

Abstract
Mammalian orthoreoviruses (reoviruses) are prototype members of the Reoviridae family of nonenveloped viruses. Reoviruses contain ten double-stranded RNA gene segments enclosed in two concentric protein shells, outer capsid and core. These viruses serve as a versatile experimental system for studies of virus cell entry, innate immunity, and organ-specific disease. Reoviruses engage cells by binding to cell-surface carbohydrates and the immunoglobulin superfamily member, junctional adhesion molecule-A (JAM-A). JAM-A is a homodimer formed by extensive contacts between its N-terminal immunoglobulin-like domains. Reovirus attachment protein σ1 disrupts the JAM-A dimer, engaging a single JAM-A molecule by virtually the same interface used for JAM-A homodimerization. Following attachment to JAM-A and carbohydrate, reovirus internalization is promoted by β1 integrins, most likely via clathrin-dependent endocytosis. In the endocytic compartment, reovirus outer-capsid protein σ3 is removed by cathepsin proteases, which exposes the viral membrane-penetration protein, μ1. Proteolytic processing and conformational rearrangements of μ1 mediate endosomal membrane rupture and delivery of transcriptionally active reovirus core particles into the host cell cytoplasm. These events also allow the φ cleavage fragment of μ1 to escape into the cytoplasm where it activates NF-κB and elicits apoptosis. This review will focus on mechanisms of reovirus cell entry and activation of innate immune response signaling pathways.
Keywords: Sialic Acid, Membrane Penetration, Sialic Acid Binding, Reovirus Infection, NPXY Motif
Introduction
The mammalian reoviruses are members of the Reoviridae family, which includes the important human pathogens rotavirus and Colorado-tick fever virus (Schiff et al. 2007). Like other Reoviridae members, reoviruses are nonenveloped, icosahedral particles that contain a segmented, double-stranded (ds) RNA genome surrounded by concentric protein shells (Schiff et al. 2007). These viruses are ubiquitous and display a broad host range, resulting in infection of wide variety of mammals including humans (Virgin et al. 1997). However, reovirus causes disease primarily in the very young (Mann et al. 2002; Tardieu et al. 1983; Tyler et al. 2004). These viruses have served in some respects as prototypes for the study of the Reoviridae due to the availability of isolates that display dissimilar phenotypes, the ability to perform genetic analysis using reassortant viruses and reverse genetics, and the existence of a murine model of virus-induced disease.
Initiation of reovirus infection requires deposition of the genome-containing inner capsid (known as the core) into the cytoplasm. Delivery of this rather large cargo (∼70 nm in diameter) requires an exquisitely timed and regulated series of events both at the host cell surface and within host endosomes. The virus must attach to host cells, internalize and traffic to cellular endosomes, undergo proteolytic disassembly to expose the viral membrane-penetration apparatus, and penetrate host cell membranes for delivery of the viral core into the cytoplasm. These early events during reovirus infection activate innate immune signaling pathways. Here, we describe our current understanding of each of these steps in the cell entry pathway used by reovirus.
Structural Analysis of Reovirus Virions and Attachment Protein σ1
Reovirus particles are approximately 850 Å in diameter and consist of two concentric protein shells, the inner core and outer capsid (Dryden et al. 1993; Schiff et al. 2007) (Fig. 1). The reovirus genome consists of ten segments of dsRNA, which range in length from ∼1.2 to ∼3.9 kilobases. The genome segments are named based on size, with three large (L), three medium (M), and four small (S) segments. Reovirus proteins are designated according to the encoding gene segments, lambda (λ) for L, mu (μ) for M, and sigma (σ) for S. The reovirus inner core has T = 1 symmetry and is primarily formed by a shell of 60 asymmetric dimers of λ1 and 150 monomers of σ2 (Reinisch et al. 2000). Pentameric turrets of λ2, a capping enzyme and conduit for viral transcripts exiting the core, are located at the icosahedral vertices of the reovirus particle and span both the inner core and the outer capsid (Bartlett et al. 1974; Cleveland et al. 1986; Dryden et al. 1993; Fausnaugh and Shatkin 1990; Furuichi et al. 1976; Gillies et al. 1971; Luongo et al. 1998, 2000; Mao and Joklik 1991; Reinisch et al. 2000). Minor core components include μ2 (∼24 copies) and λ3 (12 copies) (Coombs 1998; Dryden et al. 1998). Each copy of viral RNA-dependent RNA polymerase λ3 is associated with three monomers of λ1 and occupies a single icosahedral vertex in the inner core (Drayna and Fields 1982; Starnes and Joklik 1993; Tao et al. 2002; Zhang et al. 2003). Surrounding the core is the outer capsid, which has quasi T = 13 (laevo) icosahedral symmetry (Metcalf 1982) and is composed of 200 heterohexamers of the membrane-penetration protein, μ1, and its protective cap, σ3 (μ13σ33) (Dryden et al. 1993; Liemann et al. 2002; Metcalf 1982). Extending from a λ2 turret at each vertex of a reovirus particle is a trimer of σ1, the viral attachment protein, which is released during entry (Chappell et al. 2002; Dryden et al. 1993; Fraser et al. 1990; Furlong et al. 1988; Strong et al. 1991).
Fig. 1.
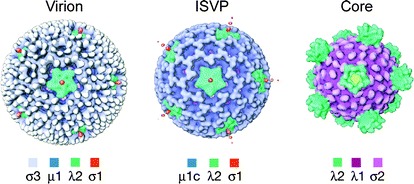
Reovirus disassembly intermediates. Surface-shaded representations of cryo-EM image reconstructions of reovirus are shown, as viewed along a twofold axis of symmetry. Density corresponding to σ1 can be seen extending from turrets of λ2 at the icosahedral axes of virions and ISVPs. Cores lack σ1. Image adapted from Dryden et al. (1993)
The reovirus σ1 protein mediates binding to cellular receptors (Barton et al. 2001b; Chappell et al. 2000) and influences target-cell selection in the infected host (Weiner et al. 1977, 1980). The 455 amino acids of strain T3D σ1 fold into a trimer approximately 480 Å long and 90 Å wide at its broadest point, with a globular C-terminal head, a central body, and a slender N-terminal tail (Chappell et al. 2002; Fraser et al. 1990; Guglielmi et al. 2006) (Fig. 2). Residues 310–455 comprise the head, which is constructed from two Greek-key motifs that assemble into an eight-stranded β-barrel (Chappell et al. 2002; Schelling et al. 2007). With the exception of the loop connecting β-strands D and E (D–E loop), which contains a 310 helix, loops connecting individual strands of the β-barrel are very short. N-terminal to the σ1 head, residues 246–309 form repeating units of two antiparallel β-strands connected by short loops. Three such units assemble into a triple β-spiral, which is a motif observed to date only in viral fibers, including the adenovirus fiber (van Raaij et al. 1999), bacteriophage PRD1 P5 (Merckel et al. 2005), avian reovirus attachment protein σC (Guardado et al. 2005), and mammalian reovirus T3D σ1 (Chappell et al. 2002). In addition to the three β-spiral repeats observed in the crystallized σ1 fragment, sequence analysis suggests that the remainder of the T3D σ1 body (residues 167–249) contains an additional five N-terminal β-spiral repeats (Chappell et al. 2002; Guglielmi et al. 2006). Alternatively, these residues may form a combination of β-spiral repeats and α-helical coiled-coil, as suggested by sequence analysis (Chappell et al. 2002; Guglielmi et al. 2006; Nibert et al. 1990) and an observed narrowing in this region in a composite negative-stain electron micrograph (EM) (Fraser et al. 1990). The structure of the N-terminal tail, residues ∼1–160, of σ1 is unknown. However, a repeating heptad sequence motif is predictive of an amphipathic α-helix, which likely assembles into an α-helical coiled-coil in the trimer (Chappell et al. 2002; Guglielmi et al. 2006; Nibert et al. 1990). The extreme N-terminus of σ1 does not contain any obvious sequence motifs. It is hydrophobic and anchors the protein into the pentameric turret formed by λ2 in the reovirus virion (Dryden et al. 1993; Furlong et al. 1988). This symmetry mismatch suggests an interaction of limited strength, which may aid in σ1 release during viral disassembly (Stehle and Dermody 2003).
Fig. 2.

Model of reovirus attachment to JAM-A on the cell surface. A ribbon-trace model of full-length T3D σ1, extending from a schematic virion, with the known structure of the C-terminus (Chappell et al. 2002) in tricolor and the prediction for the N-terminus in gray. The predicted SA-binding site (Chappell et al. 2000; Dermody et al. 1990) is marked with a hexagon. The extracellular domains D1 and D2 of JAM-A (Prota et al. 2003) and schematic representations of the transmembrane (TM) and intracellular domains are shown in green. Asterisks indicate regions of flexibility. For clarity, only two JAM-A monomers are shown bound to σ1. Figure and legend modified from Kirchner et al. (2008)
The σ1 molecule possesses discrete regions of flexibility along its length (Chappell et al. 2002; Fraser et al. 1990) (Fig. 2). One site of substantial flexibility in T3D σ1 is contributed by a four-residue insertion between the two most C-terminal β-spiral repeats (Cavalli et al. 2004; Chappell et al. 2002). Sequence alignments suggest that σ1 of reovirus prototype strains T1L and T2J each contain a six-residue insertion at the same position (Chappell et al. 2002). This insertion appears to correspond to a region of flexibility observed just below the σ1 head in EM images (Fraser et al. 1990). A second region of flexibility observed at the midpoint of σ1 may correspond to the transition between the predicted α-helical coiled-coil region of the tail and the β-spiral-containing body, and a final region of flexibility close to the N-terminus likely represents the virion insertion domain (Chappell et al. 2002; Fraser et al. 1990; Guglielmi et al. 2006; Nibert et al. 1990).
Reovirus σ1 undergoes significant conformational alterations during viral disassembly (Dryden et al. 1993; Furlong et al. 1988; Nibert et al. 1995). Some serotype 3 reoviruses, including T3D, can be cleaved by intestinal proteases (Nibert et al. 1995). Cleavage occurs in the σ1 body (Chappell et al. 1998), just N-terminal to the first β-spiral repeat in the crystal structure (Chappell et al. 2002). This proteolytic cleavage enhances viral hemagglutination capacity, suggesting an unmasking or conformational change in the sialic acid (SA)-binding region of the molecule (Nibert et al. 1995). This idea is supported by the observation of σ1 molecules with either single- or multilobed head regions (Fraser et al. 1990), which suggests that σ1 may exist in both “open” and “closed” conformations.
Although neither the precise mechanism nor the nature of σ1 conformational changes is understood, structural studies of σ1 provide clues about how these changes might occur. A cluster of six conserved aspartic acid residues on a rigid β-hairpin at the base of the σ1 head, sandwiched between hydrophobic residues that block access to solvent, forms the main contact area between monomers in the trimer (Chappell et al. 2002; Schelling et al. 2007). Of the two aspartic acid residues contributed by each monomer, one (Asp346) is neutralized by a salt-bridge interaction with a nearby residue, while the other (Asp345) is not (Chappell et al. 2002; Schelling et al. 2007). The three Asp345 side chains closely appose each other at the center of the trimer in an otherwise hydrophobic environment. Since accumulation of negative charge in this region is predicted to destabilize the trimer (Cavalli et al. 2004), and a D345N mutation results in σ1 trimers with a structure indistinguishable from wild-type (Schelling et al. 2007), it is likely that Asp345 is protonated in the σ1 crystal structure (Chappell et al. 2002), representing the “closed” conformation of σ1. This conformation might form during crystallization at near-neutral pH and physiologically in conditions of low pH, similar to those encountered in the endocytic compartment during reovirus entry (Schelling et al. 2007). Thus, the aspartic acid sandwich motif may contribute to σ1 conformational rearrangements by acting as a molecular switch that mediates the oligomeric state of the σ1 head, depending on environmental pH (Schelling et al. 2007).
