

Abstract
Metabolic syndrome is a group of obesity-related metabolic abnormalities that increase an individual’s risk of developing type 2 diabetes and cardiovascular disease. Here, we show that mice genetically deficient in Toll-like receptor 5 (TLR5), a component of the innate immune system that is expressed in the gut mucosa and that helps defend against infection, exhibit hyperphagia and develop hallmark features of metabolic syndrome, including hyperlipidemia, hypertension, insulin resistance, and increased adiposity. These metabolic changes correlated with changes in the composition of the gut microbiota, and transfer of the gut microbiota from TLR5-deficient mice to wild-type germ-free mice conferred many features of metabolic syndrome to the recipients. Food restriction prevented obesity, but not insulin resistance, in the TLR5-deficient mice. These results support the emerging view that the gut microbiota contributes to metabolic disease and suggest that malfunction of the innate immune system may promote the development of metabolic syndrome.
Humanity is facing an epidemic of interrelated metabolic diseases collectively referred to as metabolic syndrome, the hallmarks of which include hyperglycemia, hyperlipidemia, insulin resistance, obesity, and hepatic steatosis (1). The increasing incidence of metabolic syndrome is widely thought to result from nutrient excess due to increased food consumption and/or reduced levels of physical activity. Such nutrient excess results in obesity and may activate endoplasmic reticulum stress pathways resulting in chronic activation of proinflammatory kinase cascades that desensitize the metabolic response to insulin (2). Such insulin resistance can result in hyperglycemia and, in some cases, type 2 diabetes. Recent work suggests a possible role for the gut microbiota in obesity (3) and, consequently, other aspects of metabolic syndrome. In both humans and mice, the development of obesity correlates with shifts in the relative abundance of the two dominant bacterial phyla in the gut, the Bacteroidetes and the Firmicutes (4–6). In addition, it has been shown that transfer of the gut microbiota from obese (ob/ob) mice to germ-free wild-type (WT) recipients leads to an increase in fat mass in the recipients, leading to speculation that the gut microbiota promotes obesity by increasing the capacity of the host to extract energy (calories) from ingested food (7).
The gut microbiota is shaped by both environment and host genetics, with the innate immune system in particular, long appreciated for its role in defending against infection by pathogenic microbes, now suggested to play a key role in regulating the gut microbiota (8). Thus, in addition to its role in infection/inflammation, innate immunity may play a key role in promoting metabolic health. Toll-like receptor (TLR) 5 is a transmembrane protein that is highly expressed in the intestinal mucosa and that recognizes bacterial flagellin. In previous work with mice genetically deficient in TLR5 (T5KO mice), we found that 10% of the mutant mice exhibited severe colitis and an additional 30% exhibited gross and/or histopathologic evidence of colitis (9). The remaining 60% of the T5KO mice exhibited broadly elevated proinflammatory gene expression but lacked the histopathologic features that define colitis; however, we observed that, by 4 weeks of age, these mice had body masses that were on average 15% higher than those of their WT littermates. To eliminate potential opportunistic pathogens that may have been present in T5KO and WT littermates, and to make their gut microbiota similar to that of mice from the Jackson Laboratory (the world’s largest supplier of research mice), we “rederived” T5KO mice by transplanting embryos into mice purchased from this supplier (10). Such standardization of the microbiota in the T5KO mice greatly attenuated the severity of their colitis and resulted in a more uniform phenotype characterized by mild inflammation (fig. S1) and obesity (Fig. 1).
Fig. 1.
