

Abstract
Obesity is a risk factor for colorectal cancer. The accumulation of abdominal fat tissue causes abundant reactive oxygen species production through the activation of NADPH oxidase due to excessive insulin stimulation. The enzyme NADPH oxidase catalyzes the production of reactive oxygen species and evokes the initiation and progression of tumorigenesis. Apocynin is an NADPH oxidase inhibitor that blocks the formation of the NADPH oxidase complex (active form). In this study, we investigated the effects of apocynin on the development of azoxymethane‐induced colonic aberrant crypt foci in obese KK‐A y mice and on the development of intestinal polyps in Apc mutant Min mice. Six‐week‐old KK‐A y mice were injected with azoxymethane (200 μg/mouse once per week for 3 weeks) and given 250 mg/L apocynin or 500 mg/L apocynin in their drinking water for 7 weeks. Six‐week‐old Min mice were also treated with 500 mg/L apocynin for 6 weeks. Treatment with apocynin reduced the number of colorectal aberrant crypt foci in KK‐A y mice by 21% and the number of intestinal polyps in Min mice by 40% compared with untreated mice. Both groups of mice tended to show improved oxidation of serum low‐density lipoprotein and 8‐oxo‐2′‐deoxyguanosine adducts in their adipose tissues. In addition, the inducible nitric oxide synthase mRNA levels in polyp tissues decreased. Moreover, apocynin was shown to suppress nuclear factor‐κB transcriptional activity in vitro. These results suggest that apocynin and other NADPH oxidase inhibitors may be effective colorectal cancer chemopreventive agents.
Keywords: Apc mutant mice, apocynin, iNOS, KK‐Ay mice, NADPH oxidase
Obesity is a cause of diabetes mellitus type 2. Insulin resistance induces high levels of fasting glucose, insulin, and IGF1 in the blood. It is now well recognized that individuals with diabetes mellitus type 2 are at high risk for colorectal cancer development.1, 2 The factors that have been suggested to link diabetes mellitus type 2 with colorectal cancer development include hyperinsulinemia, high levels of IGF1 that accelerate cell viability and proliferation,3 and the dysregulated production of ROS and inflammatory cytokines.4 It has been shown that insulin activates NADPH oxidase, which produces superoxide and H2O2.5, 6
Phagocyte‐derived NADPH oxidase is a well‐known ROS‐producing enzyme that acts against bacterial infection and inflammation.7, 8 Additional non‐phagocyte‐derived NADPH oxidase homologs belong to the Duox family. There are seven isoforms in mammals: Nox1, Nox2, Nox3, Nox4, Nox5, Duox1, and Duox2.9 Nox1, Nox2, Nox4, and Nox5 are expressed in the endothelium, vascular smooth muscle cells, fibroblasts, or perivascular adipocytes. Nox1 is mainly expressed in differentiated colonic epithelial cells.10, 11 Additional homologs have not been identified or are expressed at such low levels that their roles have not been established. Interestingly, Nox/Duox members have been reported to be involved in cancer development. Nox1 stimulates mitogenesis, cell transformation and tumorigenesis when ectopically expressed in NIH3T3 fibroblasts and DU‐145 prostate epithelial cells.12 The overexpression of Nox1 has been observed in prostate, breast, ovarian, and colon cancers.13, 14, 15 Nox1 is overexpressed in human colon cancers and has been correlated with activating mutations in K‐ras.16 In Nox1 homozygous knockout mice, the implantation of tumorigenic B16F0 melanoma cells has been observed to result in smaller and less‐vascularized implanted tumors. This reduction is associated with the reduced expression levels of several genes, including VEGF, MMP‐2, MMP‐9, and NF‐κB.17 Thus, it is assumed that NADPH oxidase inhibitors may inhibit intestinal tumorigenesis by hindering ROS production and NF‐κB activation.
To transport electrons across membranes for the production of superoxides, the Nox/Duox family proteins must form a complex. The catalytic transmembrane protein Nox1 forms a complex with p22phox and the cytoplasmic subunits p67phox/NOXO1, p47phox/NOXA1, p40phox, and Rac1/2. Apocynin, which belongs to the methoxy‐substituted catechol family, effectively inhibits NADPH oxidase activity by blocking the formation of the NADPH oxidase complex; thus, it is used as a standard NADPH oxidase inhibitor in experimental research.9
In this study, we show the suppressive effects of apocynin on the number of AOM‐induced colorectal aberrant crypt foci (ACF) in obese KK‐A y mice and on intestinal polyp development in Min mice. Min mice have been reported to show high levels of oxidative stress.18 We showed that the suppressive effects of apocynin treatment on intestinal polyp formation in Min mice were partly explained by the suppression of iNOS. Moreover, apocynin was shown to suppress NF‐κB transcriptional activity in vitro.
Materials and Methods
Cell culture and chemicals
A human colon cancer cell line (SW48; ATCC, Manassas, VA, USA) and a murine macrophage cell line (RAW264; Riken Cell Bank, Tsukuba, Japan), were cultured in DMEM and RPMI‐1640, respectively, containing 10% FBS (HyClone Laboratories, Logan, UT, USA) and antibiotics at 37°C in a humidified incubator at 5% CO2. The apocynin, 1‐(4‐hydroxy‐3‐methoxyphenyl) ethanone, was purchased from Sigma‐Aldrich (SAFC, Buchs, Switzerland).
