

Abstract
Transforming growth factor‐β (TGF‐β) is a potent growth inhibitor in normal epithelial cells. However, a number of malignant tumors produce excessive amounts of TGF‐β, which affects the tumor‐associated microenvironment by furthering the progression of tumorigenicity. Although it is known that the tumor‐associated microenvironment often becomes hypoxic, how hypoxia influences TGF‐β signaling in this microenvironment is unknown. We investigated whether TGF‐β signaling is influenced by long‐term exposure to hypoxia in Lewis lung carcinoma (LLC) cells. When the cells were exposed to hypoxia for more than 10 days, their morphology was remarkably changed to a spindle shape, and TGF‐β‐induced Smad2 phosphorylation was enhanced. Concomitantly, TGF‐β‐induced transcriptional activity was augmented under hypoxia, although TGF‐β did not influence the activity of a hypoxia‐responsive reporter. Consistently, hypoxia influenced the expression of several TGF‐β target genes. Interestingly, the expressions of TGF‐β type I receptor (TβRI), also termed activin receptor like kinase‐5 (ALK5), and TGF‐β1 were increased under the hypoxic condition. When we monitored the hypoxia‐inducible factor‐1 (HIF‐1) transcriptional activity by use of green fluorescent protein governed by the hypoxia‐responsive element in LLC cells transplanted into mice, TGF‐β‐induced Smad2 phosphorylation was upregulated in vivo. Our results demonstrate that long‐term exposure to hypoxia might alter responsiveness to TGF‐β signaling and affected the malignancy of LLC cells.
Keywords: Hypoxia, Lewis lung carcinoma, Smad, transforming growth factor‐β, tumor microenvironment
Hypoxia, a typical feature of solid tumor microenvironments, is associated with aberrantly accelerated proliferation of cancer cells and has, therefore, received extensive attention as an inducer of metastasis.1, 2 Hypoxia is capable of stabilizing the hypoxia‐inducible factor‐1α (HIF‐1α) protein, which is ubiquitylated by an E3‐ubiquitin ligase, pVHL, under normoxic conditions. The stabilized HIF‐1α then makes a heterodimer with HIF‐1β (alternatively termed Arnt) to transcriptionally regulate hypoxia target genes involved in metastasis, angiogenesis, epithelial‐to‐mesenchymal transition (EMT), and histone demethylation.3, 4, 5 Thus, elucidating the molecular mechanisms for the physiological actions linked to hypoxia is important for understanding tumor progression.
To detect hypoxic regions, immunohistochemical analysis is performed with an anti‐HIF‐1α antibody and/or a hypoxic marker, pimonidazole, which binds to thiol‐containing proteins. However, HIF‐1α‐positive cells do not always overlap with the regions stained with pimonidazole.6, 7 Therefore, a fluorescence protein reporter combined with a hypoxia‐responsive element (HRE) might be a good tool for monitoring HIF‐1α‐positive cells. Such a system allows us to detect hypoxic regions in tumors in real time because the fluorescence intensity is inversely correlated with the oxygen tension in this system.8
The TGF‐β family is a family of multifunctional cytokines required for both normal embryonic development and homeostasis of adult tissues.9, 10 The TGF‐β family transduces its signal to the nucleus through the formation of heteromeric receptor complexes between specific type I and type II serine/threonine kinase receptors known as the TGF‐β type I (TβRI; also termed activin receptor‐like kinase‐5 [ALK5]) and type II receptors (TβRII), respectively. Acting downstream of TβRII, ALK5 phosphorylates two serine residues at the extreme C‐terminal end of receptor‐regulated Smads (R‐Smads; i.e. Smad2 and Smad3), which play crucial roles in canonical TGF‐β signaling. Subsequently, two activated R‐Smads bind to a common partner Smad, Smad4, to make a ternary complex and accumulate in the nucleus, where they act as transcriptional regulators to govern TGF‐β target genes.
The TGF‐β family potentiates cytostatic actions through the inhibition of c‐myc expression and induction of the cyclin‐dependent kinase (CDK) inhibitors p15 and p21.10 These CDK inhibitors keep the cell cycle at G1 arrest. Although loss of cytostatic actions seems to be an essential step of tumor progression, several cancer cells still possess TGF‐β signaling pathways other than that for cytostatic actions. Indeed, a number of malignant tumors produce excessive amounts of TGF‐β, which are required by the tumor in the later stages of tumorigenicity, through induction of the EMT mediated by Twist, Snail and Slug.11 Thus, growing lines of evidence indicate that TGF‐β influences tumor‐associated microenvironments so that tumor cells ultimately acquire malignant transformation.
In the present study, we examined how hypoxia affects TGF‐β signaling in LLC cells. In addition, we examined the effects of TGF‐β signaling in tumor tissue in vivo. Interestingly, TGF‐β signaling was augmented in LLC cells under the hypoxic condition.
Materials and Methods
Cell culture
Lewis lung carcinoma (LLC) cells were cultured in DMEM (Nacalai Tesque, Kyoto Japan) containing 10% FCS (Invitrogen, Carlsbad, CA, USA), 1× MEM Non‐Essential Amino Acids Solutio (Sigma, St. Louis, MO, USA) and 100 U/mL penicillin/streptomycin. To obtain stable transformants, the LLC cells were maintained in DMEM containing 10% FCS and 1200 μg/mL hygromycin B (Wako, Tokyo, Japan).
Transcriptional reporter assays
One day before transfection, the LLC cells were seeded at 1 × 105 cells/well in 12‐well plates. The cells were transfected the indicated reporter construct and pCH110 (GE Healthcare, Uppsala, Sweden) using 2.5 mg/mL polyethyleneimine (Polysciences, Warrington, PA, USA). Where indicated, 5 ng/mL TGF‐β was added to the wells 24 h after transfection. Subsequently, the cells were cultured in DMEM containing 0.3% FCS for 18 h. The lysates were prepared and the luciferase and beta‐galactosidase activities were then measured. For all experiments, the values for luciferase and β‐galactosidase activities were corrected for differences in transfection efficiency by normalizing each luciferase value to the corresponding value for beta‐galactosidase. Each transfection was carried out in triplicate and repeated at least twice. Values are presented as the means ± SD (n = 3).
