

Abstract
A recombinant hepatitis B virus (HBV) expressing NanoLuc (NL) (HBV/NL) was produced by cotransfecting a plasmid containing a 1.2‐fold HBV genome carrying the NL gene with a plasmid bearing a packaging‐defective 1.2‐fold HBV genome into a human hepatoma cell line, HepG2. We found that NL activity in HBV/NL‐infected primary hepatocytes or sodium taurocholate cotransporting polypeptide‐transduced human hepatocyte‐derived cell lines increased linearly for several days after infection and was concordant with HBV RNA levels in the cells. Treatment of the virus‐infected cells with HBV inhibitors reduced NL activity in a dose‐dependent manner. Detection of HBV/NL infection, monitored by NL activity, was highly sensitive and less expensive than detection using the conventional method to evaluate HBV infection. In addition, because we also studied host factors, this system is applicable not only for studying the HBV life cycle, but also for exploring agent(s) that regulate HBV proliferation.
Keywords: HBV, host factors, luciferase, reporter HBV, virus entry
Approximately 370 million people worldwide are infected with hepatitis B virus (HBV). Antiviral therapies consisting of HBV polymerase inhibitors and/or interferon have been used in active chronic HBV carriers,1 which may delay the progress of HBV‐related diseases, but cannot cure HBV due to the persistence of HBV covalently closed circular DNA (cccDNA) in hepatocytes. A new strategy for HBV treatment depends on intensive characterization of the HBV life cycle through development of an efficient in vitro experimental system. However, the limited host range and liver tropism of HBV has hampered efforts to establish such a system. Human liver cells reflecting a primary hepatocyte nature are now available to monitor HBV infection and replication with comparably high efficiency.2 Moreover, the discovery of sodium taurocholate cotransporting polypeptide (NTCP) as a prominent HBV receptor candidate enabled the establishment of HBV‐susceptible cells derived from cell lines such as HepG2 and HuH7 by ectopic expression of NTCP.3, 4
A foreign gene, such as a reporter or marker gene, is successfully incorporated into some viral genomes, including HIV‐1 and hepatitis C virus without the loss of replication competency.5, 6, 7 In contrast, the compact nature of the HBV genome and the presence of genomic cis‐acting elements make it difficult to introduce a foreign gene without losing competency and replication efficiency. In constructing a recombinant HBV‐reporter virus, cis‐acting control regions such as the replication control region for pre‐genomic RNA synthesis, packaging, and reverse transcription that locates from upstream of the X gene to the N‐terminal region of the core gene need to remain intact.8, 9 The size limitation of the genome restricts the length of foreign genes that often require replacement of virus DNA to accommodate the foreign gene; the destroyed function must be provided in trans to produce recombinant virus particles. Various methods have been explored to produce recombinant HBV.10, 11, 12, 13, 14, 15, 16, 17 However, these recombinant HBVs are not designed to detect HBV infection with high sensitivity to analyze the HBV life cycle, nor for high‐throughput screening of factors affecting HBV infection and replication.
Here, we constructed a reporter‐based HBV to monitor infection with high sensitivity using the luciferase gene NanoLuc (NL) as a marker gene because NL is the shortest luciferase gene commercially available. Moreover, NL is approximately 100‐fold brighter, with a linear increase of broad range greater than either firefly (Photinus pyralis) or Renilla reniformis luciferase. The use of NL or other relevant genes such as Gaussia luciferase would increase the sensitivity of HBV infection more than previously reported HBV recombinant viruses and authentic HBV.
Materials and Methods
Cells
HepG2 and HuH7 were cultured in DMEM (Life Technologies, Carlsbad, CA, USA) supplemented with 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 100 U/mL non‐essential amino acids (Life Technologies) unless otherwise described. Primary human hepatocytes (PHH), PXB cells, isolated from urokinase‐type plasminogen activator transgenic/SCID mice inoculated with PHH and HepaRG were purchased from PhoenixBio, Hiroshima, Japan and KAC, Kyoto, Japan respectively, and cultured under manufacturer's protocols. HepG2/NTCP and HuH7/NTCP cells are HepG2‐ and HuH7‐derived cell lines transduced by pCAN‐NTCP‐myc and are susceptible to HBV infection.
Plasmids
We used a 1.2‐fold HBV genome (isolate C_JPNAT, genotype C, accession number AB246345) cloned into the HindIII/EcoRI site of pUC19, termed pUC1.2xHBV.18 The plasmid pUC1.2xHBV/NL was constructed by deleting 562 nt (219–782) from the first codon of the HBV preCore coding frame and then inserting the NL gene (513 nt) from pNL2.1 N1061 NLuc (Promega, Madison, WI, USA) by BD In‐Fusion PCR cloning. Similarly, pUC1.2xHBV/NL+pol was constructed by deleting 141 nt (295–436) from the first codon of the HBV preCore coding frame and then inserting the NL gene at the 178 nt position from the first codon of preCore/Core by the In‐Fusion method. The genome sizes of HBV/NL and HBV/NL+pol were 3302 and 3731 nt, respectively. Expression of NL was designed to start from its own initiation codon. HBV/NL(‐Met) has mutations of all methionine residues converted to other amino acids or a terminator codon in the defective pol coding sequence of HBV/NL without affecting the amino acid sequence of S protein. The mutations are as follows: All ATGs at positions 330, 902, 1329, 1422, 1548, 1647, 1785, 1962, and 2142 in the polymerase gene were converted to GTG, GTG, GTG, TTG, GTG, TTG, TAG, TAG, and TAG, respectively. The plasmid pUC1.2xHBV‐D, which produces all HBV proteins, has two mutations in the encapsidation signal (CTGTGCC to CTATGTC), and, thus, does not produce progeny virus. The plasmid pUC1.2xHBV‐D/MHD is mutated in the catalytic domain, MDD, of HBV‐D pol to MHD. The plasmid pCAN‐NTCP‐myc was constructed by inserting human NTCP cDNA tagged with myc at the 3′‐end into pCAN. The plasmid pX330 was obtained from Addgene (plasmid 42230; Cambridge, MA, USA). Oligonucleotides designed for each target site were inserted into the BbsI site of pX330.
