

Abstract
2-Amino-4-oxo-6-substituted-pyrrolo[2,3-d]-pyrimidine antifolate thiophene regioisomers of AGF94 (4) with a thienoyl side chain and three-carbon bridge lengths [AGF150 (5) and AGF154 (7)] were synthesized as potential antitumor agents. These analogues inhibited proliferation of Chinese hamster ovary (CHO) sublines expressing folate receptors (FRs) α or β (IC50s < 1 nM) or the proton-coupled folate transporter (PCFT) (IC50 < 7 nM). Compounds 5 and 7 inhibited KB, IGROV1, and SKOV3 human tumor cells at subnanomolar concentrations, reflecting both FRα and PCFT uptake. AGF152 (6) and AGF163 (8), 2,4-diamino-5-substituted-furo[2,3-d]pyrimidine thiophene regioisomers, also inhibited growth of FR-expressing CHO and KB cells. All four analogues inhibited glycinamide ribonucleotide formyltransferase (GARFTase). Crystal structures of human GARFTase complexed with 5 and 7 were reported. In severe combined immunodeficient mice bearing SKOV3 tumors, 7 was efficacious. The selectivity of these compounds for PCFT and for FRα and β over the ubiquitously expressed reduced folate carrier is a paradigm for selective tumor targeting.
INTRODUCTION
Classic antifolates such as methotrexate (MTX), pemetrexed (PMX), and pralatrexate (PDX) (Figure 1) are integral components of the chemotherapy drug arsenal for cancer and other diseases.1,2 These analogues are all excellent substrates for the reduced folate carrier (RFC) and are variously substrates for the proton-coupled folate transporter (PCFT) and high affinity folate receptors (FRs) α and β. Whereas both RFC and PCFT are facilitative transporters,2–6 FRs are glycosylphosphatidylinositol-tethered proteins that mediate cellular uptake of folates by receptor-mediated endocytosis.3
Figure 1.

Structures of classical antifolates.
RFC is expressed ubiquitously in tissues and tumors and is the major mechanism of cellular uptake of classical antifolate drugs.6 FRs are expressed in malignancies such as ovarian cancer (FRα) and in myeloid leukemias (FRβ).3 FRs are also expressed in some normal tissues where, unlike FRs expressed in tumor cells, they are inaccessible to circulating (anti)folates (renal tubules and FRα) or are nonfunctional (thymus and FRβ).3 PCFT is expressed in a number of solid tumors including ovarian cancer, generally in combination with FRα, and is active at acidic pHs associated with the tumor microenvironment.2,5 Whereas normal tissues such as the proximal small intestine, liver and kidney also express PCFT5 with the exception of the small intestine, the pH of the microenvironments of most normal tissues are not conducive to high levels of PCFT transport.
PMX is currently among the most prescribed antitumor agents used in the clinic.7 The principal indications for PMX are malignant pleural mesothelioma (with cisplatin) and non-small cell lung cancer.7–11 Neutropenia is the major dose-limiting toxicity associated with PMX12 and reflects its nonselective uptake into normal cells, as PMX is transported by RFC, which is expressed in normal as well as tumor cells.2,6 In addition to its toxicity, another limitation of PMX therapy is the onset of resistance, at least in part involving thymidylate synthase (TS),13 its principal intracellular target.7,8 Although PMX has been shown to inhibit other intracellular targets including 5-aminoimidazole-4-carboxamide (AICA) ribonucleotide formyltransferase (AICARFTase) and glycinamide ribonucleotide (GAR) formyltransferase (GARFTase) in de novo purine nucleotide biosynthesis and dihydrofolate reductase, these are considered to be secondary targets.8,14
Lometrexol [(6R)5,10-dideazatetrahydrofolate; LMTX],15 (2S)-2-((5-(2-((6R)-2-amino-4-oxo-5,6,7,8-tetrahydro-1H-pyrido[2,3-d]pyrimidin-6-yl)ethyl)thiophene-2-carbonyl)-amino) pentanedioic acid (LY309887),15 and (2S)-2-((5-(2-((6S)-2-amino-4-oxo-1,6,7,8-tetrahydropyrimido[5,4-b][1,4]-thiazin-6-yl)ethyl)thiophene-2-carbonyl)amino)pentane-dioic acid (AG2034)16 were described as antipurine antifolates that inhibited GARFTase as their principal cellular targets and progressed to clinical trials.17–19 However, toxicities for these drugs were dose-limiting, likely at least in part the result of a lack of selectivity for tumor cells and their cellular uptake by RFC and metabolism to polyglutamates in normal tissues.
Reflecting the tumor expression and functional profiles of the folate transporters, the notion of selective tumor targeting cytotoxic agents via their transport specificities for FRα and/or PCFT is especially appealing.2,5,20,21 Thus, folate and pteroate conjugates have been engineered to deliver therapeutic agents to FRα-expressing tumors including ovarian cancer.21 The FR-targeted conjugate EC145 (vintafolide) successfully completed a phase II clinical trial with platinum resistant ovarian cancer22 and progressed to a phase III clinical trial. EC145 is being evaluated in a randomized phase II trial for non-small cell lung cancer. N-[4-[2-Propyn-1-yl[(6S)-4,6,7,8-tetrahydro-2-(hydroxymethyl)-4-oxo-3H-cyclopenta[g]quinazolin-6-yl]amino]-benzoyl]-L-γ-glutamyl-D-glutamic acid (ONX0801) is a TS inhibitor and selective FR substrate being tested in a clinical trial.23
We discovered 6-substituted-2-amino-4-oxo pyrrolo[2,3-d]-pyrimidine scaffolds related to PMX with thieno side chains as potent and selective GARFTase inhibitors (rather than TS inhibition as for PMX) and with a high level of selectivity for FRα, FRβ, and PCFT over RFC [3 (AGF71) and 4 (AGF94); Figure 2].24–29 With 3 as the lead, we synthesized regioisomers 1 (AGF117) and 2 (AGF118) (Figure 2) to better define the distance between the bicyclic scaffold and the glutamate.28 Transporter selectivity and potencies were tested against engineered Chinese hamster ovary (CHO) and FRα- and PCFT-expressing human tumor cells, establishing that both 1 and 2 were selective for FR and PCFT over RFC uptake. Toward human tumor cells (KB, IGROV1), potencies were equivalent or in some cases better than those for 3.28 Finally, reducing the number of bridge carbons on 3 from four to three afforded the most potent analogue (4) of this series, with
Figure 2.

Structures of 6-substituted pyrrolo[2,3-d]pyrimidine antifolates.
In this study, we report the synthesis and biological activities of the 3′,5′ (5), and 2′,4′ (7) thienoyl regioisomers of 4 (Figure 3). The synthetic methodology adopted for 5 and 7 also provided the corresponding 2,4-diamino-5-substituted-furo[2,3-d]pyrimidines 6 and 8, which were chromatographically separated from their pyrrolo[2,3-d]pyrimidine regioisomer precursors and were biologically evaluated. Using 7 as a prototype, we document its selective membrane transport by PCFT and FRs, correlating with its potent in vitro antitumor activities. Further, we report GARFTase inhibition by compounds 5–8 and present for the first time crystal structures of human GARFTase complexed with 5 and 7 which strongly corroborate our molecular modeling efforts and enzyme inhibition results. Compound 7 exhibits potent in vivo antitumor efficacy with modest toxicity, reflecting its selectivity for cellular uptake by FR and PCFT over RFC and would be expected to circumvent resistance to PMX by inhibiting GARFTase rather than TS. Compounds 6 and 8 were of interest to determine if FRα and/or PCFT transport selectivity over RFC along with GARFTase inhibitory activity could be achieved with a 2,4-diaminofuro[2,3-d]pyrimidine scaffold. To our knowledge, no 2,4-diaminofuro[2,3-d]pyrimidines as folate transport substrates, along with GARFTase inhibitory activity, have been previously reported. This series of compounds is a paradigm for selective tumor targeting, reflecting tumor-specific drug uptake.
Figure 3.

Structures of classical regioisomeric side chain thieno-2-amino-4-oxo-6-substituted pyrrolo[2,3-d]pyrimidine antifolates 5 and 7 and 2,4-diamino-5-substituted-furo[2,3-d]pyrimidine antifolates 6 and 8. antiproliferative activities at low and subnanomolar concentrations toward PCFT- and FR-expressing cells including human tumors.27 Thus, it was of considerable interest to synthesize thiophene regioisomers of 4, with the intention of providing analogues with improved antitumor potencies and transporter selectivity, compared to PMX, which would overcome toxicity and potentially resistance to PMX.
MOLECULAR MODELING STUDIES
The X-ray crystal structures of human FRα (PDB: 4LRH, 2.8 Å resolution),30 FRβ (PDB: 4KN0, 2.1 Å resolution)31, and human GARFTase (PDB: 1NJS, 1.98 Å resolution)32 are known. Thus, it was of interest to dock our proposed 6-substituted-pyrrolo[2,3-d]pyrimidine analogues 5 and 7 into each of these structures using the software LeadIT 2.1.633 and previously described methods34 to predict potential activities of the analogues for uptake by FRs and for GARFTase inhibition prior to their synthesis.
Figure 4 shows the superimposition of the docked poses of 5 (red) and 7 (green) in the human FRα (PDB ID: 4LRH)30 active site. Both compounds bind in the folate binding cleft of FRα, which is occupied by folic acid in the crystal structure (not shown). The 2-NH2 moieties of 5 and 7 interact with Asp81, while the 4-oxo moieties of both compounds interact with the side chain NH of Arg103 and His135. The pyrrolo[2,3-d]pyrimidine scaffolds are sandwiched in a hydrophobic pocket created by the side chains of Tyr60, Tyr85, and Trp171, similar to that seen with the pteroyl ring of folic acid in its bound conformation.30 The L-glutamate moieties of both compounds are similarly oriented and mimic the corresponding glutamate in folic acid. The α-carboxylic acid of the glutamate side chain forms a network of hydrogen bonds with the backbone NH of Gly137 and Trp138 and with the side chain NH of Trp140. The γ-carboxylic acid interacts with the side chain NH of Trp102 and Lys136. Differences in the docked poses of the two compounds arise in the orientation of the thiophene side chains in the hydrophobic pteroyl binding region of FRα. The docked conformation of 5 orients the thiophene moiety toward the side chain of Trp102. On the other hand, the docked conformation of the thiophene side chain of 7 is almost orthogonal relative to the thiophene side chain of 5. This change in orientation provides additional hydrophobic interactions between the thiophene moiety of 7 and Trp140, His135, and the side chain of Phe62 in the pocket and is different from those formed by the thiophene moiety of 5. The hydrophobic three-carbon linker of both compounds forms nonspecific hydrophobic interactions with Tyr60, Phe62, Trp102, Trp134, and His135. The docking scores of 5 and 7 were −37.94 and −38.61 kJ/mol compared with −44.67 kJ/mol for folic acid.
Figure 4.
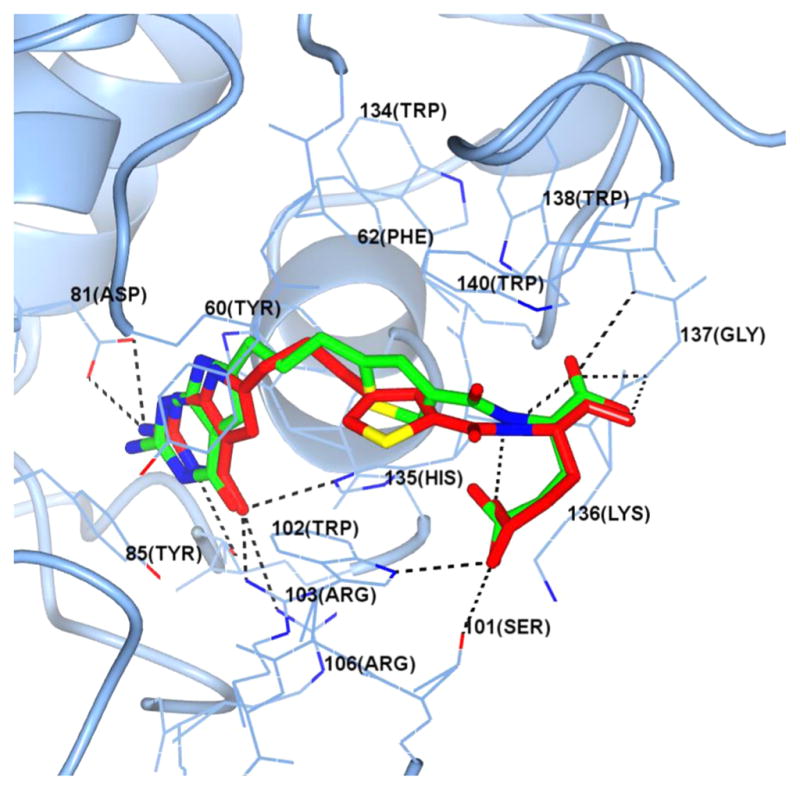
Molecular modeling of 5 and 7 with FRα. Superimposition of the docked poses of 5 (red) and 7 (green) in the folate binding site of human FRα (PDB: 4LRH).
Thus, the molecular docking studies predicted that 5 and 7 retain key interactions in the binding pockets of FRα (Figure 4). Analogous results were obtained with FRβ (Figure 1S) and GARFTase (Figure 2S) and provide substantial support for the synthesis and biological evaluation of 5 and 7 as FRα and FRβ transport substrates and as GARFTase inhibitors.
CHEMISTRY
As shown in Scheme 1, synthesis of the target compounds 5–8 started with a palladium-catalyzed Sonogashira coupling of 4-bromo-thiophene-2-carboxylic acid methyl ester or 5-bromo-thiophene-3-carboxylic acid methyl ester with but-3-yn-1-ol 9 to afford the thiophenebutynyl alcohols 10–11. Catalytic hydrogenation afforded the saturated alcohols 12–13. Subsequent oxidation using periodic acid and pyridinium chlorochromate gave the carboxylic acids 14–15. Conversion to the acid chlorides 16–17, and immediate reaction with diazomethane, followed by concentrated HCl, gave the desired α-chloromethylketones 20–21. Condensation of 2,6-diamino-3H-pyrimidin-4-one, with 20–21 at 60 °C for 3 days afforded, after chromatographic separation, the 2-amino-4-oxo-pyrrolo-[2,3-d]pyrimidines 22–23 and 2,4-diamino-furo[2,3-d]-pyrimidines 26–27, respectively.
Scheme 1.

Synthesis of Classical 2-Amino-4-oxo-6-substituted-pyrrolo[2,3-d]-pyrimidines 5 and 7 and 2,4-Diamino-5-substituted-furo[2,3-d]pyrimidines 6 and 8a
aConditions: (a) 4-bromo-thiophene-2-carboxylic acid methyl ester or 5-bromo-thiophene-3-carboxylic acid methyl ester, CuI, PdCl2, PPh3, Et3N, CH3CN, 100 °C, 6 h; (b) 10% Pd/C, H2, 55 psi, MeOH, 4 h; (c) 2.2 equiv H5IO6, 2 mol % PCC, CH3CN, 0 °C, 1 h; (d) oxalyl chloride, CH2Cl2, reflux, 1 h; (e) diazomethane, Et2O, RT, 1 h; (f) concd HCl, reflux, 1.5 h; (g) 2,6-diamino-3H-pyrimidin-4-one, DMF, 60 °C, 3 days; (h) (i) 1N NaOH, (ii) 1 N HCl; (i) N-methylmorpholine, 2-chloro-4,6-dimethoxy-1,3,5-triazine, L-glutamate diethyl ester hydrochloride, DMF, RT, 12 h.
On the basis of the report by Secrist and Lui,35 identification of the chromatographically separated structures of both the pyrrolo- and furo[2,3-d]pyrimidines obtained by the pyrimidinone cyclization reaction was possible. By 1H NMR, the magnitude of the chemical shift of the H5 protons in 22–23 at δ 5.95 can be assigned to pyrrolo[2,3-d]pyrimidines, while in 26–27 the chemical shift of δ 7.14 can be assigned to the H6 protons of furo[2,3-d]pyrimidines.35 In addition, two sets of exchangeable protons in the δ 5.97 and 6.41 regions in 26–27 confirm the 2,4-diamino pyrimidine-fused furans, whereas only one set of exchangeable protons at δ 6.43 in 22–23 confirms the 2-amino-4-oxo pyrimidine-fused pyrroles. Hydrolysis of 22–23 and 26–27 afforded the corresponding free acids 24–25 and 28–29. Subsequent coupling with L-glutamate diethyl ester using 2-chloro-4,6-dimethoxy-1,3,5-triazine as the activating agent afforded the diesters 30–31 and 32–33. Final saponification of the diesters gave the target compounds 5–8, respectively.