Antibodies
Antibodies were obtained from the following sources: mouse monoclonal anti‐GFP antibody from Wako; mouse anti‐β‐actin monoclonal antibody from Sigma; mouse anti‐Smad2/3 antibody from BD Transduction Laboratories (San Jose, CA, USA); rabbit polyclonal anti‐Smad4 and anti‐ALK5 antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA); rabbit polyclonal anti‐cleaved caspase‐3 antibody from Cell Signaling Technology (Danvers, MA USA); and rabbit polyclonal anti‐Ki‐67 antibody from Leica (Newcastle, UK). Rabbit polyclonal anti‐phosphorylated Smad2 (PS2) and anti‐Smad2 antibodies were prepared in‐house.12
Immunoprecipitation and western blot analysis
Immunoprecipitation and western blot analysis was performed as described previously.13, 14 Briefly, 1 day before the starvation, LLC cells under either normoxia or hypoxia were seeded at 5 × 105 cells/well in 6‐cm plates. Then, the cells were cultured in DMEM containing 0.3% FCS for 18 h, followed by stimulation of cells with 5 ng/mL TGF‐β for 1 h. Subsequently, cells were lysed in 500 μL TNE buffer (10 mM Tris [pH 7.4]; 150 mM NaCl; 1 mM EDTA; 1% NP‐40; 1 mM PMSF; 5 μg/mL leupeptin; 100 U/mL aprotinin; 2 mM sodium vanadate; 40 mM NaF; and 20 mM β‐glycerophosphate). The cell lysate were immunoprecipitated with anti‐Smad2/3 antibody and ProteinG‐Sepharose 4B beads, and boiled for 10 min in sample buffer. The samples were separated by SDS‐PAGE and transferred to Hybond‐C Extra membranes (GE Healthcare). The membranes were probed with the indicated antibodies. Primary antibodies were detected with HRP‐conjugated goat anti‐rabbit or anti‐mouse IgG antibody (GE Healthcare) with chemiluminescent substrate (Thermo Scientific, Rockford, IL, USA).
Detection of apoptotic cells
As a positive control, LLC cells (1 × 106 cells/10 cm dish) were stimulated with 0.4 mM H2O2 in DMEM containing 0.3% FCS for 24 h. Then, cell lysates were used for western blot analysis. To analyze apoptotic cells using a cell sorter (SH800; Sony, Tokyo, Japan), LLC cells were trypsinized and fixed with ice‐cold 70% ethanol for 30 min at 4°C. After cells were resuspended with PBS containing 40 μg/mL RNase A and 8 μg/mL propidium iodide, the apoptotic cells were measured. Data were analyzed using FlowJo software (Tree Star, San Carlos, CA, USA).
Transforming growth factor‐β1 ELISA
The amounts of TGF‐β1 in the culture supernatants were determined using the mouse TGF‐β1 ELISA kit (R&D Systems, Minneapolis, MI, USA) according to the manufacturer's instructions. After LLC cells (5 × 105 cells/6 cm dish) were cultured with 3 mL of PANSERIN 401 serum free medium (PAN Biotech, Passau, Germany) for 24 h, media were collected. For determination of the total TGF‐β1 concentrations, media were treated according to the method of an acidification procedure prior to the ELISA assay.
Hypoxic tumor studies in vivo
Lewis lung carcinoma cells carrying p5HRE‐EGFP (LLC/HIFGFP‐3 cells; 2.5 × 105 cells per 100 μL in 0.9% NaCl) were transplanted subcutaneously into C57BL/6 mice. The mice were killed 14 days after inoculation. Then, the transplanted tumor grafts were removed, embedded into NEG 50 (Thermo Scientific) and frozen in liquid nitrogen for sectioning.
Immunofluorescence and histology
Frozen sections (10 μm) were cut with a CM1850 cryostat (Leica), mounted on Cryofilm (Leica) and fixed in 100% ethanol. The films were washed three times with PBS, permeabilized with 0.1% Triton X‐100 (Sigma) for 10 min, and blocked with Blocking Reagent (PerkinElmer, Boston, MA) for 1 h at 37°C. Mouse anti‐GFP (1:200) and rat anti‐PECAM1 (1:200) antibodies in Blocking Reagent were added and incubated for 1 h at 37°C. The films were washed three times with PBS and then incubated with Alexa488‐conjugated goat anti‐mouse IgG (Molecular Probes, Eugene, OR, USA) or Alexa594‐conjugated goat anti‐rabbit IgG (Molecular Probes) antibody at 1:200 for 1 h at room temperature. After the nuclei were stained with 2 μg/mL DAPI for 10 min, the samples were washed three times with distilled water, and the fluorescence signals were visualized by confocal microscopy (Olympus, Tokyo, Japan).
RNA isolation and RT‐PCR
The LLC cells were seeded at 3 × 105 cells/dish in 6 cm dishes 1 day before the experiments. The cells were starved overnight with DMEM containing 0.3% FCS and stimulated with or without 5 ng/mL TGF‐β for the indicated times. Total RNA was isolated using the ReliaPrep RNA Cell Miniprep System (Promega, Madison, WI, USA). Reverse transcription was carried out with the PrimScript II First Strand, Osaka, Japan cDNA Synthesis Kit (Takara, Ohtsu, Japan). RT‐PCR was performed with BlendTaq (TOYOBO). β‐actin was used as an internal control. The primer sequences used are described in Table 1.
Table 1.
PCR conditions to amplify each cDNA
| Primer | Sequence (5′–3′) | Annealing temperature (°C) | Cycle | Length (bp) | |
|---|---|---|---|---|---|
| Tgf‐β1 | F: | AGGAGACGGAATACAGGGCT | 60 | 25 | 482 |
| R: | CCACGTAGTAGACGATGGGC | ||||
| Smad7 | F: | AAGTGTTCAGGTGGCCGGATCTCAG | 60 | 25 | 205 |
| R: | ACAGCATCTGGACAGCCTGCAGTTG | ||||
| Snail | F: | AGGACAGTGGCAAAAGCTCCCA | 60 | 25 | 381 |
| R: | AGCGGTCAGCAAAAGCACGGTT | ||||
| p21 | F: | CTAGGGGAATTGGAGTCAGGC | 60 | 25 | 413 |
| R: | GAGTGCAAGACAGCGACAAG | ||||
| ALK5 | F: | TTGCTGCAATCAGGACCACTGC | 60 | 27 | 189 |
| R: | ACACGGTGGTGAATGACAGTGC | ||||
| β‐Actin | F: | TGAACCCTAAGGCCAACCGTG | 60 | 22 | 396 |
| R: | GCTCATAGCTCTTCTCCAGGG | ||||
Proximity ligation assay
The proximity ligation assay (PLA) method has been described previously.15 To visualize the fluorescence, a confocal microscope (Olympus) was used.