Production of recombinant virus
The pUC1.2xHBV/NL, pUC1.2xHBV/NL+pol, or pUC1.2xHBV/NL(‐Met) plasmids were cotransfected with pUCxHBV‐D into HepG2 or HuH7 cells using Lipofectamine 3000 (Life Technologies). Medium was harvested 3 or 4 days after transfection and the virus fraction prepared by precipitation with 13% PEG6000 (Sigma‐Aldrich, St. Louis, MO, USA) containing 0.75 M NaCl. Virus was further purified by precipitation through 20% sucrose in TNE buffer (10 mM Tris [pH 7.6], 50 mM NaCl, 1 mM EDTA) at 100 000 g for 3 h. Virus was then suspended in Opti‐MEM (Life Technologies) and stored at −80°C until use.
Infection and assay of NL activity
Cells were infected with virus at a genome equivalent of 10–100 in the presence of 4% PEG8000 and 2% DMSO overnight. Activity of NL and cell viability were then measured using the NL Luciferase Assay Kit and CellTiter‐Glo Luminescent Cell Viability Assay Kit (Promega) according to the manufacturers' protocols. Human hepatitis B immunoglobulin and β‐interferon (IFNβ) were from Mitsubishi Tanabe Pharma Corporation (Osaka, Japan) and Mochida pharmaceutical (Tokyo, Japan), respectively. Entecavir was from Toronto Research Chemicals (Toronto, Canada).
Quantitative RT‐PCR and PCR
Quantitative real‐time PCR was carried out with Fast SYBR Green Master Mix (Life Technologies, Carlsbad, CA, USA), and fluorescent signals were analyzed with the Fast Real‐Time PCR system (Applied Biosystems). The primers used were 5′‐CCTCTGCCTAATCATCTCATGTTC‐3′ (forward) and 5′‐CGGTGTCGAGGAGATCTCGAATAG‐3′ (reverse) for HBV and 5′‐ATGGTCTTCACACTCGAAG‐3′ (forward) and 5′‐TCAGGACAATCCTTTGGATC‐3′ (reverse) for NL.
Cesium chloride density equilibrated centrifugation
The virus fraction was centrifuged at 150 000 g for 50 h on a CsCl gradient (in 10 mM Tris [pH 7.6], 150 mM NaCl, and 1 mM EDTA) from 1.1 to 1.6 g/mL. An aliquot from the top of the gradient tube was collected for further analysis.
Small interfering RNA transfection
The siRNA was transfected using Lipofectamine RNAiMAX Reagent (Life Technologies) according to the manufacturer's protocol. The duplex nucleotides of siRNA specific to the mRNA and the Mission siRNA Universal Negative control were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Sequences of siRNAs for hepatocyte nuclear factor 4 alpha (HNF4A) and NTCP were: HNF4A, 5′‐CGCUACUGCAGGCUCAAGATT‐3′ (sense) and 5′‐UCUUGAGCCUGCAGUAGCGTT‐3′ (antisense); and NTCP 5′‐ CAAGGCUCACUUAUGGAAGTT‐3′ (sense), and 5′‐ CUUCCAUAAGUGAGCCUUGTT‐3′ (antisense).
Screening of host factors by siRNA
siGENOME containing siRNAs targeting 18 236 human genes was purchased from Dharmacon GE Healthcare (Little Chalfont, Buckinghamshire HP7 9NA, UK). HepG2/NTCP, HuH7/NTCP, or PXB cells were transfected with 4 pmol siRNA using Lipofectamine RNAiMAX in 96‐well plates. At 48 h following transfection, cells were infected with HBV/NL in the presence of 2% DMSO and 4% PEG8000. At 96 h following infection, cells were lysed and NL activity and cell viability were measured.
Gene knockout
The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9‐based gene knockout (KO) plasmid was generated using annealed oligonucleotides and cloned into pX330. HuH7/NTCP or HepG2/NTCP cells were then transfected with each targeting plasmid derived from the pX330 plasmid along with a puromycin‐resistant plasmid. After transfection, cells were treated with 1.5 μg/mL puromycin and single cell colonies were collected. The KO of each gene was confirmed by Western blotting or sequencing of the target region.
Results
Construction of HBV and NL plasmids
We inserted the NL gene downstream of the HBV replication control region as described in “Materials and Methods”. If the size of the HBV genome is 0.7 kb over the original genome size,15 replication decreases, therefore HBV DNA similar to the size of the NL gene was deleted from the 3′‐terminal region of the core gene to the 5′‐terminal region of the polymerase gene to make pUC1.2xHBV/NL. Thus, pUC1.2xHBV/NL lacked 562 nt of HBV DNA from the 3′‐terminal half of the core coding region to the 5′‐coding region of the polymerase coding region, resulting in non‐functional preCore/Core and polymerase proteins. However, pUC1.2xHBV/NL+pol lacked part of the core coding sequence but retained the entire polymerase coding sequence. The deletion of 141 nt from the core coding sequence made this construct 3731 nt long, which is more than 450 nt longer than the original HBV genome. In pUC1.2xHBV/NL(‐Met), the internal methionine codons in the defective pol coding region of pUC1.2xHBV/NL were mutated to prevent the possible translation of any polymerase‐derived peptide that may interfere with HBV replication. These mutations were designed so as not to alter the amino acid sequence of hepatitis B surface antigen. The expression of the NL gene was expected to be under the same regulatory mechanism of authentic preCore/Core because transcriptional control elements of the HBV genome remained intact. A schema of these genomic structures is shown in Figure 1(a). HBV/NL+pol and HBV/NL(‐Met) were also examined for infectivity by assaying NL activity in the infected cells (Fig. 1B). HBV/NL+pol had less than one‐tenth the NL activity of HBV/NL. Production of HBV/NL+pol as measured by qPCR for HBV DNA in the culture medium of pUC1.2xHBV/NL+pol‐transfected HepG2 cells was less than one‐tenth that of HBV/NL (data not shown). It is likely that the genome size limitation for packaging was exceeded, resulting in less efficient virus production.15 HBV/NL(‐Met) had the same NL activity levels as HBV/NL, and virus production, as measured by HBV DNA from the cells transfected with pUC1.2xHBV/NL(‐Met), was nearly the same as that from the pUC1.2xHBV/NL‐transfected cells (data not shown). These data indicate that production of peptides from internal methionine residues in the polymerase reading frame does not interfere with recombinant virus production or influence NL activity.