Animals and chemicals
Female 5‐week‐old KK‐A y/TaJcl (KK‐A y) mice were purchased from Clea Japan (Tokyo, Japan). Male 5‐week‐old C57BL/6J‐Apc Min/+ mice (Min mice) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and genotyped according to The Jackson Laboratory's protocol. Heterozygotes of the Min strain and wild‐type (C57BL/6J) mice were acclimated to laboratory conditions for 1 week. Four to five mice were housed per plastic cage with sterilized softwood chips as bedding in a barrier‐sustained animal room at 24 ± 2°C and 55% humidity on a 12:12 h light : dark cycle. The apocynin was well dissolved in drinking water at a concentration of 250 mg/L or 500 mg/L.19
Animal experiments
Food and water were available ad libitum. The clinical characteristics and mortality rates of the animals were observed daily. The body weights and food consumption rates were measured weekly. For the induction of ACF by AOM (Sigma‐Aldrich), 6‐week‐old female KK‐A y (n = 12) mice were given i.p. injections of AOM (200 μg/mouse) once a week for 3 weeks and 250 mg/L apocynin or 500 mg/L apocynin in their drinking water for 7 weeks. At the end of the experimental period, a blood sample was collected from the abdominal vein and the colorectum was removed, opened longitudinally, and fixed flat between sheets of filter paper in 10% buffered formalin for over 24 h. The colorectum was divided into proximal and rectal segments (1.5 cm in length), and the remainder was divided into proximal (middle) and distal halves. These colorectal sections were stained with 0.2% methylene blue (Merck, Darmstadt, Germany) and PBS, and the mucosal surfaces were assessed for ACF with a stereoscopic microscope as previously reported.20
To investigate the effects of apocynin on intestinal tumor formation, seven male 6‐week‐old Min mice were given 500 mg/L apocynin in their drinking water for 6 weeks, and eight male Min mice without apocynin treatment were used as the control. The intestinal tract was removed and separated into the small intestine, cecum, and colon. The small intestine was first divided to produce a proximal segment (4 cm in length), and the remainder was split into proximal (middle) and distal halves. The tumors in the proximal segment were counted, all tumors were picked up under a stereoscopic microscope, and the remaining intestinal mucosae (non‐tumor mucosal portions) were removed by scraping. They were then both stored at −80°C for further analyses using real‐time PCR. Additional segments were opened longitudinally and fixed flat between sheets of filter paper in 10% buffered formalin. The numbers and sizes of the tumors and their distributions in the intestines were assessed with a stereoscopic microscope. A portion of the liver, visceral fat, and kidneys were placed into 10% buffered formalin, and liver and visceral fat residues were frozen using liquid nitrogen and stored at −80°C. The experiments were carried out according to the Guidelines for Animal Experiments of the National Cancer Center (Tokyo, Japan) and were approved by the Institutional Ethics Review Committee for Animal Experimentation of the National Cancer Center.
Measurements of mouse serum lipid levels in mice
The serum levels of triglycerides, total cholesterol, and LDL were measured as reported previously.21 Mouse serum oxLDL levels were measured using an ELISA kit (USCN Life Science, Hubei, China) according to the manufacturer's protocol.
Quantification of 8‐oxo‐dG using LC‐MS/MS
Mouse adipose tissue DNA (from 100 mg tissue) was extracted using the QIAamp DNA Isolation Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions with minor modifications; deferoxamine mesylate (final concentration, 0.1 mM; Sigma‐Aldrich) was added to all samples to prevent the formation of artifactual oxidative adducts during the purification steps. The extracted DNA was dissolved in 25 μL distilled water and stored at −80°C for further analyses. DNA concentration and quality were determined using the NanoDrop ND‐1000 spectrometer (Thermo Fisher Scientific, Wilmington, DE, USA). 15N5‐8‐oxo‐dG was added to the DNA solutions prior to enzymatic digestion at a concentration of 1.02 nM. The enzymatic digestion conditions were as follows: DNA (2.6–7.0 μg) samples in 5 mM Tris‐HCl buffer (pH 7.4) were incubated with DNase I for 3 h. Next, nuclease P1, 10 mM sodium acetate (pH 5.3; final concentration, 10 mM), and ZnCl2 (final concentration, 34 mM) were added, and the samples were incubated for an additional 3 h at 37°C. Finally, alkaline phosphatase, phosphodiesterase I, and Tris base (final concentration, 15.4 mM) were added, and the samples were incubated for 18 h at 37°C. The samples were purified using Vivacon 500 (10‐kDa molecular weight cut‐off filters Sartorius, Goettingen, Germany), and 15 μL of each of the flow‐through fractions was subjected to LC‐MS/MS. 8‐Oxo‐7,8‐dihydro‐2′‐deoxyguanosine and its internal standard were quantified by LC‐MS/MS. Positive ions were acquired in the multiple reaction monitoring mode. Multiple reaction monitoring transitions were monitored; the cone voltages and collision energies used were as follows: 8‐oxo‐dG, [284→168, 35 V, 14 eV], and 15N5‐8‐oxodG, [289→173, 35 V, 14 eV]. The 15N5‐8‐oxo‐dG levels were normalized to the DNA concentrations.