BIOLOGICAL EVALUATION AND DISCUSSION
Antiproliferative Activities of 6-Substituted Pyrrolo-[2,3-d]pyrimidine Thieonyl Regioisomers of Compound 4 in Relation to Major Mechanisms of Folate Transport
Our goal was to explore the antiproliferative effects of 1,3-regioisomers of 4, analogous to 1–328 (Figure 2), including 5 [3′,5′] and 7 [2′,4′] (Figure 3), in order to expand the therapeutic spectrum of this series. These analogues were initially tested against a panel of isogenic CHO sublines engineered to individually express human FRα (RT16), FRβ (D4), RFC (pC43-10), or PCFT (R2/PCFT4).24,36,37 All the CHO sublines were derived from RFC-, FR-, and PCFT-null MTXRIIOuaR2-4 CHO cells38 (hereafter designated R2). FR-binding capacities (a determinant of cellular uptake by this mechanism) and FR, RFC, and PCFT uptake characteristics for all these CHO cell lines are documented.24,37 For these experiments, cells were cultured over a range of drug concentrations and proliferation was measured after 96 h with a fluorescence-based metabolic assay (Cell Titer Blue). For the PC43-10 and R2/PCFT4 sublines, growth inhibition results were compared to those for parental R2 CHO cells and to R2 cells that were transfected with empty pCDNA3.1 expression vector [designated R2(VC)]. Results for 5 and 7 were compared to those for 4 and to classical antifolate drugs including MTX, PMX, PDX, and LMTX (Table 1). To confirm FR-mediated cellular uptake for the FRα-expressing cells, parallel incubations were performed in the presence of 200 nM folic acid.
Table 1.
IC50 Values (nM) for Inhibition of Proliferation of CHO and KB Sublinesa
| antifolate | RFC
|
FRα
|
FRβ
|
PCFT
|
FRα/PCFT/RFC
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| PC43-10 | R2 | RT16 | RT16 (+FA) | D4 | D4 (+FA) | R2/hPCFT4 | R2(VC) | KB | KB (+FA) | Ade/Thd/AICA | |
| 4 | 101.0(16.6) | 273.5(49.1) | 0.38(0.12) | 173(35) | 0.17(0.03) | 126(13) | 3.34(0.26) | 288(12) | 0.26(0.03) | 101(7) | Ade/AICA |
| 5 | 197(49) | 355(10) | 0.33(0.15) | 494(254 | 0.34(0.03) | 557(147) | 5.39(1.27) | >1000 | 0.17(0.05)* | 337(127) | Ade/AICA |
| 6 | 54.0(9.8) | >1000 | 154(50) | >1000 | 17.5(0.6) | >1000 | >1000 | >1000 | 20.7(4.9) | 275(25) | Ade/AICA |
| 7 | 189(51) | 290(8) | 0.61(0.11) | 506(118) | 0.10(0.01) | 654(169) | 6.51(1.30) | >1000 | 0.09(0.02)** | 105(3) | Ade/AICA |
| 8 | >1000 | >1000 | 148(28) | >1000 | 54.3(9.6) | >1000 | >1000 | >1000 | 70.3(13.7) | >1000 | Ade/AICA |
| MTX | 12(1.1) | 216(8.7) | 114(31) | 461(62) | 106(11) | 211(43) | 120.5(16.8) | >1000 | 6.0(0.6) | 20(2.4) | Ade/Thd |
| PDX | 0.69(0.07) | 819(94) | 43 (6) | 289 (33) | 2.5(0.1) | 951(29) | 57(12) | >1000 | 0.47(0.20) | 1.94(0.28) | Ade/Thd |
| PMX | 138(13) | 894(93) | 42(9) | 388(68) | 60(8) | 254(78) | 13.2(2.4) | 974.0(18.1) | 68(12) | 327(103) | Thd/Ade |
| LMTX | 12(2.3) | >1000 | 12(8) | 188(41) | 2.6(1.0) | 275(101) | 248.0(18.2) | >1000 | 1.2(0.6) | 31(7) | Ade/AICA |
Growth inhibition assays were performed with isogenic CHO sublines engineered to express human RFC (PC43-10), FRα (RT16), FRβ (D4), or PCFT (R2/PCFT4) from transporter-null (R2) CHO cells.24,36,37 R2(VC) were R2 cells transfected with empty PCDNA3. Results are also shown for the KB human tumor subline (expresses RFC, FRα, and PCFT).39 For the FRα experiments, growth inhibition assays were performed in the presence and absence of excess (200 nM) folic acid. Results shown are mean values from 3–10 experiments (± standard errors in parentheses) and are presented as IC50 values, representing the concentrations that inhibit growth by 50% relative to cells incubated without drug. Certain data for MTX, PDX, PMX, and LMTX were previously published. 24–27,29,37 Results are also summarized for the protective effects of adenosine (60 μM) or thymidine (10 μM), or 5-aminoimidazole-4-carboxamide (320 μM) with KB cells, as shown in Figure 9. The structures for compounds 4–8 are in Figure 3. Experimental details are provided in the Experimental Procedures. Undefined abbreviations: Ade, adenosine; AICA, 5-aminoimidazole-4-carboxamide; FA, folic acid; ND, not determined; Thd, thymidine. Statistics: *p < 0.005 and **p < 0.05 when compared to 4 in KB cells.
Compounds 5 and 7, like 4, were potently inhibitory toward both FRα- and FRβ-expressing CHO cells with IC50 values less than 1 nM, far lower (~277–1890-fold) than the values for RFC-expressing PC43-10 cells (Table 1). For RT16 and D4 cells, antiproliferative effects were reversed (~450–6500-fold) in the presence of 200 nM folic acid. The modest growth inhibitions with 5 and 7 toward RFC-expressing PC43–10 cells were also seen with R2 CHO cells, suggesting a nonmediated cellular uptake process. Similar results were obtained with 4 (Table 1). Inhibition of proliferation was also seen with R2/PCFT4 CHO cells treated with low nanomolar concentrations of 5 and 7 at levels comparable to that for 4 (Table 1). Unlike FR-expressing cells, inhibition of R2/PCFT4 cells was not affected by the addition of 200 nM folic acid (not shown).
The inhibitory potencies for 5 and 7 exceeded those for the classical antifolates MTX, PMX, and PDX toward the PCFT-and FR-expressing CHO sublines (Table 1). In addition, 5 and 7 were 8–10 times more potent against R2/PCFT4 cells than their four-carbon bridge analogues 1 and 2, respectively.28 The selectivity of these analogues for FR- and PCFT-expressing CHO cells also exceeded that for PMX. Using the IC50 values of 4, 5, 7, and PMX against the transporter-specific CHO cell lines from Table 1, “selectivity ratios” for FRα, FRβ, and PCFT over RFC were calculated, defined as the IC50 RFC/IC50 FRα, -FRβ, or -PCFT, respectively (Table 2). According to this metric, 7 is the most selective compound of the series for FRα and FRβ over RFC, with selectivities of 610-fold and 1890-fold, respectively. Thus, 7 should be transported almost exclusively by FRα or FRβ over RFC. Further, 4, 5, and 7 are substantially more selective for FRα and FRβ than PMX. For 7, the calculated selectivity ratios for FRα and FRβ were 185-fold and 822-fold, respectively, greater than those for PMX. With PMX, among the best substrates for PCFT,5 the selectivity ratios for PCFT to RFC were 2.9–3.5-fold less than those for 4, 5, and 7. PCFT selectivity for 7 compared to PMX was also seen with HeLa cells engineered to express PCFT without RFC (R1-11-PCFT4 cells)40 and expressing RFC without PCFT (R1-11-RFC2 cells) (Supporting Information Figure 3S). Collectively, these results suggest that unlike PMX, compounds such as 7 would be selectively transported into tumor cells over normal cells. This would be expected to translate into reduced toxicity to normal cells compared to PMX. In addition, because PCFT is optimally active at acidic pH values below 7 that exist in tumor microenvironment,41,42 5 and 7 should be particularly effective against solid tumors.
Table 2.
Ratios of Transporter Selectivity over RFCa
| antifolate | selectivity ratios
|
||
|---|---|---|---|
| RFC IC50/FRα IC50 |
RFC IC50/FRβ IC50 |
RFC IC50/PCFT IC50 |
|
| 4 | 326 | 594 | 30.2 |
| 5 | 277 | 579 | 36.5 |
| 7 | 610 | 1890 | 29 |
| PMX | 3.3 | 2.3 | 10.5 |
Selectivity ratios, corresponding to the IC50 values for RFC to FRα, FRβ, and PCFT, as measured in the engineered CHO sublines, PC43-10, RT16, D4, and R2/PCFT4, respectively, are shown. On the basis of this metric, high ratios reflect increased selectivity via FR- or PCFT-mediated uptake over RFC.
Compounds 5 and 7 inhibited proliferation of FRα/PCFT/RFC-expressing KB human nasopharyngeal carcinoma cells at subnanomolar concentrations lower than those for 4 (Table 1). Compounds 4 and 7 were also tested as growth inhibitors with IGROV1 and SKOV3 ovarian cancer cells with similar levels of PCFT transport (measured at pH 5.5 with [3H]MTX) but differing ~5-fold in levels of FRα (measured by surface binding with [3H]folic acid, paralleling previous results24) (Figure 5). IC50s of 0.73 (±0.11 (SE)) and 0.77 (±0.08) nM, respectively, were measured with IGROV1 cells, and IC50s of 2.2 (±0.5) and 15.5 (±2.8) nM, respectively, were recorded with SKOV3 cells. The extent of reversal of drug effects by 200 nM folic acid differed among the tumor sublines. For instance, in the presence of 200 nM folic acid, KB cells showed IC50s of 101 (±7) nM for 4 and 105 (±3) nM for 7, 388- and 1167-fold increases, respectively, over the IC50 values in the absence of excess folic acid. With IGROV1 cells, IC50s of 126 (±28) nM and 276 (±56) nM for 4 and 7, respectively, were measured in the presence of 200 nM folic acid. For SKOV3 cells incubated with 200 nM folic acid, the IC50s were 47 (±7) and 76 (±3) nM, respectively (Figure 5). These results likely reflect relative levels of FRα and PCFT among the assorted cell lines, such that in SKOV3 cells which have modest FRα levels there was greater relative drug uptake by PCFT, even in the presence of excess folic acid (above).
Figure 5.

FRα binding and PCFT uptake correlates with 4 and 7 growth inhibition toward IGROV1 and SKOV3 cells. Left panels: FRα levels were assayed by surface binding of [3H]folic acid (upper), whereas PCFT activity was measured with [3H]MTX (0.5 μM) over 5 min at 37 °C at pH 5.5 (lower). Right panels: Representative growth inhibition experiments for IGROV1 (upper) and SKOV3 (lower) cells treated with a range of concentrations of 4 or 7 in the absence (NA for no additions) or presence of 200 nM folic acid are shown. For both transport and binding assays, cell-associated radioactive substrate was expressed in units of pmol/mg protein. The methods are described in the Experimental Procedures.
Membrane Transport Characteristics for 5 and 7
The results in Table 1 strongly suggest that 5 and 7, like 4, are transport substrates for FRα and β and for PCFT. We performed experiments to assess parameters directly relevant to the cellular uptake of 5 and 7 by these transport systems.
When total surface FRs were titrated by [3H]folic acid binding in RT16 (FRα) and D4 (FRβ) CHO cells, total FRs between the cell lines were within a 2-fold range.24 To directly establish relative FR binding affinities for 5 and 7 compared to 4, we initially used [3H]folic acid binding assays with RT16 cells and D4 cells at 4 °C with unlabeled 4, 5, and 7 (0–1000 nM). Relative affinities are reflected in the extents to which the novel antifolates competed with [3H]folic acid for FR binding compared to competition by a comparable concentration range of nonradioactive folic acid (positive control) and MTX (negative control)24,26–29,37 (Figure 6). FRα-bound [3H]folic acid was quantified and normalized to total cell protein. Relative binding affinities to FRs were expressed as the inverse molar ratios of unlabeled ligands needed to reduce the level of FR-bound [3H]folic acid by 50%, with the affinity of folic acid assigned a value of 1 (Figure 6). By this assay, in RT16 cells 5 and 7 showed affinities approximately 60% of that for folic acid, whereas 4 showed an affinity approximately 70% of that for folic acid. In D4 cells binding affinities to FRβ were somewhat reduced compared to FRα in RT16 cells and ranged from approximately 35–40% of the affinity for folic acid for 5 and 7 and approximately 50% of that for folic acid for 4. Slightly decreased binding of 5 and 7 to FRα compared to folic acid were predicted from our modeling studies for FRα (Figure 4). However, for FRβ, docking scores were less predictive of the relative 5 and 7 binding affinities measured experimentally (Figure 1S, Supporting Information).
Figure 6.
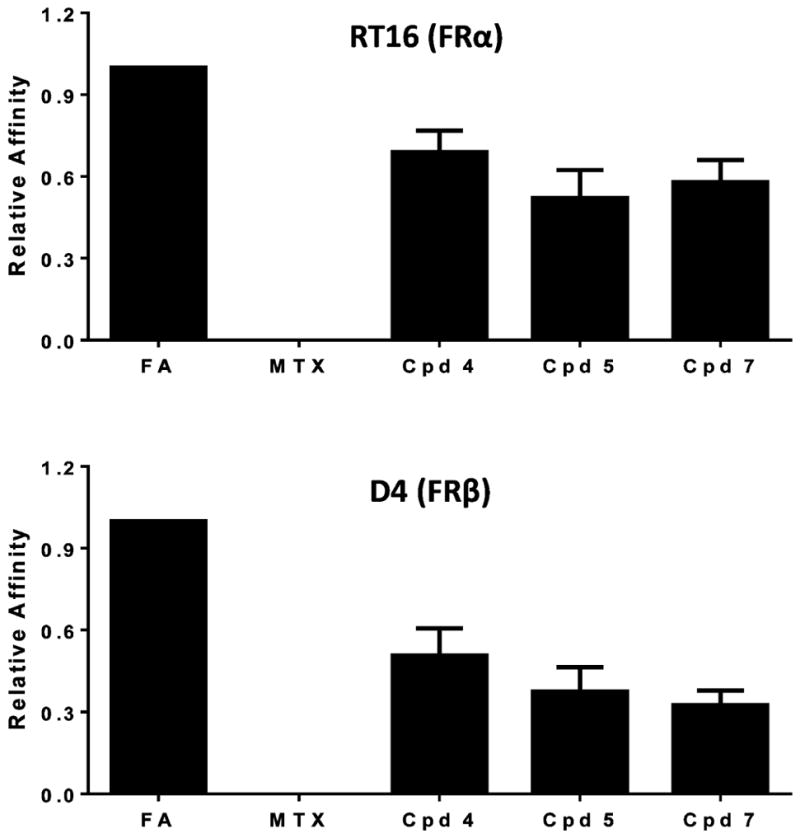
FRα and FRβ binding assays for assorted antifolates. Data are shown for the relative binding affinities of the unlabeled ligands with FRα-expressing RT16 and FRβ-expressing D4 CHO cells. Relative binding affinities for assorted folate/antifolate substrates were determined over a range of ligand concentrations and were calculated as the inverse molar ratios of unlabeled ligands required to inhibit [3Hfolic acid binding by 50%. By definition, the relative affinity of folic acid is 1. Results are presented as mean values plus/minus standard errors from 4–10 experiments. The detailed methods are provided in the Experimental Procedures. Abbreviations: Cpd, compound; FA, folic acid.
We also performed direct assays of [3H]7 binding and mediated uptake by RT-16 and D4 cells. For these experiments, cells were washed with acid-buffered saline (pH 3.5) then incubated at pH 7.4 with [3H]7 (50 nM) at 4 °C for determination of (i) surface FR-bound [3H]7, or at 37 °C for 1 h, after which (ii) total cell-associated [3H]7 (includes surface bound and internalized drug) and (iii) internalized [3H]7 were measured43 (Figure 7). In spite of a modest difference in relative binding affinities of 7 to FRα and FRβ (Figure 6), this was not accompanied by a corresponding change in levels of internalized [3H]7 between RT16 and D4 cells (2.25 ±0.24 and 3.35 ± 0.25 pmol/mg, respectively) during a 1 h exposure (Figure 6). Further, the slight differences in 7 growth inhibitions measured between RT16 and D4 cells were not accompanied by corresponding differences in 7 binding affinities for FRα and FRβ (Figure 6) or cellular uptake by FRα and FRβ (Figure 7).
Figure 7.

Uptake of [3H]7 by FRα and FRβ. RT16 (FRα) and D4 (FRβ) CHO cells were washed with acid (pH 3.5) buffered saline to remove FR-bound folates and then incubated at pH 7.4 with 50 nM [3H]7 at 0 °C (“Surface”) or at 37 °C for 60 min (“Total”). An additional condition involved treatment with 50 nM [3H]7 at 37 °C for 60 min, followed by an acid buffer wash to remove the extracellular FR-bound [3H]7 fraction (“Intracellular”). Cellular proteins were solubilized with NaOH for determinations of cell-associated [3H]7 and proteins. Experimental details are provided in the Experimental Procedures. Results are presented as mean values plus/minus SEM from three experiments.