Reovirus Attachment Is Mediated by Cell-Surface Sialic Acid and Junctional Adhesion Molecule-A
Similar to viruses from a broad array of families that use carbohydrates as receptors (Olofsson and Bergstrom 2005), cell-surface SA serves as a receptor for several serotype 3 reovirus strains, including prototype strain T3D (Barton et al. 2001a; Chappell et al. 2000; Gentsch and Pacitti 1985, 1987; Paul et al. 1989). T3D exhibits a reduced capacity to agglutinate erythrocytes following treatment with neuraminidase, which removes terminal SA moieties (Gentsch and Pacitti 1987). Preincubation of either L cells with neuraminidase or virus with sialosides also significantly diminishes T3D binding (Gentsch and Pacitti 1985; Paul et al. 1989). SA residues linked in either α2,3 or α2,6 configurations effectively block serotype 3 reovirus binding to L cells (Paul et al. 1989). Reovirus T3D binds to sialoglycophorin, but not to asialoglycophorin, with an avidity of ∼5 × 10−9 M (Barton et al. 2001a), which is a property mediated by the σ1 protein (Chappell et al. 2000). Thus, SA functions as a serotype 3 reovirus receptor in cultured cells. In addition, SA binding also serves an important role in reovirus tropism and pathogenesis in vivo (Barton et al. 2003). An SA-binding strain of reovirus, but not a non-SA-binding strain, causes bile duct injury in newborn mice and exhibits 1,000-fold greater binding capacity for human cholangiocarcinoma cells, which are derived from bile duct epithelium.
Although the structure of σ1 in complex with SA is not yet available, studies using expressed proteins indicate that the region of T3D σ1 required for SA binding resides near the midpoint of the body, while a region just N-terminal to the head domain of T1L σ1 binds carbohydrate (Chappell et al. 2000). For both T1L and T3D, interactions with carbohydrate are mediated by a region of predicted β-spiral (Chappell et al. 2002). The capacity to bind SA is essential for reovirus infection of murine erythroleukemia (MEL) cells (Chappell et al. 1997; Rubin et al. 1992). Adaptation of non-SA-binding reoviruses to growth in MEL cells results in amino acid substitutions at residues 198, 202, and 204 of σ1 that confer SA-binding capacity on the resultant viruses (Chappell et al. 1997). Molecular modeling of the σ1 body, based on available structure and sequence data, suggests that these residues are surface-exposed and proximal to one another in the predicted β-spiral region (Chappell et al. 2002). Thus, residues 198, 202, and 204 are likely to contribute to an SA-binding site in T3D σ1.
In addition to SA, reovirus also binds junctional adhesion molecule-A (JAM-A, also known as F11R/JAM/JAM1), a member of the immunoglobulin superfamily (Barton et al. 2001b; Martin-Padura et al. 1998; Williams et al. 1999). JAM-A was identified as a reovirus receptor using a genetic screen and subsequently shown to bind directly to the σ1 head domain with nanomolar affinity (Barton et al. 2001b; Schelling et al. 2007). Human and murine homologs of JAM-A, but not JAM family members JAM-B or JAM-C, serve as receptors for all reovirus serotypes and strains tested to date (Barton et al. 2001b; Campbell et al. 2005; Prota et al. 2003). The role of JAM-A as a reovirus receptor in vivo has been examined using JAM-A-null mice (Antar et al. 2009). Following peroral inoculation, JAM-A is dispensable for reovirus growth in the intestine. However, it is required for infection of vascular endothelial cells and promotes efficient hematogenous dissemination of reovirus to sites of secondary infection. Thus, JAM-A serves as a high-affinity reovirus receptor in cultured cells and in vivo.
Structural and biochemical studies highlight the regions and specific interactions that mediate reovirus engagement of JAM-A (Campbell et al. 2005; Chappell et al. 2002; Forrest et al. 2003; Guglielmi et al. 2007; Kirchner et al. 2008; Prota et al. 2003) (Fig. 3). The largest area of conserved residues in σ1 forms the D–E and F–G loops in the head domain (Campbell et al. 2005; Chappell et al. 2002). The crystal structure of the T3D σ1 head domain in complex with the JAM-A D1 domain reveals that residues in this region, centered at the D–E loop and its 310 helix, form the largest area of JAM-A contact (Kirchner et al. 2008). Interactions in this area are highly polar and involve residues Thr380, Gly381, and Asp382. A second, unpredicted area of JAM-A contact resides within the σ1 body, just N-terminal to the head domain. Interactions in this region are largely hydrophobic and involve β-spiral residues Tyr298 and Arg316, the α-helical turn that connects the β-spiral with the β-barrel, and Pro377.
Fig. 3.
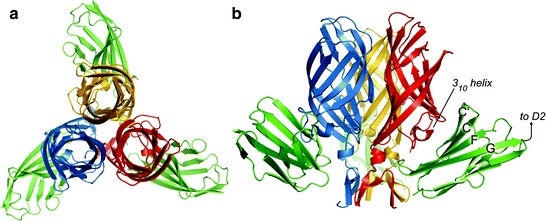
Crystal structure of the σ1-JAM-A complex. (a and b) Ribbon drawings of a complex formed between the trimeric σ1 head domain and monomeric JAM-A D1, viewed along the threefold symmetry axis (a) and from the side (b). Monomers of the σ1 head are shown in blue, red, and yellow; JAM-A D1 is shown in green. Secondary structure elements are labeled. Figure and legend modified from Kirchner et al. (2008)
The D1 domain of JAM-A is required for high-affinity binding to σ1 (Forrest et al. 2003; Guglielmi et al. 2007; Prota et al. 2003). Mutation of individual JAM-A D1 domain residues Arg59, Glu61, Lys63, Leu72, Tyr75, and Asn76, which lie in or adjacent to the dimer interface, diminishes or abolishes σ1 binding and reovirus infectivity (Guglielmi et al. 2007). Concordantly, the structure of the σ1-JAM-A complex shows that each σ1 trimer binds three independent JAM-A monomers (Kirchner et al. 2008). Contacts primarily involve the JAM-A dimer interface and a conserved region at the base of the σ1 head (Chappell et al. 2002; Kirchner et al. 2008) (Fig. 3). In addition, the structure of the σ1-JAM-A complex also identifies residues bound by σ1 that are found just outside the dimer interface of JAM-A (Kirchner et al. 2008). These residues may serve as initial contact points for σ1 and facilitate disruption of the JAM-A homodimer to allow interaction of σ1 with the JAM-A dimer interface. It is also possible that a cavity in the JAM-A dimer interface renders the homodimer intrinsically unstable, thereby promoting its disruption by σ1. Regardless of the mechanism, the σ1-JAM-A interaction is thermodynamically favored, as the K D is approximately 1,000-fold lower than the K D of the JAM-A homodimer interaction (Guglielmi et al. 2007; Kirchner et al. 2008).
Reovirus employs a multistep mechanism of viral attachment in which a low-affinity interaction with SA serves to tether the virion to target cells and precedes a high-affinity interaction with JAM-A (Barton et al. 2001a). This strategy for adhesion to host cells is used by members of unrelated virus families (Berger et al. 1999; Dragic et al. 1996; Montgomery et al. 1996; Ugolini et al. 1999). In some cases, such as with HIV, initial receptor engagement leads to conformational changes in the viral attachment protein that permit coreceptor engagement (Sattentau and Moore 1991). It is not known whether binding to SA induces structural changes in σ1, which affect its capacity to interact with JAM-A. However, it is clear that SA binding is not a necessary prerequisite for JAM-A binding, as non-SA-binding reoviruses are capable of binding JAM-A (Barton et al. 2001b).
Internalization of Reovirus Virions into the Endocytic Pathway Is Mediated by β1 Integrins
Following attachment to cell-surface carbohydrate and JAM-A, reovirus is internalized by receptor-mediated endocytosis (Borsa et al. 1979, 1981; Ehrlich et al. 2004; Maginnis et al. 2006, 2008; Sturzenbecker et al. 1987) (Fig. 4). Expression of a JAM-A truncation mutant lacking a cytoplasmic tail allows reovirus to infect nonpermissive cells (Maginnis et al. 2006), suggesting that molecules other than JAM-A mobilize the internalization apparatus that promotes reovirus cell entry. Based on similarities in the structures of the reovirus and adenovirus attachment proteins and receptors (Stehle and Dermody 2004), it was hypothesized that reovirus and adenovirus employ similar integrin-dependent internalization mechanisms to enter cells. In keeping with this hypothesis, reovirus λ2 protein contains conserved integrin-binding motifs, RGD and KGE (Breun et al. 2001; Seliger et al. 1987). These sequences are displayed on surface-exposed loops of λ2 (Reinisch et al. 2000), where they could interact with integrins. Interestingly, the λ2-encoding L2 gene segment is genetically linked to viral shedding in infected mice and spread to littermates (Keroack and Fields 1986), suggesting a role for λ2 in reovirus-induced disease.
Fig. 4.

The reovirus cell entry pathway. (a) Following attachment to cell-surface carbohydrate (α-linked sialic acid for serotype 3 reoviruses) and JAM-A, reovirus virions enter cells by receptor-mediated endocytosis. (b) Within the endocytic compartment, the viral outer capsid undergoes acid-dependent proteolysis. (c) The first disassembly intermediate is the ISVP, which is characterized by loss of σ3 and cleavage of μ1C into particle-associated fragments δ and φ. (d) The ISVP then undergoes further conformational changes to form the ISVP*. The ISVP* is characterized by conformational rearrangements of the μ1 fragments to expose hydrophobic residues, release of μ1N, and loss of attachment protein σ1. (e) The μ1 cleavage fragments mediate penetration through the endosomal membrane, releasing the transcriptionally active core into the cytoplasm
Treatment of cells with antibodies specific for β1 integrin reduces reovirus infection, while antibodies specific for the other integrin subunits expressed on permissive cells, including those specific for α integrin subunits, have no effect (Maginnis et al. 2006). However, antibodies specific for β1 integrin do not alter infection by in-vitro generated infectious subvirion particles (ISVPs) (Maginnis et al. 2006), which directly penetrate the plasma membrane and do not require endocytosis (Hooper and Fields 1996; Lucia-Jandris et al. 1993). These findings suggest that β1 integrin blockade inhibits endocytic uptake of virions. In comparison to β1 integrin-expressing cells, β1-null cells are substantially less susceptible to infection by reovirus virions, while infection by ISVPs is equivalent in both cell types (Maginnis et al. 2006). Diminished reovirus replication in β1-null cells correlates with diminished viral uptake (Fig. 5), indicating that β1 integrin is required for efficient reovirus cell entry.
Fig. 5.