Immunohistochemical staining
The small intestines of Min mice with intestinal polyps were fixed, embedded and then sectioned for further immunohistochemical examinations using the avidin–biotin complex immunoperoxidase technique with mouse monoclonal IgG anti‐PCNA and anti‐cyclin D1 antibodies (Merck Millipore, Billerica, MA, USA) at a 200 × and 100 × dilution, respectively. As the secondary antibody, biotinylated horse anti‐rabbit IgG affinity‐purified antibody was used at a 200 × dilution. Staining was carried out using avidin–biotin reagents (Vectastain ABC reagents; Vector Laboratories, Inc. Burlingame, CA, USA), 3,3′‐diaminobenzidine (Sigma‐Aldrich), and hydrogen peroxide, and the sections were counterstained with hematoxylin to facilitate orientation. As a negative control, consecutive sections were immunostained without exposure to the primary antibody. The ratio of PCNA‐positive cells was calculated by the formula: % = number of PCNA positive cells in polyp / number of whole cells in polyp (magnification, ×100).
Real‐time PCR analysis
The tissue samples from the intestinal mucosae or from the polyps of the mice were rapidly deep‐frozen in liquid nitrogen and stored at −80°C. Total RNA was isolated from the tissues using TRIzol reagent (Sigma‐Aldrich), and 1 μg aliquots in final volumes of 10 μL were used for the synthesis of cDNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real‐time PCR was carried out using the CFX96 thermal cycler (Bio‐Rad, Hercules, CA, USA) with the FastStart Universal SYBR Green Mix (2 ×) (Roche, Basel, Switzerland) according to the manufacturer's instructions. The primers used included mouse c‐Myc (5′‐GCTCGCCCAAATCCTGTACCT and 3′‐TCTCCACAGACACCACATCAATTTC), cyclin D1 (5′‐CCATGGAACACCAGCTCCTG and 3′‐CGGTCCAGGTAGTTCATGGC), GAPDH (5′‐TGTCAGCAATGCATCCTGCA and 3′‐TTACTCCTTGGAGGCCATGT), iNOS (5′‐CCGGCAAACCCAAGGTCTACGTT and 3′‐CACATCCCGAGCCATGCGCACAT), Nox1 (5′‐TCCCTTTGCTTCCTTCTTGA and 3′‐CCAGCCAGTGAGGAAGAGTC), p22phox (5′‐CGTGGCTACTGCTGGACGTT and 3′‐TGGACCCCTTTTTCCTCTTT), and Pai‐1 (5′‐GACACCCTCAGCATGTTCATC and 3′‐AGGGTTGCACTAAACATGTCAG). Cycling conditions were as follows: 95°C for 15 s, annealing at 60°C for 10 s, and 42 cycles at 72°C for 20 s after an initial step of 95°C for 10 min followed by a final elongation step at 72°C for 5 s. To assess the specificity of each primer set, melting curves were constructed for the amplicons generated by the PCR.
Luciferase assay of NF‐κB transcriptional activity
To measure NF‐κB transcriptional activity, the colon cancer cell line SW48 and rodent macrophage cell line RAW264 were seeded in 96‐well plates (2 × 104 cells/well). After 24 h of incubation, the cells were transiently transfected with 100 ng/well of the pGL4.32 (luc2P/NF‐κB RE/Hygro) (Promega, Madison, WI, USA) reporter plasmid and pGL4.73 (hRluc/SV40) (Promega) control plasmid using the FuGENE 6 Transfection Reagent (Roche) according to the manufacturer's instructions and cultured for 24 h. The cells were then treated with 200 μM apocynin for 24 h and, finally, firefly luciferase and Renilla luciferase activities were determined by the Luciferase Assay Systems and Renilla Luciferase Assay Systems (Promega), respectively. The values were normalized according to Renilla luciferase activity levels. The basal luciferase activity of the untreated cells was set as 1.0. The percent of luciferase activity for each treatment was calculated using data from triplicate wells.
Statistical analysis
Statistical analysis was carried out using Student's t‐test. Differences were considered to be statistically significant at P < 0.05.
Results
Suppression of AOM‐induced colorectal ACF in KK‐A y mice by apocynin
To determine the effects of apocynin on colorectal ACF development in KK‐A y mice, the KK‐A y mice were treated with AOM, with or without apocynin. Treatment with apocynin did not significantly change the food intake rates, behaviors, or body weights of the mice during the experimental period. The final body weights of the untreated 13‐week‐old female KK‐A y mice and those that were treated with 250 mg/L apocynin and 500 mg/L apocynin were 49.1 ± 3.3, 47.9 ± 2.4, and 45.0 ± 8.5 g, respectively. During the experimental period, no significant differences in body weights were observed between the groups.
Table 1 shows the numbers and distributions of colorectal ACF in the KK‐A y mice treated with or without apocynin. All KK‐A y mice treated with AOM developed colorectal ACF at 13 weeks. The total number of ACF in the group treated with 250 mg/L apocynin was reduced to 68.5% (P < 0.01) of the control value. The numbers of ACF in the middle and distal portions of the colons of the mice treated with 250 mg/L apocynin were reduced significantly (P < 0.01, 0.05). There were no significant differences in the number of aberrant crypts per focus between the groups. However, treatment with 250 mg/L apocynin reduced the numbers of small‐sized ACF (Fig. S1A).
Table 1.