We assayed PCFT-mediated uptake of 0.5 μM [3H]7 by R2/hPCFT4 cells at pH 5.5 at 37 °C over 5 min. Controls included PCFT-null R2 cells and parallel treatments with unlabeled 4 (10 μM) as a competitive inhibitor of [3H]7 uptake by PCFT. Results with [3H]7 were compared to those for 0.5 μM [3H]PMX. By these assays, high levels of [3H]7 and [3H]PMX uptake were measured in R2/PCFT4 cells (but not in R2 cells) that were potently inhibited by unlabeled 4 (Figure 8). Uptake of [3H]7 exceeded that for [3H]PMX by ~40%.
Figure 8.

PCFT uptake assays of PMX and 7. Uptake of [3H]PMX and [3H]7 (0.5 μM) was measured at pH 5.5 in R2 and R2/PCFT4 CHO cells over 2 min at 37 °C in the absence (NA for no additions) and in the presence of nonradioactive 4 (10 μM) as a competitive transport inhibitor. Experimental details are provided in the Experimental Procedures. Abbreviation: Cpd, compound. Results are presented as mean values plus/minus SEM from three experiments.
We measured transport kinetics for [3H]7 and [3H]PMX in R2/PCFT4 cells at pH 5.5 and pH 6.8. At pH 5.5, Kt values for PMX and 7 were 0.044 (±0.14) and 0.22 (+/0.04) μM, respectively, whereas Vmax values were 13.42 (± 4.22) and 24.62 (±6.60) pmol/mg/min, respectively. At pH 6.8, the Kt values were 0.27 (±0.05) μM for PMX and 0.17 (± 0.02) μM for 7; the Vmax values were 9.2 (±1.7) pmol/mg/min for PMX and 3.0 (±0.3) pmol/mg/min for 7. These results demonstrate differences in PCFT binding affinities (as reflected in the Kt values) and maximal transport rates in response to pH between these structurally related antifolates.
Identification of GARFTase as the Intracellular Enzyme Target of 5 and 7 in KB Tumor Cells
As with 4,27 in KB cells 5 and 7 inhibited de novo purine nucleotide biosynthesis, as adenosine (60 μM) rather than thymidine (10 μM) abolished the antiproliferative effects of these compounds (Table 1 and Figure 9). AICA, a precursor of AICA ribonucleotide (ZMP), the substrate for AICARFTase which circumvents the GARFTase step,5 also reversed the inhibitory effects of 5 and 7 along with 4. This strongly suggested that the primary intracellular enzyme target of 5 and 7 is GARFTase, which catalyzes the first folate-dependent step leading to the purines.
Figure 9.

Growth inhibition of KB cells by 4 and 6–8 and protection by excess folic acid, nucleosides, or 5-aminoimidazole-4-carboxamide (AICA). KB cells were plated (4000 cells/well) in folate-free RPMI 1640 medium with 10% FBS, antibiotics, L-glutamine, and 2 nM LCV with a range of concentrations of 4, 6, 7, or 8 in the presence of folic acid (200 nM), adenosine (60 μM), and/or thymidine (10 μM) or in the presence of AICA (320 μM). Cell proliferation was assayed with Cell Titer Blue using a fluorescence plate reader. Data are representative of at least triplicate experiments. Analogous results were obtained for 5 (not shown) and are summarized along with those for 4, 6, 7, and 8 in Table 1.
To directly confirm GARFTase inhibition in KB cells treated with 5 and 7, we used in situ and in vitro GARFTase assays. The in situ GARFTase assay measures incorporation of [14C]glycine into [14C]GAR and subsequently [14C]formyl GAR, the GARFTase product which accumulates in the presence of azaserine.24,26–29,37 For these experiments, KB cells were treated with a range of concentrations of 5 and 7 for 16 h in the presence of [14C]glycine under conditions identical to those for the cell proliferation experiments (Table 1). Results were compared to those for 4, previously shown to inhibit GARFTase.27 Our results established that 5 and 7 were essentially equipotent to 4 in inhibiting intracellular GARFTase with IC50s of ~2–3 nM (Figure 10).
Figure 10.

In situ GARFTase assay in KB tumor cells treated with compounds 4–8. GARFTase activity and inhibition were evaluated in situ with KB cells. KB cells were treated with drug and [14C]glycine for 16 h. Radioactive metabolites were extracted and fractionated on anion exchange columns so that the accumulation of [14C]formyl GAR could be quantified. Results are expressed as a percent of untreated controls over a range of drug concentrations. The detailed methods are described in the Experimental Procedures. Results are presented as mean values ± standard errors from three experiments. IC50s were as follows: 3.5 nM, 4; 2.4 nM, 5; 2.6 nM, 7; 82 nM, 6; 101 nM, 8. For comparison, the IC50 for GARFTase inhibition in KB cells by PMX was previously reported as 30 nM and inhibition by LMTX was reported as 14 nM.37 Abbreviation: Cpd, compound.
An in vitro GARFTase assay was performed with recombinant human GARFTase. This involved a spectrophotometric assay at 285.5 nm which measures one-carbon transfer from 10-formyl-5,8-dideazafolic acid to GAR, forming 5,8-dideazafolic acid and formyl GAR.44 Assays were performed over a range of concentrations of 4, 5, 7, or PMX. Graphs of the initial rates versus antifolate concentrations were fit to a hyperbola to determine Ki values (Figure 4S, Supporting Information). Ki values are summarized in Table 3. By this analysis, 5 and 7 were ~6- and ~7-fold, respectively, more potent as inhibitors of human GARFTase than 4, and were ~79- and ~114-fold, respectively, more potent than PMX.
Table 3.
GARFTase Kis by 6-Pyrrolo[2,3-d]pyrimidine Thienoyl Antifolatesa
| antifolate | Ki (nM) (±SEM) |
|---|---|
| PMX | 1000 (±160) |
| 4 | 68 (±11) |
| 5 | 13 (±2.9) |
| 6 | 5500 (±1000) |
| 7 | 9.1 (±1.2) |
| 8 | 6500 (±980) |
GARFTase activity was determined by measuring formation of 5,8-dideazafolate from 10-formyl-5,8-dideazafolic acid using a spectrophotometric assay (295 nm) in the presence of the indicated antifolate. Detailed methods are described in the Experimental Procedures. Results are shown as mean values ± standard errors from three replicate assays.
Collectively, these results identify GARFTase as the principal intracellular target for 5 and 7, analogous to 4. While these results parallel those for the in situ GARFTase assays, the magnitude of these differences in relative GARFTase inhibitions by 5 and 7 vis á vis 4 or PMX between the in vitro GARFTase enzyme assay and the in situ cell-based GARFTase assay likely reflect differences in cellular uptake between the drugs and the impact of antifolate polyglutamylation on drug binding and inhibition at GARFTase.8
Crystal Structures of 5, 7, and PMX in Complex with Human GARFTase
To visualize the binding of monoglutamyl 5, 7, and 4 with human GARFTase, crystal structures of human GARFTase in complexes with each 6-substituted pyrrolo[2,3-d]pyrimidine antifolate and β-GAR substrate were determined (Figure 11). Compounds 5 and 7, with Ki values of 13 and 9.1 nM, respectively (Table 3), bind to human GARFTase in identical orientations. On the basis of the crystallographic models, 5 and 7 form six hydrogen bond interactions with GARFTase peptide backbone atoms via the pyrrolo[2,3-d]pyrimidine moiety. In addition, two hydrogen bonds with the peptide backbone, a bidentate polar interaction with Arg871, and three long distance charge–charge interactions with Arg897 and Lys844 are mediated via the glutamyl tail (Figure 11A,B and Table 4S, Supporting Information). In this binding pose, 5 and 7 appear to bind in an orientation to maximize contacts with GARFTase. In contrast, 4, which exhibits an increased Ki for GARFTase (Ki = 68 nM), assumes an orientation that preserves the pyrrolo[2,3-d]pyrimidine ring contacts but lacks many of the glutamyl contacts due to a relative 180° rotation of the glutamyl tail (Figure 11C). Notably, the bidentate interaction with Arg871 and the hydrogen bond with the carbonyl of Met896 are no longer present, while the α-carboxylate is too far from Arg897 and Lys844 to provide stabilizing contacts. Furthermore, the electron density map (Figure 5S, Supporting Information) shows that Arg871 exists in two orientations in the 4 complex crystal structure, implying greater flexibility as a reflection of decreased inhibitor contact with this residue. Thus, the crystal structures for 4, 5, and 7 bound to GARFTase explain the increased inhibitory potencies of the 1,3-disubstituted thiophene regioisomers (5 and 7) over the 2,5-disubstituted regioisomer (4).
Figure 11.

Structural analyses of 5, 7, and PMX conformations in ternary complexes with human GARFTase and β-GAR. Crystal structures of the most potent GARFTase inhibitors, 5 (A) and 7 (B), are shown alongside overlays of 7 (pink) with 4 (C, green) or the less potent PMX (D, green) in the 10-formyl tetrahydrofolate binding pocket of human GARFTase with interacting residues from GARFTase shown in stick representation. (C,D) GARFTase ribbon is shown in blue for the 7 complex and in dark gray for either the 4 or PMX complex, with contacts to the latter indicated with dashed lines. Details including interaction distances and ligand electron density maps for 5, 7, and PMX complexes are included in in Table 4S and Figure 5S, Supporting Information.
Previous reports suggested that PMX is a modest inhibitor of GARFTase in addition to its primary target, TS.8 By in situ GARFTase activity assays, PMX was less potent in inhibiting synthesis of [14C]formyl GAR in KB cells than 6-subsitututed pyrrolo[2,3-d]pyrimidine antifolates.45 Likewise, our in vitro enzyme inhibition data (Table 3) show a low inhibitory potency of PMX relative to 5 and 7, where 7 is >100-fold more potent than PMX. We determined a crystallographic structure of GARFTase bound to PMX and β-GAR to better understand the weaker potency of this 5-substituted pyrrolo[2,3-d]-pyrimidine antifolate. In contrast to 4, 5, and 7, for which changes in Ki values correlate with differences in the binding mode of the glutamyl tail, the weaker inhibition of GARFTase via PMX is likely due to changes in the enzyme conformation required to accommodate the 5-substituted pyrrolo[2,3-d]pyrimidine ring. Specifically, the pyrrolo[2,3-d]pyrimidine ring in PMX is shifted relative to that of 5 and 7, resulting in a change in the position of the loop containing residues 896–899, as well as the α-helix containing Arg871 (Figure 11D). Because of the conformational changes in GARFTase, PMX cannot make hydrogen bond contacts with the two peptide backbone atoms seen to interact with the three-atom bridge compounds 5 and 7. Additionally, the region of GARFTase including Lys844 and Arg897 that provides interacting residues for γ-carboxylate stabilization of 5 and 7 has moved such that no interactions are possible with PMX. The α-carboxylate of PMX has only one polar contact and two longer distance charge–charge interactions, which would be predicted to be less stable than the binding mode of 4, 5, or 7.
We expect that the stronger potency of the 6-pyrrolo[2,3-d]pyrimidine antifolates 5 and 7 compared to PMX for in vitro GARFTase inhibition can be attributed to alterations in the GARFTase conformation, arising from a change in the position of pyrrolo[2,3-d]pyrimidine substitution position and the shorter bridge length in PMX. A comparison of the docked pose of 7 and its X-ray crystal structure pose in the 10-formyl tetrahydrofolate binding site of human GARFTase is shown (Figure 6S, Supporting Information).
In Vivo Antitumor Efficacy with 7
On the basis of the in vitro proliferation inhibition of SKOV3 ovarian cancer cells by 7 (Figure 5), an in vivo drug efficacy trial was performed. Ten-week-old female NCR SCID mice were implanted bilaterally with subcutaneous human SKOV3 ovarian tumors (express RFC, PCFT, and FRα). Mice were maintained ad libitum on a folate-deficient diet so as to reduce serum folates to levels approximating those in humans.46 A control cohort of mice was maintained on a standard folate-replete diet. Both dietary groups were independently pooled, implanted with SKOV3 tumor fragments, and then nonselectively randomized into their respective control and treatment groups (5 mice/group). Compounds 4 (28 mg/kg) and 7 (16 mg/kg) were intravenously administered on a Q4dx3 schedule on days 3, 7, and 11 postimplantation. Tumors were measured twice weekly, and mice were weighed daily for the duration of the study. For mice maintained on the folate-deficient diet, antitumor activities (T/C) on day 21 for 4 and 7 were 8% and 15%, respectively [median tumor burden of no treatment controls on day 21 was 966 mg (range 550–1098 mg)]. For both 4 and 7, treatment produced 1.9 logs of gross cell kill (Figure 12). Overall, the treatment regimen was well tolerated and the only dose-limiting symptom observed was reversible body weight loss (7.9% for compound 4 and 12.1% for compound 7). Antitumor activities for 4 and 7 were ablated in mice maintained on standard folate-replete diet, with T/C values of 79% and 69%, respectively [median tumor burden for folate-replete no treatment controls on day 21 was 973 mg (range 766–1171 mg)]. No weight loss or other symptoms were observed.
Figure 12.

In vivo efficacy trial of compounds 4 and 7 against SKOV3 xenografts. Female NCR SCID mice (10 weeks old; 19.9 g average body weight) were maintained on a folate-deficient diet ad libitum for 13 days prior to subcutaneous tumor implantation. Human SKOV3 ovarian tumor fragments were implanted bilaterally, and mice were nonselectively randomized into 5 mice/group. Compounds 4 (28 mg/mg) and 7 (16 mg/kg) [dissolved in 5% ethanol (v/v), 1% Tween-80 (v/v), 0.5% NaHCO3, and deionized H2O; pH 6.5] were administered on a Q4dx3 schedule intravenously (0.2 mL/injection) on days 3, 7, and 11. Mice were observed and weighed daily; tumors were measured twice per week. For the experiment shown, treatment resulted in a significant growth delay (T–C value) of 13 days and nearly identical antitumor activities for both compounds 4 and 7. Abbreviation: Cpd, compound; Rx, treatment.
Our results from the in vivo efficacy trial with early stage FRα- and PCFT-expressing SKOV3 human ovarian tumors establish that under the conditions tested, the antitumor efficacy of 7 was roughly equivalent to that for 4, reflecting its cellular uptake by FRα and PCFT, resulting in inhibition of GARFTase.
Antiproliferative Activities and GARFTase Inhibition by the 5-Substituted Furo[2,3-d]pyrimidine Thienoyl Analogues 6 and 8
Important (minor) byproducts from the synthesis of the major 2-amino-4-oxo-pyrrolo[2,3-d]-pyrimidines 5 and 7 include the 2,4-diamino-furo[2,3-d]-pyrimidines 6 and 8 (Figure 3 and Scheme 1). Compounds 6 and 8 also inhibited cell proliferation, albeit less so than either 5 or 7 (Table 1). Toward the FRα-expressing RT16 CHO subline, IC50 values were ~150 nM with both compounds; for FRβ-expressing D4 CHO cells, IC50 values were ~18 and 54 nM for 6 and 8, respectively. Compound 6 modestly inhibited PC43-10 CHO cells which express RFC, a result consistent with those previously reported for 2-amino-4-oxo-5-substituted heterocyclic compounds (i.e., 5-substituted pyrrolo[2,3-d]-pyrimidine antifolates45). However, 8 did not inhibit PC43-10 cells. This, to our knowledge, is the first 2,4-diamino analogue with complete selectivity for FRs over RFC. These results with the engineered CHO sublines were largely recapitulated with KB human tumor cells with the potency for 6 exceeding that for 8, likely reflecting both FRα and RFC uptake (Table 1). Interestingly, with KB cells, 6 and 8, like 5 and 7, inhibited de novo purine nucleotide biosynthesis at GARFTase, as reflected in the ability of adenosine and AICA to completely protect from growth inhibition (Figure 9). Compounds 6 and 8 also inhibited GARFTase activity in situ (Figure 10) and in vitro (Table 3). Relative GARFTase inhibitions were greatly reduced compared to 5 and 7 and paralleled the relative inhibitions of KB cell proliferation (Table 1). Neither 6 nor 8 were potent inhibitors of DHFR or TS, as demonstrated from the nucleoside protection results (Figure 9), as well as results of isolated enzyme assays (not shown).
We were interested in explaining the GARFTase inhibitory activities (Figures 9 and 10) of these 2,4-diamino-furo[2,3-d]pyrimidine compounds, as known GARFTase inhibitors are more commonly 2-amino-4-oxo analogues (e.g., LMTX, 7) rather than 2,4-diamino compounds. On the basis of the excellent agreement between our computational model and crystallographic structure of GARFTase in complex with 7 which validates our modeling protocols (Figure 6S, Supporting Information), we performed molecular modeling with 6 and 8 docked into our crystal structure of 7 in GARFTase. The most stable poses obtained for 6 and 8 docked into our GARFTase structure are shown in Figure 13. Our results suggest that both 6 and 8 have the best docked pose in a 180° flipped 2,4-diamino-5-substituted-furo[2,3-d]pyrimidine orientation compared to the 2-amino-4-oxo-6-substituted-pyrrolo[2,3-d]-pyrimidine structure of 5 and 7. This can be easily achieved by a simple 180° rotation about the C2–NH2 bond, generating a different conformer (Scheme 2). In this flipped orientation, the furan oxygen atom of the conformers of 6 and 8 mimics the 4-oxo moiety of 5 and 7.
Figure 13.