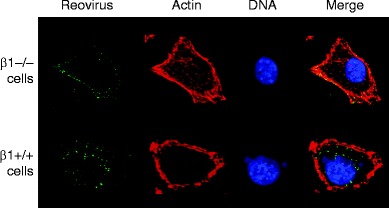
β1 Integrin enhances reovirus entry into cells. GD25 (β1−/−) and GD25β1A (β1+/+) cells were chilled, adsorbed with strain T1L virions, and incubated at 4°C for 1 h. Nonadherent virus was removed, warm medium was added, and cells were incubated at 37°C for the times shown. Cells were fixed, stained for reovirus (green), actin (red), and DNA (blue), and imaged using confocal immunofluorescence microscopy. Representative digital fluorescence images of the same field are shown in each row. Figure and legend modified from Maginnis et al. (2006)
Most available evidence suggests that reovirus is internalized by a clathrin-dependent pathway (Borsa et al. 1979, 1981; Ehrlich et al. 2004; Maginnis et al. 2008; Sturzenbecker et al. 1987). Reovirus virions are observed to colocalize with clathrin in living cells (Ehrlich et al. 2004), and treatment of cells with chlorpromazine, a clathrin-specific chemical inhibitor, inhibits reovirus internalization and infection (Maginnis et al. 2008). However, both clathrin- and caveolin-dependent mechanisms can be employed by some viruses to enter host cells (Laniosz et al. 2008; Querbes et al. 2006). Although there are no published reports of clathrin-independent uptake strategies for reovirus, a role for caveolae in reovirus cell entry has not been conclusively excluded.
NPXY motifs in the β1 integrin cytoplasmic tail play a key role in sorting reovirus within the endocytic compartment. NPXY motifs are found in the cytoplasmic domains of many receptors (Chen et al. 1990; Davis et al. 1986; Oleinikov et al. 2000) and recruit adaptor protein 2 or disabled protein 2 (Morris and Cooper 2001; Oleinikov et al. 2000) to initiate clathrin assembly at the plasma membrane. Substitution of a tyrosine with a phenylalanine residue in either or both β1 integrin NPXY motifs (NPXF) results in inefficient internalization of reovirus virions and diminished infectivity (Maginnis et al. 2008). Infection of cells expressing NPXF β1 integrin results in distribution of virions to lysosomes where they are degraded, suggesting that the β1 integrin NPXY motifs target reovirus to the precise endocytic organelle that permits functional disassembly. Cellular signaling networks that respond to reovirus and facilitate its uptake and endocytic transport are unknown.
Removal of Outer-Capsid Protein σ3 by Cathepsin Proteases Initiates the Reovirus Disassembly Cascade
In cellular endosomes, reovirus virions undergo stepwise disassembly to form discrete intermediates, the first of which is the ISVP (Borsa et al. 1981; Chang and Zweerink 1971; Silverstein et al. 1972; Sturzenbecker et al. 1987) (Figs. 1 and 4). ISVPs are characterized by the loss of σ3, a conformational change in σ1, and cleavage of μ1 to form δ and φ. The rate-limiting step in reovirus disassembly is the proteolytic removal of σ3 (Baer and Dermody 1997; Sturzenbecker et al. 1987). Proteolysis of σ3 is dependent on acidic pH in some cell types (Dermody et al. 1993; Sturzenbecker et al. 1987) and endocytic cysteine proteases (Baer and Dermody 1997). Cathepsins B and L catalyze reovirus disassembly in fibroblasts (Ebert et al. 2002). Both enzymes are optimally active at acidic pH and serve functions in extracellular matrix formation, antigen presentation, and apoptosis (Chapman et al. 1997). These enzymes also mediate cell entry of several other viruses, including Ebola virus (Chandran et al. 2005), Hendra virus (Pager and Dutch 2005), and SARS coronavirus (Huang et al. 2006). Cathepsin S, a neutral pH cysteine protease required for processing internalized antigens (Riese et al. 1996), mediates uncoating of some reovirus strains in a macrophage cell line (Golden et al. 2004). It is possible that the broad tissue tropism displayed by reovirus is determined in part by the multiple host proteases capable of mediating its disassembly, analogous to highly pathogenic influenza viruses that disseminate systemically by utilization of alternative proteases for hemagglutinin processing (Goto and Kawaoka 1998; Stieneke-Grober et al. 1992).
Proteolytic enzymes also are required for reovirus infection following peroral inoculation of mice (Bass et al. 1990; Bodkin et al. 1989). Reovirus virions are converted to ISVPs in the intestinal lumen by the resident serine proteases chymotrypsin and trypsin. ISVPs produced in this fashion infect intestinal M cells to allow systemic dissemination of reovirus in the host (Amerongen et al. 1994). ISVPs generated by chymotrypsin or trypsin in vitro or in the gut lumen (Bass et al. 1990; Bodkin et al. 1989) are indistinguishable from ISVPs generated by cathepsin B or cathepsin L in vitro or in the endocytic compartment of cells (Baer et al. 1999; Ebert et al. 2002).
Sequences in σ3 that influence its susceptibility to proteolysis have been identified through studies of viruses selected during persistent infection (PI viruses) or mutant viruses selected for resistance to either cysteine protease inhibitor E64 (D-EA viruses) (Ebert et al. 2001) or ammonium chloride (ACA-D viruses) (Clark et al. 2006). These viruses exhibit accelerated kinetics of disassembly and harbor a Tyr→His mutation at amino acid 354 near the C-terminus of the protein (Clark et al. 2006; Ebert et al. 2001; Wetzel et al. 1997) (Fig. 6). Cryo-EM image analysis of a PI virus with an isolated Y354H mutation reveals a structural alteration in σ3 at a hinge region located between its two major domains (Wilson et al. 2002). These findings suggest that the C-terminus of σ3 regulates susceptibility of the protein to cleavage.
Fig. 6.
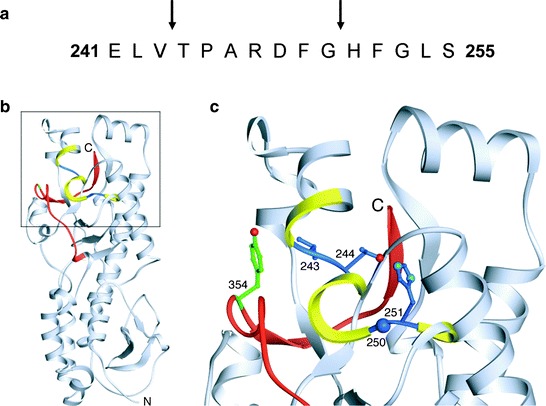
The σ3 protein is a target for cathepsin proteolysis. (a) The primary amino acid sequence of σ3 from amino acids 241 to 255 is shown. Arrows highlight cathepsin L cleavage sites identified by N-terminal sequencing of σ3 cleavage products following treatment of reovirus strain T1L with cathepsin L in vitro. (b) Cathepsin L cleavage sites are highlighted in the crystal structure of σ3. A ribbon diagram of the crystal structure of T3D σ3 (Olland et al. 2001) is displayed on the left. The cathepsin L cleavage sites in T1L are depicted in blue between amino acids 243 and 244 and between 250 and 251. Surrounding residues, from amino acids 241 to 253, are shown in yellow. The C-terminal residues of σ3, from amino acids 340 to 365, are colored red. Amino acid 354, which is altered in PI, D-EA, and ACA-D viruses, is colored green. The virion-distal end of σ3 is at the top of the figure, and the virion-proximal end and N-terminus are at the bottom. (c) An enlarged view of the boxed region of σ3 indicated in panel B is shown using the same color scheme. Amino acids 243, 244, 250, 251, and 354 are depicted in ball-and-stick representation. Figure and legend modified from Ebert et al. (2002)
The σ3 C-terminus also dictates strain-specific differences in the susceptibility of σ3 to proteolytic attack (Jané-Valbuena et al. 1999, 2002). The σ3 protein of strain T1L is cleaved more rapidly than that of T3D. Analysis of ISVPs recoated with chimeric σ3 proteins generated from T1L and T3D revealed that the C-terminus is primarily responsible for the rate of σ3 proteolysis. Moreover, sequence polymorphisms at residues 344, 347, and 353 in σ3 contribute to this effect (Jané-Valbuena et al. 2002).
Treatment of reovirus virions in vitro with either cathepsin B or cathepsin L leads to an initial cleavage of σ3 at a terminus (Ebert et al. 2002). Since sequence polymorphisms in the σ3 C-terminus determine susceptibility to proteolysis, the initial cleavage of σ3 probably occurs in this region. During proteolysis by cathepsin L, subsequent cleavages occur between residues 243–244 and 250–251 (Ebert et al. 2002) (Fig. 6a). These cleavage sites are physically located near the C-terminus in the σ3 crystal structure (Olland et al. 2001) (Fig. 6b, c). Because of this proximity, the small end fragment released following initial cathepsin L cleavage likely exposes the cleavage sites between residues 243–244 and 250–251, rendering them sensitive to proteolysis. The C-terminus therefore appears to control access to internal, proteolytically sensitive sites in σ3. Because reovirus disassembly in some cell types is acid-dependent (Dermody et al. 1993; Sturzenbecker et al. 1987), the C-terminus might be primed for movement at acidic pH. Mutations near the C-terminus, like Y354H, may alter the conformation of the protein to allow improved access to these cleavage sites and thus accelerate outer capsid disassembly (Wilson et al. 2002). High-resolution structural analysis of Y354H-σ3, which is currently ongoing, will enhance an understanding of σ3 proteolysis.
Penetration of Endosomal Membranes by Reovirus Is Mediated by Outer-Capsid Protein μ1
Studies to assess the capacity of reovirus entry intermediates to penetrate artificial lipid bilayers, model membranes of erythrocytes, or membranes of cells that support reovirus infection indicate that ISVPs but not virions or cores mediate membrane penetration (Borsa et al. 1979; Chandran and Nibert 1998; Chandran et al. 1999, 2001; Hooper and Fields 1996; Lucia-Jandris et al. 1993; Tosteson et al. 1993). Such studies led to the idea that ISVPs or a related subviral particle is the membrane-active intermediate in the reovirus entry pathway. Since ISVPs differ from cores by the presence of outer-capsid proteins σ1 and μ1 (Coombs 1998; Dryden et al. 1993), and because cores recoated in vitro with μ1 alone are capable of membrane penetration (Chandran et al. 1999), these findings point to a role for the μ1 protein in membrane penetration. This biochemical evidence is also supported by several genetic studies. Differences in membrane-penetration efficiency displayed by reovirus strains T1L and T3D segregate with the μ1-encoding M2 gene segment (Chandran et al. 2002; Lucia-Jandris et al. 1993). Additionally, viruses selected for resistance to denaturants such as ethanol contain mutations within the M2 gene segment and display alterations in membrane penetration capacity (Chandran et al. 2002; Danthi et al. 2008b; Hooper and Fields 1996; Wessner and Fields 1993). Together, these data demonstrate a function for the μ1 protein in membrane penetration.
The μ1 protein folds into four distinct domains (Fig. 7a). Domains I, II, and III are primarily α-helical and show no homology with other proteins. Domain IV forms a jelly-roll β-barrel commonly found in the capsid proteins of many nonenveloped viruses (Harrison 2001). This domain interacts extensively with similar domains of the neighboring μ1 molecules and with σ3. The μ1 protein also contains three proteolytic cleavage sites (Fig. 7b). These include an autocatalytic cleavage site at amino acid 42, which separates μ1N and μ1C, a cleavage site at approximately amino acid 580, which releases the δ and φ fragments, and a cleavage site at the C-terminus that releases an ∼10 amino acid peptide (Chandran et al. 2003; Mendez et al. 2003; Nibert and Fields 1992; Odegard et al. 2004). While the physiologic roles of both the δ–φ and the C-terminal cleavages are unclear, studies using reovirus cores recoated with a μ1N–μ1C cleavage-resistant μ1 mutant indicate that cleavage of μ1 to generate μ1N and μ1C is required for membrane penetration and virion infectivity (Odegard et al. 2004). Since μ1N is released from viral particles, it is postulated that cleavage of μ1 is required for release of μ1N, which then interacts with membranes as a function of its myristate moiety to effect membrane penetration (Ivanovic et al. 2008). The requirement for the release of a small hydrophobic peptide for membrane penetration is strikingly similar to the entry mechanisms employed by other nonenveloped viruses such as adenoviruses (Wiethoff et al. 2005), nodaviruses (Schneemann et al. 1992; Walukiewicz et al. 2008), and picornaviruses (Danthi et al. 2003).