Statistical analyses
Statistically significant differences were determined using the t‐test. Probability values below 0.05 were considered significant.
Ethical conduct of animal experiments
All animal experiments were approved by the Animal Research Committee of Tokyo University of Pharmacy and Life Sciences (Tokyo, Japan) and performed in accordance with the university's animal experiment guidelines.
Results
Effect of hypoxia on Lewis lung carcinoma cell morphology
To test whether hypoxia affects the LLC cell morphology, LLC cells were cultured under hypoxic conditions (1% O2) for the indicated times. While under hypoxia, the cells were passaged every other day at 3 × 105 cells/6 cm dish. Photographs were taken 0, 2, 6 and 10 days after subjection to hypoxia (Fig. 1a). Under hypoxia, the morphology of the LLC cells gradually altered to become spindle‐shaped, the shape typically exhibited by malignant cells (Fig. 1b). Because it has been reported that hypoxia increase apoptosis,16, 17 we investigated whether hypoxia affects survival of LLC cells. However, we could not detect any apoptotic responses in either normoxic or hypoxic condition (Fig. 1c,d). Because it took 10 days for the LLC cells to become completely spindled, the following experiments were performed using LLC cells kept under hypoxia for more than 14 days.
Figure 1.
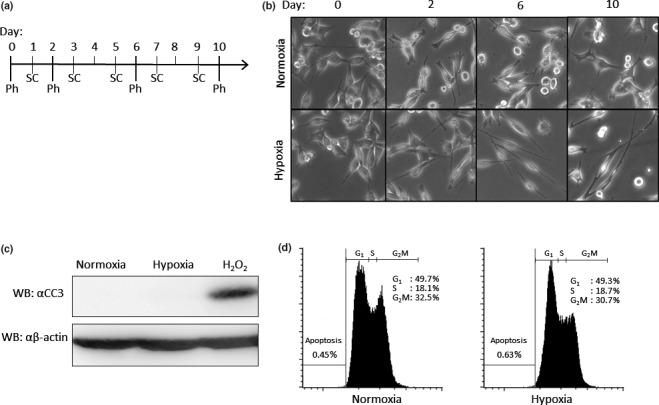
Effect of hypoxia on cell morphology. (a) Time schedule of experiment. (b) Photographs of morphology under either normoxia or hypoxia. Lewis lung carcinoma (LLC) cells were cultured under the normoxic or hypoxic condition. Cells were passaged every other day at 3 × 105 cells/6 cm dish. For each condition, photographs were taken. Ph, date when each photo was taken; SC, date when cells were subcultured. (c) Expression of cleaved caspase‐3 (CC3) in LLC cells. Cells were cultured in either normoxic or hypoxic conditions. As a positive control, LLC cells were treated with 0.4 mM H2O2 in DMEM containing 0.3% FCS for 24 h in normoxic condition. Anti‐CC3 (upper panel) and anti‐β‐actin antibodies (lower panel) were used for western blot analysis. (d) Quantification of apoptotic cells. LLC cells cultured under normoxia (left panel) and hypoxia (right panel) were stained with propidium iodide before analysis by FACS.
Effect of hypoxia on transforming growth factor‐β/Smad signaling
To examine the effect of hypoxia on TGF‐β signaling, Smad phosphorylation induced upon TGF‐β stimulation was investigated. After LLC cells cultured under hypoxia had been starved for 16 h, they were stimulated with TGF‐β for the indicated times (Fig. 2a,b). Phosphorylation of Smad2 was seen 1 h after the TGF‐β treatment and then decreased over time under both normoxic and hypoxic conditions. Interestingly, the cells under hypoxia strongly responded to TGF‐β as compared with those under normoxia. Furthermore, we explored the effect of hypoxia on BMP signaling as well (Fig. S1a). Like TGF‐β, BMP‐induced phosphorylation of Smad1/5 in hypoxic condition was stronger than that in normoxic condition. Therefore, we tested the effect of hypoxia on the activity of the Smad‐dependent (CAGA)12‐luc reporter.18 LLC cells under hypoxia showed higher induction of luciferase activity than did those under normoxia (Fig. 2c). Consistent with these results, LLC cells transfected with the (SBE)4‐luc reporter19 also exhibited increased induction of reporter activity upon TGF‐β and BMP stimulation under hypoxia in comparison with those under normoxia (Figs 2d,S1b). In contrast, Heikkinen et al.20 report opposite results using the (CAGA)12‐luc reporter. Curiously, they cultured cells in 1% oxygen for 16 h before TGF‐β stimulation for the Smad phosphorylation and reporter assay. Thus, we analyzed the effect of short‐term hypoxia exposure on LLC cells. After the cells had been exposed to hypoxia for 16 h, they were stimulated with TGF‐β for the indicated times (Fig. 2e,f). To our surprise, TGF‐β induced Smad2 phosphorylation was decreased under hypoxia. Similarly, when we performed the reporter assay using LLC cells exposed to hypoxia for 16 h before 8 h treatment of cells with TGF‐β, the hypoxic condition showed an inhibitory effect on TGF‐β‐induced transcriptional activity, unlike long‐term exposure of the cells to hypoxia (Fig. 2g). These results suggested that hypoxia maintained over a long time led LLC cells to enhance TGF‐β‐mediated responses via Smad phosphorylation.
Figure 2.