Superimposition of the docked pose of 6 (green), 7 (blue), and 8 (pink) in the 10-formyl tetrahydrofolate binding site of GARFTase.
Scheme 2.

Schematic Showing the 180° Flipped 2,4-Diamino-5-substituted-furo[2,3-d]pyrimidine Orientation of 6 and 8a
aIn this flipped orientation, the furan oxygen atom of 6 and 8 mimics the 4-oxo moiety of 5 and 7.
In this mode, hydrogen bonds can be formed between the backbone carbonyl atoms of Leu92 and Glu141 with the 2-amino moieties, between the carbonyl of Arg90 and the 4-amino moiety, and between the backbone NH of Leu92 and the N3 atoms of 6 or 8. Hydrophobic interactions can occur between the bicyclic scaffold and hydrophobic interactions with Leu85, Leu92, and Val139. This binding orientation enables the thienoyl moieties of 6 and 8 to occupy the benzoyl binding region of GARFTase, where they can form hydrophobic interactions with Met89, Ile91, and Val143. The terminal glutamate chains of 6 and 8 are oriented similarly to the glutamate side chains of 5 and 7 and can form hydrogen bonds between the α-carboxyl and the backbone NH of Ile91 and electrostatic interactions with Arg90. The docking scores of 6 and 8 were −49.30 and −48.46 kJ/mol, respectively, compared with −57.77 kJ/mol for 7. Clearly, 6 and 8 were predicted to be less potent GARFTase inhibitors compared with 7, as observed in Table 3.
Thus, the flipped orientations of 6 and 8 provide a reasonable explanation for the GARFTase inhibitory activities of these 2,4-diamino analogues which mimic the 2-amino-4-oxo analogues 5 and 7.
CONCLUSIONS
In this report, we describe a comprehensive investigation that explores the structure–activity relationships for antitumor efficacies of 6-substituted pyrrolo[2,3-d]pyrimidine antifolates, reflecting selective cellular uptakes by FRα and -β and by PCFT vis á vis RFC, and inhibition of de novo purine nucleotide biosynthesis at GARFTase. We describe the unique biological characteristics and therapeutic potentials of 5 and 7, thienoyl regioisomers of 4, previously the most potent analogue of this series toward human tumors, including KB, IGROV1, and H2452 tumors.27,47,48
We established potent antiproliferative effects of 5 and 7, at least equivalent to those for 4. From patterns of growth inhibition in engineered CHO cell lines expressing FRα and FRβ, PCFT, or RFC, 5 and 7, like 4 previously,27 showed a highly selective cellular uptake by FRs α and β, and by PCFT over RFC, which far exceeded that for PMX. Compound 7 was custom radiolabeled and used as a prototype substrate for FRs and PCFT. Compound 7 was confirmed as a bona fide substrate for both FRα and -β and for PCFT, for which uptake was inhibited by competitive inhibitors (folic acid and 4, respectively). PCFT uptake of 7 exceeded that of PMX. Thus, 7 would be expected to be much more selective toward tumor cells over normal cells and less toxic toward normal cells than standard antifolates such as PMX, reflecting its selective transport by FRs and PCFT. In addition, because PCFT is optimally active at acidic pH values below 7 as exist in the tumor microenvironment,41,42 compound 7 should be especially effective against solid tumors.
We tested the activity of 5 and 7 against human KB tumor cells. Exposure to subnanomolar concentrations of these drugs was accompanied by suppression of KB cell proliferation due to potent inhibition of de novo purine nucleotide biosynthesis at the step catalyzed by GARFTase, as measured in situ at concentrations only slightly greater than those required to inhibit cell proliferation. This was further corroborated by the demonstration of direct inhibition of isolated human GARFTase with monoglutamyl 5 and 7, as reflected in Ki values, and by the crystal structures of human GARFTase complexed with monoglutamyl 5 and 7. The latter results provided compelling evidence that differences in inhibition of GARFTase by 5 and 7 versus 4 and PMX are due to differences in the orientations of the pyrrolo[2,3-d]pyrimidine or thienoyl moieties, or the length of the side chain bridge, when bound to GARFTase. While these results establish that 5 and 7, in particular, can potently inhibit GARFTase without the need for polyglutamylation, the striking differences in relative inhibitions in cell-based (in situ) versus isolated enzyme assays for the 6-pyrrolo[2,3-d]pyrimidine antifolates nonetheless suggest important roles for membrane transport and metabolism to polyglutamates in determining drug efficacies in intact cells.26,27
We expanded our studies with KB tumor cells to include IGROV1 and SKOV3 ovarian cancer cells, more challenging tumor models with progressively lower levels of FRs and lower levels of PCFT activity. In IGROV1 and SKOV3 cells, 4 and 7 inhibited cell proliferation at concentrations only slightly higher than those that inhibited growth of KB cells. In spite of lower levels of FR, FRα-mediated effects prevailed, as reflected in appreciable losses of growth inhibition in the presence of excess folic acid which selectively blocks the FR uptake fraction. The decreased impact of excess folic acid in reversing the inhibitory effects of 4 and 7 on SKOV3 cells compared to IGROV1 cells likely reflects the greater amount of FRα in IGROV1 cells and the increased relative contribution of PCFT to overall drug uptake in the SKOV3 tumor subline. These results demonstrate the clear therapeutic benefit of dual transport of 7 and related agents by both FRα and PCFT for tumor targeting ovarian and non-small cell lung cancers that express both these uptake systems. Our in vitro findings with SKOV3 tumors with 4 and 7 were extended in vivo using SKOV3 xenografts. Against subcutaneous SKOV3 xenografts in SCID mice, treatment with 4 and 7 at the respective doses tested resulted in similar efficacies, as reflected by T/C, tumor growth delay, and gross log cell kill values.
Finally, we reported the antiproliferative effects of two important synthetic byproducts, 6 and 8, of the major 2-amino-4-oxo-pyrrolo[2,3-d]pyrimidines 5 and 7, attributable to cellular uptake by FRs (6,8) and by RFC (6). Most surprising was the finding that these 2,4-diamino-furo[2,3-d]pyrimidine antifolates were primarily inhibitors of GARFTase, as the 2,4-diamino substitution pattern is typically associated with DHFR inhibition.49,50 We hypothesized that the furan oxygen of 6 and 8 mimics the 4-oxo moiety of 5 and 7 to facilitate drug binding via different conformations. To our knowledge, this is the first report of 2,4-diaminofuro[2,3-d]pyrimidines inhibiting GARFTase as the principal mechanism of tumor cell inhibition and of transport selectivity for FRs over RFC. For the 2-amino-4-oxo-6-substituted pyrrolo[2,3-d]pyrimdines 4, 5, and 7, the regioisomeric nature of the side chain thiophene ring is relatively unimportant. This contrasts with the biological activity of the 2,4-diamino-5-substituted-furo[2,3-d]pyrimidines 6 and 8 for which the regioisomeric nature of the thiophene ring dictates differences in biological activities.
Collectively, our results establish that the pyrrolo[2,3-d]pyrimidine thienoyl analogues including the three-carbon bridge typified by 4, 5, and 7 are particularly effective for targeting tumors via selective FR and PCFT transport and for potently inhibiting de novo purine nucleotide biosynthesis at GARFTase. While inhibition of intracellular GARFTase by 6-substituted pyrrolo[2,3-d]pyrimidine inhibitors such as compound 4 results in decreased pools of purine nucleotides27,47 (analogous to LMTX15,51) and likely explains the antitumor effects of this series, it is the transport selectivity for FRs and PCFT over RFC, combined with selective expression and/or transport function of FRs and PCFT in tumors, that primarily confers tumor specificity. Loss of RFC uptake for this series would confer decreased toxicity to normal tissues. GARFTase targeting by compounds such as 5 and 7 could be extraordinarily useful in treating tumor cells resistant to PMX where TS is the principal intracellular target.
EXPERIMENTAL PROCEDURES
All solvents and chemicals were purchased from Aldrich Chemical Co. or Fisher Scientific and were used as received. All evaporations were carried out with a rotary evaporator under vacuum. Analytical samples were dried in a CHEM-DRY drying apparatus over P2O5 at 80 °C under vacuum (0.2 mmHg). Whatman Sil G/UV254 silica gel plates with a fluorescent indicator were used to perform thin layer chromatorgraphy (TLC), and the spots were visualized under 254 and 365 nm illumination. All analytical samples were homogeneous on TLC in three different solvent systems. Proton nuclear magnetic resonance spectra (1H NMR) were recorded on either a Bruker WH-400 (400 MHz) spectrometer or a Bruker WH-500 (500 MHz) spectrometer. The chemical shift values are expressed in ppm (parts per million) using tetramethylsilane as an internal standard: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad singlet. A VG-7070 double-focusing mass spectrometer or a LKB-9000 instrument in the electron ionization (EI) mode were used to record mass spectra. A MEL-TEMP II melting point apparatus with a FLUKE 51 K/J electronic thermometer was used to determine melting points. Reported melting points are uncorrected. Chemical names follow IUPAC nomenclature. Proportions of solvents used for TLC are by volume. Column chromatography was performed using 230–400 mesh silica gel (Fisher, Somerville, NJ). Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA. Element compositions are within 0.4% of the calculated values. In spite of 24–48 h of drying in vacuo, fractional moles of water or solvent frequently found in the analytical sample of target compounds could not be prevented and were confirmed where possible by their presence in the 1H NMR spectra. For all the compounds submitted for biological evaluation, elemental analysis (C, H, N) was performed to confirm >95% purity.
4-(4-Hydroxy-but-1-ynyl)-thiophene-2-carboxylic Acid Methyl Ester (10)
To a solution 4-bromo-thiophene-2-carboxylic acid methyl ester (4.42 g, 20 mmol) in anhydrous acetonitrile (20 mL) was added palladium chloride (142 mg, 0.8 mmol), triphenylphosphine (261 mg, 0.8 mmol), copper iodide (608 mg, 3.2 mmol), triethylamine (20.2 g, 0.2 mol), and but-3-yn-1-ol, 9 (2.1 g, 30 mmol). The reaction mixture was heated to 100 °C for 6 h. Then silica gel (10 g) was added and the solvent was evaporated under reduced pressure to afford a plug which was loaded on to a silica gel column (3.5 cm × 12 cm) and eluted with hexane followed by 50% EtOAc in hexane. The desired fractions (TLC) were pooled and evaporated to afford 2.8 g (67%) of 10 as a light-yellow oil. TLC Rf 0.33 (hexane/EtOAc 1:1). 1H NMR (DMSO-d6): δ 2.52 2.55 (t, J = J = 6.8 Hz, 2H, CH2), 3.56–3.59 (t, J = 6.8 Hz, 2H, CH2), 3.83 (s, 3H, COOCH3), 7.73 (s, 1H, Ar), 8.03 (s, 1H, Ar).
5-(4-Hydroxy-but-1-ynyl)-thiophene-3-carboxylic Acid Methyl Ester (11)
To a solution 5-bromo-thiophene-3-carboxylic acid methyl ester (4.42 g, 20 mmol) in anhydrous acetonitrile (20 mL) was added palladium chloride (142 mg, 0.8 mmol), triphenylphosphine (261 mg, 0.8 mmol), copper iodide (608 mg, 3.2 mmol), triethylamine (20.2 g, 0.2 mol), and but-3-yn-1-ol, 9 (2.1 g, 30 mmol). The reaction mixture was heated to 100 °C for 6 h. Then silica gel (10 g) was added and the solvent was evaporated under reduced pressure to afford a plug which was loaded on to a silica gel column (3.5 cm × 12 cm) and eluted with hexane followed by 50% EtOAc in hexane. The desired fractions (TLC) were pooled and evaporated to afford 2.96 g (71%) of 11 as a light-yellow oil. TLC Rf 0.33 (hexane/EtOAc 1:1). 1H NMR (DMSO-d6): δ 2.58–2.61 (t, J = 6.5 Hz, 2H, CH2), 3.56–3.59 (t, J = 6.5 Hz, 2H, CH2), 3.80 (s, 3H, COOCH3), 4.93–4.95 (t, J = 5.5 Hz, 1H, OH, exch), 7.47 (s, J = 1.5 Hz, 1H, Ar), 8.28 (d, J = 1.5 Hz, 1H, Ar).
4-(4-Hydroxy-butyl)-thiophene-2-carboxylic Acid Methyl Ester (12)
Compound 10 (2.8 g, 13 mmol), 10% palladium on activated carbon (1.4 g), and MeOH (50 mL) were added to a Paar flask, and hydrogenation was carried out at 55 psi of H2 for 4 h. The reaction mixture was filtered through Celite, washed with MeOH/CHCl3 (1:1) (100 mL), passed through a short silica gel column (3 cm × 5 cm) and concentrated under reduced pressure to give 2.78 g (99%) of 12 as a yellow oil. TLC Rf 0.34 (hexane/EtOAc 1:1). 1H NMR (DMSO-d6): δ 1.38–1.45 (m, 2H,CH2), 1.56–1.64 (m, 2H, CH2), 2.58–2.62 (t, J = 7.2 Hz, 2H, CH2), 3.39–3.42 (t, J = 6.8 Hz, 2H, CH2), 3.81 (s, 3H, COOCH3), 7.57 (s, 1H, Ar), 7.67 (s, 1H, Ar).
5-(4-Hydroxy-butyl)-thiophene-3-carboxylic Acid Methyl Ester (13)
Compound 11 (2.96 g, 13.7 mmol), 10% palladium on activated carbon (1.5 g), and MeOH (50 mL) were added to a Paar flask, and hydrogenation was carried out at 55 psi of H2 for 4 h. The reaction mixture was filtered through Celite, washed with MeOH/CHCl3 (1:1) (100 mL), passed through a short silica gel column (3 cm × 5 cm), and concentrated under reduced pressure to give 2.78 g (91%) of 13 as a yellow oil. TLC Rf 0.34 (hexane/EtOAc 1:1). 1H NMR (DMSO-d6): δ 1.43–1.49 (m, 2H, CH2), 1.60–1.68 (m, 2H, CH2), 2.79–2.82 (t, J = 7.2 Hz, 2H, CH2), 3.40–3.42 (t, J = 6.5 Hz, 2H, CH2), 3.78 (s, 3H, COOCH3), 4.41 (s, 1H, OH, exch), 7.18 (s, 1H, Ar), 8.13 (s, 1H, Ar).
4-(3-Carboxy-propyl)-thiophene-2-carboxylic Acid Methyl Ester (14)
Periodic acid (6.52 g, 28.6 mmol) was added to acetonitrile (40 mL), and the mixture was stirred vigorously for 15 min. Compound 12 (2.78 g, 13 mmol) was then added (in ice–water bath), followed by addition of PCC (56 mg, 0.26 mmol), and the reaction mixture was stirred for 1 h. The solvent was evaporated under reduce pressure to afford a residue which was diluted with EtOAc (100 mL), washed consequtively with brine–water, satd aq NaHSO3 solution, and brine, dried over anhydrous Na2SO4, and concentrated to give 2.34 g (79%) of 14 as a light-yellow oil. TLC Rf 0.58 (hexane/ EtOAc1:1). 1H NMR (DMSO-d6): δ 1.78–1.84 (m, 2H, CH2), 2.20–2.23 (t, J = 7.5 Hz, 2H, CH2), 2.60–2.63 (t, J = 7.5 Hz, 2H, CH2), 3.81 (s, 3H, COOCH3), 7.59 (s, 1H, Ar), 7.68 (s, 1H, Ar), 12.07 (br, 1H, COOH, exch).
5-(3-Carboxy-propyl)-thiophene-3-carboxylic Acid Methyl Ester (15)
Periodic acid (6.52 g, 28.6 mmol) was added to acetonitrile (40 mL), and the mixture was stirred vigorously for 15 min. Compound 13 (2.78 g, 13 mmol) was then added (in ice–water bath), followed by addition of PCC (56 mg, 0.26 mmol), and the reaction mixture was stirred for 1 h. Evaporation of the solvent under reduced pressure provided a residue which was diluted with EtOAc (100 mL) and washed sequentially with brine–water, satd aq NaHSO3 solution, and brine, dried over anhydrous Na2SO4, and concentrated to give 3.01 g (99%) of 15 as a light-yellow oil. TLC Rf 0.58 (hexane/ EtOAc 1:1). 1H NMR (DMSO-d6): δ 1.80–1.87 (m, 2H, CH2), 2.25–2.28 (t, J = 7.2 Hz, 2H, CH2), 2.81–2.84 (t, J = 7.2 Hz, 2H, CH2), 3.78 (s, 3H, COOCH3), 7.20 (s, 1H, Ar), 8.15 (s, 1H, Ar), 12.15 (br, 1H, COOH, exch).