Fig. 7.
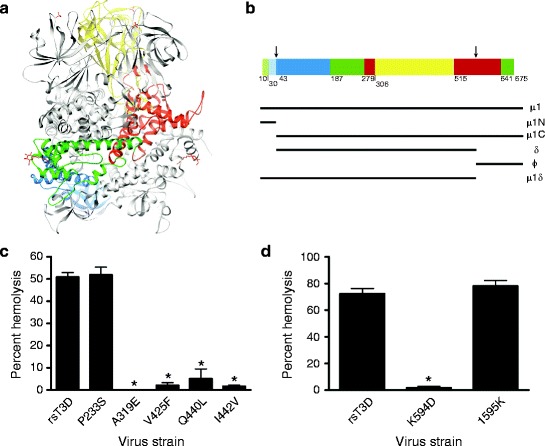
The μ1 protein mediates membrane penetration. (a) Ribbon diagram of the crystal structure of the T1L μ1 trimer without bound σ3. One μ1 subunit is colored by domain (domain I, light and dark blue [μ1N, μ1C]; domain II, light and dark green [μ1N, μ1C]; domain III, red; domain IV, yellow); the other two μ1 subunits are shown in gray. The β-octyl glucosides and sulfate ions present in the structure are shown in red and yellow. (b) Domain segmentation of the amino acid sequence as determined from the three-dimensional structure. The domain color code is as depicted in (a). Cleavage sites are indicated by arrows. Figure modified from Liemann et al. (2002). (c and d) A 3% v/v solution of bovine erythrocytes was incubated with 5.4 × 1010 ISVPs of wild-type rsT3D or the indicated μ1 δ (c) or μ1 φ (d) mutant at 37°C for 1 h. Hemolysis was quantified by determining absorbance of the supernatant at 415 nm. Hemolysis following treatment of an equal number of cells with virion-storage buffer or virion-storage buffer containing 1% TX-100 was considered to be 0 or 100%, respectively. Results are expressed as mean percent hemolysis for triplicate samples. Error bars indicate SD. *, P < 0.05 as determined by Student’s t-test in comparison to rsT3D. Figure modified from Danthi et al. (2008a, b)
In the native μ1 structure present in virions and ISVPs, the myristoylated μ1N fragment is buried inside a hydrophobic cavity in the α-helical pedestal formed by portions of domains I, II, and III (Liemann et al. 2002; Zhang et al. 2005). Based on these studies, massive conformational rearrangements resulting in unwinding of the μ1 trimer must be required to release μ1N during cell entry (Liemann et al. 2002; Zhang et al. 2006). Evidence for conformational changes in particle-associated μ1 following interaction of ISVPs with membranes or when exposed to high salt concentrations has led to the identification of an ISVP-like entry intermediate in the reovirus cell entry pathway (Chandran et al. 2002). This intermediate, referred to as ISVP*, is characterized by changes in the conformation of the μ1 δ fragment, loss of the σ1 protein, and an increase in the overall hydrophobicity of the particle (Chandran et al. 2002). Thus, the μ1 protein associated with ISVPs is in a metastable state primed to undergo conformational changes to assume a more hydrophobic structure capable of interaction with membranes. While it is not understood how these conformational changes in μ1 are triggered, it is thought that interaction of an anion-binding site in domain IV with phospholipid head groups in endosomal membranes might trigger the requisite rearrangements in μ1 that reveal the myristoylated μ1N and the internal hydrophobic residues (Liemann et al. 2002). At high particle concentrations, ISVP* conversion is regulated by a positive feedback mechanism in which μ1N, which is released during ISVP* formation, promotes ISVP-to-ISVP* conversion of the remaining particles (Agosto et al. 2008). Acceleration of ISVP* formation by μ1N is dependent on temperature and target flexibility, suggesting that particle dynamics are required to expose a μ1N interaction domain (Agosto et al. 2008). Since such particle concentrations are unlikely to be achieved following a low multiplicity viral infection, it remains unclear how these findings translate to ISVP* formation in cellular endosomes during viral entry.
Genetic studies using ethanol-resistant or thermostable mutants indicate that μ1 residues affecting the overall stability of the virus also regulate membrane-penetration efficiency (Chandran et al. 2002; Danthi et al. 2008b; Hooper and Fields 1996; Wessner and Fields 1993). These and other stability-altering residues identified in thermostable reovirus mutants (Middleton et al. 2007) are located between residues 383 and 612 of μ1 and map to either domain IV that forms the jelly-roll β-barrel or the α-helical portions of domain III that lie just below the β-barrel structure. Since these μ1 domains participate in interactions between neighboring μ1 monomers, these residues are thought to modulate viral stability by preventing unwinding of the μ1 trimer (Liemann et al. 2002). Consistent with an increase in μ1 protein rigidity in ethanol-resistant and thermostable mutants, viral cores recoated with mutant μ1 proteins, or recombinant reoviruses containing single amino acid substitutions in μ1 in an otherwise wild-type background, display diminished ISVP-to-ISVP* conversion and have defects in membrane penetration (Wessner and Fields 1993; Hooper and Fields 1996; Chandran et al. 2002; Middleton et al. 2007; Danthi et al. 2008a, b) (Fig. 7c, d). These studies suggest that a central region of μ1 involved in intermolecular interactions is an important regulator of the ISVP-to-ISVP* transition. In addition to these residues, changes in the C-terminal φ fragment also control viral stability (Middleton et al. 2007) and affect membrane penetration by reducing the efficiency of ISVP-to-ISVP* conversion (Danthi et al. 2008a). While it is not clear how φ residues modulate these properties, since both μ1N and φ are released from the virus particle during ISVP* formation (Ivanovic et al. 2008), it is likely that conformational rearrangements in μ1 during ISVP* formation are not restricted to the δ domain but also involve the μ1N and φ domains. Therefore, amino acid substitutions within φ that negatively affect its conformational flexibility would likely prevent the μ1 reorganization required for ISVP* formation. Biochemical and structural characterization of additional mutant viruses that may be affected to varying degrees in the capacity to undergo μ1 conformational changes may identify as yet unknown intermediates during ISVP-to-ISVP* conversion and offer insights into mechanisms that promote the elaborate remodeling of μ1 required for membrane penetration.
Analogous to the picornaviruses (Danthi et al. 2003), reovirus forms small, size-selective pores in erythrocyte model membranes (Agosto et al. 2006). Both μ1N and ISVP*s associate with erythrocyte membranes (Agosto et al. 2006; Ivanovic et al. 2008), but μ1N is capable of pore formation in the absence of other viral components (Ivanovic et al. 2008). While φ also associates with membranes (Ivanovic et al. 2008), its recruitment does not result in membrane penetration. These findings are consistent with the observation that viruses incapable of δ–φ cleavage can penetrate membranes and are fully infectious (Chandran and Nibert 1998; Chandran et al. 1999). Since pore formation by μ1N is enhanced by the presence of φ, it is possible that φ functions as a μ1N chaperone and facilitates membrane penetration by reovirus (Ivanovic et al. 2008). Pores formed by released μ1N fragments are considerably smaller than those required to allow the viral intermediate to traverse the membrane (Agosto et al. 2006). Therefore, it is not clear how pore formation in model membranes relates to membrane penetration during cell entry. Analogous to erythrocyte membrane rupture, pore formation may result in osmotic lysis of endosomes in which viral particles are present. Alternatively, the initial small pore formed by the virus might recruit cellular factors that produce larger pores or channels through which the viral intermediate can translocate.
Both the viral core and the δ fragment of μ1 are found in the cytoplasm following reovirus entry into host cells (Chandran et al. 2003). While δ is found distributed diffusely throughout the cytosol, viral cores display a more punctuate cytoplasmic localization (Chandran et al. 2003). These observations suggest that the δ fragment disassociates from the ISVP* either during or immediately after membrane penetration. This idea is supported by the evidence that reovirus cores are transcriptionally active in the cytoplasm and that activation of transcription requires complete removal of the μ1 fragments. Removal of δ from cores is thought to be accomplished by direct interaction of δ with the host chaperone Hsc70 via an ATP-dependent process (Ivanovic et al. 2007). Based on the finding that chaperones can translocate proteins across membranes (Young et al. 2004), it is possible that concomitant with removal of particle-associated δ, Hsc70 also aids in transport of the viral core across membranes (Ivanovic et al. 2007). Additional experiments are required to reveal the precise mechanism by which host membranes are breached by reovirus.
Reovirus Entry Evokes Innate Immune Responses that Trigger Cell Death
Reovirus infection elicits apoptosis of cultured cells and in vivo. Apoptosis induction by reovirus requires activation of innate immune transcription factors NF-κB and IRF-3 (Connolly et al. 2000; Hansberger et al. 2007; Holm et al. 2007) (Fig. 8). In cultured cells, reovirus-induced apoptosis does not require de novo synthesis of viral RNA and protein (Connolly and Dermody 2002; Danthi et al. 2006), indicating that the proapoptotic stimulus is contained within infecting viral capsids. Consistent with these findings, strain-specific differences in the capacity of reovirus to induce apoptosis segregate genetically with the viral S1 and M2 gene segments (Connolly et al. 2001; Tyler et al. 1995, 1996), which encode σ1 and μ1, respectively (McCrae and Joklik 1978; Mustoe et al. 1978). Antibody-dependent uptake of reovirus virions in an entry process that does not require JAM-A and SA leads to apoptotic cell death, indicating that signaling pathways triggered by σ1-receptor interactions are dispensable for reovirus-induced apoptosis (Danthi et al. 2006). Regardless of the receptors used to mediate attachment, initiation of prodeath signaling following reovirus infection requires viral disassembly in cellular endosomes (Danthi et al. 2006), suggesting an essential function for the μ1 protein in apoptosis induction.
Fig. 8.
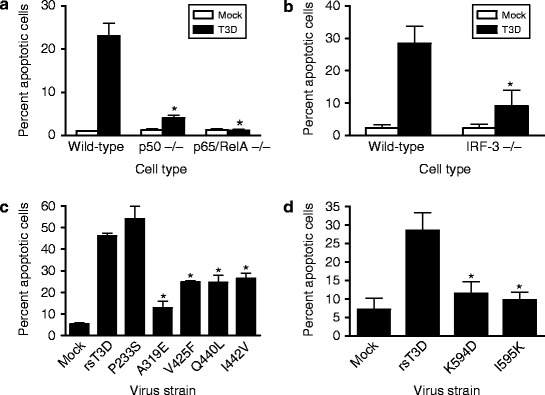
Reovirus entry triggers apoptosis dependent on NF-κB and IRF-3. (a and b) Wild-type cells or cells lacking NF-κB p50, NF-κB p65/RelA, or IRF-3 were either mock-infected or infected with T3D at an MOI of 100 PFU/cell. After incubation at 37°C for 48 h, cells were stained with acridine orange. The results are expressed as the mean percentage of cells undergoing apoptosis for three independent experiments. Error bars indicate SD. *, P < 0.05 as determined by Student’s t-test in comparison to T3D-infected wild-type cells. Figure modified from Connolly et al. (2000) and Holm et al. (2007). (c and d) HeLa cells were infected with rsT3D or each μ1 δ (c) or μ1 φ (d) mutant at an MOI of 100 PFU/cell. Following 48 h incubation, the percentage of apoptotic cells was determined by staining with acridine orange. Results are expressed as the mean percentage of apoptotic cells for triplicate samples. Error bars indicate SD. *, P < 0.05 as determined by Student’s t-test in comparison to rsT3D. Figure modified from Danthi et al. (2008a, b)
Introduction of single amino acid substitutions into the δ region of μ1 decreases the capacity of the resultant mutant viruses to effect membrane penetration, mobilize NF-κB, and evoke apoptosis (Danthi et al. 2008b) (Fig. 8c). These findings suggest that the membrane-penetration and apoptosis induction-functions of μ1 are linked and that the δ region of μ1 is an essential modulator of both processes (Danthi et al. 2008b). It is possible that membrane penetration directly initiates proapoptotic signals. Alternatively, membrane penetration might allow delivery of the μ1 cleavage fragments into the cytoplasm where prodeath signaling is elicited. Two lines of evidence support the latter possibility. First, plasmid-driven expression of the μ1 φ domain in the cytoplasm is sufficient to induce apoptosis (Coffey et al. 2006). Second, recombinant viruses with engineered substitutions within φ are diminished in NF-κB activation and apoptosis (Danthi et al. 2008a) (Fig. 8d). Importantly, a membrane-penetration-proficient φ mutant is impaired in the capacity to activate prodeath signaling, indicating that φ modulates apoptosis independent of an effect on membrane penetration (Danthi et al. 2008a). Based on these findings, it appears likely that cytoplasmic delivery of φ subsequent to membrane penetration initiates prodeath signaling following reovirus infection (Danthi et al. 2008a).