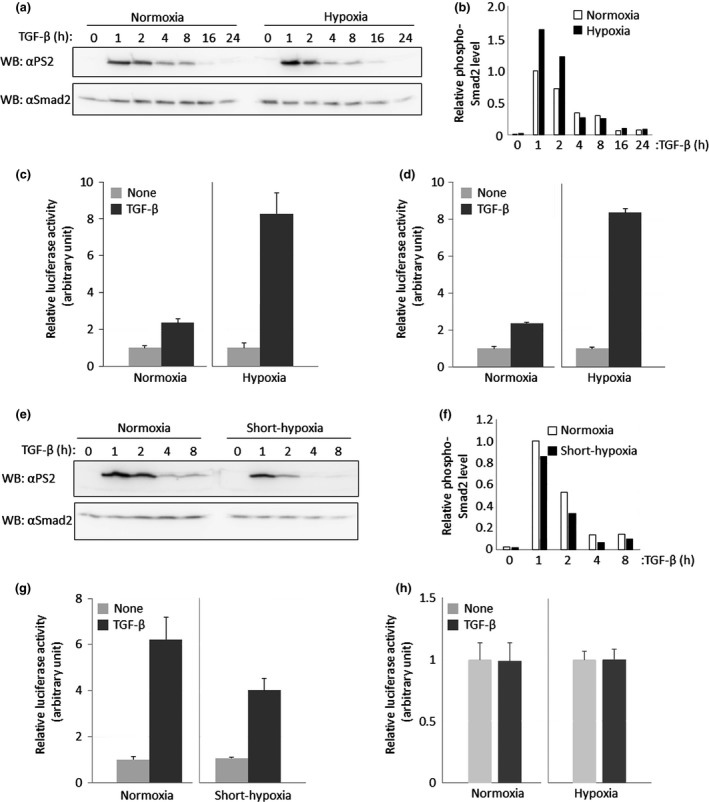
Effect of hypoxia on transforming growth factor‐β (TGF‐β)/Smad signaling. (a) Effect of long‐term hypoxia on TGF‐β‐induced Smad2 phosphorylation. Lewis lung carcinoma (LLC) cells maintained under hypoxia for 14 days were stimulated with 5 ng/mL TGF‐β for 1, 2, 4, 8, 12, 16 and 24 h. These cells were lysed and subjected to western blot analysis using PS2 (upper panel) and anti‐Smad2 antibody (lower panel). (b) Quantification for TGF‐β‐induced Smad2 phosphorylation under long‐term hypoxia. The intensity of Smad2 phosphorylation (a; upper panel) was normalized using the intensity of the band corresponding to Smad2 (a; lower panel). Each relative intensity was calculated by comparing it with the value for cells which are treated with TGF‐β for 1 h under normoxia. (c) Comparison of TGF‐β‐mediated (CAGA)12‐luc reporter activity between normoxia and long‐term hypoxia. LLC cells maintained under hypoxia for more than 14 days were transiently transfected with the (CAGA)12‐luc reporter and stimulated with 5 ng/mL TGF‐β for 16 h. The value from cells in the presence of TGF‐β in each condition was normalized using that in the absence of TGF‐β in each condition. (d) Comparison of TGF‐β‐mediated (SBE)4‐luc reporter activity between normoxia and long‐term hypoxia. All experiments except for (SBE)4‐luc reporter instead of (CAGA)12‐luc reporter were carried out according to (c). (e) Effect of short‐term hypoxia on TGF‐β‐induced Smad2 phosphorylation. LLC cells maintained under hypoxia for 16 h were stimulated with 5 ng/mL TGF‐β for 1, 2, 4 and 8 h. These cells were lysed and subjected to western blot analysis using PS2 (upper panel) and anti‐Smad2 antibody (lower panel). (f) Quantification for TGF‐ β‐induced Smad2 phosphorylation under short‐term hypoxia. Normalization was performed according to (b). (g) Comparison of TGF‐β‐mediated (CAGA)12‐luc reporter activity between normoxia and short‐term hypoxia. LLC cells maintained under hypoxia for 16 h were transiently transfected with (CAGA)12‐luc and stimulated with 5 ng/mL TGF‐β for 16 h. Experiments were performed according to (c). (h) Comparison of TGF‐β‐mediated p5HRE‐luc reporter activity between normoxia and long‐term hypoxia. All experiments except for p5HRE‐luc instead of (CAGA)12‐luc were carried out according to (c).
Because stabilized HIF‐1α under hypoxia accumulates in the nucleus where HIF‐1α makes a heteromeric complex with HIF‐1β to bind to an HIF‐1 responsive element (HRE) within HIF‐1 target genes such as VEGF, we supposed that TGF‐β signaling might affect HIF‐1 signaling. To explore this possibility, LLC cells transfected with a p5HRE‐luc reporter21 were stimulated with TGF‐β. However, no obvious enhancement was seen in the LLC cells upon TGF‐β stimulation under hypoxia, although this reporter activity under hypoxia was remarkably higher than that under normoxia (Fig. 2h). Taken together, these results indicate that hypoxia potentiates TGF‐β/Smad signaling, although TGF‐β does not affect HIF‐1α signaling.
It is known that R‐Smads can make a complex with Smad4 upon TGF‐β stimulation.22, 23 Thus, we examined the endogenous interaction between Smad2/3 with Smad4 in LLC cells under either normoxia or hypoxia upon TGF‐β stimulation. As expected, endogenous interaction between Smad2/3 and Smad4 in LLC cells under hypoxia could be obviously detected compared with that under normoxia (Fig. 3a). We used the in situ PLA method to further confirm the endogenous interaction between Smad2/3 and Smad4.24 Without TGF‐β stimulation, the Smad2/Smad4 (Fig. 3b) complexes were hardly detected, as reflected by red dots in the LLC cells under normoxia, whereas a few red dots could be detected in LLC cells under hypoxia without TGF‐β stimulation. In contrast, more red dots could be observed in the LLC cells under hypoxia 30 min after TGF‐β stimulation than in those under normoxia.
Figure 3.