Figure 1.
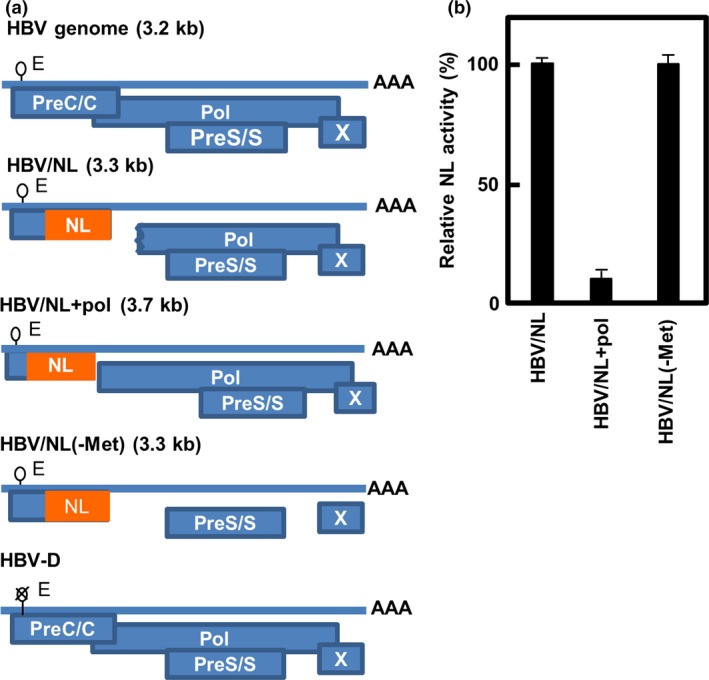
Pregenomic structure of the reporter hepatitis B viruses (HBVs) and relative NanoLuc (NL) activity in cells infected by the viruses. (a) Pregenomes of wild‐type and reporter HBV, and putative ORFs of the virus are shown. The indicated sizes (kb) are of the pregenomes. A stretch of “A”s indicates a poly A tail of the putative pregenomic RNA. A lariat rope with “E” indicates an encapsidation signal and “X” on that indicates defect of encapsidation. The NL gene is inserted into the genome so as to be translated from its own initiator methionine. Virus was produced by co‐transfecting with the pUC1.2xHBV/NL, pUC1.2xHBV/NL+pol, or pUC1.2xHBV/NL(‐Met) and pUCxHBV‐D into HepG2 or HuH7 cells and the same volume of virus fractions was infected into PXB cells. (b) PXB cells were harvested 7 days after infection and NL activity in cell lysates was measured. The NL activity in the cell lysates was quantified (mean ± SD; n = 3). The data shown are of three independent experiments.
Production and characterization of recombinant viruses
Recombinant viruses were produced from the plasmids described above and then characterized. HepG2 or HuH7 cells were transfected with the reporter plasmid, pUC1.2xHBV/NL, pUC1.2xHBV/NL+pol, or pUC1.2xHBV/NL(‐Met) together with the plasmid pUC1.2xHBV‐D. HBV‐D is a replication‐defective virus genome that produces the complementary proteins required for recombinant virus production. As a positive control, pUC1.2xHBV, which produces wild‐type HBV, was also transfected. Culture medium from cells was harvested 4 days after transfection and the virus was partially purified as described in “Materials and Methods”. Production of HBV and its derivatives was then analyzed by CsCl density equilibrated centrifugation (Fig. 2). The medium from the cells transfected with the plasmid producing wild‐type HBV yielded an HBV DNA profile with two peaks, consistent with the Southern blot data (Fig. 2a). Upon analysis, fractions 4–7 were found to be infectious, with the highest infectivity from fraction 6. Fraction 6 had a density of 1.23 g/mL and coincided with the lighter peak of the HBV DNA profile. The DNA peak with the high density appeared to be derived from the unenveloped capsid particles of HBV, as described by others.16 A profile of HBV/NL was observed through analysis of the enriched virus fraction of pUC1.2xHBV/NL plus pUC1.2xHBV‐D‐transfected cell culture medium. After infection of each fraction to HepG2/NTCP, NL activity was detectable only in fractions 4–7, peaking at fraction 6 with the highest buoyant density similar to wild‐type HBV (Fig. 2b), indicating that the infectious HBV/NL was produced. We also observed two peaks by DNA titration analyzed by quantitative PCR (qPCR) using primers for the NL gene as well as Southern blot using HBV DNA as a probe (Fig. 2b). As no peak of HBV DNA was observed in culture medium from the cells transfected with pUC1.2xHBV‐D, virus‐like particles were not produced from the plasmid bearing HBV‐D DNA (Fig. 2c). Moreover, no virus with NL activity was produced from culture medium of cells transfected with pUC1.2xHBV/NL and pUC1.2xHBV‐D/MHD, both of which lack pol activity (data not shown). This observation strongly suggests that generation of wild HBV by genetic conversion between these plasmids is less likely.