IRF-3 activation following reovirus infection requires the RIG-I pathogen sensor and the IPS-1 adaptor protein (Holm et al. 2007). Interestingly, unlike other viral systems, these host proteins are dispensable for reovirus-induced NF-κB activation (Holm et al. 2007). Since activation of IRF-3 also does not require viral RNA synthesis and occurs during viral entry, it is thought that viral genomic dsRNA triggers these signaling pathways (Holm et al. 2007). Empty reovirus particles devoid of genome can stimulate NF-κB but not IRF-3, providing additional support for the idea that NF-κB and IRF-3 are activated following reovirus infection via distinct mechanisms (Connolly et al. 2000; Holm et al. 2007). Since reovirus empty particles are capable of eliciting apoptosis (Connolly and Dermody 2002) but do not lead to IRF-3 activation (Holm et al. 2007), IRF-3 appears to play a contributory but nonessential role in reovirus-induced apoptosis. Precise mechanisms by which the products of reovirus disassembly activate innate immune response signaling networks are unknown.
Conclusions and Future Directions
The process of cell entry is poorly understood for many pathogenic viruses. This gap in knowledge has been a significant impediment to the rational design of antiviral agents and vaccines that target distinct steps in the entry process. Studies of mammalian reovirus have uncovered discrete attachment and internalization receptors, a function for cathepsin proteases in disassembly, an intricate mechanism for protein-membrane interactions, and a framework for activation of innate immune response signaling. Many of these functions are shared by other viral pathogens, suggesting conserved mechanisms of cell entry that should be amenable to common therapeutic approaches. However, there is much more to learn.
The current model of σ1-JAM-A interactions at the cell surface suggests that structural characteristics of σ1 may facilitate concurrent engagement of JAM-A and carbohydrates by appropriately positioning the receptor-binding domains. Indeed, studies of adenovirus fiber, which is structurally homologous to σ1 (Stehle and Dermody 2003), have highlighted the importance of length and flexibility in viral tropism (Wu et al. 2003). However, the contributions of σ1 length and flexibility to reovirus receptor engagement have not been explored. In the structure of a σ1-JAM-A complex, the σ1 head forms a trimer. Yet, there is evidence to suggest that σ1 may at times exist in a partially detrimerized conformation (Fraser et al. 1990; Schelling et al. 2007). An improved understanding of the role of the unusual aspartic acid sandwich trimerization motif may lend insight into the contributions of trimer instability to reovirus attachment and entry.
Reovirus serotypes display striking differences in pathogenesis that are directly linked to σ1 (Tyler et al. 1986; Weiner et al. 1977, 1980). However, each of the reovirus serotypes uses JAM-A as a receptor (Campbell et al. 2005). Considering previous studies of reovirus interactions with JAM-A and carbohydrates, we think there are three possible explanations of σ1-mediated serotype-specific differences in reovirus tropism and disease. First, the carbohydrate specificity of a particular strain of reovirus might direct infection to specific cells or tissues. In support of this idea, serotype 3 reovirus strains that vary in SA utilization also vary in disease pathogenesis in the hepatobiliary system (Barton et al. 2003). While there is some evidence that T1L binds SA in intestinal loops (Helander et al. 2003), the exact nature of the carbohydrate coreceptors used by reovirus serotypes 1 and 2 remains undefined. Second, variations in the interaction kinetics or affinity of a particular σ1 serotype for JAM-A might contribute to differences in tropism. While there is overlap among JAM-A residues required for reovirus T1L, T2J, and T3D binding, there is evidence that the binding sites in JAM-A for these viruses differ (Guglielmi et al. 2007). Furthermore, sequence alignments reveal that the residues in T3D σ1 that contact JAM-A in the complex (Kirchner et al. 2008) are not entirely conserved among reoviruses of all serotype (Campbell et al. 2005; Chappell et al. 2002). On a cell that expresses only low levels of JAM-A, differences in receptor interaction kinetics or affinity might determine whether or not reovirus can initiate infection. Third, JAM-A might serve as a serotype-independent reovirus receptor at some sites within the host, and other, as yet unidentified, receptors confer serotype-dependent tropism in the central nervous system. Indeed, studies using non-SA-binding reovirus to infect JAM-A-null mice provide support for this hypothesis and point specifically to the existence of unidentified receptors in both the intestine and the central nervous system (Antar et al. 2009). Future exploration of each of these possibilities will help clarify the role of σ1 in reovirus pathogenesis.
Internalization of reovirus requires β1 integrin (Maginnis et al. 2006), but it is not known whether reovirus directly engages β1 integrin to initiate internalization or induces interactions between JAM-A (or other receptors) and β1 integrin to activate the uptake machinery. Furthermore, it is unclear whether activation of signaling pathways is required to trigger reovirus internalization by β1 integrin. Studies using mutant β1 integrin constructs suggest that NPXY motifs within β1 integrin direct transport of reovirus to the subcellular compartment for disassembly and membrane penetration (Maginnis et al. 2008). However, the composition of the endocytic machinery recruited by the NPXY motifs that directs reovirus to the appropriate endocytic organelle for disassembly is yet to be identified. Since early steps in reovirus replication influence several stages of reovirus–host interactions (Virgin et al. 1997), it is possible that engagement of β1 integrin influences reovirus pathogenesis. However, a function for β1 integrin in reovirus disease is unproven.
Disassembly of reovirus, which results in proteolytic removal of the σ3 outer-capsid protein, is essential for exposure of the viral membrane-penetration apparatus. While this process must be precisely controlled to ensure efficient infection, mechanisms that underlie this regulation are not understood. It is not known whether the low pH environment of the endocytic compartment is merely required for optimal activity of endocytic proteases that catalyze reovirus disassembly or also functions to trigger the conformational changes in σ3 that lead to its degradation. The σ3 protein contains multiple cathepsin protease cleavage sites that may be sequentially employed to facilitate its timely removal (Ebert et al. 2002). However, it is not known whether a temporal pattern of cleavage site utilization exists for σ3. Cathepsins B and L are expressed in the intestine, liver, heart, and brain (Turk et al. 2001), which serve as sites for reovirus infection in newborn mice (Barton et al. 2003; O’Donnell et al. 2005). Cathepsin S is largely restricted to cells and tissues of the immune system (Chapman et al. 1997), which may influence reovirus replication in Peyer’s patches during enteric infection (Fleeton et al. 2004; Morrison et al. 1991). Definition of cathepsin function in reovirus pathogenesis awaits the results of ongoing studies of reovirus infection using cathepsin-deficient mice.
Recent studies have provided important insights into mechanisms by which reovirus mediates membrane penetration. While a few residues within μ1 δ and φ that regulate conformational changes required for membrane penetration have been identified by analysis of mutant viruses (Chandran et al. 2002; Danthi et al. 2008a, b), it is not known how the conformational alterations of the membrane-penetration apparatus liberate μ1N, which mediates membrane penetration (Ivanovic et al. 2008). Additionally, the domains of μ1 reorganized during disassembly are only partially identified. The mechanism by which interaction of μ1N with the membrane results in pore formation and how its pore-forming capacity is enhanced by its interaction with φ also remain to be elucidated (Agosto et al. 2006; Ivanovic et al. 2008). Finally, it is not apparent how formation of small pores in membranes results in translocation of the reovirus core across the membrane.
Early steps in reovirus infection activate innate immune response transcription factors NF-κB and IRF-3 (Connolly et al. 2000; Holm et al. 2007), which drive the apoptotic response following reovirus infection (Connolly et al. 2000; Hansberger et al. 2007; Holm et al. 2007; O’Donnell et al. 2006). The activation of NF-κB is modulated by the μ1 φ domain subsequent to membrane penetration (Danthi et al. 2008a). However, the precise mechanism by which μ1 φ evokes NF-κB activation is unclear. Neither the fate of the φ fragment following entry of reovirus into host cells nor the cellular sensors that detect φ to trigger the prodeath function of NF-κB is known. Activation of IRF-3 following reovirus infection is dependent on the recognition of viral genomic dsRNA by RIG-I and IPS-1 (Holm et al. 2007), but how the genomic dsRNA escapes from the viral core for detection by RIG-I is not evident.
In addition to enhancing an understanding of fundamental aspects of entry mechanisms employed by nonenveloped viruses, studies of reovirus cell entry are also pertinent to the development of optimal reovirus-based oncolytic and vaccine vectors. Reovirus infects transformed cells much more efficiently than it does nontransformed cells (Duncan et al. 1978). Based on initial success in using reovirus for tumor killing in animal models (Coffey et al. 1998; Hirasawa et al. 2002), reovirus is currently undergoing clinical trials as a virotherapeutic for aggressive and refractory human tumors (Stoeckel and Hay 2006). Since reovirus undergoes primary replication in intestinal tissue with few or no symptoms in humans (Tai et al. 2005) and is now amenable to genetic modification (Kobayashi et al. 2007), it also is an excellent candidate for development of a multifunctional vaccine modality to elicit mucosal immunity. Therefore, understanding the precise mechanisms by which reovirus attaches to host cells and initiates an infectious cycle will allow reovirus to be strategically engineered to facilitate retargeting to distinct host cells or enhance the efficiency of cell entry for a variety of therapeutic applications.
Acknowledgments
We acknowledge support from Public Health Service awards T32 GM08554 (K.M.G.), T32 HL07751 and F32 AI080108 (B.A.M.), R37 AI32539 (T.S.D.), R01 AI38296 (T.S.D.), R01 AI50080 (T.S.D.), and R01 GM67853 (T.S. and T.S.D.) and the Elizabeth B. Lamb Center for Pediatric Research.