Development of colorectal aberrant crypt foci (ACF) in KK‐A y mice treated with azoxymethane and apocynin
| Apocynin, mg/L | No. of mice with ACF | No. of ACF/colorectum | Mean no. of AC/focus | ||||
|---|---|---|---|---|---|---|---|
| Proximal | Middle | Distal | Rectum | Total | |||
| 0 | 12/12 | 1.1 ± 1.5 | 16.9 ± 4.8 | 38.0 ± 10.0 | 14.5 ± 8.3 | 70.5 ± 17.2 | 1.3 ± 0.1 |
| 250 | 12/12 | 0.3 ± 0.7 | 7.8 ± 5.9** | 28.0 ± 10.9* | 12.1 ± 6.3 | 48.3 ± 20.6** | 1.2 ± 0.1 |
| 500 | 12/12 | 0.2 ± 0.6 | 10.8 ± 9.8 | 32.6 ± 12.8 | 12.1 ± 4.7 | 55.8 ± 22.3 | 1.3 ± 0.1 |
*P < 0.05, **P < 0.01 versus 0 mg/L. Data are expressed as mean ± SD. AC, aberrant crypt.
Suppression of intestinal polyp formation in Min mice by apocynin treatment
Treatment of Min mice with 500 mg/L apocynin for 6 weeks also did not alter body weights, food intake rates, or clinical signs compared with the untreated mice throughout the experimental period. Table 2 summarizes the numbers and distributions of intestinal tumors in the untreated control group and 500 mg/L apocynin‐treated group. Almost all tumors developed in the small intestine, with only a few forming in the colon. Treatment with 500 mg/L apocynin led to a decrease in the total number of tumors to 60.7% (P < 0.05) of the untreated control value. The total number of tumors in the distal segment decreased by 42.2% in the apocynin group. The majority of tumors were observed to be between 0.5 and 3.0 mm in diameter. Treatment with 500 mg/L apocynin reduced the numbers of tumors of most sizes and significantly reduced the numbers of tumors that were 1.0–1.5 mm in size (Fig. S1B).
Table 2.
Number of intestinal tumors in Min mice treated with apocynin
| Apocynin, mg/L | No. of mice | No. of tumors/mouse | ||||
|---|---|---|---|---|---|---|
| Small intestine | Colon | Total | ||||
| Proximal | Middle | Distal | ||||
| 0 | 8 | 3.0 ± 0.6 | 12.1 ± 2.1 | 34.1 ± 4.6 | 1.1 ± 0.4 | 50.4 ± 6.1 |
| 500 | 7 | 2.6 ± 0.6 | 8.0 ± 1.7 | 19.7 ± 1.3* | 0.3 ± 0.8 | 30.6 ± 3.3* |
*P < 0.05 versus 0 mg/L. Data are mean ± SD.
To investigate the effects of apocynin on intestinal epithelial cell growth, the intestinal tumor sections of Min mice were immunohistochemically stained with anti‐PCNA antibody. The proportions of PCNA‐positive cells in the intestinal tumor sections significantly decreased by 48.2% in the mice treated with 500 mg/L apocynin (P < 0.05) (Fig. 1a). Immunohistopathological analysis for cyclin D1 in intestinal tumors in Min mice also showed a significant decrease of cyclin D1‐positive cells in the intestinal tumor sections (Fig. S2). To assess the mechanisms underlying the inhibition of cell growth by apocynin, several cell growth‐related genes were analyzed by real‐time PCR. The downregulation of the expression levels of c‐Myc and cyclin D1 in the small intestinal tumors of Min mice were apparent compared with those of the untreated group (Fig. 1b,c).
Figure 1.
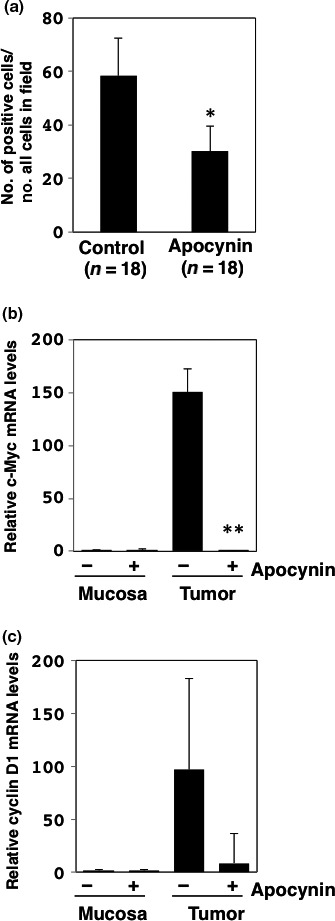
Changes in cell cycle‐related factors in intestinal tumors treated with or without apocynin. (a) Immunohistochemistry was performed for determination of proliferating cell nuclear antigen (PCNA)‐positive cell numbers in tumor sections (n = 18) of small intestines of Min mice treated with 500 mg/L apocynin (n = 7) and untreated controls (n = 8). Ratio of the number of PCNA‐positive cells per whole cell in field (100 ×) is shown. Data are represented by mean ± SD. *P < 0.05 versus untreated control. Real‐time PCR analysis was carried out to obtain c‐Myc (b) and cyclin D1 (c) mRNA levels. Values were set at 1.0 in untreated controls, and relative levels were expressed as mean ± SD (n = 4, a pair of mucosa and tumor samples for apocynin or untreated controls). **P < 0.01 versus untreated control. GAPDH mRNA levels were used to normalize data.