4-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-2-carboxylic Acid Methyl Ester (22)
To 14 (2.28 g, 10 mmol) in a 100 mL flask were added oxalyl chloride (7.6 g, 60 mmol) and anhydrous CH2Cl2 (20 mL). The resulting solution was heated at reflux for 1 h and then cooled to room temperature. After evaporating the solvent under reduced pressure, the residue was dissolved in 20 mL of Et2O. The resulting solution was added dropwise to an ice-cooled diazomethane (generated in situ from 10 g of diazald by using Aldrich Mini Diazald apparatus) in an ice bath over 10 min. The resulting mixture was allowed to stand for 30 min and then stirred for an additional 1 h. Concd HCl (20 mL) was added to this solution, and the resulting mixture was heated at reflux for 1.5 h. After cooling to room temperature, the organic layer was separated and the aqueous layer extracted with Et2O (50 mL × 2). The combined organic layer and Et2O extract was washed with two portions of 10% Na2CO3 solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure afforded a light-yellow residue. To this residue in anhydrous DMF (15 mL) was added 2,6 diamino-3H-pyrimidin-4-one, (1.26 g, 10 mmol). The resulting mixture was stirred under N2 at 50–60 °C for 3 days. Silica gel (2 g) was then added, and the solvent was evaporated under reduced pressure. Chromatographic separation of the resulting plug was performed on a silica gel column (2.5 cm × 12 cm) and elution with CHCl3, 2% MeOH in CHCl3, and then 4% MeOH in CHCl3. Fractions with an Rf 0.17 (CHCl3/MeOH 10:1) were pooled and evaporated to afford 920 mg (28%) of 22 as a light-yellow powder; mp 182–183 °C. 1H NMR (DMSO-d6): δ 1.87–1.94 (m, 2H, CH2), 2.49–2.52 (t, J = 7.6 Hz, 2H, CH2), 2.60–2.64 (t, J = 7.6 Hz, 2H, CH2), 3.81 (s, 3H, COOCH3), 5.95 (d, J = 2.0 Hz, 1H, C5-CH), 6.43 (s, 2H, 2-NH2, exch), 7.60 (d, J = 1.6 Hz, 1H, Ar), 7.70 (d, J = 1.6 Hz, 1H, Ar), 10.55 (s, 1H, 3-NH, exch), 11.08 (s, 1H, 7-NH, exch).
4-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-2-carboxylic Acid Methyl Ester (26)
Fractions with an Rf 0.25 (CHCl3/MeOH 10:1) were pooled and evaporated to afford 320 mg (9.6%) of 26 as a light-yellow powder; mp 156–157 °C. 1H NMR (DMSO-d6): δ 1.81–1.88 (m, 2H, CH2), 2.63–2.69 (m, 4H, CH2, CH2), 3.81 (s, 3H, COOCH3), 5.97 (s, 2H, 2-NH2, exch), 6.41 (s, 2H, 4-NH2, exch), 7.14 (s, 1H, C6-CH), 7.61 (d, J = 1.2 Hz, 1H, Ar), 7.71 (d, J = 1.2 Hz, 1H, Ar).
5-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-3-carboxylic Acid Methyl Ester (23)
To 15 (2.28 g, 10 mmol) in a 100 mL flask were added oxalyl chloride (7.6 g, 60 mmol) and anhydrous CH2Cl2 (20 mL). The resulting solution was heated at for 1 h and then cooled to room temperature. After evaporating the solvent under reduced pressure, the residue was dissolved in 20 mL of Et2O. The resulting solution was added dropwise to an ice-cooled diazomethane (generated in situ from 10 g of diazald by using Aldrich Mini Diazald apparatus) in an ice bath over 10 min. The resulting mixture was allowed to stand for 30 min and then stirred for an additional 1 h. Concentrated HCl (20 mL) was added to this solution, and the resulting mixture was heated at reflux for 1.5 h. After cooling to room temperature, the organic layer was separated and the aqueous layer extracted with Et2O (50 mL × 2). The combined organic layer and Et2O extract was washed with two portions of 10% Na2CO3 solution and dried over Na2SO4. Evaporation of the solvent under reduced pressure afforded a light-yellow residue. To this residue in anhydrous DMF (15 mL) was added 2,6-diamino-3H-pyrimidin-4-one, (1.26 g, 10 mmol). The resulting mixture was stirred under N2 at 50–60 °C for 3 days. Silica gel (2 g) was then added, and the solvent was evaporated under reduced pressure. Chromatographic separation of the resulting plug was performed on a silica gel column (2.5 cm × 12 cm) and elution with CHCl3, 2% MeOH in CHCl3, and then 4% MeOH in CHCl3. Fractions with an Rf 0.17 (CHCl3/MeOH 10:1) were pooled and evaporated to afford 986 mg (30%) of 23 as a light-yellow powder; mp 182–183 °C. 1H NMR (DMSO-d6): δ 1.90–1.97 (m, 2H, CH2), 2.51–2.56 (t, J = 7.2 Hz, 2H, CH2), 2.80–2.83 (t, J = 7.2 Hz, 2H, CH2), 3.78 (s, 3H, COOCH3), 5.90 (d, J = 1.6 Hz, 1H, C5-CH), 5.98 (s, 2H, 2-NH2, exch), 7.21 (s, 1H, Ar), 8.14 (s, 1H, Ar), 10.15 (s, 1H, 3-NH, exch), 10.84 (s,1H, 7-NH, exch).
5-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-3-carboxylic Acid Methyl Ester (27)
Fractions with an Rf 0.25 (CHCl3/MeOH 10:1) were pooled and evaporated to afford 327 mg (9.8%) of 25 as a light-yellow powder; mp 156–157 °C. 1H NMR (DMSO-d6): δ 1.83–1.92 (m, 2H, CH2), 2.66–2.70 (t, J = 7.6 Hz, 2H, CH2), 2.86–2.89 (t, J = 7.6 Hz, 2H, CH2), 3.78 (s, 3H, COOCH3), 5.99 (s, 2H, 2-NH2, exch), 6.44 (s, 2H, 4-NH2, exch), 7.15 (s, 1H, C6-CH), 7.22 (d, J = 1.2 Hz, 1H, Ar), 8.14 (d, J = 1.2 Hz, 1H, Ar).
4-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-2-carboxylic Acid (24)
First, 1 N NaOH (10 mL) was added to a solution of 22 (460 mg, 1.4 mmol) in MeOH (10 mL) and the mixture was stirred under N2 at 40 °C for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.16) and generation of one major spot at the baseline (CHCl3/MeOH 10:1).The reaction mixture was then evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1 N HCl. The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried under reduced pressure using P2O5 to afford 367 mg (83%) of 24 as a yellow powder; mp 181–182 °C. 1H NMR (DMSO-d6): δ 1.86–1.93 (m, 2H, CH2), 2.48–2.52 (t, J = 7.6 Hz, 2H, CH2), 2.59–2.62 (t, J = 7.6 Hz, 2H, CH2), 5.90 (s, 1H, C5-CH), 6.01 (s, 2H, 2-NH2, exch), 7.53 (s, 1H, Ar), 7.62 (s, 1H, Ar), 10.17 (s, 1H, 3-NH,exch), 10.84 (s, 1H, 7-NH, exch), 12.98 (br, 1H, COOH, exch).
5-[3(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-3-carboxylic Acid (25)
To a solution of 23 (460 mg, 1.4 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at 40 °C for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.16) and generation of one major spot at the baseline (CHCl3/MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1 N HCl. The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried under reduced pressure using P2O5 to afford 376 mg (85%) of 25 as a yellow powder; mp 181–182 °C. 1H NMR (DMSO-d6): δ 1.89–1.97 (m, 2H, CH2), 2.51–2.56 (t, J = 7.6 Hz, 2H, CH2), 2.78–2.82 (t, J = 7.6 Hz, 2H, CH2), 5.90 (s, 1H, C5-CH), 6.00 (s, 2H, 2-NH2, exch), 7.17 (s, J = 1.2 Hz, 1H, Ar), 8.05 (s, J = 1.2 Hz, 1H, Ar), 10.17 (s, 1H, 3-NH, exch), 10.85 (s, 1H, 7-NH, exch), 12.62 (br, 1H, COOH, exch).
4-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-2-carboxylic Acid (28)
To a solution of 26 (320 mg, 0.96 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at 40 °C for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.25) and generation of one major spot at the baseline (CHCl3/ MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1 N HCl. The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried under reduced pressure using P2O5 to afford 280 mg (92%) of 28 as a light-yellow powder; mp 156–157 °C. 1H NMR (DMSO-d6): δ 1.80–1.88 (m, 2H, CH2), 2.63–2.68 (m, 4H, CH2, CH2), 5.97 (s, 2H, 2-NH2, exch), 6.41 (s, 2H, 4-NH2, exch), 7.14 (s, 1H, C6-CH), 7.52 (d, J = 1.6 Hz, 1H, Ar), 7.61 (d, J = 1.6 Hz, 1H, Ar), 12.99 (br, 1H, COOH, exch).
4-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-2-carboxylic Acid (29)
To a solution of 27 (320 mg, 0.96 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at 40 °C for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.25) and generation of one major spot at the baseline (CHCl3/ MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), the resulting solution was cooled in an ice bath, and the pH was adjusted to 3–4 with dropwise addition of 1 N HCl. The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried under reduced pressure using P2O5 to afford 283 mg (93%) of 29 as a light-yellow powder; mp 156–157 °C. 1H NMR (DMSO-d6): δ 1.81–1.91 (m, 2H, CH2), 2.66–2.70 (t, J = 7.6 Hz, 2H, CH2), 2.85–2.88 (t, J = 7.6 Hz, 2H, CH2), 5.98 (s, 2H, 2-NH2, exch), 6.44 (s, 2H, 4-NH2, exch), 7.15 (s, 1H, C6-CH), 7.17 (d, J = 1.6 Hz, 1H, Ar), 8.03 (d, J = 1.2 Hz, 1H, Ar), 12.63 (br, 1H, COOH, exch).
(S)-2-({4-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-2-carbonyl}-amino)-pentanedioic Acid Diethyl Ester (30)
To a solution of 24 (159 mg, 0.5 mmol) in anhydrous DMF (10 mL) were added N-methylmorpholine (91 mg, 0.9 mmol) and 2-chloro-4,6-dimethoxy-1,3,5 triazine (158 mg, 0.9 mmol). The resulting mixture was stirred at room temperature for 2 h. To this mixture were added N-methylmorpholine (91 mg, 0.9 mmol) and L-glutamate diethyl ester hydrochloride (180 mg, 0.75 mmol). The reaction mixture was stirred for an additional 4 h at room temperature and then evaporated to dryness under reduced pressure. The residue was dissolved in the minimum amount of CHCl3/MeOH (4:1) and chromatographed on a silica gel column (1.5 cm × 15 cm) and with 5% CHCl3 in MeOH as the eluent. Fractions that showed the desired spot (TLC) were pooled and the solvent evaporated to dryness under reduced pressure to afford 220 mg (87%) of 30 as a yellow powder; mp 81–82 °C. TLC Rf 0.13 (CHCl3/MeOH 10:1). 1H NMR (DMSO-d6): δ 1.15–1.21 (m, 6H, COOCH2CH3), 1.87–2.14 (m, 4H, β-CH2,CH2), 2.41–2.45 (t, J = 7.6 Hz, 2H, γ-CH2), 2.51–2.55 (t, J = 7.6 Hz, 2H, CH2), 2.59–2.62 (t, J = 7.6 Hz, 2H, CH2), 4.02–4.14 (m, 4H, COOCH2CH3), 4.36–4.41 (m, 1H, α-CH), 5.92–5.93 (d, J = 2.0 Hz, 1H, C5-CH), 6.14 (s, 2H, 2-NH2, exch), 7.44 (d, J = 1.2 Hz, 1H, Ar), 7.78 (d, J = 1.2 Hz, 1H, Ar), 8.68–8.70 (d, J = 7.2 Hz, 1H, CONH, exch), 10.28 (s, 1H, 3-NH, exch), 10.90 (s, 1H, 7-NH, exch).
(S)-2-({5-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl) propyl]-thiophene-3-carbonyl}-amino)-pentanedioic Acid Diethyl Ester (31)
N-Methylmorpholine (91 mg, 0.9 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (158 mg, 0.9 mmol) were added to a solution of 25 (159 mg, 0.5 mmol) in anhydrous DMF (10 mL). The resulting mixture was stirred at room temperature for 2 h. To this mixture were added N-methylmorpholine (91 mg, 0.9 mmol) and L-glutamate diethyl ester·HCl (180 mg, 0.75 mmol). The reaction mixture was stirred for an additional 4 h at room temperature and then evaporated to dryness under reduced pressure. The residue was dissolved in the minimum amount of CHCl3/MeOH (4:1) and chromatographically separated on a silica gel column (1.5 cm × 15 cm) with 5% CHCl3 in MeOH as the eluent. Fractions that showed the desired spot (TLC) were pooled and the solvent evaporated to dryness to afford 223 mg (88%) of 31 as a yellow powder; mp 81–82 °C. TLC Rf 0.13 (CHCl3/MeOH 10:1). 1H NMR (DMSO-d6): δ 1.15–1.21 (m, 6H, COOCH2CH3), 1.87–2.11 (m, 4H, β-CH2, CH2), 2.41–2.44 (t, J = 7.6 Hz, 2H, γ-CH2), 2.54–2.58 (t, J = 7.6 Hz, 2H, CH2), 2.78–2.82 (t, J = 7.6 Hz, 2H, CH2), 4.02–4.13 (m, 4H, COOCH2CH3), 4.36–4.41 (m, 1H, α-CH), 5.94 (d, J = 2.0 Hz, 1H, C5-CH), 6.34 (s, 2H, 2-NH2, exch), 7.29 (d, J = 1.2 Hz, 1H, Ar),8.01 (d, J = 1.2 Hz, 1H, Ar), 8.49–8.51 (d, J = 7.6 Hz, 1H, CONH, exch), 10.46 (s, 1H, 3-NH, exch), 1.18 (s, 1H, 7-NH, exch).
(S)-2-({4-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-2-carbonyl}-amino)-pentanedioic Acid Diethyl Ester (32)
N-Methylmorpholine (91 mg, 0.9 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (158 mg, 0.9 mmol) were added to a solution of 28 (159 mg, 0.5 mmol) in anhydrous DMF (10 mL). The resulting mixture was stirred at room temperature for 2 h. To this mixture were added N-methylmorpholine (91 mg, 0.9 mmol) and L-glutamate diethyl ester hydrochloride (180 mg, 0.75 mmol). The reaction mixture was stirred for an additional 4 h at room temperature and then evaporated to dryness under reduced pressure. The residue was dissolved in the minimum amount of CHCl3/MeOH (4:1) and chromatographically separated on a silica gel column (1.5 cm × 15 cm) with 2% CHCl3 in MeOH as the eluent. Fractions that showed the desired spot (TLC) were pooled and the solvent evaporated to dryness to afford 200 mg (80%) of 32 as a yellow powder; mp 79–80 °C. TLC Rf 0.26 (CHCl3/MeOH 10:1). 1H NMR (DMSO-d6): δ 1.15–1.21 (m, 6H, COOCH2CH3), 1.82–2.12 (m, 4H, β-CH2, CH2), 2.41–2.45 (t, J = 7.2 Hz, 2H, γ-CH2), 2.64–2.70 (m, 4H, CH2, CH2), 4.02–4.14 (m, 4H, COOCH2CH3), 4.36–4.42 (m, 1H, α-CH), 5.97 (s, 2H, 2-NH2, exch), 6.41 (s, 2H, 4-NH2, exch), 7.16 (s, 1H, C6-CH), 7.44 (d, J = 1.2 Hz, 1H, Ar), 7.76 (d, J = 1.2 Hz, 1H, Ar), 8.65–8.67 (d, J = 7.6 Hz, 1H, CONH, exch).
(S)-2-({5-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-3-carbonyl}-amino)-pentanedioic Acid Diethyl Ester (33)
N-Methylmorpholine (91 mg, 0.9 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (158 mg, 0.9 mmol) were added to a solution of 29 (159 mg, 0.5 mmol) in anhydrous DMF (10 mL). The resulting mixture was stirred at room temperature for 2 h. To this mixture were added N-methylmorpholine (91 mg, 0.9 mmol) and L-glutamate diethyl ester hydrochloride (180 mg, 0.75 mmol). The reaction mixture was stirred for an additional 4 h at room temperature and then evaporated to dryness under reduced pressure. The residue was dissolved in the minimum amount of CHCl3/MeOH (4:1) and chromatographically separated on a silica gel column (1.5 cm × 15 cm) with 2% CHCl3 in MeOH as the eluent. Fractions that showed the desired spot (TLC) were pooled and the solvent evaporated to dryness to afford 205 mg (80%) of 33 as a yellow powder; mp 79–80 °C. TLC Rf 0.26 (CHCl3/MeOH 10:1). 1H NMR (DMSO-d6): δ 1.15–1.21 (m, 6H, COOCH2CH3), 1.82–2.13 (m, 4H, β-CH2, CH2), 2.40–2.44 (t, J = 7.6 Hz, 2H, γ-CH2), 2.68–2.71 (t, J = 7.2 Hz, 2H, CH2), 2.84–2.88 (t, J = 7.2 Hz, 2H, CH2), 4.02–4.13 (m, 4H, COOCH2CH3), 4.36–4.41 (m, 1H, α-CH), 5.99 (s, 2H, 2-NH2, exch),
(S)-2-({4-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-2-carbonyl}-amino)-pentanedioic Acid (5)
To a solution of 30 (220 mg, 0.45 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at room temperature for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.13) and generation of one major spot at the baseline (CHCl3/MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), and the resulting solution was cooled in an ice bath. The pH of the solution was then adjusted to 3–4 with 1 N HCl (dropwise addition). The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford 125 mg (62%) of 5 as yellow powder; mp 191–192 °C. 1H NMR (DMSO-d6): δ 1.87–2.12 (m, 4H, β-CH2, CH2), 2.33–2.37 (t, J = 7.2 Hz, 2H, γ-CH2), 2.51–2.53 (t, J = 7.2 Hz, 2H, CH2), 2.59–2.62 (t, J = 7.2 Hz, 2H, CH2), 4.32–4.37 (m, 1H, α-CH), 5.92 (d, J = 2.0 Hz, 1H, C5-CH), 5.98 (s, 2H, 2-NH2, exch), 7.43 (d, J = 1.2 Hz, 1H, Ar), 7.77 (d, J = 1.2 Hz, 1H, Ar), 8.55–8.57 (d, J = 7.6 Hz, 1H, CONH, exch), 10.14 (s, 1H, 3-NH, exch), 10.84 (s, 1H, 7-NH, exch) 12.45 (br, 2H, COOH, exch). Anal. Calcd for (C19H21N5O6S·1.7H2O): C, H, N, S.