Contributor Information
John E. Johnson, Phone: 858784-8643, FAX: 858784-7979, Email: jackj@scripps.edu
Terence S. Dermody, Email: terry.dermody@vanderbilt.edu
References
- Agosto MA, Ivanovic T, Nibert ML. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc Natl Acad Sci USA. 2006;103:16496–16501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agosto MA, Myers KS, Ivanovic T, Nibert ML. A positive-feedback mechanism promotes reovirus particle conversion to the intermediate associated with membrane penetration. Proc Natl Acad Sci USA. 2008;105:10571–10576. doi: 10.1073/pnas.0802039105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amerongen HM, Wilson GAR, Fields BN, Neutra MR. Proteolytic processing of reovirus is required for adherence to intestinal M cells. J Virol. 1994;68:8428–8432. doi: 10.1128/jvi.68.12.8428-8432.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar AAR, Konopka JL, Campbell JA, Henry RA, Perdigoto AL, Carter BD, Pozzi A, Abel TW, Dermody TS. Junctional adhesion molecule-A is required for hematogenous dissemination of reovirus. Cell Host Microbe. 2009;5:59–71. doi: 10.1016/j.chom.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer GS, Dermody TS. Mutations in reovirus outer-capsid protein σ3 selected during persistent infections of L cells confer resistance to protease inhibitor E64. J Virol. 1997;71:4921–4928. doi: 10.1128/jvi.71.7.4921-4928.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer GS, Ebert DH, Chung CJ, Erickson AH, Dermody TS. Mutant cells selected during persistent reovirus infection do not express mature cathepsin L and do not support reovirus disassembly. J Virol. 1999;73:9532–9543. doi: 10.1128/jvi.73.11.9532-9543.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett NM, Gillies SC, Bullivant S, Bellamy AR. Electron microscope study of reovirus reaction cores. J Virol. 1974;14:315–326. doi: 10.1128/jvi.14.2.315-326.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem. 2001;276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell F, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Barton ES, Youree BE, Ebert DH, Forrest JC, Connolly JL, Valyi-Nagy T, Washington K, Wetzel JD, Dermody TS. Utilization of sialic acid as a coreceptor is required for reovirus-induced biliary disease. J Clin Invest. 2003;111:1823–1833. doi: 10.1172/JCI16303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass DM, Bodkin D, Dambrauskas R, Trier JS, Fields BN, Wolf JL. Intraluminal proteolytic activation plays an important role in replication of type 1 reovirus in the intestines of neonatal mice. J Virol. 1990;64:1830–1833. doi: 10.1128/jvi.64.4.1830-1833.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev lmmunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- Bodkin DK, Nibert ML, Fields BN. Proteolytic digestion of reovirus in the intestinal lumens of neonatal mice. J Virol. 1989;63:4676–4681. doi: 10.1128/jvi.63.11.4676-4681.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsa J, Morash BD, Sargent MD, Copps TP, Lievaart PA, Szekely JG. Two modes of entry of reovirus particles into L cells. J Gen Virol. 1979;45:161–170. doi: 10.1099/0022-1317-45-1-161. [DOI] [PubMed] [Google Scholar]
- Borsa J, Sargent MD, Lievaart PA, Copps TP. Reovirus: evidence for a second step in the intracellular uncoating and transcriptase activation process. Virology. 1981;111:191–200. doi: 10.1016/0042-6822(81)90664-4. [DOI] [PubMed] [Google Scholar]
- Breun LA, Broering TJ, McCutcheon AM, Harrison SJ, Luongo CL, Nibert ML. Mammalian reovirus L2 gene and λ2 core spike protein sequences and whole-genome comparisons of reoviruses type 1 Lang, type 2 Jones, and type 3 Dearing. Virology. 2001;287:333–348. doi: 10.1006/viro.2001.1052. [DOI] [PubMed] [Google Scholar]
- Campbell JA, Shelling P, Wetzel JD, Johnson EM, Wilson GAR, Forrest JC, Aurrand-Lions M, Imhof B, Stehle T, Dermody TS. Junctional adhesion molecule-A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli A, Prota AE, Stehle T, Dermody TS, Recanatini M, Folkers G, Scapozza L. A molecular dynamics study of reovirus attachment protein σ1 reveals conformational changes in σ1 structure. Biophys J. 2004;86:3423–3431. doi: 10.1529/biophysj.103.030825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Nibert ML. Protease cleavage of reovirus capsid protein μ1/μ1C is blocked by alkyl sulfate detergents, yielding a new type of infectious subvirion particle. J Virol. 1998;72:467–475. doi: 10.1128/jvi.72.1.467-475.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Walker SB, Chen Y, Contreras CM, Schiff LA, Baker TS, Nibert ML. In vitro recoating of reovirus cores with baculovirus-expressed outer-capsid proteins μ1 and σ3. J Virol. 1999;73:3941–3950. doi: 10.1128/jvi.73.5.3941-3950.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Zhang X, Olson NH, Walker SB, Chappell JD, Dermody TS, Baker TS, Nibert ML. Complete in vitro assembly of the reovirus outer capsid produces highly infectious particles suitable for genetic studies of the receptor-binding protein. J Virol. 2001;75:5335–5342. doi: 10.1128/JVI.75.11.5335-5342.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Farsetta DL, Nibert ML. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein μ1 mediates membrane disruption. J Virol. 2002;76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. The delta region of outer-capsid protein μ1 undergoes conformational change and release from reovirus particles during cell entry. J Virol. 2003;77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CT, Zweerink HJ. Fate of parental reovirus in infected cell. Virology. 1971;46:544–555. doi: 10.1016/0042-6822(71)90058-4. [DOI] [PubMed] [Google Scholar]
- Chapman HA, Riese RJ, Shi GP. Emerging roles for cysteine proteases in human biology. Annu Rev Physiol. 1997;59:63–88. doi: 10.1146/annurev.physiol.59.1.63. [DOI] [PubMed] [Google Scholar]
- Chappell JD, Gunn VL, Wetzel JD, Baer GS, Dermody TS. Mutations in type 3 reovirus that determine binding to sialic acid are contained in the fibrous tail domain of viral attachment protein σ1. J Virol. 1997;71:1834–1841. doi: 10.1128/jvi.71.3.1834-1841.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JD, Barton ES, Smith TH, Baer GS, Duong DT, Nibert ML, Dermody TS. Cleavage susceptibility of reovirus attachment protein σ1 during proteolytic disassembly of virions is determined by a sequence polymorphism in the σ1 neck. J Virol. 1998;72:8205–8213. doi: 10.1128/jvi.72.10.8205-8213.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JD, Duong JL, Wright BW, Dermody TS. Identification of carbohydrate-binding domains in the attachment proteins of type 1 and type 3 reoviruses. J Virol. 2000;74:8472–8479. doi: 10.1128/JVI.74.18.8472-8479.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell JD, Prota A, Dermody TS, Stehle T. Crystal structure of reovirus attachment protein σ1 reveals evolutionary relationship to adenovirus fiber. EMBO J. 2002;21:1–11. doi: 10.1093/emboj/21.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WJ, Goldstein JL, Brown MS. NPXY, a sequence often found in cytoplasmic tails, is required for coated pit-mediated internalization of the low density lipoprotein receptor. J Biol Chem. 1990;265:3116–3123. [PubMed] [Google Scholar]
- Clark KM, Wetzel JD, Bayley J, Ebert DH, McAbee SA, Stoneman EK, Baer GS, Zhu Y, Wilson GJ, Prasad BVV, Dermody TS. Reovirus variants selected for resistance to ammonium chloride have mutations in viral outer-capsid protein σ3. J Virol. 2006;80:671–681. doi: 10.1128/JVI.80.2.671-681.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleveland DR, Zarbl H, Millward S. Reovirus guanylyltransferase is L2 gene product lambda 2. J Virol. 1986;60:307–311. doi: 10.1128/jvi.60.1.307-311.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- Coffey CM, Sheh A, Kim IS, Chandran K, Nibert ML, Parker JS. Reovirus outer capsid protein μ1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J Virol. 2006;80:8422–8438. doi: 10.1128/JVI.02601-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Dermody TS. Virion disassembly is required for apoptosis induced by reovirus. J Virol. 2002;76:1632–1641. doi: 10.1128/JVI.76.4.1632-1641.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. Reovirus-induced apoptosis requires activation of transcription factor NF-κB. J Virol. 2000;74:2981–2989. doi: 10.1128/JVI.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly JL, Barton ES, Dermody TS. Reovirus binding to cell surface sialic acid potentiates virus-induced apoptosis. J Virol. 2001;75:4029–4039. doi: 10.1128/JVI.75.9.4029-4039.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs KM. Stoichiometry of reovirus structural proteins in virus, ISVP, and core particles. Virology. 1998;243:218–228. doi: 10.1006/viro.1998.9061. [DOI] [PubMed] [Google Scholar]
- Danthi P, Tosteson M, Li QH, Chow M. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J Virol. 2003;77:5266–5274. doi: 10.1128/JVI.77.9.5266-5274.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Hansberger MW, Campbell JA, Forrest JC, Dermody TS. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. J Virol. 2006;80:1261–1270. doi: 10.1128/JVI.80.3.1261-1270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. Independent regulation of reovirus membrane penetration and apoptosis by the μ1 φ domain. PLoS Pathog. 2008;4:e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. Reovirus apoptosis and virulence are regulated by host cell membrane-penetration efficiency. J Virol. 2008;82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CG, Lehrman MA, Russell DW, Anderson RG, Brown MS, Goldstein JL. The J.D. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell. 1986;45:15–24. doi: 10.1016/0092-8674(86)90533-7. [DOI] [PubMed] [Google Scholar]
- Dermody TS, Nibert ML, Bassel-Duby R, Fields BN. A sigma 1 region important for hemagglutination by serotype 3 reovirus strains. J Virol. 1990;64:5173–5176. doi: 10.1128/jvi.64.10.5173-5176.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermody TS, Nibert ML, Wetzel JD, Tong X, Fields BN. Cells and viruses with mutations affecting viral entry are selected during persistent infections of L cells with mammalian reoviruses. J Virol. 1993;67:2055–2063. doi: 10.1128/jvi.67.4.2055-2063.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Drayna D, Fields BN. Activation and characterization of the reovirus transcriptase: genetic analysis. J Virol. 1982;41:110–118. doi: 10.1128/jvi.41.1.110-118.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryden KA, Farsetta DL, Wang G, Keegan JM, Fields BN, Baker TS, Nibert ML. Internal structures containing transcriptase-related proteins in top component particles of mammalian orthoreovirus. Virology. 1998;245:33–46. doi: 10.1006/viro.1998.9146. [DOI] [PubMed] [Google Scholar]
- Duncan MR, Stanish SM, Cox DC. Differential sensitivity of normal and transformed human cells to reovirus infection. J Virol. 1978;28:444–449. doi: 10.1128/jvi.28.2.444-449.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Wetzel JD, Brumbaugh DE, Chance SR, Stobie LE, Baer GS, Dermody TS. Adaptation of reovirus to growth in the presence of protease inhibitor E64 segregates with a mutation in the carboxy terminus of viral outer-capsid protein σ3. J Virol. 2001;75:3197–3206. doi: 10.1128/JVI.75.7.3197-3206.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Deussing J, Peters C, Dermody TS. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
- Ehrlich M, Boll W, Van Oijen A, Hariharan R, Chandran K, Nibert ML, Kirchhausen T. Endocytosis by random initiation and stabilization of clathrin-coated pits. Cell. 2004;118:591–605. doi: 10.1016/j.cell.2004.08.017. [DOI] [PubMed] [Google Scholar]