Figure 2.
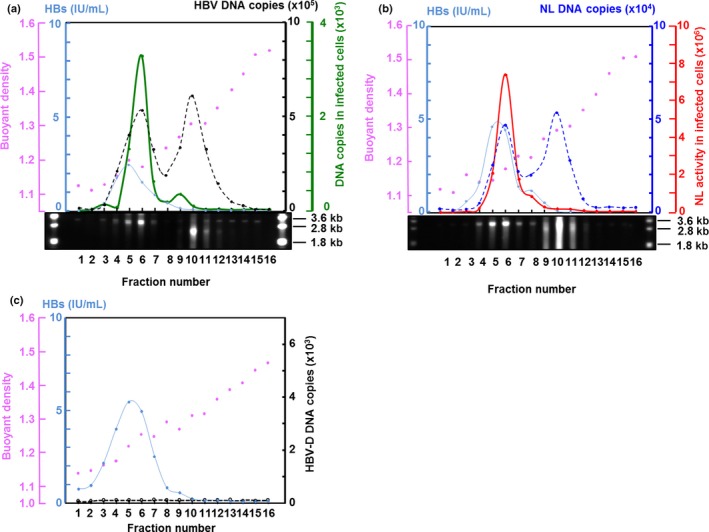
Cesium chloride (CsCl) density equilibrated centrifugation of wild‐type hepatitis B virus (HBV) and recombinant HBV expressing NanoLuc (HBV/NL). Supernatants of HepG2 cells transfected with pUC1.2xHBV (a), pUC1.2xHBV/NL plus pUC1.2xHBV‐D (b), and pUC1.2xHBV‐D (c) were centrifuged at 150 000 g for 50 h in a CsCl gradient. An aliquot from the top of the centrifuged tube was collected. Hepatitis B surface antigen (HBsAg; blue) in 10% of each aliquot was quantified by automated ELISA. The amount of NL DNA (dotted blue line) (b) and HBV DNA (dotted black line) (a, c) in 10% of each fraction was measured by quantitative PCR using a primer set for the NL gene and HBV DNA, respectively. Infectivity of 25% of each fraction in a and b was assayed by infecting 2 × 105 HepG2/NTCP cells and measuring HBV DNA by quantitative PCR for wild HBV (green) (a) and NL activity for HBV/NL (red) (b) of 10% of the total cell lysate. Pink dots denote buoyant density. Southern blot analysis of each fraction was carried out by probing with digoxigenin‐labeled HBV DNA.
Activity of NL in HBV/NL‐infected cells reflects HBV RNA levels
In order to analyze the HBV life cycle as well as to screen for factors affecting the HBV life cycle, we analyzed how faithfully the NL activity of HBV/NL reflected the authentic HBV life cycle in infected cells and how sensitively NL activity could evaluate the HBV life cycle. We carried out a time course of NL activity in HBV/NL‐infected PXB cells, a PHH susceptible to HBV infection (Fig. 3).19 The HBV DNA and RNA levels increased in a time‐dependent manner in PXB cells following HBV infection (Fig. 3a); NL activity became detectable 3 days after HBV/NL infection and increased for 6 days along with HBV RNA levels (Fig. 3b). Although HBV DNA was detectable throughout these time points, it stayed unchanged at a low level (Fig. 3b). Similar levels of NL activity and RNA were observed in HBV/NL‐infected HepG2/NTCP cells (Fig. 3c). A slight decline in both markers 9 or 13 days after the infection of PXB or HepG2/NTCP cells, respectively, coincided with cell loss (Fig. 3b,c and data not shown). These data suggest that NL activity was a consequence of transcription from the HBV recombinant genome in cells after infection. Hepatitis B virus transcription begins on virus cccDNA,9 therefore NL expression indicates the completion of an early event in the HBV life cycle, from entry to transcription/translation through cccDNA formation. Consistently low levels of HBV DNA in PXB cells from 3 days after infection suggest that neither de novo synthesis of the relaxed circular form of HBV DNA, nor the amplification process of cccDNA occurred due to the lack of active polymerase from the recombinant virus (Fig. 3b,c). However, cccDNA in HBV/NL‐infected HuH7/NTCP cells was detectable as early as 4 days following infection (Fig. 3d). This cccDNA could be the product converted from the relaxed circular form of HBV DNA by HBV polymerase residing in the virus particles that penetrated the cells.
Figure 3.
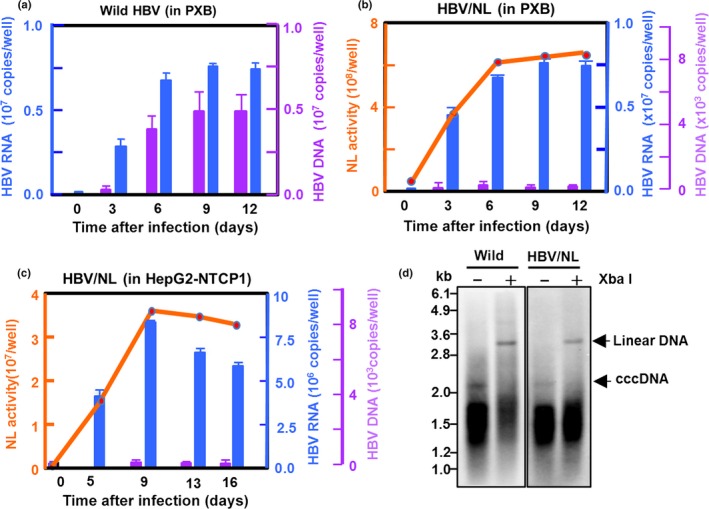
Time course of hepatitis B virus (HBV) RNA, DNA, and NanoLuc (NL) activity in wild HBV‐ and HBV/NL‐infected cells. (a) PXB cells (2 × 105/well) were infected with approximately 2 × 107 copies wild‐type HBV and HBV RNA (blue) and DNA (purple) in 10% of the cell lysates measured by quantitative RT‐PCR and quantitative PCR, respectively, at the indicated days after infection. (b, c) PXB or HepG2/NTCP cells (2 × 105/well) were infected with approximately 2 × 107 HBV/NL and NL activity (orange), HBV RNA (blue), and DNA (purple) in 10% of cell lysates measured at the indicated days after infection. The data shown are indicative of three independent experiments. (d) Southern blot analysis of covalently closed circular DNA (cccDNA) fractions obtained from 1 × 107 HuH7/NTCP cells infected with approximately 1000 equivalent copies of wild HBV or HBV/NL DNA per cell. The cccDNA fraction was prepared by the method previously reported.29 Digestion with XbaI, which has a single cutting site in the genomes, converts the cccDNA to a single linear form.