Suppressive effects of apocynin on oxidative stress in mice
To clarify the suppression of ROS production by NADPH oxidase inhibition, we examined the effects of apocynin on serum oxLDL‐cholesterol levels in the KK‐A y and Min mice. The treatment of the KK‐A y mice with 250 mg/L apocynin significantly suppressed serum oxLDL‐cholesterol levels (Table S1). The suppression of serum oxLDL‐cholesterol levels was observed in KK‐A y and Min mice at a dose of 500 mg/L apocynin, although the results were not statistically significant (Table S2). Moreover, serum triglyceride, total cholesterol, and LDL‐cholesterol levels were measured to assess their effects on the oxLDL‐cholesterol levels. The apocynin treatments did not show the serum triglyceride, total cholesterol, or LDL‐cholesterol levels to decrease in a dose‐dependent manner (Tables S1,S2) in KK‐A y mice; decreases were only observed in Min mice.
To evaluate whether treatment with apocynin improved the oxidative status in other parameters, DNA was extracted from the adipose tissues of both KK‐A y and Min mice, and 8‐oxo‐dG adducts were quantified per nucleoside by LC‐MS/MS. The 8‐oxo‐dG levels in the adipose tissues of KK‐A y and Min mice tended to decrease following the 500 mg/L apocynin treatment (Fig. S3).
Suppression of iNOS mRNA levels in intestinal tumor sections from Min mice treated with apocynin
To confirm the expression levels of the target molecules of apocynin in Min mice, the levels of NADPH oxidase (Nox1, p22phox, and Nox2) mRNA were examined by quantitative RT‐PCR. The levels of Nox1 and p22phox increased by 2.2‐fold and 10.4‐fold, respectively, in the intestinal tumor sections compared with those of the non‐tumor mucosal sections in Min mice (Fig. 2a,b). However, the expression levels of Nox2 did not differ between the normal mucosal and tumor sections (Fig. 2c). Nox1, p22phox, and Nox2 mRNA levels decreased by 14.3%, 61.2%, and 65.0%, respectively, in the tumor sections following treatment with 500 mg/L apocynin compared with the untreated group (Fig. 2).
Figure 2.
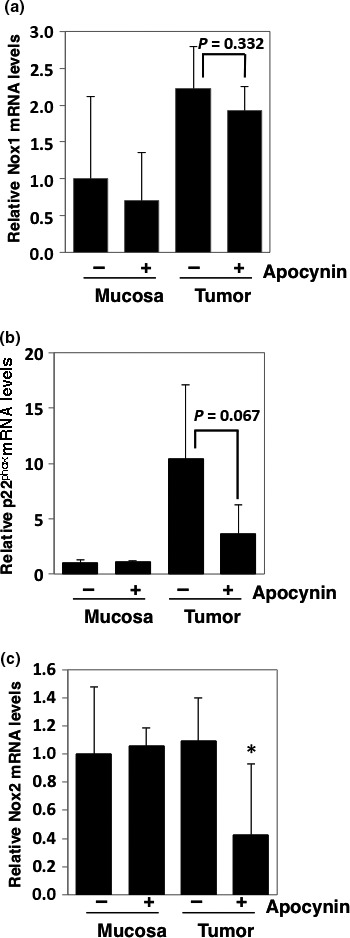
Relative expression levels of NADPH oxidase‐associated genes in intestinal mucosae and tumors of Min mice. Real‐time PCR analysis was used to obtain Nox1 (a), p22phox (b), and Nox2 (c) mRNA expression levels. Values were set at 1.0 in untreated controls, and relative levels were expressed as mean ± SD (n = 4, a pair of mucosa and tumor samples for apocynin or untreated controls). *P < 0.05 versus untreated control. GAPDH mRNA levels were used to normalize data.
Intestinal epithelial cell growth is generally affected by inflammation‐related factors, including cytokines and growth factors. Thus, we next focused on the expression levels of interleukin‐6, iNOS, and Pai‐1 in the intestinal tissues. In the tumor sections, the Pai‐1 and iNOS mRNA levels were upregulated compared with the levels observed in the non‐tumor mucosal sections, and these levels decreased following 500 mg/L apocynin treatment (Fig. 3a,b). However, interleukin‐6 mRNA levels decreased only in the non‐tumor mucosal sections following apocynin treatment (Fig. 3c).
Figure 3.
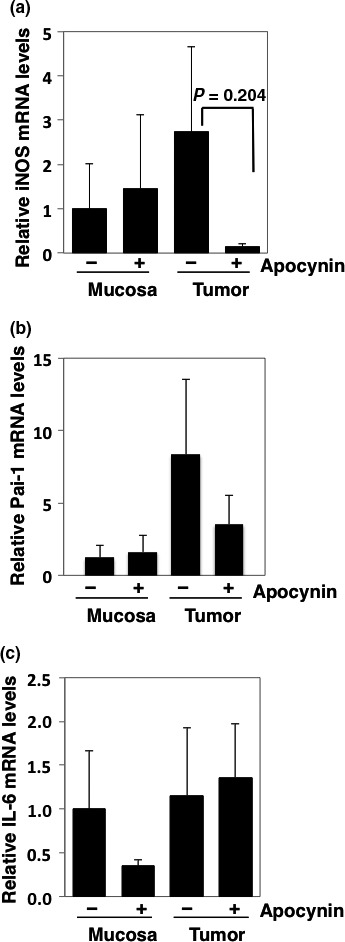
Relative expression levels of inflammation‐ and carcinogenesis‐related genes in intestinal mucosae and tumors of Min mice treated with or without 500 mg/L apocynin. Real‐time PCR analysis was carried out to obtain iNOS (a), Pai‐1 (b), and interleukin‐6 (IL‐6) (c) mRNA levels. Values were set at 1.0 in untreated controls, and relative levels were expressed as mean ± SD (n = 4, a pair of mucosa and tumor samples for apocynin or untreated controls). GAPDH mRNA levels were used to normalize data.