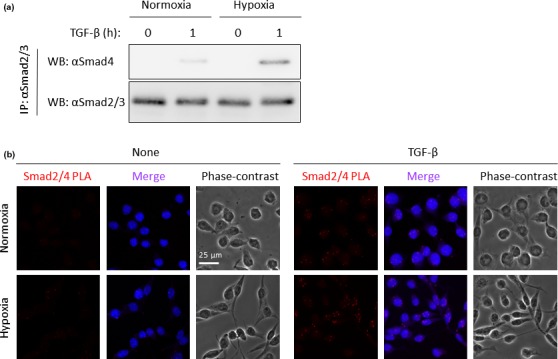
Enhancement of the R‐Smads/Smad4 complex under hypoxia. (a) Endogenous interaction between Smad2/3 and Smad4 upon transforming growth factor‐β (TGF‐β) stimulation under hypoxia. Lewis lung carcinoma (LLC) cells cultured under either normoxia or hypoxia for 14 days were stimulated with or without 5 ng/mL TGF‐β for 1 h and cell extracts were used for immunoprecipitation (IP) with anti‐Smad2/3 antibody. After blotting the samples on the membrane, the anti‐Smad4 antibody was used for western blot analysis. (upper panel) Interaction between Smad2/3 and Smad4, (lower panel) expression of Smad2/3 in immunoprecipitates with anti‐Smad2/3 antibody. (b) Interaction between Samd2 and Samd4 using proximity ligation assay (PLA) method. LLC cells cultured under hypoxia for 14 days were stimulated with 5 ng/mL TGF‐β for 30 min. After fixation, Smad2/Smad4 complex formation was determined by PLA method. The heteromeric complex was visualized using hybridization probes labeled with Alexa 555 (red). The nuclei were stained with DAPI (blue). Photos taken with phase contrast microscopy are also displayed.
Upregulation of transforming growth factor‐β/Smad target genes under hypoxia
Since it is already known that hypoxia upregulates the expression of the TGF‐β1 gene in hematopoietic stem cells,25 we attempted to confirm the induction of mRNA expression of the TGF‐β1 gene under hypoxic conditions. Hypoxia increased basal TGF‐β1 mRNA expression, whereas TGF‐β did not induce its expression (Fig. 4a). When we investigated the expression of Smad7, Snail and p21, which are known to be direct target genes of the TGF‐β/Smad pathway, hypoxia augmented the TGF‐β‐induced transcription of the Smad7 and Snail genes more potently than did normoxia. In contrast, we were unable to detect induction of p21 mRNA by TGF‐β under hypoxia because of considerably high basal expression of p21 mRNA under hypoxia (Fig. 4a).
Figure 4.
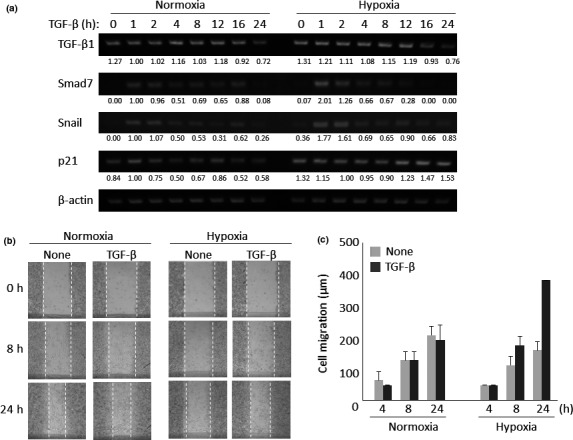
Effect of hypoxia on transforming growth factor‐β (TGF‐β)‐mediated responses. (a) Expression of TGF‐β target genes under either normoxia or hypoxia. Lewis lung carcinoma (LLC) cells cultured under hypoxia for 14 days were stimulated with 5 ng/mL TGF‐β for 1, 2, 4, 8, 12, 16 and 24 h. Total RNA from these cells were subjected to quantitative RT‐PCR for TGF‐β1 (upper panel), Smad7 (second panel), Snail (third panel), p21 (fourth panel) and β‐actin mRNAs (internal control; bottom panel). Each expression was normalized using the intensity of the band corresponding to β‐actin. Inducibility was calculated relative to the value for cells in the absence of TGF‐β. (b) Wound healing assay in the presence or absence of TGF‐β under hypoxia. LLC cells cultured under normoxia or hypoxia for 14 days were stimulated with or without 5 ng/mL TGF‐β for 24 h. Photos were taken 8 and 24 h after TGF‐β stimulation. Results representative of three independent experiments are shown. (c) Quantification for TGF‐β‐mediated cell migration under long‐term hypoxia. Each migration distance in (b) was measured. Data are presented as means ± SD (n = 3).
Next, we used a wound healing assay to examine whether hypoxia potentiates TGF‐β‐mediated cell motility. Under normoxia, LLC cells moved to fill in the wound area, but TGF‐β stimulation did not alter the velocity of cell motility. In contrast, LLC cells cultured under hypoxia showed higher migratory potency upon TGF‐β stimulation (Fig. 4b,c).
Increased ALK5 expression under hypoxia
To investigate the mechanism for the enhancement of TGF‐β signaling under hypoxia, we explored the expression of ALK5 and TGF‐β1 mRNA at several time points after LLC cells were kept in hypoxic condition. As shown in Figure 5(a), the RNA expressions of ALK5 and TGF‐β1 were increased over time. We also observed increased expression of ALK5 (Fig. 5b) and TGF‐β1 proteins (Fig. 5c) in LLC cells under hypoxia by western blot analysis and ELISA assay, respectively. Collectedly, long‐term culture under hypoxia augments ALK5 as well as TGF‐β1 expression in LLC cells to potentiate TGF‐β/Smad signaling.
Figure 5.