(S)-2-({5-[3-(2-Amino-4-oxo-4,7-dihydro-3H-pyrrolo[2,3-d]-pyrimidin-6-yl)-propyl]-thiophene-3-carbonyl}-amino)-pentanedioic Acid (7)
To a solution of 31 (220 mg, 0.45 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at room temperature for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.13) and generation of one major spot at the baseline (CHCl3/MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), and the resulting solution was cooled in an ice bath. The pH of the solution was then adjusted to 3–4 with 1 N HCl (dropwise addition). The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford 131 mg (65%) of 7 as yellow powder; mp 166–167 °C. 1H NMR (DMSO-d6): δ 1.87–2.10 (m, 4H, β-CH2,CH2), 2.33–2.36 (t, J = 7.2 Hz, 2H, γ-CH2), 2.55–2.57 (t, J = 7.2 Hz, 2H, CH2), 2.79–2.82 (t, J = 7.2 Hz, 2H,CH2), 4.35 (m, 1H, α-CH), 5.92 (s, 1H, C5-CH), 6.03 (s, 2H, 2-NH2, exch), 7.30 (s, 1H, Ar), 7.99 (s, 1H, Ar), 8.35–8.37 (d, J = 7.2 Hz, 1H, CONH, exch), 10.19 (s, 1H, 3-NH, exch), 10.87 (s, 1H, 7-NH, exch) 12.41 (br, 2H, COOH, exch). Anal. Calcd for (C19H21N5O6S·1.4H2O): C, H, N, S.
(S)-2-({4-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-2 carbonyl}-amino)-pentanedioic Acid (6)
To a solution of 32 (200 mg, 0.41 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at room temperature for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.26) and generation of one major spot at the baseline (CHCl3/MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), and the resulting solution was cooled in an ice bath. The pH of the solution was then adjusted to 3–4 with 1 N HCl (dropwise addition) The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford 145 mg (79%) of 6 as a light-yellow powder; mp 148–149 °C. 1H NMR (DMSO-d6): δ 1.82–2.12 (m, 4H, β-CH2, CH2), 2.33–2.37 (t, J = 7.6 Hz, 2H, γ-CH2), 2.64–2.70 (m, 4H, CH2, CH2), 4.32–4.37 (m, 1H, α-CH), 6.01 (s, 2H, 2-NH2, exch), 6.45 (s, 2H, 4-NH2, exch), 7.17 (s, 1H, C6-CH), 7.43 (d, J = 1.2 Hz, 1H, Ar), 7.77 (d, J = 1.2 Hz, 1H, Ar), 8.54–8.56 (d, J = 7.6 Hz, 1H, CONH, exch), 12.44 (br, 2H, COOH, exch). Anal. Calcd for (C19H21N5O6S·1.2HCOOH): C, H, N, S. HRMS calcd for C19H21N5O6S (M + H)+, 448.1285; found, 448.1280.
(S)-2-({5-[3-(2,4-Diamino-furo[2,3-d]pyrimidin-5-yl)-propyl]-thiophene-3-carbonyl}-amino)-pentanedioic Acid (8)
To a solution of 33 (200 mg, 0.41 mmol) in MeOH (10 mL) was added 1 N NaOH (10 mL), and the mixture was stirred under N2 at room temperature for 16 h. Monitoring the reaction using TLC indicated consumption of the starting material (Rf 0.26) and generation of one major spot at the baseline (CHCl3/MeOH 10:1). The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (10 mL), and the resulting solution was cooled in an ice bath. The pH of the solution was then adjusted to 3–4 with 1 N HCl (dropwise addition). The resulting suspension was frozen in a dry ice–acetone bath, thawed to 4–5 °C in the refrigerator, and filtered. The residue was washed with a small amount of cold water and dried in vacuum using P2O5 to afford 149 mg (81%) of 8 as a light-yellow powder; mp 150–151 °C. 1H NMR (DMSO-d6): δ 1.82–2.11 (m, 4H, β-CH2,CH2), 2.32–2.36 (t, J = 7.6 Hz, 2H, γ-CH2), 2.68–2.71 (t, J = 7.6 Hz, 2H,CH2), 2.84–2.88 (t, J = 7.6 Hz, 2H,CH2), 4.32–4.38 (m, 1H, α-CH), 5.99 (s, 2H, 2-NH2, exch), 6.44 (s, 2H, 4-NH2, exch), 7.16 (s, 1H, C6–CH), 7.30 (d, J = 1.2 Hz, 1H, Ar), 7.98 (d, J = 1.2 Hz, 1H, Ar), 8.34–8.36 (d, J = 7.6 Hz, 1H, CONH, exch), 12.42 (br, 2H, COOH, exch). Anal. Calcd for (C19H21N5O6S·0.3CHCl3): C, H, N, S.
Molecular Modeling and Computational Studies
The X-ray crystal structures of human FRα bound to folic acid (PDB: 4LRH, 2.80 Å),30 FRβ bound to PMX (PDB: 4KN2, 2.60 Å),31 and human GARFTase bound to 7 (described in this manuscript) or trifluoroacetyl-5,10-dideaza-acyclic-5,6,7,8-tetrahydrofolic acid (10-CF3CO–DDACTHF) (PDB: 1NJS, 1.98 Å)32 were obtained from the protein database.
Docking studies were performed using LeadIT 2.1.6.33 Default settings were used to calculate the protonation state of the proteins, and the ligands and free rotation of water molecules in the active site (defined by amino acids within 6.5 Å from the crystal structure ligand) were permitted. Ligands for docking were sketched using MOE 2013.0852 and energy minimized using the MMF94X force field (limit of 0.05 kcal/mol). Molecules were docked using the triangle matching placement method and scored using default settings. The docked poses were visualized using CCP4MG.53
To validate the docking process using LeadIT 2.1.6, the crystallized ligands (folic acid for FRα, PMX for FRβ, and 10-CF3CO–DDACTHF for GARFTase) were sketched using MOE, energy minimized, and docked as described. Deviation of the best docked poses from the crystal structure conformation was calculated using an RMSD SVL code obtained from the ChemComp Web site.54
The best pose of folic acid in FRα had an RMSD of 0.81 Å. PMX in FRβ had an RMSD of 1.01 Å, and 10-CF3CO–DDACTHF in the human GARFTase had an RMSD of 1.04 Å. Thus, LeadIT 2.1.6 was validated for our docking purposes in FRα, FRβ, and human GARFTase.
Reagents for Biological Studies
[3′,5′,7,9-3H]Folic acid (25 Ci/ mmol), [3′,5′,7-3H]MTX (20 Ci/mmol), [3H]PMX (1.3 Ci/mmol), and [14C(U)]-glycine (87mCi/mmol) were purchased from Moravek Biochemicals (Brea, CA). [3H]7 (12.3 Ci/mmol) was custom radiolabeled by Moravek Biochemicals. Unlabeled folic acid was purchased from the Sigma Chemical Co. (St. Louis, MO). LCV [(6R,S)5-formyl tetrahydrofolate] was provided by the Drug Development Branch, National Cancer Institute, Bethesda, MD. The sources of the classical antifolate drugs were as follows: MTX, Drug Development Branch, National Cancer Institute (Bethesda, MD); PDX [N-(4-{1-[(2,4-diaminopteridin-6-yl)methyl]but-3-yn-1-yl}-benzoyl)-L-glutamic acid], Allos Therapeutics (Westminster, CO); and PMX [(4-(2-(2-amino-4-oxo-4,7-dihydro-1H-pyrrolo[2,3-d]-pyrimidin-5-yl)ethyl)benzoyl)-L-glutamic acid] (Alimta), Eli Lilly and Co. (Indianapolis, IN). Other chemicals were obtained from commercial sources in the highest available purity.
Cell Lines and Assays of Antitumor Drug Activities
The engineered CHO sublines including RFC-, PCFT-, and FRα-null MTXRIIOuaR2-4 (R2), and RFC-(pC43-10), PCFT-(R2/PCFT4), or FRα-(RT16) and FRβ-(D4) expressing CHO sublines were previously described.24,36–38 The CHO cells were cultured in α-minimal essential medium (MEM) supplemented with 10% bovine calf serum (Invitrogen, Carlsbad, CA), penicillin (1000 U/mL) streptomycin (1000 μg/mL), and 2 mM L-glutamine at 37 °C with 5% CO2. All the transfected CHO cells [PC43-10, RT16, R2/hPCFT4, R2(VC)] were cultured in complete α-MEM media plus 1 mg/mL G418. RT16 and D4 cells were cultured for 3 days in complete folatefree RPMI 1640 (without added folate) prior to the cytotoxicity assays. R2/PCFT4 and R2(VC) cells were cultured in complete folate-free RPMI 1640 supplemented with dialyzed FBS (Invitrogen) and 25 nM LCV plus 1 mg/mL G418 prior to the drug sensitivity assays. KB human nasopharengeal carcinoma cells and SKOV3 ovarian cancer cells were purchased from the American Type Culture Collection (Manassas, VA). IGROV1 ovarian carcinoma cells were a gift of Dr. Manohar Ratnam (Karmanos Cancer Institute). IGROV1, SKOV3, and KB cells were cultured in folate-free RPMI 1640 medium, supplemented with 10% FBS (Invitrogen), penicillin–streptomycin solution, and 2 mM L-glutamine at 37 °C with 5% CO2. PCFT- and RFC-null R1-11 HeLa cells and R1-11-PCFT4 HeLa cells engineered to express PCFT without RFC were previously described.40 R1-11-RFC2 cells were generated from R1-11 cells by transfection with human RFC with a hemagglutinin epitope at position 591 cloned in pZeoSV2(+) vector, using Lipofectamine Plus reagent (Life Technologies). All the R1-11 HeLa sublines were cultured in complete RPMI 1640 medium (Sigma-Aldrich), containing 10% calf serum, 2 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL zeocin, plus 1 mg/mL G418, in a humidified atmosphere at 37 °C in the presence of 5% CO2.
For growth inhibition studies, cells (CHO, KB, SKOV3, IGROV1, HeLa) were plated in 96 well dishes (~2500–5000 cells/well, total volume of 200 μL medium) over a range of antifolate concentrations. 27,47 The medium was standard RPMI 1640 with 10% dialyzed FBS and antibiotics for experiments with R2 and PC43-10 cells. For experiments with RT16, D4, KB, SKOV3, and IGROV1 cells, cells were cultured in folate-free RPMI medium with 10% dialyzed FBS and antibiotics; the medium was supplemented with 2 nM LCV. FR-mediated drug uptake in these experiments was established by adding 200 nM folic acid to parallel incubations, conditions which minimally affected the growth inhibition with R2/PCFT4 cells. For studies with R2/PCFT4 and the R1-11-PCFT4 and R1-11-RFC2 cells, cells were cultured in folate-free RPMI 1640 (pH 7.2) containing 25 nM LCV, supplemented with 10% dialyzed FBS and antibiotics. Incubations were routinely for up to 96 h. Viable cells (reflecting cell viability) were assayed with Cell Titer Blue reagent (Promega, Madison, WI). A fluorescence plate reader was used to measure fluorescence (590 nm emission, 560 nm excitation). Relative fluorescence data were analyzed for calculations of IC50s, corresponding to the drug concentrations that resulted in 50% loss of cell proliferation.
For some experiments with KB tumor cells, in vitro growth inhibition was measured in the presence of thymidine (10 μM) and/or adenosine (60 μM) to establish the inhibitory effects of the antifolate drugs on de novo thymidylate synthase versus de novo purine nucleotide biosynthesis (GARFTase and AICARFTase).24,26,27,37 For de novo purine biosynthesis, additional protection experiments used AICA (320 μM) to distinguish inhibitory effects at GARFTase from those at AICARFTase.24,26,27,37
FR Binding Assay
Total FRα levels were measured for KB, IGROV1, and SKOV3 cells by determining [3H]folic acid binding to surface FRs.24 Briefly, cells (~(2–4) × 106) in a 60 mm culture dish were rinsed (3×) with Dulbecco’s phosphate-buffered saline (DPBS), then with acetate buffer (10 mM sodium acetate, 150 mM NaCl, pH 3.5) (2×) to remove FR-bound folates, and finally with HEPES-buffered saline (20 mM HEPES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, 5 mM glucose, pH 7.4) (HBS) (3×). Cells were incubated in HBS with [3H]folic acid (50 nM, specific activity 0.5 Ci/mmol) in the presence and absence of unlabeled folic acid (10 μM) for 15 min at 0 °C. The dishes were rinsed three times with ice-cold HBS, after which the cells were solubilized with 0.5 N sodium hydroxide and aliquots measured for radioactivity, and proteins. Proteins were measured with Folin-phenol reagent.55 Bound [3H]folic acid was calculated as pmol/ mg protein. Total FR levels of different cell lines were calculated from the differences in pmol/mg of bound [3H]folic acid in the absence and presence of excess unlabeled folic acid.
To measure relative binding affinities for FR ligands,24,26,27,37 FRα-expressing RT16 and FRβ-expressing D4 CHO cells in 60 mM dishes ((2–4) × 106 cells) were sequentially rinsed at 4 °C with DPBS (3×), acidic buffer (10 mM sodium acetate, 150 mM NaCl, pH 3.5) (2×) (to remove FR-bound folates), and HBS (pH 7.4). Cells were incubated in HBS with [3H]folic acid (50 nM, specific activity 0.5 Ci/ mmol) in the presence and absence of a range of concentrations of unlabeled folic acid, MTX (negative control), or the 6-pyrrolo[2,3-d]pyrimidine antifolates for 15 min at 0 °C. The dishes were rinsed (3×) with ice-cold HBS, after which the cells were solubilized with 0.5 N NaOH. Aliquots of the alkaline homogenate were measured for radioactivity and protein contents. Protein concentrations were measured with Folin-phenol reagent.55 Bound [3H]folic acid was calculated as pmol/mg protein, and relative binding affinities were calculated as the inverse molar ratios of unlabeled ligands required to inhibit [3H]folic acid binding by 50%. For both FRα and β, the affinity for folic acid was assigned a value of 1.
Transport Assays
For assays of [3H]7 uptake by FRα- and FRβ-expressing CHO sublines (RT16 and D4, respectively), cells were seeded at ~1.5 × 106 cells/60 mm dish 1 day prior to experiment. For the uptake assays, the cells were washed 3× with room temperature DPBS, then washed 2× with acidic buffer (pH 3.5). The cells were then washed 2× with Hank’s Balanced Salts Solution (HBSS) (pH 7.4, 1.5 mL, 37 °C) containing 50 nM [3H]7 in the absence and presence of 10 μM folic acid. Cells were incubated at 0 °C (surface fraction) or at 37 °C for 60 min (total fraction). An additional condition involved treatment with 50 nM [3H]7 at 37 °C for 60 min, followed by an acid wash to remove [3H]7 bound to surface FRs (“intracellular”). Cellular proteins were solubilized with NaOH for determinations of cell-associated [3H]7 and proteins. Results were presented as pmol/mg cell protein.
For PCFT transport assays in monolayer cultures (KB, IGROV1, SKOV3), uptake was measured with [3H]MTX (0.5 μM) over 5 min at 37 °C at pH 5.5 in 4-morpholinepropanesulfonic acid (MES)-buffered saline (20 mM MES, 140 mM NaCl, 5 mM KCl, 2 mM MgCl2, and 5 mM glucose).25,37 For PCFT uptake in R2/hPCFT4 and R2 CHO cells, cells grown as monolayers were used to seed spinner flasks. Uptakes in CHO cell suspensions (5–10 × 107 cells) were measured over 2 min at 37 °C at pH 5.5 in MES-buffered saline with [3H]PMX or [3H]7 (both at 0.5 μM) in the absence and presence of unlabeled 4 (10 μM). Radiolabeled drug fluxes were quenched with ice-cold DPBS, cells washed with ice-cold DPBS (3×) and solubilized with 0.5 N NaOH. Levels of intracellular radioactivity were expressed as pmol/mg protein, calculated from direct measurements of radioactivity and protein contents of cell homogenates. Protein concentrations were measured with Folin-phenol reagent.55 Transport kinetics were measured over 2 min at 37 °C at pH 5.5 in MES-buffered saline or at pH 6.8 in HBS over a range of concentrations of [3H]PMX or [3H]7 (0.02–0.33 μM). Kinetic constants (Kt, Vmax) were calculated from Lineweaver–Burke plots.