Nuclear factor‐κB transcription is inhibited by apocynin treatment
To examine the effects of apocynin on the transcriptional factors that regulate the expression of iNOS, we examined NF‐κB transcriptional activity in SW48 (human colon cancer cell line) and RAW264 (murine macrophage cell line) cells. The NF‐κB transcriptional activities in both cell lines were reduced by 24 h of 200 μM apocynin treatment (Fig. 4).
Figure 4.
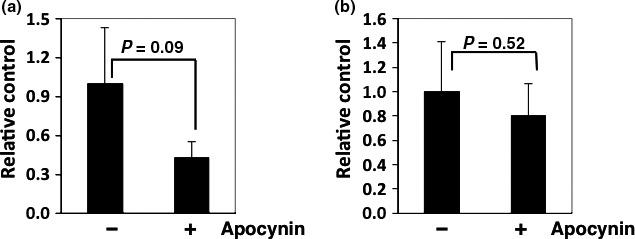
Nuclear factor‐κB (NF‐κB) transcriptional activity levels in human colon cancer cells and rodent macrophage cells. SW48 (a) and RAW264 (b) cells were seeded in 96‐well plates (2 × 104 cells/well) and were transiently transfected with pGL4.32 (luc2P/NF‐kB RE/Hygro) reporter plasmid and pGL4.73 (hRluc/SV40) control plasmid for 24 h. Cells were then treated with 200 μM apocynin for 24 h, and firefly luciferase and Renilla luciferase activities were determined by luciferase assay systems and Renilla luciferase assay systems, respectively. Basal luciferase activity of untreated cells was set at 1.0. Percentage of luciferase activity was calculated from data obtained from triplicate wells for each treatment. Values were normalized by Renilla luciferase activity levels. Data are expressed as mean ± SD (n = 3).
Discussion
In the present study, we showed the suppressive effects of apocynin on AOM‐induced colorectal ACF formation in obese KK‐A y mice and on intestinal tumor development in Min mice. Moreover, the clear suppression of c‐Myc and cyclin D1 mRNA levels and a reduced ratio of PCNA‐positive epithelial cells were observed in the intestinal tumor sections of Min mice. Treatment with apocynin tended to decrease levels of oxidative stress in both KK‐A y and Min mice. The mechanism involved in the suppressive effects of apocynin treatment on oxidative stress was partially related to the inhibition of NADPH oxidase, resulting in reduced ROS production, and the suppression of iNOS mRNA levels. In addition, the reporter gene assay revealed that apocynin inhibited NF‐κB transcriptional activity in human colon cancer cells.
It has been reported that Nox1 knockdown induces G0/G1 arrest in a human colon cell line, HT29, whereas the same knockdown treatment in Caco2 cells strongly induces apoptosis.22 In addition, apocynin also blocks cell growth by inducing G0/G1 arrest and downregulating cyclin D1 in a human prostate cancer cell line, LNCaP.19 We obtained similar results, showing that apocynin suppressed c‐Myc and cyclin D1 mRNA levels and reduced the ratio of PCNA‐positive epithelial cells in the intestinal polyp sections of Min mice. This inhibition of cell growth factors may result in a reduction in AOM‐induced colorectal ACF formation in obese KK‐A y mice and intestinal tumor development in Min mice.
Because apocynin is known to be an NADPH oxidase inhibitor, we examined ROS activity by measuring serum oxLDL levels, and the number of 8‐oxo‐dG adducts per nucleoside in the adipose tissue was quantified by LC‐MS/MS. Both assessments indicated a tendency toward reduction by the apocynin treatment. Compared with other serum lipids, serum oxLDL levels were not affected by the amount of total serum cholesterol or LDL. Because ROS are generally produced by mitochondria and peroxisomes and the enzymes cytochrome P450 and NADPH oxidase,9, 23, 24 the targeting of only NADPH oxidase does not sufficiently reduce whole‐body ROS production. We speculated that apocynin did not affect whole‐body ROS production, but may play an important role in the local tumor parts of the intestine in which NADPH oxidase is overexpressed. Neoplastic lesions with characteristically elevated levels of NADPH oxidase, such as human colon adenomas and well‐differentiated adenocarcinomas,15 may be good targets for NADPH oxidase inhibitors.
The present study is the first to show that Nox1 and p22 phox mRNA levels tend to be elevated in the intestinal tumors of Min mice. It has been reported that PhIP‐induced colon tumors show increased Nox1 expression and NF‐κB activation.22 PhIP is a heterocyclic amine that has been associated with colon cancer in rodents. In addition, our results imply that increased Nox/Duox expression is not specific to heterocyclic amines or to other colon carcinogens, such as AOM. It has been reported that Nox1 overexpression in human colon cancers correlate with activating mutations in K‐ras.16 However, considering that genetic alterations in β‐catenin or APC have been detected in over 80% of human colorectal cancers and that Min mice possessed Apc mutations in this study, β‐catenin signaling may also be influenced by the expression of Nox1. Further research is needed to confirm this possibility.