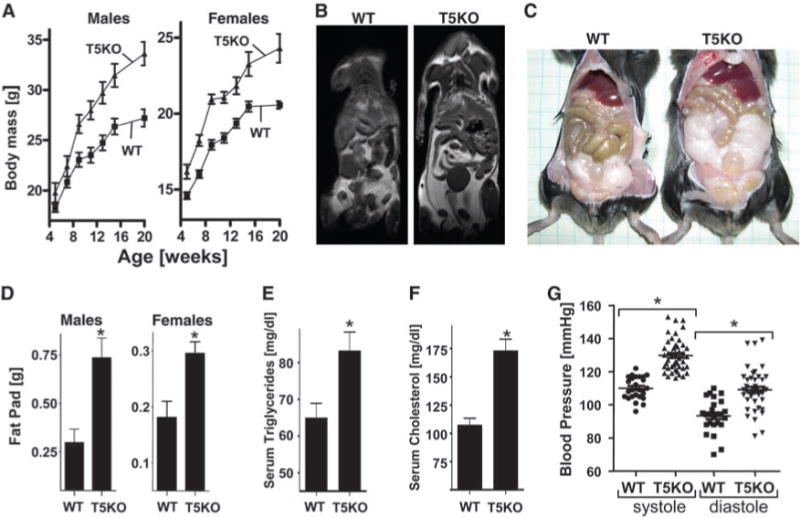
T5KO mice develop obesity. T5KO mice and WT littermates were monitored for 20 weeks. (A) Body mass. (B) MRI image showing fat distribution. (C) Abdominal photograph of representative 20-week-old male mice. (D) Fat-pad mass. (E) Serum triglycerides. (F) Serum cholesterol. (G) Blood pressure. Results in (A) to (C) are from an individual experiment (n = 20, 10 males and 10 females) and representative of more than six distinct groups of mice. Results in (B) are representative of three separate analyses performed on mice that had median body mass of their litters. Results in (C) to (E) are from a single experiment (n = 6) and representative of several experiments. Results in (G) compare groups (n = 4) of mice with 5 to 10 independent measurements per mouse. *, P < 0.05.
Analysis of these rederived mice showed that, at 20 weeks of age, both male and female T5KO mice had body masses that were 20% greater than those of WT mice (Fig. 1A). Magnetic resonance imaging (MRI) revealed increased fat mass throughout the body of the T5KO mice, with a particular increase in visceral fat (Fig. 1B). T5KO mice had epididymal fat pads that were about twice as large as those in WT littermates at 20 weeks of age (Fig. 1, C and D). This increase in fat mass correlated with a substantial increase in serum levels of triglycerides and cholesterol and with an increase in blood pressure (Fig. 1, E to G), features often associated with metabolic syndrome. Ex vivo analysis revealed higher production of proinflammatory cytokines interferon-γ (IFN-γ) and interleukin-1β(IL-1β) in T5KO adipose tissue versus WT adipose tissue (fig. S2), which suggests that increased adiposity may play a role in perpetuating the low-grade chronic inflammation exhibited by the mutant mice.
We next examined blood glucose levels of T5KO and WT littermates. After an overnight (15-hour) fast, T5KO mice exhibited mild but statistically significant elevations in blood glucose relative to WT littermates (Fig. 2A). Consistent with mild loss of glycemic control, when administered a bolus of glucose, T5KO mice showed impaired ability to restore blood glucose to baseline levels (Fig. 2B). In contrast to the modest effects of T5KO deficiency on glucose homeostasis, basal insulin levels were substantially elevated in T5KO mice (Fig. 2C), as was the amount of insulin produced in response to glucose challenge (Fig. 2D). The T5KO mice also exhibited a reduced response to exogenous insulin relative to WT mice (Fig. 2E), all features consistent with a state of insulin resistance. Accordingly, T5KO mice showed elevated serum levels of the adipokine lipocalin-2 (fig. S1E), which promotes insulin resistance (11), and exhibited an increase in the number and size of pancreatic islets that immunostained positive for insulin (Fig. 2F and fig. S3). These data suggest that T5KO mice have mild loss of glycemic control that is likely driven by insulin resistance and partially compensated for by increased insulin production—conditions typically seen in humans with metabolic syndrome.
Fig. 2.