In Situ GARFTase Inhibition Assays
To determine the extent of inhibition of intracellular GARFTase inhibition by the antifolate inhibitors, we measured the metabolic incorporation of [14C(U)] glycine into [14C]formyl GAR.26,28,34,45,56 KB cells were seeded in complete media (above) and after 24 h were washed with folate-free RPMI 1640 (L-glutamine-free) with 10% dialyzed FBS, penicillin–streptomycin, and 2 nM LCV. The cells were incubated at 37 °C with 5% CO2 for 1 h with folate- and L-glutamine-depleted media including 2 nM LCV, with a range of concentrations of 4–7 or 8. Control cells were treated with an equal volume of DMSO. Azaserine (4 μM final) was added to the cells and allowed to incubate for 30 min. This was followed by addition of L-glutamine (2 mM) and [14C(U)]glycine (final specific activity 0.1 mCi/L). The cells were incubated 16 h, followed by washing with ice-cold DPBS. The cells were trypsinized, collected, and treated with 5% trichloroacetic acid (TCA) at 0 °C. Samples were centrifuged (4 °C, 14000 rpm), and the precipitated proteins were solubilized with 0.5 N NaOH. Proteins were quantified with the Folin-phenol protein method.55 The TCA supernatants were extracted with ether at 4 °C. The aqueous layer was gravity filtered through a 1 cm chromatography column (Bio-Rad) of AG1x8 (chloride form). The columns were washed with 0.5 N formic acid (10 mL), then with 4 N formic acid (10 mL), and finally eluted with 1 N HCl (8 mL). Eight 1 mL fractions were collected. The eluate was measured for radioactivity, and the percentages of radioactivity in the [14C]formyl GAR and nonspecific [14C]fractions were calculated. The results were calculated as cpm [14C]formyl GAR/mg cell protein. Data were graphed and the IC50s calculated for the drug-treated samples. The results for the drug-treated samples were normalized to those for the untreated controls.
GARFTase Expression and Purification
Two constructs pSD002 and pSD009 in pHis-parallel plasmids57 were used to express the formyltransferase domain of human GARFTase. pSD002 encoding human HisGARFTase, containing an N-terminal, cleavable hexahistidine tag for purification, was used for the enzyme inhibition assays. pSD009 encoding human GARFTaseHis, a fusion protein containing GARFTase with a noncleavable C-terminal hexahistidine tag for purification, was used in crystallization and subsequent structure determinations. All constructs were confirmed by DNA sequencing prior to enzymatic or structural studies.
Both pSD002 and pSD009 plasmids were transformed and expressed in Rosetta (DE3)pLysS cells. Cultures (1 L) were grown in LB media in the presence of 34 μg/mL chloramphenicol and 100 μg/mL ampicillin at 37 °C until the absorbance at 600 nm reached 0.6. Expression was induced via the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside, and cells were incubated overnight at 20 °C. Cultures were pelleted and then suspended in 40 mL of 25 mM Tris, pH 8, 300 mM NaCl, 5 mM β-mercaptoethanol (β-ME), 10 mM CaCl2, and 10 mM MgCl2 before cell lysis and pressurized cell cracking was performed. Both GARFTase proteins were purified from lysates by immobilized metal affinity chromatography over a nickelnitrilotriacetic acid (Ni-NTA) column (Qiagen). Ni-NTA purification involved passing the lysate over a 8 mL column on an AKTA FPLC (GE Healthcare), washing the column with 10 column volumes of 25 mM Tris, pH 8.0, 300 mM NaCl, 10% glycercol, and 5 mM β-ME (“wash buffer”), and eluting protein with a 0–75% gradient over 10 column volumes in elution buffer containing all components of the wash buffer and 300 mM imidazole. For further purification, samples were loaded onto on a Superdex 75 16/60 (GE Healthcare) column equilibrated with 25 mM Tris (pH 8.0), 300 mM NaCl, and 10 mM β-ME. To prevent loss of activity, HisGARTfase was stored at 90 μM in 20% glycerol at –80 °C.
In Vitro GARFTase Inhibition Assays
GARFTase activity was determined via measurement of the formation of 5,8-dideazafolate in the presence of varying concentrations of antifolate based on an established spectrophotometric protocol.44 Assays included 30 μM α,β GAR, 5.4 μM 10-formyl-5,8-dideazafolate, and a range of concentrations for 5–8 (dissolved in DMSO) or PMX, in 0.1 M Hepes, pH 7.5 (150 μL total volume). Reactions were preincubated at 37 °C in a UV transparent 96-well plate (Costar). To initiate the enzyme assay, 150 μL of 20 nM GARFTase or buffer (reference wells) were added and the plate was shaken for 5 s. Measurements were recorded at 295 nm every 15 s over 20 min using a BioTek Synergy H1M plate reader. For each antifolate, triplicate assays were performed at 20 different concentrations of antifolate. To determine an initial slope for each concentration, the average absorbance value of the reference well was subtracted to remove the effect of the antifolate on the raw absorbance. Absorbance values were converted to changes in absorbance over time by subtracting the absorbance at 295 nm at the first measurement from all subsequent measurements. Finally, correlation coefficients were compared over time spans to identify regions over which a consistent, linear increase in absorbance was observed for all replicates. Initial slopes were graphed against concentration of antifolate and a hyperbola fit [y = (–bx/(Ki + x)) + b, where “b” is the y-intercept] was used to determine a Ki for each antifolate (KaleidaGraph version 4.1).
In Vivo Efficacy Study with SKOV3 Human Ovarian Tumor Xenografts
Methods for determining the biological significance of the drug treatment results with transplantable tumors have been described previously.26,27,58–61 Cultured SKOV3 human ovarian tumor cells were implanted subcutaneously (5 × 106 cells/flank) to establish a solid tumor xenograft model in female NCR SCID mice (obtained from the NCI Animal Production Program). For the efficacy trial, mice were 10 weeks old on day 0 (tumor implant) with an average body weight of 19.9 g. Mice were supplied food and water ad libitum. Study mice were maintained on either a folate-deficient diet from Harlan-Teklad (TD.00434) or a folate-replete diet from Lab Diet (5021; autoclavable mouse breeder diet) starting 11 days before subcutaneous tumor implant to ensure serum folate levels would approximate those of humans. Serum folate levels were determined prior to tumor implant and post study via a Lactobacillus casei bioassay.62 For the trial, animals on both diets were respectively pooled and implanted bilaterally subcutaneously with 30–60 mg tumor fragments using a 12 gauge trocar and again respectively pooled before unselective distribution to the various treatment and control groups. Chemotherapy began 3 days after tumor implantation when the number of cells was small (below 63 mg, the established limit of palpation). Tumors were measured with a caliper 2–3 times weekly (depending on the doubling time of the tumor). Mice were sacrificed when the cumulative tumor burden reached 1500 mg. Tumor weights were estimated from two-dimensional measurements [i.e., tumor mass (in mg) = (a × b2)/2, where a and b are the tumor length and width in mm, respectively]. For calculating end points, both tumors on each mouse were added together, and the total mass per mouse was used. The following end-points were used to assess antitumor activity: (i) tumor growth delay [T – C, where T is the median time in days required for the treatment group tumors to reach a predetermined size (e.g., 1000 mg) and C is the median time in days for the control group tumors to reach the same size; tumor-free survivors are excluded from these calculations] and (ii) calculation of tumor cell kill [log10 cell kill total (gross) = (T – C)/(3.32) (Td), where T – C is the tumor growth delay, as described above, and Td is the tumor volume doubling time in days, estimated from the best fit straight line from a log–linear growth plot of control group tumors in exponential growth (100–800 mg range)]. The T/C value in percent was also determined for each treatment group. Treatment (T) and control (C) groups were measured when the control groups reach 700–1200 mg in size (exponential growth phase). The median of each group was then determined (including zeros). The T/C value is a nonquantitative determination of antitumor activity and the inverse of tumor growth inhibition (TGI). It is a measure of antitumor effectiveness based on the specific day of caliper measurement.
Crystallization of Human GARFTase and X-ray Data Collection and Structure Determination
GARFTaseHis was buffer-exchanged to 25 mM Tris (pH 8), 200 mM NaCl, and 10 mM β-ME and 3-fold molar excess α,β-GAR and 6-pyrrolo[2,3-d]-pyrimidine antifolate (dissolved in DMSO). The final protein concentration was 10 mg/mL. The protein–ligand solution was incubated at 4 °C for 2 h before setting up crystallization trays. Using 0.1 M Tris (pH 7.5), 0.333 mM NaCl, 18% polyethylene glycol (PEG) 3350, and 2% PEG 400 precipitant solution, sitting plates were set up with 1 μL of protein, 1 μL of precipitant and 0.2 μL of either 32 mM N-nonyl-β-D-thiomaltoside or 9 mM N-decyl-β-D-thiomaltoside (Hampton Research). Plates were incubated at 4 °C, and multifaceted cube and obelisk shaped crystals were obtained within a few days. Crystals were frozen in liquid nitrogen after being transferred through a cryoprotectant gradient that increased the PEG 400 concentration of the mother liquid to 14% via 2% increases. Cyroprotectant solutions also contained α,β-GAR and antifolate in the same concentrations as the crystal drop. These conditions were used for all GARFTase/ antifolate complex crystals.
Data collection was performed at the Lawrence Berkeley National Laboratory Advanced Light Source Beamline 4.2.2 using a CMOS detector. All data sets were processed to a P32 space group (HKL2000).63 Molecular replacement was performed using one chain of 4X72 with ligands and waters removed as a search model (PHENIX).64,65 Subsequent model building and refinement was performed using Coot and PHENIX.66,67
Supplementary Material
Acknowledgments
This work was supported in part by grants from the National Institutes of Health, CA125153 (A.G.), CA152316 (L.H.M. and A.G.), CA53535 (L.H.M.), CA166711 (A.G., L.H.M., and C.E.D.), and GM094472 (C.E.D.), and the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence (A.G.). M.R.W. and S.M.-R. were supported by T32 CA009531 (L.H.M.). S.M.-R. was also supported by F31 CA165853 (S.M.-R.). S.M.D. was supported by T32 GM007757. The Animal Model and Therapeutics Evaluation Core (AMTEC; L.P., K.W., J.K.) was supported, in part, by NIH Center grant P30 CA022453 to the Karmanos Cancer Institute at Wayne State University. We thank T. S. Widlanski for providing the α, β-GAR substrate for crystallography and enzyme inhibition experiments. All crystallization experiments were carried out in the Indiana University METACyt Crystallization Automation Facility. Crystallographic data were collected with remote assistance provided by Dr. Jay Nix on beamline 4.2.2 at the Advanced Light Source at the Lawrence Berkeley National Laboratory, part of the Molecular Biology Consortium supported by the US Department of Energy and Indiana University.
ABBREVIATIONS USED
- AICA
5-aminoimidazole-4-carboxamide
- AICARFTase
5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase
- CHO
Chinese hamster ovary
- FBS
fetal bovine serum
- DPBS
Dulbecco’s phosphate-buffered saline
- FR
folate receptor
- GAR
glycinamide ribonucleotide
- GARFTase
glycinamide ribonucleotide formyltransferase
- HBSS
Hank’s Balanced Salts Solution
- HBS
HEPES-buffered saline
- LCV
leucovorin
- β-ME
β-mercaptoethanol
- MTX
methotrexate
- MEM
minimal essential media
- Ni-NTA
nickel nitrilotriacetic acid
- PMX
pemetrexed
- PDX
pralatrexate
- PCFT
protoncoupled folate transporter
- PEG
polyethylene glycol
- RFC
reduced folate carrier
- SCID
severe combined immunodeficient
- TCA
trichloroacetic acid
- 10-CF3CO-DDACTHF
trifluoroacetyl-5,10-dideaza-acyclic-5,6,7,8-tetrahydrofolic acid
- TS
thymidylate synthase
Footnotes
Accession Codes
The coordinate and structure factor files for GARFTase complexes with 5, 7, and PMX are deposited in the Protein Data Bank with assigned codes 4ZZ1, 4ZZ2, and 4ZZ3.
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00801.
