain,10 differing from FpA in its loss of the first alanine at the amino terminus. Previous studies showed that a linear form of FpA was inactive in vitro, but the first 24 amino terminal amino acids (ADSGEGDFLAEGGGVRGPRVVERH) of the fibrinogen α chain (named alphastatin) tested as a synthetic peptide showed anti‐angiogenesis activity.31 Neither the FpA portion (1‐16aa) (ADSGEGDFLAEGGGVR) nor the 17–24 amino acids (GPRVVERH) of alphastatin could mimic the inhibitory effect of alphastatin.31 In alphastatin, a fragment of 11 amino acids (DFLAEGGGVRG) was identified to be a key sequence containing the activity (termed AHN419).32 In our study, we synthesized a series of deleted peptides of fibrinostatin and tested their inhibition of tumor growth in mice. We found that the six amino acids (DFLAEG) of fibrinostatin (DSGEGDFLAEGGGVR) comprised the key inhibitory sequence of fibrinostatin, which was included in AHN419. These results suggest that the addition of extra amino acids at either the amino or the carboxyl terminus of the key sequence (DFLAEG) may contribute to the conformation or stability of the core sequence that subsequently contributes to the biological activity of the key sequence. We speculate that a proper structure may be required for the interaction and stabilization of binding of fibrinostatin with its cellular target protein.
VEGF and its receptors play important roles in tumor angiogenesis.33 However, we did not find fibrinostatin to interfere with the VEGF signal pathway. Together with fluorescence visualization results, we propose that fibrinostatin binds to its target protein in cytoplasm and induces apoptosis. Increased p53 and Bax expressions were observed when treated with fibrinostatin, but the mechanism underlying these two molecules' upregulation and their contributions to fibrinostatin‐induced apoptosis remain to be determined. The target of fibrinostatin and the mechanism of fibrinostatin's specific effect on endothelial cells also deserve further investigation.
Taken together, our study provides evidence for development of a new therapeutic candidate, fibrinostatin, as an anti‐angiogenesis inhibitor targeting endothelial cells. Fibrinostatin blocks angiogenesis by inhibiting endothelial cell proliferation, adhesion, migration and tubule formation. We have also demonstrated the anti‐tumor effects of fibrinostatin on three cancer types (gastric cancer, lung cancer and colon cancer) in mouse xenograft models, supporting the notion that fibrinostatin may have a broad spectrum of anti‐tumor/anti‐angiogenesis activities. The toxicity studies demonstrated the good safety profiles in rats and monkeys. These findings support further investigation of fibrinostatin in preclinical and clinical studies as a drug candidate for treatment of a variety of cancers.
Disclosure Statement
The authors declare no competing financial interests.
Supporting information
Fig. S1. Fibrinostatin inhibits proliferation of HMEC, but not WI‐38 or HEK‐293.
Acknowledgments
We thank Dr Ullrich Schwertschlag (Tufts University), Dr Xiaojia Chang (Attogen Biocompany) and Dr Sonya Wei Song (Capital Medical Science University) for their critical comments on the manuscript. This research was supported by the Major Project on Drug Research and Development for the 12th Five‐Year Plan of China (2011ZX09102‐001‐16), the National 973 Program of China (2015CB553906), the National Natural Science Foundation of China (81301966, 30672418) and the New Teacher's Fund from the Chinese Ministry of Education (20130001120120).
Cancer Sci 106 (2015) 1596–1606
Funding Information
This research was supported by the Major Project on Drug Research and Development for the 12th Five‐Year Plan of China (2011ZX09102‐001‐16), the National 973 Program of China (2015CB553906), the National Natural Science Foundation of China (81301966, 30672418) and the New Teacher's Fund from Chinese Ministry of Education (20130001120120).
References
- 1. Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med 1995; 1: 27–31. [DOI] [PubMed] [Google Scholar]
- 2. Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000; 6: 389–95. [DOI] [PubMed] [Google Scholar]
- 3. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86: 353–64. [DOI] [PubMed] [Google Scholar]
- 4. Folkman J. Angiogenesis. Annu Rev Med 2006; 57: 1–18. [DOI] [PubMed] [Google Scholar]
- 5. Browder T, Folkman J, Pirie‐Shepherd S. The hemostatic system as a regulator of angiogenesis. J Biol Chem 2000; 275: 1521–4. [DOI] [PubMed] [Google Scholar]
- 6. Costantini V, Zacharski LR, Memoli VA, Kisiel W, Kudryk BJ, Rousseau SM. Fibrinogen deposition without thrombin generation in primary human breast cancer tissue. Cancer Res 1991; 51: 349–53. [PubMed] [Google Scholar]
- 7. Zacharski LR, Memoli VA, Rousseau SM. Coagulation‐cancer interaction in situ in renal cell carcinoma. Blood 1986; 68: 394–9. [PubMed] [Google Scholar]
- 8. Bootle‐Wilbraham CA, Tazzyman S, Marshall JM, Lewis CE. Fibrinogen E‐fragment inhibits the migration and tubule formation of human dermal microvascular endothelial cells in vitro. Cancer Res 2000; 60: 4719–24. [PubMed] [Google Scholar]