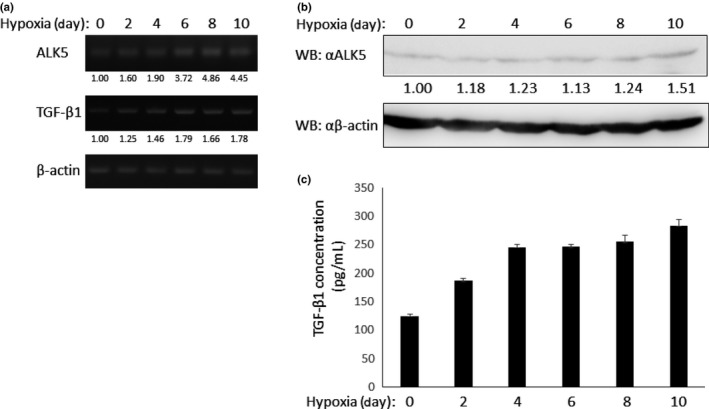
Enhancement of ALK5 and transforming growth factor‐β (TGF‐β) expression under hypoxia. (a) Expression of ALK5 and TGF‐β1 mRNA under hypoxia. Lewis lung carcinoma (LLC) cells were exposed to hypoxia for indicated periods. Then, total RNA from cells at each time point was prepared. RT‐PCR was performed using specific primers. (upper panel) ALK5 (middle panel), TGF‐β1 and (lower panel) β‐actin mRNA. (b) Protein expression of ALK5 under hypoxia. LLC cells were exposed to hypoxia for indicated periods. Then, they were lysed and subjected to western blot analysis using anti‐ALK5 (upper panel) and anti‐β‐actin antibodies (lower panel). (c) Secretion of TGF‐β1 from LLC cells under hypoxia. LLC cells were exposed to hypoxia for indicated periods. Then, concentration of TGF‐β1 in medium was measured using ELISA assay.
Upregulation of transforming growth factor‐β/Smad signaling in vivo
To visualize the activity of HIF‐1α, LLC cells were stably transfected with p5HRE‐EGFP, which has five hypoxia‐responsive elements upstream of the reporter gene.26 Thus, these transformants become EGFP‐positive when HIF‐1α becomes stabilized in the nucleus upon hypoxia. After selection with hygromycin B, these transformants were cultured under hypoxia for 16 h. As shown in Figure 6(a), clone 3 (termed LLC/HIF‐EGFP‐3) showed the highest GFP expression among the six stable transformants. Therefore, the LLC/HIF‐EGFP‐3 clone was used for the subsequent experiments. To confirm the GFP expression induced by HIF‐1α signaling, LLC/HIF‐EGFP‐3 was cultured either under hypoxia or in the presence of 100 μM CoCl2, which can activate HIF‐1α (Fig. 6b).
Figure 6.
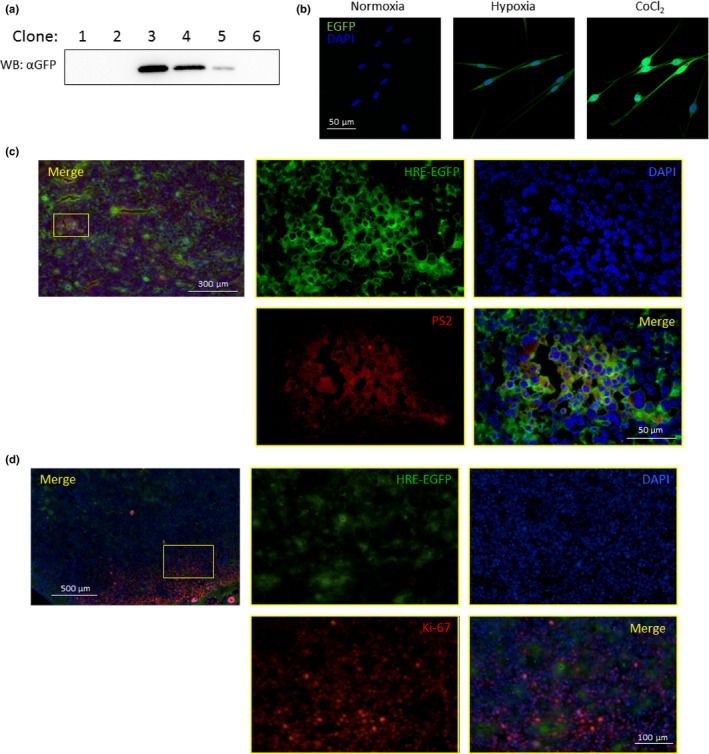
Detection of hypoxia‐mediated enhancement of transforming growth factor‐β (TGF‐β) signaling in vivo. (a) Stable expression of p5HRE‐GFP in Lewis lung carcinoma (LLC) cells. The p5HRE‐EGFP reporter was stably transfected into LLC cells. The expression of GFP under hypoxia was subjected to western blot analysis using anti‐GFP antibody. LLC/HIF‐EGFP‐3 cells showed the highest expression of GFP among the clones examined. (b) Expression of GFP under hypoxia. LLC/HIF‐EGFP‐3 cells were cultured under hypoxia for 14 days or in the presence of CoCl2 for 24 h. Nuclei were stained blue. (c) Overlap of hypoxic area with high active region of TGF‐β signaling in implanted LLC/HIF‐EGFP‐3 cells. LLC/HIF‐EGFP‐3 cells were transplanted subcutaneously into C57/BL6 mice. Fourteen days after the transplantation, the tumors were removed. Subsequently, the sections from the tumors were stained with anti‐GFP antibody (green), anti‐phosphorylated Smad2 (PS2) (red) and DAPI (blue). (d) Less overlap of hypoxic area with proliferative region in implanted LLC/HIF‐EGFP‐3 cells. The sections shown in (c) were stained with anti‐GFP antibody (green), anti‐Ki‐67 antibody (red) and DAPI (blue).
Then, we implanted LLC/HIFGFP‐3 cells subcutaneously into C57BL/6 mice. Fourteen days after implantation, the subcutaneous tumors were excised for preparation of fresh‐frozen sections. When stained with anti‐GFP and anti‐phosphorylated Smad2 (PS2) antibodies, the GFP‐positive area overrode the PS2‐positive area. These results indicate that hypoxia is involved in Smad2 phosphorylation (Fig. 6c).
The expression of p21 under hypoxia was enhanced under the hypoxic condition (Fig. 4a). Thus, we next examined whether hypoxia affects cell growth in vivo. The transplanted tumor sections shown in Figure 6(c) were stained with anti‐Ki‐67 antibody, which can detect proliferating cells. The areas in which high GFP expression could be seen (green) exhibited low Ki‐67 expression (red) (Fig. 6d).