Molecular formula strings (CSV)
Elemental analysis, high-resolution mass spectra (HRMS) (ESI), crystallographic statistics, and ligand contacts PDF)
References
- 1.Gonen N, Assaraf YG. Antifolates in Cancer Therapy: Structure, Activity and Mechanisms of Drug Resistance. Drug Resist Updates. 2012;15:183–210. doi: 10.1016/j.drup.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Matherly LH, Wilson MR, Hou Z. The Major Facilitative Folate Transporters Solute Carrier 19A1 and Solute Carrier 46A1: Biology and Role in Antifolate Chemotherapy of Cancer. Drug Metab Dispos. 2014;42:632–649. doi: 10.1124/dmd.113.055723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elnakat H, Ratnam M. Distribution, Functionality and Gene Regulation of Folate Receptor Isoforms: Implications in Targeted Therapy. Adv Drug Delivery Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Zhao R, Matherly LH, Goldman ID. Membrane Transporters and Folate Homeostasis: Intestinal Absorption and Transport Into Systemic Compartments and Tissues. Expert Rev Mol Med. 2009;11:e4. doi: 10.1017/S1462399409000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desmoulin SK, Hou Z, Gangjee A, Matherly LH. The Human Proton-Coupled Folate Transporter: Biology and Therapeutic Applications to Cancer. Cancer Biol Ther. 2012;13:1355–1373. doi: 10.4161/cbt.22020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matherly LH, Hou Z, Deng Y. Human Reduced Folate Carrier: Translation of Basic Biology to Cancer Etiology and Therapy. Cancer Metastasis Rev. 2007;26:111–128. doi: 10.1007/s10555-007-9046-2. [DOI] [PubMed] [Google Scholar]
- 7.Chattopadhyay S, Moran RG, Goldman ID. Pemetrexed: Biochemical and Cellular Pharmacology, Mechanisms, and Clinical Applications. Mol Cancer Ther. 2007;6:404–417. doi: 10.1158/1535-7163.MCT-06-0343. [DOI] [PubMed] [Google Scholar]
- 8.Shih C, Thornton DE. Preclinical Pharmacology Studies and the Clinical Development of a Novel Multitargeted Antifolate, MTA (LY231514) In: Jackman AL, editor. Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press, Inc; Totowa, NJ: 1999. pp. 183–201. [Google Scholar]
- 9.Hazarika M, White RM, Jr, Booth BP, Wang YC, Ham DY, Liang CY, Rahman A, Gobburu JV, Li N, Sridhara R, Morse DE, Lostritto R, Garvey P, Johnson JR, Pazdur R. Pemetrexed in Malignant Pleural Mesothelioma. Clin Cancer Res. 2005;11:982–992. [PubMed] [Google Scholar]
- 10.Cohen MH, Justice R, Pazdur R. Approval Summary: Pemetrexed in the Initial Treatment of Advanced/Metastatic Non-Small Cell Lung Cancer. Oncologist. 2009;14:930–935. doi: 10.1634/theoncologist.2009-0092. [DOI] [PubMed] [Google Scholar]
- 11.Cohen MH, Cortazar P, Justice R, Pazdur R. Approval Summary: Pemetrexed Maintenance Therapy of Advanced/Metastatic Nonsquamous, Non-Small Cell Lung Cancer (NSCLC) Oncologist. 2010;15:1352–1358. doi: 10.1634/theoncologist.2010-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ettinger DS. Pemetrexed (Alimta): A New Antifolate for Non-Small-Cell Lung Cancer. Clin Lung Cancer. 2002;3(Suppl 1):S22–S25. [PubMed] [Google Scholar]
- 13.Ozasa H, Oguri T, Uemura T, Miyazaki M, Maeno K, Sato S, Ueda R. Significance of Thymidylate Synthase for Resistance to Pemetrexed in Lung Cancer. Cancer Sci. 2010;101:161–166. doi: 10.1111/j.1349-7006.2009.01358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by Cytotoxic Metabolite Accumulation: Pemetrexed Causes ZMP Accumulation, AMPK Activation, and Mammalian Target of Rapamycin Inhibition. Cancer Res. 2009;69:5467–5474. doi: 10.1158/0008-5472.CAN-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mendelsohn LG, Worzalla JF, Walling JM. Preclinical and Clinical Evaluation of the Glycinamide Ribonucleotide Formyltransferase Inhibitors Lometrexol and LY309887. In: Jackman AL, editor. Anticancer Drug Development Guide: Antifolate Drugs in Cancer Therapy. Humana Press, Inc; Totowa, NJ: 1999. pp. 261–280. [Google Scholar]
- 16.Boritzki TJ, Barlett CA, Zhang C, Howland EF, Margosiak SA, Palmer CL, Romines WH, Jackson RC. AG2034: A Novel Inhibitor of Glycinamide Ribonucleotide Formyltransferase. Invest New Drugs. 1996;14:295–303. doi: 10.1007/BF00194533. [DOI] [PubMed] [Google Scholar]
- 17.Bissett D, McLeod HL, Sheedy B, Collier M, Pithavala Y, Paradiso L, Pitsiladis M, Cassidy J. Phase I Dose-Escalation and Pharmacokinetic Study of a Novel Folate Analogue AG2034. British journal of cancer. 2001;84:308–312. doi: 10.1054/bjoc.2000.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Budman DR, Johnson R, Barile B, Bowsher RR, Vinciguerra V, Allen SL, Kolitz J, Ernest CS, 2nd, Kreis W, Zervos P, Walling J. Phase I and Pharmacokinetic Study of LY309887: A Specific Inhibitor of Purine Biosynthesis. Cancer Chemother Pharmacol. 2001;47:525–531. doi: 10.1007/s002800000272. [DOI] [PubMed] [Google Scholar]
- 19.Ray MS, Muggia FM, Leichman CG, Grunberg SM, Nelson RL, Dyke RW, Moran RG. Phase I Study of (6R)-5,10-dideazatetrahydrofolate: A Folate Antimetabolite Inhibitory to De Novo Purine Synthesis. J Natl Cancer Inst. 1993;85:1154–1159. doi: 10.1093/jnci/85.14.1154. [DOI] [PubMed] [Google Scholar]
- 20.Salazar MD, Ratnam M. The Folate Receptor: What Does It Promise In Tissue-Targeted Therapeutics? Cancer Metastasis Rev. 2007;26:141–152. doi: 10.1007/s10555-007-9048-0. [DOI] [PubMed] [Google Scholar]
- 21.Xia W, Low PS. Folate-targeted therapies for cancer. J Med Chem. 2010;53:6811–6824. doi: 10.1021/jm100509v. [DOI] [PubMed] [Google Scholar]
- 22.Reddy JA, Dorton R, Westrick E, Dawson A, Smith T, Xu LC, Vetzel M, Kleindl P, Vlahov IR, Leamon CP. Preclinical Evaluation of EC145, A Folate-Vinca Alkaloid Conjugate. Cancer Res. 2007;67:4434–4442. doi: 10.1158/0008-5472.CAN-07-0033. [DOI] [PubMed] [Google Scholar]
- 23.Gibbs DD, Theti DS, Wood N, Green M, Raynaud F, Valenti M, Forster MD, Mitchell F, Bavetsias V, Henderson E, Jackman AL. BGC 945, A Novel Tumor-Selective Thymidylate Synthase Inhibitor Targeted to Alpha-Folate Receptor-Overexpressing Tumors. Cancer Res. 2005;65:11721–11728. doi: 10.1158/0008-5472.CAN-05-2034. [DOI] [PubMed] [Google Scholar]
- 24.Deng Y, Wang Y, Cherian C, Hou Z, Buck SA, Matherly LH, Gangjee A. Synthesis and Discovery of High Affinity Folate Receptor-Specific Glycinamide Ribonucleotide Formyltransferase Inhibitors With Antitumor Activity. J Med Chem. 2008;51:5052–5063. doi: 10.1021/jm8003366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kugel Desmoulin S, Wang Y, Wu J, Stout M, Hou Z, Fulterer A, Chang MH, Romero MF, Cherian C, Gangjee A, Matherly LH. Targeting the Proton-Coupled Folate Transporter for Selective Delivery Of 6-Substituted Pyrrolo[2,3-d]pyrimidine Antifolate Inhibitors of De Novo Purine Biosynthesis in the Chemotherapy of Solid Tumors. Mol Pharmacol. 2010;78:577–587. doi: 10.1124/mol.110.065896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang L, Cherian C, Kugel Desmoulin SK, Polin L, Deng Y, Wu J, Hou Z, White K, Kushner J, Matherly LH, Gangjee A. Synthesis and Antitumor Activity of a Novel Series of 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Antifolate Inhibitors of Purine Biosynthesis with Selectivity For High Affinity Folate Receptors and the Proton-Coupled Folate Transporter Over the Reduced Folate Carrier for Cellular Entry. J Med Chem. 2010;53:1306–1318. doi: 10.1021/jm9015729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang L, Kugel Desmoulin S, Cherian C, Polin L, White K, Kushner J, Fulterer A, Chang MH, Mitchell-Ryan S, Stout M, Romero MF, Hou Z, Matherly LH, Gangjee A. Synthesis, Biological, and Antitumor Activity of a Highly Potent 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Antifolate Inhibitor with Proton-Coupled Folate Transporter and Folate Receptor Selectivity Over the Reduced Folate Carrier that Inhibits Beta-Glycinamide Ribonucleotide Formyltransferase. J Med Chem. 2011;54:7150–7164. doi: 10.1021/jm200739e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang L, Cherian C, Kugel Desmoulin S, Mitchell-Ryan S, Hou Z, Matherly LH, Gangjee A. Synthesis and Biological Activity of 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Regioisomers as Inhibitors of De Novo Purine Biosynthesis with Selectivity for Cellular Uptake by High Affinity Folate Receptors and the Proton-Coupled Folate Transporter Over the Reduced Folate Carrier. J Med Chem. 2012;55:1758–1770. doi: 10.1021/jm201688n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang Y, Cherian C, Orr S, Mitchell-Ryan S, Hou Z, Raghavan S, Matherly LH, Gangjee A. Tumor-Targeting with Novel Non-Benzoyl 6-Substituted Straight Chain Pyrrolo[2,3-d]-pyrimidine Antifolates via Cellular Uptake by Folate Receptor Alpha and Inhibition of De Novo Purine Nucleotide Biosynthesis. J Med Chem. 2013;56:8684–8695. doi: 10.1021/jm401139z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J, Yong EL, Xu HE, Melcher K. Structural Basis for Molecular Recognition of Folic Acid by Folate Receptors. Nature. 2013;500:486–489. doi: 10.1038/nature12327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wibowo AS, Singh M, Reeder KM, Carter JJ, Kovach AR, Meng W, Ratnam M, Zhang F, Dann CE., 3rd Structures of Human Folate Receptors Reveal Biological Trafficking States and Diversity in Folate and Antifolate Recognition. Proc Natl Acad Sci U S A. 2013;110:15180–15288. doi: 10.1073/pnas.1308827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Desharnais J, Marsilje TH, Li C, Hedrick MP, Gooljarsingh LT, Tavassoli A, Benkovic SJ, Olson AJ, Boger DL, Wilson IA. Rational Design, Synthesis, Evaluation, and Crystal Structure of a Potent Inhibitor of Human GAR Tfase: 10-(trifluoroacetyl)-5,10-dideazaacyclic-5,6,7,8-tetrahydrofolic acid. Biochemistry. 2003;42:6043–6056. doi: 10.1021/bi034219c. [DOI] [PubMed] [Google Scholar]
- 33.LeadIT 216. BioSolveIT GmbH; Sankt Augustin, Germany: 2014. [Google Scholar]
- 34.Golani LK, George C, Zhao S, Raghavan S, Orr S, Wallace A, Wilson MR, Hou Z, Matherly LH, Gangjee A. Structure-Activity Profiles of Novel 6-Substituted Pyrrolo[2,3-d]-pyrimidine Thienoyl Antifolates with Modified Amino Acids for Cellular Uptake by Folate Receptors Alpha and Beta and the Proton-Coupled Folate Transporter. J Med Chem. 2014;57:8152–8166. doi: 10.1021/jm501113m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Secrist JAL, Liu PS. Studies Directed Toward a Total Synthesis of Nucleoside Q. Annulation of 2,6-diaminopyrimidin-4-one with.alpha.-halo Carbonyls to Form Pyrrolo[2,3-d]pyrimidines and Furo[2,3-d]pyrimidines. J Org Chem. 1978;43:3937–3941. [Google Scholar]
- 36.Wong SC, Proefke SA, Bhushan A, Matherly LH. Isolation of Human cDNAs That Restore Methotrexate Sensitivity and Reduced Folate Carrier Activity in Methotrexate Transport-Defective Chinese Hamster Ovary Cells. J Biol Chem. 1995;270:17468–17475. doi: 10.1074/jbc.270.29.17468. [DOI] [PubMed] [Google Scholar]
- 37.Deng Y, Zhou X, Kugel Desmoulin S, Wu J, Cherian C, Hou Z, Matherly LH, Gangjee A. Synthesis and Biological Activity of a Novel Series of 6-Substituted Thieno[2,3-d]pyrimidine Antifolate Inhibitors of Purine Biosynthesis with Selectivity for High Affinity Folate Receptors Over the Reduced Folate Carrier and Proton-Coupled Folate Transporter for Cellular Entry. J Med Chem. 2009;52:2940–2951. doi: 10.1021/jm8011323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flintoff WF, Davidson SV, Siminovitch L. Isolation and Partial Characterization of Three Methotrexate-Resistant Phenotypes from Chinese Hamster Ovary Cells. Somatic Cell Genet. 1976;2:245–261. doi: 10.1007/BF01538963. [DOI] [PubMed] [Google Scholar]
- 39.Kugel Desmoulin S, Wang L, Hales E, Polin L, White K, Kushner J, Stout M, Hou Z, Cherian C, Gangjee A, Matherly LH. Therapeutic Targeting of a Novel 6-Substituted Pyrrolo [2,3-d]pyrimidine Thienoyl Antifolate to Human Solid Tumors Based on Selective Uptake by the Proton-Coupled Folate Transporter. Mol Pharmacol. 2011;80:1096–1107. doi: 10.1124/mol.111.073833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao R, Qiu A, Tsai E, Jansen M, Akabas MH, Goldman ID. The Proton-Coupled Folate Transporter: Impact on Pemetrexed Transport and on Antifolates Activities Compared with the Reduced Folate Carrier. Mol Pharmacol. 2008;74:854–862. doi: 10.1124/mol.108.045443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the Tumor Microenvironment. J Magn Reson Imaging. 2002;16:430–450. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- 42.Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat Rev Cancer. 2011;11:671–677. doi: 10.1038/nrc3110. [DOI] [PubMed] [Google Scholar]
- 43.Elnakat H, Gonit M, D’Alincourt Salazar M, Zhang J, Basrur V, Gunning W, Kamen B, Ratnam M. Regulation of Folate Receptor Internalization by Protein Kinase C Alpha. Biochemistry. 2009;48:8249–8260. doi: 10.1021/bi900565t. [DOI] [PubMed] [Google Scholar]
- 44.Caperelli CA, Giroux EL. The Human Glycinamide Ribonucleotide Transformylase Domain: Purification, Characterization, and Kinetic Mechanism. Arch Biochem Biophys. 1997;341:98–103. doi: 10.1006/abbi.1997.9947. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell-Ryan S, Wang Y, Raghavan S, Ravindra MP, Hales E, Orr S, Cherian C, Hou Z, Matherly LH, Gangjee A. Discovery of 5-Substituted Pyrrolo[2,3-d]pyrimidine Antifolates as Dual-Acting Inhibitors of Glycinamide Ribonucleotide Formyltransferase and 5-Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase in De Novo Purine Nucleotide Biosynthesis: Implications of Inhibiting 5-Aminoimidazole-4-carboxamide Ribonucleotide Formyltransferase to ampk Activation and Antitumor Activity. J Med Chem. 2013;56:10016–10032. doi: 10.1021/jm401328u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alati T, Worzalla JF, Shih C, Bewley JR, Lewis S, Moran RG, Grindey GB. Augmentation of the Therapeutic Activity of Lometrexol-(6-R)5,10-dideazatetrahydrofolate-by Oral Folic Acid. Cancer Res. 1996;56:2331–2335. [PubMed] [Google Scholar]
- 47.Kugel Desmoulin S, Wang L, Polin L, White K, Kushner J, Stout M, Hou Z, Cherian C, Gangjee A, Matherly LH. Functional Loss of the Reduced Folate Carrier Enhances the Antitumor Activities of Novel Antifolates with Selective Uptake by the Proton-coupled Folate Transporter. Mol Pharmacol. 2012;82:591–600. doi: 10.1124/mol.112.079004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cherian C, Kugel Desmoulin S, Wang L, Polin L, White K, Kushner J, Stout M, Hou Z, Gangjee A, Matherly LH. Therapeutic Targeting Malignant Mesothelioma with a Novel 6-Substituted Pyrrolo[2,3-d]pyrimidine Thienoyl Antifolate via Its Selective Uptake by the Proton-Coupled Folate Transporter. Cancer Chemother Pharmacol. 2013;71:999–1011. doi: 10.1007/s00280-013-2094-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gangjee A, Jain HD, Kurup S. Recent Advances in Classical and Non-Classical Antifolates as Antitumor and antiopportunistic infection Agents: Part I. Anti-Cancer Agents Med Chem. 2007;7:524–542. doi: 10.2174/187152007781668724. [DOI] [PubMed] [Google Scholar]
- 50.Gangjee A, Jain HD, Kurup S. Recent Advances in Classical and Non-Classical Antifolates as Antitumor and Antiopportunistic Infection Agents: Part II. Anti-Cancer Agents Med Chem. 2008;8:205–231. doi: 10.2174/187152008783497064. [DOI] [PubMed] [Google Scholar]
- 51.Pizzorno G, Moroson BA, Cashmore AR, Beardsley GP. (6R)-5,10-Dideaza-5,6,7,8-tetrahydrofolic Acid Effects on Nucleotide Metabolism in CCRF-CEM Human T-lymphoblast Leukemia Cells. Cancer Res. 1991;51:2291–2295. [PubMed] [Google Scholar]
- 52.Molecular Operating Environment (MOE) Chemical Computing Group Inc; 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2013. [Google Scholar]
- 53.McNicholas S, Potterton E, Wilson KS, Noble MEM. Presenting Your Structures: The CCP4mg Molecular-Graphics Software. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.MOE 2013.08 Scientific Vector Language (SVL) source code. Chemical Computing Group Inc; 1010 Sherbooke St., West, Suite #910, Montreal, QC, Canada, H3A 2R7: 2015. [Google Scholar]
- 55.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein Measurement with the Folin Phenol Reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 56.Friedkin M, Roberts D. Conversion of Uracil Deoxyriboside to Thymidine of Deoxyribonucleic Acid. J Biol Chem. 1956;220:653–660. [PubMed] [Google Scholar]
- 57.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, 3rd, Smith HO. Enzymatic Assembly of DNA Molecules Up to Several Hundred Kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 58.Corbett TH, Valeriote FA, Demchik L, Lowichik N, Polin L, Panchapor C, Pugh S, White K, Kushner J, Rake J, Wentland M, Golakoti T, Hetzel C, Ogino J, Patterson G, Moore R. Discovery of Cryptophycin-1 and BCN-183577: Examples of Strategies and Problems in the Detection of Antitumor Activity in Mice. Invest New Drugs. 1997;15:207–218. doi: 10.1023/a:1005875015011. [DOI] [PubMed] [Google Scholar]
- 59.Padmanabhan N, Fichtner L, Dickmanns A, Ficner R, Schulz JB, Braus GH. The Yeast HtrA Orthologue Ynm3 is a Protease with Chaperone Activity that Aids Survival Under Heat Stress. Mol Biol Cell. 2009;20:68–77. doi: 10.1091/mbc.E08-02-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Polin LCT, Roberts BJ, Lawson AJ, Leopold WR, III, White K, Kusner J, Paluch J, Hazeldine S, Moore R, Rake J, Horwitz JP. Tumor Models in Cancer Research. Humana Press, Inc; Totowa, NJ: 2011. Transplantable Syngeneic Tumors: Solid Tumors of Mice; pp. 43–78. [Google Scholar]
- 61.Polin L, Valeriote F, White K, Panchapor C, Pugh S, Knight J, LoRusso P, Hussain M, Liversidge E, Peltier N, Golakoti T, Patterson G, Moore R, Corbett TH. Treatment of Human Prostate Tumors PC-3 and TSU-PR1 with Standard and Investigational Agents in SCID Mice. Invest New Drugs. 1997;15:99–108. doi: 10.1023/a:1005856605726. [DOI] [PubMed] [Google Scholar]
- 62.Varela-Moreiras G, Selhub J. Long-Term Folate Deficiency Alters Folate Content and Distribution Differentially in Rat Tissues. J Nutr. 1992;122:986–991. doi: 10.1093/jn/122.4.986. [DOI] [PubMed] [Google Scholar]
- 63.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 64.Adams PD, Gopal K, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Pai RK, Read RJ, Romo TD, Sacchettini JC, Sauter NK, Storoni LC, Terwilliger TC. Recent Developments in the PHENIX Software for Automated Crystallographic Structure Determination. J Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- 65.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: Building New Software for Automated Crystallographic Structure Determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 66.Emsley P, Cowtan K. Coot: Model-Building Tools for Molecular Graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 67.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative Model Building, Structure Refinement and Density Modification with the PHENIX AutoBuild Wizard. Acta Crystallogr, Sect D: Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.