We then examined the effect of HBV inhibitors, including human hepatitis B immunoglobulin (HBIG), entecavir, and IFNβ on NL activity in HBV/NL‐infected cells. We first examined the effect of these inhibitors on the infection and replication of wild‐type HBV. PXB cells were infected with HBV in the presence of each inhibitor for 7 days and the level of HBV DNA and RNA was measured (Fig. 4a,b). Entecavir, an HBV polymerase inhibitor, and IFNβ suppressed HBV DNA levels by <15% at 0.1 μM and 250 IU/mL, respectively; HBIG suppressed HBV DNA levels by <10% at 1 U/mL. Both IFNβ and HBIG, at higher concentrations of 750 IU/mL and 5 U/mL, respectively, decreased HBV DNA even further (Fig. 4a). We simultaneously determined the effect of these inhibitors on HBV RNA levels and found that RNA levels in the IFNβ‐ and HBIG‐treated cells was reduced to <10% (Fig. 4b), whereas entecavir reduced RNA levels to 30% (Fig. 4b). Next, we analyzed NL activity and HBV RNA and DNA levels in PXB, HepG2/NTCP, and HepaRG cells infected with HBV/NL in the presence of the inhibitors (Figs 4c,d,S1). In these cells, entecavir, at a dose of 0.1 μM, suppressed NL activity by 30% whereas IFNβ and HBIG decreased NL activity by <10% at 250 IU/mL and 1 U/mL, respectively, 7 days after infection. The HBV RNA levels correlated well with NL activity (Figs 4d,S1), indicating that NL activity reflects HBV RNA levels.
Figure 4.
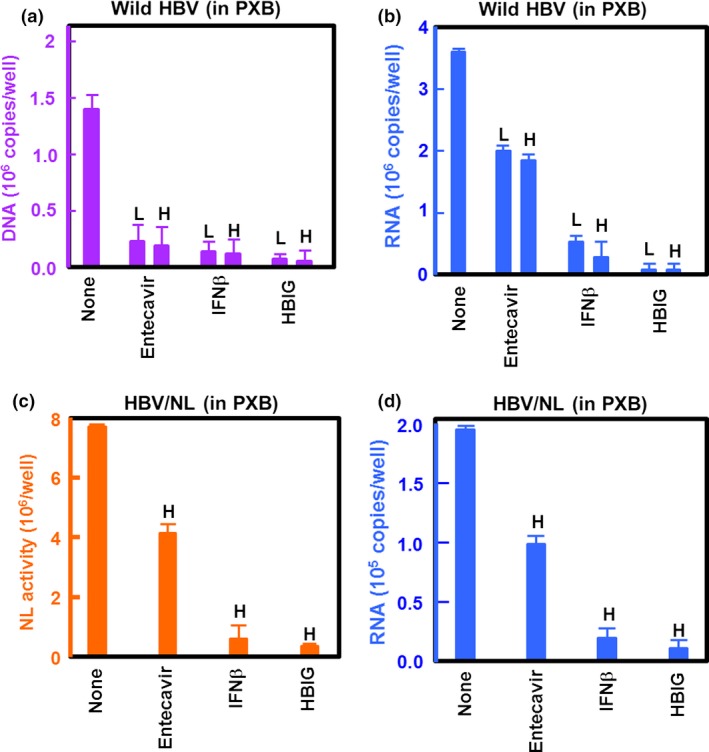
Effect of hepatitis B virus (HBV) inhibitors on recombinant HBV expressing NanoLuc (HBV/NL) infection and replication. PXB cells (2 × 105/well) were infected with wild‐type HBV at a genome equivalent of 10 in the presence of the indicated inhibitor. Virus DNA (a) and RNA (b) in 10% of the cell lysate were measured by quantitative PCR and quantitative RT‐PCR, respectively, 7 days after infection. (c, d) PXB cells were infected with HBV/NL at an approximate genome equivalent of 10. Seven days after infection, NL activity (c) as well as HBV RNA (d) was measured in 10% of the cell lysate. The concentration of drugs used was 0.1 μM (low [L]) and 0.5 μM (high [H]) entecavir, 250 IU/mL (L) and 750 IU/mL (H) β‐interferon (IFNβ), and 1 U (L) and 5 U (H) human hepatitis B immunoglobulin (HBIG).
Screening of host factors affecting early stages of the HBV life cycle
We applied the HBV/NL system to host factor screening by sequence‐specific RNAi gene silencing. We determined the feasibility of this system to evaluate host factors involved in HBV proliferation by targeting HNF4A and NTCP, which are important for HBV replication and infection, respectively.3, 20 Both HNF4A and NTCP were knocked down in HepG2/NTCP cells by specific siRNAs by 50% or less (Fig. S2a). The cells were then infected with HBV/NL to evaluate NL activity 5 days after infection. We found that NL activity was reduced to levels half that of the control cells (Fig. S2b), indicating that host factors influencing HBV infection/replication also affect NL activity of HBV/NL.