Reactive oxygen species affect cancer cell proliferation through the activation of NF‐κB transcription factors,25 which results in the induction of cyclin D1 and cyclin‐dependent kinase. Moreover, NF‐κB induces inflammatory cytokines, growth factors, and inflammation‐related enzymes, such as iNOS. In cultured rodent macrophage cells and human colon cancer cells, apocynin has been shown to inhibit NF‐κB transcriptional activity in this study. Decreased production of intracellular ROS by apocynin could be involved in the inhibition of NF‐κB transcriptional activity. Moreover, inhibition of Akt phosphorylation by apocynin26 could partly inhibit NF‐κB transcriptional activity through IKK activation. These mechanisms may partially explain the lowered expression levels of c‐Myc, cyclin D1, and iNOS, which are downstream targets of NF‐κB, observed in the intestinal polyps of Min mice in this study.
In summary, we have provided the first evidence of the involvement of Nox/Duox in AOM‐induced colon carcinogenesis in obese mice and in intestinal tumor formation in Min mice using an NADPH oxidase inhibitor, apocynin. The chemopreventive effect of apocynin on non‐obese ordinary mice is another concern. Although Min mice are not obese mice, AOM‐induced non‐obese ordinary mouse colorectal cancer will be a more suitable model to demonstrate the effect of apocynin on carcinogenesis in non‐obese ordinary mice. In addition, clarifying the correlation between Nox/Duox family members and NF‐κB–iNOS during colon cancer development warrants further investigation. We conclude that apocynin and other NADPH oxidase inhibitors may be effective colorectal cancer chemopreventive agents. Further evidence, such as that obtained from human trials, is required.
Disclosure Statement
The authors have no conflicts of interest.
Abbreviations
- ACF
aberrant crypt foci
- AOM
azoxymethane
- IGF
insulin‐like growth factor
- Duox
dual oxidase
- iNOS
inducible nitric oxide synthase
- LC‐MS/MS
liquid chromatography tandem mass spectrometry
- LDL
low‐density lipoprotein
- NF‐κB
nuclear factor‐κB
- Nox
NADPH oxidase
- oxLDL
oxidized LDL
- 8‐oxo‐dG
8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine
- Pai
plasminogen activator inhibitor
- PCNA
proliferating cell nuclear antigen
- ROS
reactive oxygen species
Supporting information
Fig. S1. Effect of apocynin on aberrant crypt foci size and intestinal tumor size distribution in the mice.
Fig. S2. Histopathological and immunohistopathological analysis of intestinal tumors in Min mice.
Fig. S3. Oxidative adduct qualification in DNA extracted from adipose tissues of mice. Mouse DNA was extracted from adipose tissues of KK‐A y and Min mice treated with or without 500 mg/L apocynin. The numbers of 8‐oxodG adducts per nucleoside were quantified by liquid chromatograph tandam mass spectrometry (LC‐MS/MS). Data are expressed as mean ±SD (N=4).
Table S1. Serum lipid levels in KK‐A y mice with or without apocynin treatment.
Table S2. Serum lipid levels in Min mice with or without apocynin treatment.
Acknowledgments
We received technical support from Ms. Shimura M. We thank Mr. Uchiya N. and the National Cancer Center Research Core Facility for some of the analyses performed in this study. The Core Facility was supported by the National Cancer Center Research and Development Fund (23‐A‐7). This work was supported by Grants‐in‐Aid for Cancer Research, the Third‐Term Comprehensive 10‐Year Strategy for Cancer Control from the Ministry of Health, Labor, and Welfare of Japan, and the Yakult Bio‐science Foundation, and Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development (15ck0106098h0002).
Cancer Sci 106 (2015) 1499–1505
Funding Information
National Cancer Center Research and Development Fund; Ministry of Health, Labor, and Welfare of Japan; Yakult Bio‐science Foundation; Japan Agency for Medical Research and Development.