- Fausnaugh J, Shatkin AJ. Active site localization in a viral mRNA capping enzyme. J Biol Chem. 1990;265:7669–7672. [PubMed] [Google Scholar]
- Fleeton M, Contractor N, Leon F, Wetzel JD, Dermody TS, Kelsall B. Peyer’s patch dendritic cells process viral antigen from apoptotic epithelial cells in the intestine of reovirus-infected mice. J Exp Med. 2004;200:235–245. doi: 10.1084/jem.20041132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest JC, Campbell JA, Schelling P, Stehle T, Dermody TS. Structure-function analysis of reovirus binding to junctional adhesion molecule 1. Implications for the mechanism of reovirus attachment. J Biol Chem. 2003;278:48434–48444. doi: 10.1074/jbc.M305649200. [DOI] [PubMed] [Google Scholar]
- Fraser RDB, Furlong DB, Trus BL, Nibert ML, Fields BN, Steven AC. Molecular structure of the cell-attachment protein of reovirus: correlation of computer-processed electron micrographs with sequence-based predictions. J Virol. 1990;64:2990–3000. doi: 10.1128/jvi.64.6.2990-3000.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furlong DB, Nibert ML, Fields BN. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol. 1988;62:246–256. doi: 10.1128/jvi.62.1.246-256.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuichi Y, Muthukrishnan S, Tomasz J, Shatkin AJ. Mechanism of formation of reovirus mRNA 5′-terminal blocked and methylated sequence M7 GpppGm pC. J Biol Chem. 1976;251:5043–5053. [PubMed] [Google Scholar]
- Gentsch JR, Pacitti AF. Effect of neuraminidase treatment of cells and effect of soluble glycoproteins on type 3 reovirus attachment to murine L cells. J Virol. 1985;56:356–364. doi: 10.1128/jvi.56.2.356-364.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentsch JR, Pacitti AF. Differential interaction of reovirus type 3 with sialylated receptor components on animal cells. Virology. 1987;161:245–248. doi: 10.1016/0042-6822(87)90192-9. [DOI] [PubMed] [Google Scholar]
- Gillies S, Bullivant S, Bellamy AR. Viral RNA polymerases: electron microscopy of reovirus reaction cores. Science. 1971;174:694–696. doi: 10.1126/science.174.4010.694. [DOI] [PubMed] [Google Scholar]
- Golden JW, Bahe JA, Lucas WT, Nibert ML, Schiff LA. Cathepsin S supports acid-independent infection by some reoviruses. J Biol Chem. 2004;279:8547–8557. doi: 10.1074/jbc.M309758200. [DOI] [PubMed] [Google Scholar]
- Goto H, Kawaoka Y. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc Natl Acad Sci USA. 1998;95:10224–10228. doi: 10.1073/pnas.95.17.10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardado CP, Fox GC, Hermo Parrado XL, Llamas-Saiz AL, Costas C, Martinez-Costas J, Benavente J, van Raaij MJ. Structure of the carboxy-terminal receptor-binding domain of avian reovirus fibre sigmaC. J Mol Biol. 2005;354:137–149. doi: 10.1016/j.jmb.2005.09.034. [DOI] [PubMed] [Google Scholar]
- Guglielmi KM, Johnson EM, Stehle T, Dermody TS. Attachment and cell entry of mammalian orthoreovirus. Curr Top Microbiol Immunol. 2006;309:1–38. doi: 10.1007/3-540-30773-7_1. [DOI] [PubMed] [Google Scholar]
- Guglielmi KM, Kirchner E, Holm GH, Stehle T, Dermody TS. Reovirus binding determinants in junctional adhesion molecule-A. J Biol Chem. 2007;282:17930–17940. doi: 10.1074/jbc.M702180200. [DOI] [PubMed] [Google Scholar]
- Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. IκB kinase subunits α and γ are required for activation of NF-κB and induction of apoptosis by mammalian reovirus. J Virol. 2007;81:1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison S. Principles of virus structure. In: Knipe DM, Howley PM, editors. Fields virology. 4. Philadelphia: Lippincott-Raven; 2001. pp. 53–85. [Google Scholar]
- Helander A, Silvey KJ, Mantis NJ, Hutchings AB, Chandran K, Lucas WT, Nibert ML, Neutra MR. The viral σ1 protein and glycoconjugates containing α2-3-linked sialic acid are involved in type 1 reovirus adherence to M cell apical surfaces. J Virol. 2003;77:7964–7977. doi: 10.1128/JVI.77.14.7964-7977.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa K, Nishikawa SG, Norman KL, Alain T, Kossakowska A, Lee PW. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62:1696–1701. [PubMed] [Google Scholar]
- Holm GH, Zurney J, Tumilasci V, Danthi P, Hiscott J, Sherry B, Dermody TS. Retinoic acid-inducible gene-I and interferon-β promoter stimulator-1 augment proapoptotic responses following mammalian reovirus infection via interferon regulatory factor-3. J Biol Chem. 2007;282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- Hooper JW, Fields BN. Role of the μ1 protein in reovirus stability and capacity to cause chromium release from host cells. J Virol. 1996;70:459–467. doi: 10.1128/jvi.70.1.459-467.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang I-C, Bosch BJ, Li F, Li W, Lee KH, Ghiran S, Vasilieva N, Dermody TS, Harrison SC, Dormitzer PR, Farzan M, Rottier PJ, Choe H. SARS coronavirus, but not human coronavirus NL63, utilizes cathepsin L to infect ACE2-expressing cells. J Biol Chem. 2006;281:3198–3203. doi: 10.1074/jbc.M508381200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic T, Agosto MA, Chandran K, Nibert ML. A role for molecular chaperone Hsc70 in reovirus outer-capsid disassembly. J Biol Chem. 2007;282:12210–12219. doi: 10.1074/jbc.M610258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J. 2008;27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jané-Valbuena J, Nibert ML, Spencer SM, Walker SB, Baker TS, Chen Y, Centonze VE, Schiff LA. Reovirus virion-like particles obtained by recoating infectious subvirion particles with baculovirus-expressed σ3 protein: an approach for analyzing σ3 functions during virus entry. J Virol. 1999;73:2963–2973. doi: 10.1128/jvi.73.4.2963-2973.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jané-Valbuena J, Breun LA, Schiff LA, Nibert ML. Sites and determinants of early cleavages in the proteolytic processing pathway of reovirus surface protein σ3. J Virol. 2002;76:5184–5197. doi: 10.1128/JVI.76.10.5184-5197.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keroack M, Fields BN. Viral shedding and transmission between hosts determined by reovirus L2 gene. Science. 1986;232:1635–1638. doi: 10.1126/science.3012780. [DOI] [PubMed] [Google Scholar]
- Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. Structure of reovirus σ1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog. 2008;4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Antar AAR, Boehme KW, Danthi P, Eby EA, Guglielmi KM, Holm GH, Johnson EM, Maginnis MS, Naik S, Skelton WB, Wetzel JD, Wilson GJ, Chappell JD, Dermody TS. A plasmid-based reverse genetics system for animal double-stranded RNA viruses. Cell Host Microbe. 2007;1:147–157. doi: 10.1016/j.chom.2007.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laniosz V, Holthusen KA, Meneses PI. Bovine papillomavirus type 1: from clathrin to caveolin. J Virol. 2008;82:6288–6298. doi: 10.1128/JVI.00569-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. Structure of the reovirus membrane-penetration protein, μ1, in a complex with its protector protein, σ3. Cell. 2002;108:283–295. doi: 10.1016/S0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucia-Jandris P, Hooper JW, Fields BN. Reovirus M2 gene is associated with chromium release from mouse L cells. J Virol. 1993;67:5339–5345. doi: 10.1128/jvi.67.9.5339-5345.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo CL, Contreras CM, Farsetta DL, Nibert ML. Binding site for S-adenosyl-l-methionine in a central region of mammalian orthoreovirus λ2 protein. Evidence for activities in mRNA cap methylation. J Biol Chem. 1998;273:23773–23780. doi: 10.1074/jbc.273.37.23773. [DOI] [PubMed] [Google Scholar]
- Luongo CL, Reinisch KM, Harrison SC, Nibert ML. Identification of the mRNA guanylyltransferase region and active site in reovirus λ2 protein. J Biol Chem. 2000;275:2804–2810. doi: 10.1074/jbc.275.4.2804. [DOI] [PubMed] [Google Scholar]
- Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. β1 Integrin mediates internalization of mammalian reovirus. J Virol. 2006;80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maginnis MS, Mainou BA, Derdowski AM, Johnson EM, Zent R, Dermody TS. NPXY motifs in the β1 integrin cytoplasmic tail are required for functional reovirus entry. J Virol. 2008;82:3181–3191. doi: 10.1128/JVI.01612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann MA, Knipe DM, Fischbach GD, Fields BN. Type 3 reovirus neuroinvasion after intramuscular inoculation: direct invasion of nerve terminals and age-dependent pathogenesis. Virology. 2002;303:222–231. doi: 10.1006/viro.2002.1699. [DOI] [PubMed] [Google Scholar]
- Mao ZX, Joklik WK. Isolation and enzymatic characterization of protein λ2, the reovirus guanylyltransferase. Virology. 1991;185:377–386. doi: 10.1016/0042-6822(91)90785-A. [DOI] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrae MA, Joklik WK. The nature of the polypeptide encoded by each of the ten double-stranded RNA segments of reovirus type 3. Virology. 1978;89:578–593. doi: 10.1016/0042-6822(78)90199-X. [DOI] [PubMed] [Google Scholar]
- Mendez II, She YM, Ens W, Coombs KM. Digestion pattern of reovirus outer capsid protein σ3 determined by mass spectrometry. Virology. 2003;311:289–304. doi: 10.1016/S0042-6822(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merckel MC, Huiskonen JT, Bamford DH, Goldman A, Tuma R. The structure of the bacteriophage PRD1 spike sheds light on the evolution of viral capsid architecture. Mol Cell. 2005;18:161–170. doi: 10.1016/j.molcel.2005.03.019. [DOI] [PubMed] [Google Scholar]
- Metcalf P. The symmetry of the reovirus outer shell. J Ultrastruct Res. 1982;78:292–301. doi: 10.1016/S0022-5320(82)80004-X. [DOI] [PubMed] [Google Scholar]
- Middleton JK, Agosto MA, Severson TF, Yin J, Nibert ML. Thermostabilizing mutations in reovirus outer-capsid protein μ1 selected by heat inactivation of infectious subvirion particles. Virology. 2007;361:412–425. doi: 10.1016/j.virol.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/S0092-8674(00)81363-X. [DOI] [PubMed] [Google Scholar]
- Morris SM, Cooper JA. Disabled-2 colocalizes with the LDLR in clathrin-coated pits and interacts with AP-2. Traffic. 2001;2:111–123. doi: 10.1034/j.1600-0854.2001.020206.x. [DOI] [PubMed] [Google Scholar]
- Morrison LA, Sidman RL, Fields BN. Direct spread of reovirus from the intestinal lumen to the central nervous system through vagal autonomic nerve fibers. Proc Natl Acad Sci USA. 1991;88:3852–3856. doi: 10.1073/pnas.88.9.3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustoe TA, Ramig RF, Sharpe AH, Fields BN. Genetics of reovirus: identification of the dsRNA segments encoding the polypeptides of the μ and σ size classes. Virology. 1978;89:594–604. doi: 10.1016/0042-6822(78)90200-3. [DOI] [PubMed] [Google Scholar]