Next, siRNA libraries for 18 236 genes were used to determine host factors that regulate the HBV life cycle. Candidate genes with more than a 40% reduction or 200% activation of NL activity in HepG2/NTCP cells were further analyzed by a second screening in HuH7/NTCP cells. Of these, genes with more than a 35% reduction or 200% activation with <20% non‐toxicity to cells were included (Table 1). These genes were significantly expressed in HepG2, HuH7, and PHH cells in the presence of 2% DMSO as evaluated by microarray (data not shown). Consistent with previous reports,21, 22, 23, 24 we also identified host factors such as CEBPA, BclxL, KPNB1, and CHORDC1.
Table 1.
Identification of host factors involved in the early stages of hepatitis B virus (HBV) replication in Huh7/NTCP cells
| Gene symbol | HBV/NL infection, %† | Cell viability, %‡ | Gene symbol | HBV/NL infection, %† | Cell viability, %‡ |
|---|---|---|---|---|---|
| CD2BP2 | 62 | 92 | PON2 | 57 | 85 |
| FBP1 | 62 | 92 | TRAF2 | 64 | 95 |
| GORASP2 | 44 | 86 | MCL1 | 56 | 104 |
| DUSP8 | 56 | 82 | DUSP5 | 49 | 97 |
| IL1RAP | 54 | 98 | PIK3R1 | 60 | 95 |
| RASA4 | 60 | 102 | IQCB1 | 57 | 91 |
| MTA2 | 51 | 106 | SART3 | 59 | 97 |
| GNAO1 | 51 | 94 | RLBP1L2 | 45 | 88 |
| RARA | 53 | 83 | IGFBPL1 | 53 | 96 |
| SNX9 | 36 | 94 | SAFB | 41 | 93 |
| CC2D2A | 56 | 95 | LOC146909 | 55 | 93 |
| NUP155 | 55 | 90 | KRTAP2‐4 | 51 | 93 |
| MFSD3 | 55 | 96 | CASP4 | 53 | 110 |
| TRIM68 | 54 | 85 | EPB41L3 | 58 | 86 |
| SH3YL1 | 57 | 96 | SEL1L | 46 | 90 |
| LY6E | 47 | 82 | NEU2 | 40 | 92 |
| OBSCN | 57 | 100 | STRN | 59 | 110 |
| WDR26 | 49 | 95 | UNC93B1 | 52 | 85 |
| ZMAT3 | 59 | 102 | C12orf54 | 57 | 99 |
| SPCS1 | 54 | 97 | TRIM26 | 58 | 109 |
| TGFBR2 | 58 | 92 | NUDT15 | 57 | 93 |
| KAT5 | 212 | 88 | NPM3 | 202 | 98 |
| HNF4A § (ref. 3) | 56 | 95 | BclxL § (ref. 20) | 54 | 96 |
| NTCP1 § (ref. 18) | 55 | 102 | KPNB1 § (ref. 21) | 52 | 96 |
| CEBPA §(ref. 19) | 37 | 100 | CHORDC1 § (ref. 22) | 57 | 99 |
†NanoLuc (NL) activity level (%) in siRNA‐treated cells compared to control cells (transfected with non‐targeting siRNA). ‡Cell viability level in siRNA‐treated cells compared to control cells (transfected with non‐targeting siRNA). §Host factors previously implicated in HBV replication. Ref., reference.
We then randomly chose several of these genes (CD2BP2, PON2, FBP1, TRAF2, GORASP2, MCL1, KAT5, and NPM3) and established KO cells of each by the CRISPR‐Cas9 system (Fig. 5a) to further evaluate their role in HBV infection and replication. Infection of HBV/NL in MCL1‐, PON2‐, TRAF2‐, or GORASP2‐KO cells clearly reduced NL activity (Fig. 5b). As expected, HBV/NL replication was significantly enhanced by KO of KAT5 or NPM3. Infectivity of authentic HBV, as measured by virus RNA levels, correlated well with HBV/NL results (Fig. 5c). The difference in HBV infectivity between cell types may reflect the degree to which the knocked‐out gene contributed to HBV infection/replication. These results clearly indicate the usefulness of the HBV/NL system to explore factors that regulate the HBV life cycle.
Figure 5.
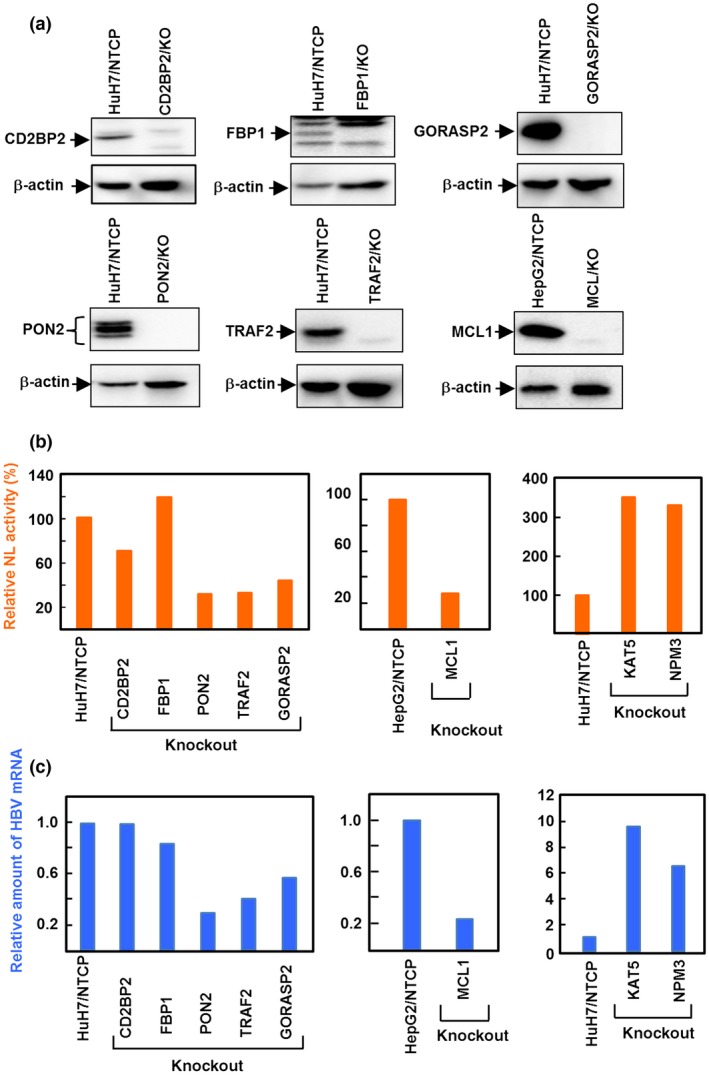
Effect of hepatitis B virus (HBV) infection and replication in HuH7/NTCP or HepG2/NTCP cells with knocked‐out anti‐HBV candidate genes. The genes were knocked‐out by a clustered regularly interspaced short palindromic repeats/Cas9 system. (a) Knockout (KO) of each gene was confirmed by Western blot analysis using the antibody specific to each gene product, except KAT5 (also known as TIP60). Knockout of KAT5 was confirmed by genomic sequencing due to the unavailability of a specific antibody. (b) Cells were infected with recombinant HBV expressing NanoLuc (HBV/NL), cell lysates harvested 7 days after infection, and NL activity measured. (c) Cells were infected with wild‐type HBV and then viral RNA were harvested 7 days after infection and analyzed by quantitative RT‐PCR. Values relative to control cells are shown.