Discussion
A vascular system is necessary for solid tumors to grow more than 2 mm in diameter. Thus, solid tumors not supplied by a vascular system often become hypoxic because oxygen diffusion is limited. To protect against cell death due to oxygen depletion, cancer cells stabilize HIF‐1, which induces expression of numerous target genes related to angiogenesis and metastasis. Therefore, tumor hypoxia represents a pivotal angiogenic signal for proliferation of tumors. Among angiogenic factors, VEGF, which is frequently secreted from not only cancer cells but also normal tissues, is a main actor implicated in tumor angiogenesis. Besides VEGF, TGF‐β is also involved in angiogenic responsiveness and malignant transformation.27, 28, 29, 30 However, the relationship between the hypoxic and TGF‐β/Smad signals has not yet been understood clearly. In the present study, we showed that long‐term exposure of LLC cells to hypoxia elevated both TGF‐β‐mediated and BMP‐mediated R‐Smad phosphorylations to affect transcriptional activity upon TGF‐β family stimulation. Furthermore, single cell analysis indicated that the TGF‐β‐induced association between R‐Smad and Smad4 in LLC cells could be detected under the hypoxic condition at a higher extent than under the normoxic condition. Thus, these data indicate that hypoxia definitely enhances TGF‐β/Smad signaling. Since it has been reported that hypoxia induces the expression of ALK5,31 we investigated the expression of ALK5 in LLC cells under hypoxia. As expected, ALK5 expression was increased in LLC cells under hypoxia over time. Because the hypoxic condition could enhance TGF‐β1 secretion from LLC cells, we supposed that increased ALK5 and TGF‐β1 under hypoxia might potentiate the TGF‐β/Smad signaling pathway in LLC cells. In contrast, TGF‐β stimulation did not influence the hypoxia signal, although several reports have been published showing that the TGF‐β/Smad pathway regulates the hypoxia signal. These contradictory results might be due to cell‐type dependency. We will explore this possibility in future investigations.
In contrast to our findings, Heikkinen et al.20 demonstrate that hypoxia inhibits TGF‐β signaling via dephosphorylation of Smad3 by the protein phosphatase 2A (PP2A). However, Heikkinen et al. kept cells under hypoxia for only 16 h before stimulation of the cells with TGF‐β for 3 h, whereas we performed all experiments using cells that were maintained under hypoxia for 14 days. It has been postulated that the TGF‐β/Smad pathway might be differently regulated dependent on the length of time when cells are exposed to hypoxia owing to direct or indirect control via the hypoxia signaling pathway.32 In our experiments using LLC cells, LLC cells had to be cultured under hypoxia for over 10 days to reveal high responsiveness to TGF‐β. It is possible that hypoxia invokes epigenetic alteration to change the expression pattern of transcription factors in cells.32, 33, 34, 35 Alternatively, responsiveness to hypoxia might differ among the cells investigated. We will try to solve these issues in future, although long‐term exposure of LLC cells to hypoxia seems to mimic the tumor microenvironment.
Lewis lung carcinoma cells carrying an EGFP expression vector controlled under HRE could become a useful tool for detecting hypoxic areas in vivo when LLC cells are implanted in mice (Fig. 6). When we indeed confirmed that the hypoxia signal could enhance phosphorylation of Smad2, most of the red areas overlapped the green areas, indicating that TGF‐β/Smad signaling is possibly augmented upon hypoxia signaling. VEGF, a target gene for hypoxia signaling, can induce TGF‐β expression in tumors.36, 37 Thus, there is another possibility that TGF‐β secreted via VEGF production from tumors coordinately activates the TGF‐β/Smad pathway in concert with enhanced expression of ALK5 in LLC cells.
Disclosure Statement
The authors have no conflict of interest to declare.
Supporting information
Fig. S1. Long‐term hypoxia potentiates BMP signaling.
Acknowledgment
This research was supported by the Japanese Ministry of Education, Culture, Sports, Science and Technology (23689023 and 26460479 to F.I. and 26305006 to S.I.), the Naito Foundation (F.I.), the Sagawa Cancer Foundation (F.I.), the Core‐to‐Core program “Cooperative International Framework in TGF‐β Family Signaling” of the Japan Society for the Promotion of Science (F.I. and S.I.), the MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (2013–2017 to S.I. and 2015–2019 to F.I.) and the Vehicle Racing Commemorative Foundation (S.I.). We thank Dr H. Harada for plasmids, Ms F. Miyamasu for excellent English proofreading and Mr T. Inagawa for his support.
Cancer Sci 106 (2015) 1524–1533
Funding Information This research was supported by the Japanese Ministry of Education, Culture, Sports, Science, and Technology (23689023 and 26460479 to F.I. and 26305006 to S.I.); Naito Foundation (F.I.); Sagawa Cancer Foundation (F.I.); the Core‐to‐Core program “Cooperative International Framework in TGF‐β Family Signaling” of the Japan Society for the Promotion of Science (F.I. and S.I.); MEXT‐Supported Program for the Strategic Research Foundation at Private Universities (2013–2017 to S.I. and 2015–2019 to F.I.), the Vehicle Racing Commemorative Foundation.