- Nibert ML, Fields BN. A carboxy-terminal fragment of protein μ1/μ1C is present in infectious subvirion particles of mammalian reoviruses and is proposed to have a role in penetration. J Virol. 1992;66:6408–6418. doi: 10.1128/jvi.66.11.6408-6418.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Dermody TS, Fields BN. Structure of the reovirus cell-attachment protein: a model for the domain organization of σ1. J Virol. 1990;64:2976–2989. doi: 10.1128/jvi.64.6.2976-2989.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibert ML, Chappell JD, Dermody TS. Infectious subvirion particles of reovirus type 3 Dearing exhibit a loss in infectivity and contain a cleaved σ1 protein. J Virol. 1995;69:5057–5067. doi: 10.1128/jvi.69.8.5057-5067.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell SM, Hansberger MW, Connolly JL, Chappell JD, Watson MJ, Pierce JM, Wetzel JD, Han W, Barton ES, Forrest JC, Valyi-Nagy T, Yull FE, Blackwell TS, Rottman JN, Sherry B, Dermody TS. Organ-specific roles for transcription factor NF-κB in reovirus-induced apoptosis and disease. J Clin Invest. 2005;115:2341–2350. doi: 10.1172/JCI22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell SM, Holm GH, Pierce JM, Tian B, Watson MJ, Chari RS, Ballard DW, Brasier AR, Dermody TS. Identification of an NF-κB-dependent gene network in cells infected by mammalian reovirus. J Virol. 2006;80:1077–1086. doi: 10.1128/JVI.80.3.1077-1086.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard AL, Chandran K, Zhang X, Parker JS, Baker TS, Nibert ML. Putative autocleavage of outer capsid protein μ1, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J Virol. 2004;78:8732–8745. doi: 10.1128/JVI.78.16.8732-8745.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleinikov AV, Zhao J, Makker SP. Cytosolic adaptor protein Dab2 is an intracellular ligand of endocytic receptor gp600/megalin. Biochem J. 2000;347:613–621. doi: 10.1042/0264-6021:3470613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olland AM, Jané-Valbuena J, Schiff LA, Nibert ML, Harrison SC. Structure of the reovirus outer capsid and dsRNA-binding protein σ3 at 1.8 Å resolution. EMBO J. 2001;20:979–989. doi: 10.1093/emboj/20.5.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson S, Bergstrom T. Glycoconjugate glycans as viral receptors. Ann Med. 2005;37:154–172. doi: 10.1080/07853890510007340. [DOI] [PubMed] [Google Scholar]
- Pager CT, Dutch RE. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J Virol. 2005;79:12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul RW, Choi AH, Lee PWK. The α-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology. 1989;172:382–385. doi: 10.1016/0042-6822(89)90146-3. [DOI] [PubMed] [Google Scholar]
- Prota AE, Campbell JA, Schelling P, Forrest JC, Peters TR, Watson MJ, Aurrand-Lions M, Imhof B, Dermody TS, Stehle T. Crystal structure of human junctional adhesion molecule 1: implications for reovirus binding. Proc Natl Acad Sci USA. 2003;100:5366–5371. doi: 10.1073/pnas.0937718100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querbes W, O’Hara BA, Williams G, Atwood WJ. Invasion of host cells by JC virus identifies a novel role for caveolae in endosomal sorting of noncaveolar ligands. J Virol. 2006;80:9402–9413. doi: 10.1128/JVI.01086-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinisch KM, Nibert ML, Harrison SC. Structure of the reovirus core at 3.6 Å resolution. Nature. 2000;404:960–967. doi: 10.1038/35010041. [DOI] [PubMed] [Google Scholar]
- Riese RJ, Wolf PR, Bromme D, Natkin LR, Villadangos JA, Ploegh HL, Chapman HA. Essential role for cathepsin S in MHC class II-associated invariant chain processing and peptide loading. Immunity. 1996;4:357–366. doi: 10.1016/S1074-7613(00)80249-6. [DOI] [PubMed] [Google Scholar]
- Rubin DH, Wetzel JD, Williams WV, Cohen JA, Dworkin C, Dermody TS. Binding of type 3 reovirus by a domain of the σ1 protein important for hemagglutination leads to infection of murine erythroleukemia cells. J Clin Invest. 1992;90:2536–2542. doi: 10.1172/JCI116147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sattentau QJ, Moore JP. Conformational changes induced in the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J Exp Med. 1991;174:407–415. doi: 10.1084/jem.174.2.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schelling P, Guglielmi KM, Kirchner E, Paetzold B, Dermody TS, Stehle T. The reovirus σ1 aspartic acid sandwich: a trimerization motif poised for conformational change. J Biol Chem. 2007;282:11582–11589. doi: 10.1074/jbc.M610805200. [DOI] [PubMed] [Google Scholar]
- Schiff LA, Nibert ML, Tyler KL. Orthoreoviruses and their replication. In: Knipe DM, Howley PM, editors. Fields virology. 5. Philadelphia: Lippincott Williams & Wilkins; 2007. pp. 1853–1915. [Google Scholar]
- Schneemann A, Zhong W, Gallagher TM, Rueckert RR. Maturation cleavage required for infectivity of a nodavirus. J Virol. 1992;66:6728–6734. doi: 10.1128/jvi.66.11.6728-6734.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seliger LS, Zheng K, Shatkin AJ. Complete nucleotide sequence of reovirus L2 gene and deduced amino acid sequence of viral mRNA guanylyltransferase. J Biol Chem. 1987;262:16289–16293. [PubMed] [Google Scholar]
- Silverstein SC, Astell C, Levin DH, Schonberg M, Acs G. The mechanism of reovirus uncoating and gene activation in vivo. Virology. 1972;47:797–806. doi: 10.1016/0042-6822(72)90571-5. [DOI] [PubMed] [Google Scholar]
- Starnes MC, Joklik WK. Reovirus protein λ3 is a poly(C)-dependent poly(G) polymerase. Virology. 1993;193:356–366. doi: 10.1006/viro.1993.1132. [DOI] [PubMed] [Google Scholar]
- Stehle T, Dermody TS. Structural evidence for common functions and ancestry of the reovirus and adenovirus attachment proteins. Rev Med Virol. 2003;13:123–132. doi: 10.1002/rmv.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle T, Dermody TS. Structural similarities in the cellular receptors used by adenovirus and reovirus. Viral Immunol. 2004;17:129–143. doi: 10.1089/0882824041310621. [DOI] [PubMed] [Google Scholar]
- Stieneke-Grober A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992;11:2407–2414. doi: 10.1002/j.1460-2075.1992.tb05305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoeckel J, Hay JG. Drug evaluation: Reolysin-wild-type reovirus as a cancer therapeutic. Curr Opin Mol Ther. 2006;8:249–260. [PubMed] [Google Scholar]
- Strong JE, Leone G, Duncan R, Sharma RK, Lee PW. Biochemical and biophysical characterization of the reovirus cell attachment protein sigma 1: evidence that it is a homotrimer. Virology. 1991;184:23–32. doi: 10.1016/0042-6822(91)90818-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturzenbecker LJ, Nibert ML, Furlong DB, Fields BN. Intracellular digestion of reovirus particles requires a low pH and is an essential step in the viral infectious cycle. J Virol. 1987;61:2351–2361. doi: 10.1128/jvi.61.8.2351-2361.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE, Dermody TS. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis. 2005;191:1221–1224. doi: 10.1086/428911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y, Farsetta DL, Nibert ML, Harrison SC. RNA synthesis in a cage – structural studies of reovirus polymerase λ3. Cell. 2002;111:733–745. doi: 10.1016/S0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- Tardieu M, Powers ML, Weiner HL. Age-dependent susceptibility to reovirus type 3 encephalitis: role of viral and host factors. Ann Neurol. 1983;13:602–607. doi: 10.1002/ana.410130604. [DOI] [PubMed] [Google Scholar]
- Tosteson MT, Nibert ML, Fields BN. Ion channels induced in lipid bilayers by subvirion particles of the nonenveloped mammalian reoviruses. Proc Natl Acad Sci USA. 1993;90:10549–10552. doi: 10.1073/pnas.90.22.10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk V, Turk B, Turk D. Lysosomal cysteine proteases: facts and opportunities. EMBO J. 2001;20:4629–4633. doi: 10.1093/emboj/20.17.4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, McPhee DA, Fields BN. Distinct pathways of viral spread in the host determined by reovirus S1 gene segment. Science. 1986;233:770–774. doi: 10.1126/science.3016895. [DOI] [PubMed] [Google Scholar]
- Tyler KL, Squier MK, Rodgers SE, Schneider SE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein σ1. J Virol. 1995;69:6972–6979. doi: 10.1128/jvi.69.11.6972-6979.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, Squier MKT, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol. 1996;70:7984–7991. doi: 10.1128/jvi.70.11.7984-7991.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler KL, Barton ES, Ibach ML, Robinson C, Valyi-Nagy T, Campbell JA, Clarke P, O’Donnell SM, Wetzel JD, Dermody TS. Isolation and molecular characterization of a novel type 3 reovirus from a child with meningitis. J Infect Dis. 2004;189:1664–1675. doi: 10.1086/383129. [DOI] [PubMed] [Google Scholar]
- Ugolini S, Mondor I, Sattentau QJ. HIV-1 attachment: another look. Trends Microbiol. 1999;7:144–149. doi: 10.1016/S0966-842X(99)01474-2. [DOI] [PubMed] [Google Scholar]
- van Raaij MJ, Mitraki A, Lavigne G, Cusack S. A triple β-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature. 1999;401:935–938. doi: 10.1038/44880. [DOI] [PubMed] [Google Scholar]
- Virgin HW, Tyler KL, Dermody TS. Reovirus. In: Nathanson N, editor. Viral pathogenesis. New York: Lippincott-Raven; 1997. pp. 669–699. [Google Scholar]
- Walukiewicz HE, Banerjee M, Schneemann A, Johnson JE. Rescue of maturation-defective flock house virus infectivity with noninfectious, mature, viruslike particles. J Virol. 2008;82:2025–2027. doi: 10.1128/JVI.02278-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Drayna D, Averill DR, Jr, Fields BN. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci USA. 1977;74:5744–5748. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner HL, Powers ML, Fields BN. Absolute linkage of virulence and central nervous system tropism of reoviruses to viral hemagglutinin. J Infect Dis. 1980;141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- Wessner DR, Fields BN. Isolation and genetic characterization of ethanol-resistant reovirus mutants. J Virol. 1993;67:2442–2447. doi: 10.1128/jvi.67.5.2442-2447.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel JD, Wilson GJ, Baer GS, Dunnigan LR, Wright JP, Tang DSH, Dermody TS. Reovirus variants selected during persistent infections of L cells contain mutations in the viral S1 and S4 genes and are altered in viral disassembly. J Virol. 1997;71:1362–1369. doi: 10.1128/jvi.71.2.1362-1369.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiethoff CM, Wodrich H, Gerace L, Nemerow GR. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol. 2005;79:1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LA, Martin-Padura I, Dejana E, Hogg N, Simmons DL. Identification and characterisation of human junctional adhesion molecule (JAM) Mol Immunol. 1999;36:1175–1188. doi: 10.1016/S0161-5890(99)00122-4. [DOI] [PubMed] [Google Scholar]
- Wilson GJ, Nason EL, Hardy CS, Ebert DH, Wetzel JD, Prasad BVV, Dermody TS. A single mutation in the carboxy terminus of reovirus outer-capsid protein σ3 confers enhanced kinetics of σ3 proteolysis, resistance to inhibitors of viral disassembly, and alterations in σ3 structure. J Virol. 2002;76:9832–9843. doi: 10.1128/JVI.76.19.9832-9843.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu E, Pache L, Von Seggern DJ, Mullen TM, Mikyas Y, Stewart PL, Nemerow GR. Flexibility of the adenovirus fiber is required for efficient receptor interaction. J Virol. 2003;77:7225–7235. doi: 10.1128/JVI.77.13.7225-7235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- Zhang X, Walker SB, Chipman PR, Nibert ML, Baker TS. Reovirus polymerase λ 3 localized by cryo-electron microscopy of virions at a resolution of 7.6 Å. Nat Struct Biol. 2003;10:1011–1018. doi: 10.1038/nsb1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. Features of reovirus outer capsid protein μ1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure. 2005;13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chandran K, Nibert ML, Harrison SC. Reovirus μ1 structural rearrangements that mediate membrane penetration. J Virol. 2006;80:12367–12376. doi: 10.1128/JVI.01343-06. [DOI] [PMC free article] [PubMed] [Google Scholar]