Discussion
We developed an HBV reporter system to analyze virus replication and screen anti‐HBV agents with a simple, high‐efficiency procedure. We produced a recombinant virus, HBV/NL, that mimics early events of the HBV life cycle. The HBV/NL virus had the same buoyant density as authentic HBV and showed NL activity following infection into various HBV‐susceptible cells (Figs 1, 2, 3). Through analysis of HBV DNA and RNA as well as NL activity in HBV/NL‐infected cells, we found that NL activity reflected RNA levels. Both NL activity and HBV RNA levels increased linearly for at least 6 days after infection, whereas HBV DNA levels remained low, likely due to the non‐functional polymerase in HBV/NL. The HBV DNA that was detected by PCR specific for cccDNA was likely derived from the incorporated virus after infection (data not shown) as Southern blot analysis showed cccDNA was present as early as 4 days following infection (Fig. 3d). Hepatitis B virus inhibitors, such as HBIG, entecavir, and IFNβ, suppressed NL activity in a dose‐dependent manner (Figs 4,S1). Suppression of NL activity by HBIG was likely an inhibition of virus entry into cells, whereas suppression of NL activity by entecavir or IFNβ was likely an inhibition of HBV reverse transcription or virus replication, respectively, as previously described.25, 26, 27 Inhibition of NL activity by entecavir in cells infected with HBV/NL that does not encode functional pol is likely to be by inhibition of polymerase that resides in the penetrated virus and is required for conversion from virion‐associated HBV DNA to the ccc form. Inhibition of NL activity by these reagents correlated with HBV RNA suppression (Fig. 4), indicating that the NL activity of HBV/NL‐infected cells reflects early events of the HBV life cycle.
Evaluation of HBV infection using the HBV/NL system is simple and more sensitive than quantitating HBV DNA or RNA by qPCR or qRT‐PCR. Furthermore, this system could be used for large‐scale screening of agents and host factors that regulate the HBV life cycle. Taking advantage of the high sensitivity of the HBV/NL system to evaluate early stages of HBV infection and replication, we searched for host factors that affect the HBV life cycle using siRNA targeting 18 236 human genes. Approximately 50 genes that affected the NL activity of HBV/NL were identified, some of which have been previously identified to regulate the HBV life cycle (Table 1). Those HBV/NL‐infected cells with siRNA knocked‐out genes CD2BP2, PON2, FBP1, TRAF2, GORASP2, MCL1, KAT5, or NPM3 (Table 1) produced similar results to the infectivity assay. However, there was no effect on HBV replication in CD2BP2 or FBP1 KO cells; this may be due to false negatives and positives that occur frequently in siRNA screening.28 Taken together, the results of the HBV/NL infectivity assay, as measured by NL activity, and the infectivity of authentic HBV, as assayed by virus RNA levels, correlated well (Fig. 5).
In summary, we successfully produced a recombinant HBV expressing NL for evaluation of the HBV early life cycle, from entry to transcription. We also showed the utility of this system by identifying cellular factors that affect HBV infection and replication.9 Due to the high sensitivity of our assay in characterizing infection, this system would be applicable for identifying low molecular weight agents that affect HBV proliferation and antibodies that inhibit HBV infection, as well as for exploring host factors affecting HBV when combined with siRNA techniques.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Inhibitory effect of known hepatitis B virus (HBV) inhibitors on recombinant HBV expressing NanoLuc (HBV/NL).
Fig. S2. Suppression of NanoLuc (NL) activity in hepatitis B virus (HBV)/NL‐infected HuH7/NTCP cells by siRNA treatment specific to HNF4A and NTCP.
Acknowledgments
We thank Ms. Hiromi Yamamoto and Ritsuko Shiina for technical assistance. This work was partly supported by Grants‐in‐Aid for Scientific Research from the Ministry of Health, Labor, and Welfare of Japan, by Grants‐in‐Aids for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan and by the Research Program on Hepatitis from Japan Agency For Medical Research and Development, AMED.