References
- 1. Keith B, Simon MC. Hypoxia‐inducible factors, stem cells, and cancer. Cell 2007; 129: 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gordan JD, Simon MC. Hypoxia‐inducible factors: central regulators of the tumor phenotype. Curr Opin Genet Dev 2007; 17: 71–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Semenza GL. Regulation of cancer cell metabolism by hypoxia‐inducible factor 1. Semin Cancer Biol 2009; 19: 12–6. [DOI] [PubMed] [Google Scholar]
- 4. Liao SH, Zhao XY, Han YH et al Proteomics‐based identification of two novel direct targets of hypoxia‐inducible factor‐1 and their potential roles in migration/invasion of cancer cells. Proteomics 2009; 15: 3901–12. [DOI] [PubMed] [Google Scholar]
- 5. Krieg AJ, Rankin EB, Chan D et al Regulation of the histone demethylase JMJD1A by hypoxia‐inducible factor 1α enhances hypoxic gene expression and tumor growth. Mol Cell Biol 2010; 30: 344–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sobhanifar S, Aquino‐Parsons C, Stanbridge EJ et al Reduced expression of hypoxia‐inducible factor‐1alpha in perinecrotic regions of solid tumors. Cancer Res 2005; 65: 7259–66. [DOI] [PubMed] [Google Scholar]
- 7. Kizaka‐Kondoh S, Konse‐Nagasawa H. Significance of nitroimidazole compounds and hypoxia‐inducible factor‐1 for imaging tumor hypoxia. Cancer Sci 2009; 100: 1366–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu J, Qu R, Ogura M et al Real‐time imaging of hypoxia‐inducible factor‐1 activity in tumor xenografts. J Radiat Res 2005; 46: 93–102. [DOI] [PubMed] [Google Scholar]
- 9. Heldin CH, Landstrom M, Moustakas A. Mechanism of TGF‐β signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr Opin Cell Biol 2009; 21: 166–76. [DOI] [PubMed] [Google Scholar]
- 10. Massagué J, Blain SW, Lo RS. TGFβ signaling in growth control, cancer, and heritable disorders. Cell 2000; 103: 295–309. [DOI] [PubMed] [Google Scholar]
- 11. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 2009; 9: 265–73. [DOI] [PubMed] [Google Scholar]
- 12. Carvalho RL, Itoh F, Goumans MJ et al Compensatory signaling induced in the yolk sac vasculature by deletion of TGFβ receptors in mice. J Cell Sci 2007; 120: 4269–77. [DOI] [PubMed] [Google Scholar]
- 13. Itoh F, Itoh S, Adachi T et al Smad2/Smad3 in endothelium is indispensable for vascular stability via S1PR1 and N‐cadherin expressions. Blood 2012; 119: 5320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Itoh F, Asao H, Sugamura K et al Promoting bone morphogenetic protein signaling through negative regulation of inhibitory Smads. EMBO J 2001; 20: 4132–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakano N, Maeyama K, Sakata N et al C18 ORF1, a novel negative regulator of transforming growth factor–β signaling. J Biol Chem 2014; 289: 12680–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carmeliet P, Dor Y, Herbert JM et al Role of HIF‐1α in hypoxia‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998; 394: 485–90. [DOI] [PubMed] [Google Scholar]
- 17. Steinbach JP, Wolburg H, Klumpp A, Probst H, Weller M. Hypoxia‐induced cell death in human malignant glioma cells: energy deprivation promotes decoupling of mitochondrial cytochrome c release from caspase processing and necrotic cell death. Cell Death Differ 2003; 10: 823–32. [DOI] [PubMed] [Google Scholar]
- 18. Dennler S, Itoh S, Vivien D et al Direct binding of Smad3 and Smad4 to critical TGF β‐inducible elements in the promoter of human plasminogen activator inhibitor‐type 1 gene. EMBO J 1998; 17: 3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jonk LJ, Itoh S, Heldin CH et al Identification and functional characterization of a Smad binding element (SBE) in the JunB promoter that acts as a transforming growth factor‐β, activin, and bone morphogenetic protein‐inducible enhancer. J Biol Chem 1998; 273: 21145–52. [DOI] [PubMed] [Google Scholar]
- 20. Heikkinen PT, Nummela M, Leivonen SK et al Hypoxia‐activated Smad3‐specific dephosphorylation by PP2A. J Biol Chem 2010; 285: 3740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harada H, Inoue M, Itasaka S et al Cancer cells that survive radiation therapy acquire HIF‐1 activity and translocate towards tumour blood vessels. Nat Commun 2012; 3: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shi Y, Massagué J. Mechanisms of TGF‐β signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700. [DOI] [PubMed] [Google Scholar]
- 23. ten Dijke P, Hill CS. New insights into TGF‐β‐Smad signalling. Trends Biochem Sci 2003; 29: 265–73. [DOI] [PubMed] [Google Scholar]
- 24. Söderberg O, Leuchowius KJ, Gullberg M et al Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods 2008; 45: 227–32. [DOI] [PubMed] [Google Scholar]
- 25. Shi YF, Fong CC, Zhang Q et al Hypoxia induces the activation of human hepatic stellate cells LX‐2 through TGF‐β signaling pathway. FEBS Lett 2007; 581: 203–10. [DOI] [PubMed] [Google Scholar]
- 26. Harada H, Kizaka‐Kondoh S, Hiraoka M. Optical imaging of tumor hypoxia and evaluation of efficacy of a hypoxia‐targeting drug in living animals. Mol Imaging 2005; 4: 182–93. [DOI] [PubMed] [Google Scholar]
- 27. ten Dijke P, Goumans MJ, Itoh F et al Regulation of cell proliferation by Smad proteins. J Cell Physiol 2002; 191: 1–16. [DOI] [PubMed] [Google Scholar]
- 28. Semenza GL. Hypoxia‐inducible factors in physiology and medicine. Cell 2012; 148: 399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flavell RA, Sanjabi S, Wrzesinski SH et al The polarization of immune cells in the tumour environment by TGFβ. Nat Rev Immunol 2010; 10: 554–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nicolás FJ, Hill CS. Attenuation of the TGF‐beta‐Smad signaling pathway in pancreatic tumor cells confers resistance to TGF‐beta‐induced growth arrest. Oncogene 2003; 22: 3698–711. [DOI] [PubMed] [Google Scholar]
- 31. Zhao X, Wang K, Liao Y et al MicroRNA‐101a inhibits cardiac fibrosis induced by hypoxia via targeting TGFβRI on cardiac fibroblasts. Cell Physiol Biochem 2015; 35: 213–26. [DOI] [PubMed] [Google Scholar]
- 32. Watson JA, Watson CJ, McCann A et al Epigenetics, the epicenter of the hypoxic response. Epigenetics 2010; 5: 293–6. [DOI] [PubMed] [Google Scholar]
- 33. Lee HY, Choi K, Oh H, Park YK, Park H. HIF‐1‐dependent induction of Jumonji domain‐containing protein (JMJD) 3 under hypoxic conditions. Mol Cell 2014; 37: 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 2013; 73: 2936–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase JMJD2C is a coactivator for hypoxia‐inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A 2012; 109: E3367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Park HY, Kim JH, Park CK. VEGF induces TGF‐β1 expression and myofibroblast transformation after glaucoma surgery. Am J Pathol 2013; 182: 2147–54. [DOI] [PubMed] [Google Scholar]
- 37. Lee KS, Park SJ, Kim SR et al Inhibition of VEGF blocks TGF‐β1 production through a PI3K/Akt signalling pathway. Eur Respir J 2008; 31: 523–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Long‐term hypoxia potentiates BMP signaling.