- 9. Brown NJ, Staton CA, Rodgers GR, Corke KP, Underwood JC, Lewis CE. Fibrinogen E fragment selectively disrupts the vasculature and inhibits the growth of tumours in a syngeneic murine model. Br J Cancer 2002; 86: 1813–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Su Y, Shen J, Qian H et al Diagnosis of gastric cancer using decision tree classification of mass spectral data. Cancer Sci 2007; 98: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Villanueva J, Shaffer DR, Philip J et al Differential exoprotease activities confer tumor‐specific serum peptidome patterns. J Clin Invest 2006; 116: 271–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ebert MP, Niemeyer D, Deininger SO et al Identification and confirmation of increased fibrinopeptide a serum protein levels in gastric cancer sera by magnet bead assisted MALDI‐TOF mass spectrometry. J Proteome Res 2006; 5: 2152–8. [DOI] [PubMed] [Google Scholar]
- 13. Bergen HR 3rd, Vasmatzis G, Cliby WA, Johnson KL, Oberg AL, Muddiman DC. Discovery of ovarian cancer biomarkers in serum using NanoLC electrospray ionization TOF and FT‐ICR mass spectrometry. Dis Markers 2003; 19: 239–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Orvisky E, Drake SK, Martin BM et al Enrichment of low molecular weight fraction of serum for MS analysis of peptides associated with hepatocellular carcinoma. Proteomics 2006; 6: 2895–902. [DOI] [PubMed] [Google Scholar]
- 15. Theodorescu D, Wittke S, Ross MM et al Discovery and validation of new protein biomarkers for urothelial cancer: a prospective analysis. Lancet Oncol 2006; 7: 230–40. [DOI] [PubMed] [Google Scholar]
- 16. Villanueva J, Martorella AJ, Lawlor K et al Serum peptidome patterns that distinguish metastatic thyroid carcinoma from cancer‐free controls are unbiased by gender and age. Mol Cell Proteomics 2006; 5: 1840–52. [DOI] [PubMed] [Google Scholar]
- 17. Jaffe EA, Nachman RL, Becker CG, Minick CR. Culture of human endothelial cells derived from umbilical veins. Identification by morphologic and immunologic criteria. J Clin Invest 1973; 52: 2745–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Snyder EL, Dowdy SF. Cell penetrating peptides in drug delivery. Pharm Res 2004; 21: 389–93. [DOI] [PubMed] [Google Scholar]
- 19. Gao Y, Su Y, Qu L et al Mitochondrial apoptosis contributes to the anti‐cancer effect of Smilax glabra Roxb. Toxicol Lett 2011; 207: 112–20. [DOI] [PubMed] [Google Scholar]
- 20. Vermeulen PB, Gasparini G, Fox SB et al Second international consensus on the methodology and criteria of evaluation of angiogenesis quantification in solid human tumours. Eur J Cancer 2002; 38: 1564–79. [DOI] [PubMed] [Google Scholar]
- 21. An P, Lei H, Zhang J et al Suppression of tumor growth and metastasis by a VEGFR‐1 antagonizing peptide identified from a phage display library. Int J Cancer 2004; 111: 165–73. [DOI] [PubMed] [Google Scholar]
- 22. Liu J, Kolath J, Anderson J et al Positive interaction between 5‐FU and FdUMP[10] in the inhibition of human colorectal tumor cell proliferation. Antisense Nucleic Acid Drug Dev 1999; 9: 481–6. [DOI] [PubMed] [Google Scholar]
- 23. Kogan NM, Blazquez C, Alvarez L et al A cannabinoid quinone inhibits angiogenesis by targeting vascular endothelial cells. Mol Pharmacol 2006; 70: 51–9. [DOI] [PubMed] [Google Scholar]
- 24. Tozer GM, Bhujwalla ZM, Griffiths JR, Maxwell RJ. Phosphorus‐31 magnetic resonance spectroscopy and blood perfusion of the RIF‐1 tumor following X‐irradiation. Int J Radiat Oncol Biol Phys 1989; 16: 155–64. [DOI] [PubMed] [Google Scholar]
- 25. Toro A, Grunebaum E. TAT‐mediated intracellular delivery of purine nucleoside phosphorylase corrects its deficiency in mice. J Clin Invest 2006; 116: 2717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zacharski LR. Basis for selection of anticoagulant drugs for therapeutic trials in human malignancy. Haemostasis 1986; 16: 300–20. [DOI] [PubMed] [Google Scholar]
- 27. Zacharski LR, Memoli VA, Costantini V, Wojtukiewicz MZ, Ornstein DL. Clotting factors in tumour tissue: implications for cancer therapy. Blood Coagul Fibrinolysis 1990; 1: 71–8. [DOI] [PubMed] [Google Scholar]
- 28. Scott CF, Brandwein H, Whitbread J, Colman RW. Lack of clinically significant contact system activation during platelet concentrate filtration by leukocyte removal filters. Blood 1998; 92: 616–22. [PubMed] [Google Scholar]
- 29. Rhim TY, Park CS, Kim E, Kim SS. Human prothrombin fragment 1 and 2 inhibit bFGF‐induced BCE cell growth. Biochem Biophys Res Commun 1998; 252: 513–6. [DOI] [PubMed] [Google Scholar]
- 30. O'Reilly MS, Pirie‐Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science 1999; 285: 1926–8. [DOI] [PubMed] [Google Scholar]
- 31. Staton CA, Brown NJ, Rodgers GR et al Alphastatin, a 24‐amino acid fragment of human fibrinogen, is a potent new inhibitor of activated endothelial cells in vitro and in vivo. Blood 2004; 103: 601–6. [DOI] [PubMed] [Google Scholar]
- 32. Staton CA, Stribbling SM, Garcia‐Echeverria C et al Identification of key residues involved in mediating the in vivo anti‐tumor/anti‐endothelial activity of Alphastatin. J Thromb Haemost 2007; 5: 846–54. [DOI] [PubMed] [Google Scholar]
- 33. Ellis LM, Hicklin DJ. VEGF‐targeted therapy: mechanisms of anti‐tumour activity. Nat Rev Cancer 2008; 8: 579–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Fibrinostatin inhibits proliferation of HMEC, but not WI‐38 or HEK‐293.