Cancer Sci 106 (2015) 1616–1624
Funding Information
Ministry of Health, Labor, and Welfare of Japan; Ministry of Education, Culture, Sports, Science and Technology of Japan
References
- 1. Ganem D, Prince AM. Hepatitis B virus infection–natural history and clinical consequences. N Engl J Med 2004; 350: 1118–29. [DOI] [PubMed] [Google Scholar]
- 2. Gripon P, Rumin S, Urban S et al Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA 2002; 99: 15655–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yan H, Zhong G, Xu G et al Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012; 1: e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ni Y, Lempp FA, Mehrle S et al Hepatitis B and D viruses exploit sodium taurocholate co‐transporting polypeptide for species‐specific entry into hepatocytes. Gastroenterology 2014; 146: 1070–83. [DOI] [PubMed] [Google Scholar]
- 5. Muller B, Daecke J, Fackler OT, Dittmar MT, Zentgraf H, Krausslich HG. Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J Virol 2004; 78: 10803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Terahara K, Yamamoto T, Mitsuki YY et al Fluorescent reporter signals, EGFP, and DsRed, encoded in HIV‐1 facilitate the detection of productively infected cells and cell‐associated viral replication levels. Front Microbiol 2012; 2: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999; 285: 110–3. [DOI] [PubMed] [Google Scholar]
- 8. Nassal M, Schaller H. Hepatitis B virus replication–an update. J Viral Hepat 1996; 3: 217–26. [DOI] [PubMed] [Google Scholar]
- 9. Seeger C, Zoulim F, Mason W. Hepadonaviruses In: Knipe D, Howley P, eds. FIELDS Virology, 6th edn New York, NY: Wolters Kluwer, 2013; 2185–221. [Google Scholar]
- 10. Chaisomchit S, Tyrrell DL, Chang LJ. Development of replicative and nonreplicative hepatitis B virus vectors. Gene Ther 1997; 4: 1330–40. [DOI] [PubMed] [Google Scholar]
- 11. Yoo J, Rho J, Lee D, Shin S, Jung G. Hepatitis B virus vector carries a foreign gene into liver cells in vitro. Virus Genes 2002; 24: 215–24. [DOI] [PubMed] [Google Scholar]
- 12. Protzer U, Nassal M, Chiang PW, Kirschfink M, Schaller H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild‐type virus infection. Proc Natl Acad Sci USA 1999; 96: 10818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Untergasser A, Protzer U. Hepatitis B virus‐based vectors allow the elimination of viral gene expression and the insertion of foreign promoters. Hum Gene Ther 2004; 15: 203–10. [DOI] [PubMed] [Google Scholar]
- 14. Ho TC, Jeng KS, Hu CP, Chang C. Effects of genomic length on translocation of hepatitis B virus polymerase‐linked oligomer. J Virol 2000; 74: 9010–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang Z, Wu L, Cheng X et al Replication‐competent infectious hepatitis B virus vectors carrying substantially sized transgenes by redesigned viral polymerase translation. PLoS ONE 2013; 8: e60306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hong R, Bai W, Zhai J et al Novel recombinant hepatitis B virus vectors efficiently deliver protein and RNA encoding genes into primary hepatocytes. J Virol 2013; 87: 6615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shin EC, Protzer U, Untergasser A et al Liver‐directed gamma interferon gene delivery in chronic hepatitis C. J Virol 2005; 79: 13412–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sugiyama M, Tanaka Y, Kato T et al Influence of hepatitis B virus genotypes on the intra‐ and extracellular expression of viral DNA and antigens. Hepatology 2006; 44: 915–24. [DOI] [PubMed] [Google Scholar]
- 19. Murakami Y, Hayakawa M, Yano Y et al Discovering novel direct acting antiviral agents for HBV using in silico screening. Biochem Biophys Res Commun 2015; 456: 20–8. [DOI] [PubMed] [Google Scholar]
- 20. Yu X, Mertz JE. Distinct modes of regulation of transcription of hepatitis B virus by the nuclear receptors HNF4alpha and COUP‐TF1. J Virol 2003; 77: 2489–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Choi BH, Park GT, Rho HM. Interaction of hepatitis B viral X protein and CCAAT/enhancer‐binding protein alpha synergistically activates the hepatitis B viral enhancer II/pregenomic promoter. J Biol Chem 1999; 274: 2858–65. [DOI] [PubMed] [Google Scholar]
- 22. Geng X, Huang C, Qin Y et al Hepatitis B virus X protein targets Bcl‐2 proteins to increase intracellular calcium, required for virus replication and cell death induction. Proc Natl Acad Sci USA 2012; 109: 18471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. Phosphorylation‐dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol 1999; 145: 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao F, Xu G, Zhou Y et al MicroRNA‐26b inhibits hepatitis B virus transcription and replication by targeting the host factor CHORDC1 protein. J Biol Chem 2014; 289: 35029–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu F, Campagna M, Qi Y et al Alpha‐interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog 2013; 9: e1003613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Janahi EM, McGarvey MJ. The inhibition of hepatitis B virus by APOBEC cytidine deaminases. J Viral Hepat 2013; 20: 821–8. [DOI] [PubMed] [Google Scholar]
- 27. Lucifora J, Xia Y, Reisinger F et al Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 2014; 343: 1221–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhu J, Davoli T, Perriera JM et al Comprehensive identification of host modulators of HIV‐1 replication using multiple orthologous RNAi reagents. Cell Rep 2014; 9: 752–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhou T, Guo H, Guo JT, Cuconati A, Mehta A, Block TM. Hepatitis B virus e antigen production is dependent upon covalently closed circular (ccc) DNA in HepAD38 cell cultures and may serve as a cccDNA surrogate in antiviral screening assays. Antiviral Res 2006; 72: 116–24. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Inhibitory effect of known hepatitis B virus (HBV) inhibitors on recombinant HBV expressing NanoLuc (HBV/NL).
Fig. S2. Suppression of NanoLuc (NL) activity in hepatitis B virus (HBV)/NL‐infected HuH7/NTCP cells by siRNA treatment specific to HNF4A and NTCP.