References
- 1. Moghaddam AA, Woodward M, Huxley R. Obesity and risk of colorectal cancer: a meta‐analysis of 31 studies with 70,000 events. Cancer Epidemiol Biomarkers Prev 2007; 16: 2533–47. [DOI] [PubMed] [Google Scholar]
- 2. Khandekar MJ, Cohen P, Spiegelman BM. Molecular mechanisms of cancer development in obesity. Nat Rev Cancer 2011; 11: 886–95. [DOI] [PubMed] [Google Scholar]
- 3. Ewton DZ, Kansra S, Lim S, Friedman E. Insulin‐like growth factor‐I has a biphasic effect on colon carcinoma cells through transient inactivation of forkhead1, initially mitogenic then mediating growth arrest and differentiation. Int J Cancer 2002; 98: 665–73. [DOI] [PubMed] [Google Scholar]
- 4. Ishino K, Mutoh M, Totsuka Y, Nakagama H. Metabolic syndrome: a novel high‐risk state for colorectal cancer. Cancer Lett 2013; 334: 56–61. [DOI] [PubMed] [Google Scholar]
- 5. Lawrece JC Jr, Larner J. Activation of glycogen synthase in rat adipocytes by insulin and glucose involves increased glucose transport and phosphorylation. J Biol Chem 1978; 253: 2104–13. [PubMed] [Google Scholar]
- 6. Mukherjee SP, Lane RH, Lynn WS. Endogenous hydrogen peroxide and peroxidative metabolism in adipocytes in response to insulin and sulfhydryl reagents. Biochem Pharmacol 1978; 27: 2589–94. [DOI] [PubMed] [Google Scholar]
- 7. Gloire G, Legrand‐Poels S, Piette J. NF‐kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol 2006; 72: 1493–505. [DOI] [PubMed] [Google Scholar]
- 8. Quinn MT, Ammons MC, Deleo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci 2006; 111: 1–20. [DOI] [PubMed] [Google Scholar]
- 9. Bedard K, Krause KH. The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007; 87: 245–313. [DOI] [PubMed] [Google Scholar]
- 10. Geiszt M, Witta J, Baffi J, Lekstrom K, Leto TL. Dual oxidases represent novel hydrogen peroxide sources supporting mucosal surface host defense. FASEB J 2003; 17: 1502–4. [DOI] [PubMed] [Google Scholar]
- 11. Szanto I, Rubbia‐Brandt L, Kiss P et al Expression of NOX1, a superoxide‐generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J Pathol 2005; 207: 164–76. [DOI] [PubMed] [Google Scholar]
- 12. Suh YA, Arnold RS, Lassegue B et al Cell transformation by the superoxide‐generating oxidase Mox1. Nature 1999; 401: 79–82. [DOI] [PubMed] [Google Scholar]
- 13. Lim SD, Sun C, Lambeth JD et al Increased Nox1 and hydrogen peroxide in prostate cancer. Prostate 2005; 62: 200–7. [DOI] [PubMed] [Google Scholar]
- 14. Desouki MM, Kulawiec M, Bansal S, Das GM, Singh KK. Cross talk between mitochondria and superoxide generating NADPH oxidase in breast and ovarian tumors. Cancer Biol Ther 2005; 4: 1367–73. [DOI] [PubMed] [Google Scholar]
- 15. Fukuyama M, Rokutan K, Sano T, Miyake H, Shimada M, Tashiro S. Overexpression of a novel superoxide‐producing enzyme, NADPH oxidase 1, in adenoma and well differentiated adenocarcinoma of the human colon. Cancer Lett 2005; 221: 97–104. [DOI] [PubMed] [Google Scholar]
- 16. Laurent E, McCoy JW 3rd, Macina RA et al Nox1 is over‐expressed in human colon cancers and correlates with activating mutation in K‐Ras. Int J Cancer 2008; 123: 100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Garrido‐Urbani S, Ugur T, Schleussner E et al Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS One 2011; 6: e14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ikeda K, Mutoh M, Teraoka N, Nakanishi H, Wakabayashi K, Taguchi R. Increase of oxidant‐related triglycerides and phosphatidylcholines in serum and small intestinal mucosa during development of intestinal polyp formation in Min mice. Cancer Sci 2011; 102: 79–87. [DOI] [PubMed] [Google Scholar]
- 19. Suzuki S, Shiraga K, Sato S et al Apocynin, an NADPH oxidase inhibitor, suppresses rat prostate carcinogenesis. Cancer Sci 2013; 104: 1711–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mutoh M, Watanabe K, Kitamura T et al Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res 2002; 62: 28–32. [PubMed] [Google Scholar]
- 21. Usui S, Nakamura M, Jitsukata K, Nara M, Hosaki S, Okazaki M. Assessment of between‐instrument variations in a HPLC method for serum lipoproteins and its traceability to reference methods for total cholesterol and HDL‐cholesterol. Clin Chem 2000; 46: 63–72. [PubMed] [Google Scholar]
- 22. Wang R, Dashwood WM, Nian H et al NADPH oxidase overexpression in human colon cancers and rat colon tumors induced by 2‐amino‐1‐methyl‐6‐phenylimidazo[4,5‐b]pyridine (PhIP). Int J Cancer 2011; 128: 2581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu WS. The signaling mechanism of ROS in tumor progression. Cancer Metastasis Rev 2006; 25: 695–705. [DOI] [PubMed] [Google Scholar]
- 24. Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Oxidative stress in prostate cancer. Cancer Lett 2009; 282: 125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sen CK, Packer L. Antioxidant and redox regulation of gene transcription. FASEB J 1996; 10: 709–20. [DOI] [PubMed] [Google Scholar]
- 26. Lirdprapamongkol K, Kramb JP, Suthiphongchai T et al Vanillin suppresses metastatic potential of human cancer cells through PI3K inhibition and decreases angiogenesis in vivo. J Agric Food Chem 2009; 57: 3055–63. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Effect of apocynin on aberrant crypt foci size and intestinal tumor size distribution in the mice.
Fig. S2. Histopathological and immunohistopathological analysis of intestinal tumors in Min mice.
Fig. S3. Oxidative adduct qualification in DNA extracted from adipose tissues of mice. Mouse DNA was extracted from adipose tissues of KK‐A y and Min mice treated with or without 500 mg/L apocynin. The numbers of 8‐oxodG adducts per nucleoside were quantified by liquid chromatograph tandam mass spectrometry (LC‐MS/MS). Data are expressed as mean ±SD (N=4).
Table S1. Serum lipid levels in KK‐A y mice with or without apocynin treatment.
Table S2. Serum lipid levels in Min mice with or without apocynin treatment.