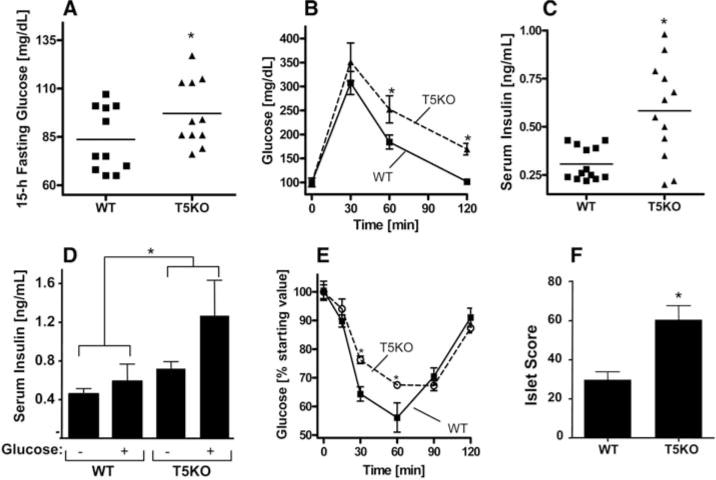
T5KO exhibit hyperglycemia/insulin resistance. Twenty-week-old T5KO and WT littermate mice were assayed for various parameters of glucose homeostasis. (A) Fifteen-hour fasting blood glucose. (B) Glucose tolerance test. Mice were intraperitoneally injected with glucose (2 g per kg of body mass). (C) Serum insulin after a 5-hour fast. (D) Serum insulin levels before and 2.5 min after challenge with glucose (3 g/kg body mass). (E) Insulin sensitivity. Mice were fasted for 5 hours and intraperitoneally injected with insulin (1.0 U/kg body mass). (F) Hematoxylin and eosin (H&E) stained pancreata were blindly scored for size and number of pancreatic islets. Results in (A), (C), and (F) are from an individual experiment (n = 12 to 13) and representative of more than six distinct groups of mice. Results in (B), (D), and (E) are from a single experiment (n = 6) and representative of several experiments. *, P < 0.05.
Development of metabolic syndrome in humans is thought to be promoted by a diet high in saturated fats. To investigate the effect of such a diet on metabolic syndrome in T5KO mice, we fed the mice a high-fat diet for 8 weeks. As expected, both WT and T5KO mice on this diet showed significant increases in body mass and fat-pad mass (fig. S4, A and B). Mice of both genotypes also had increased serum levels of triglycerides, cholesterol, leptin, and insulin (fig. S4, C to F). In contrast to WT mice, most T5KO mice on a high-fat diet had fasting glucose concentrations >120 mg/dL, indicating that they had become diabetic (Fig. 3A). High-fat-fed T5KO mice also exhibited inflammatory infiltrates in the pancreatic islets (Fig. 3B) and displayed hepatic steatosis, which is a severe manifestation of metabolic syndrome (Fig. 3B). Thus, the metabolic syndrome exhibited by T5KO mice was exacerbated by a high-fat diet.
Fig. 3.
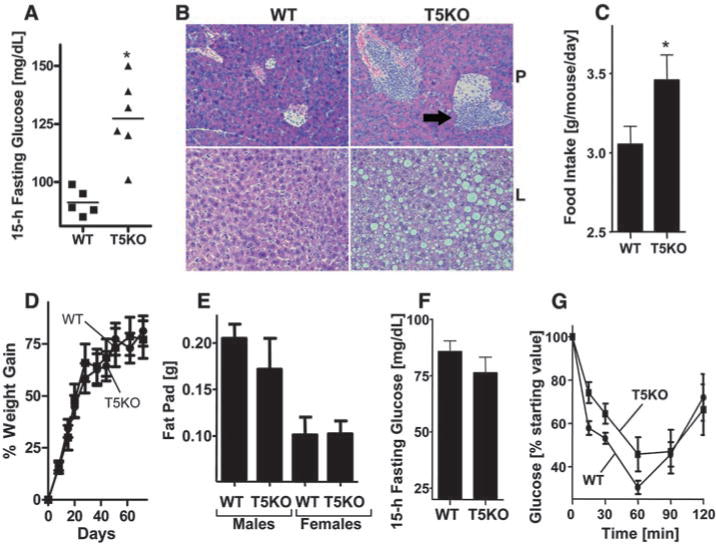
T5KO metabolic syndrome depends on hyperphagia. (A and B) Four-week-old WT and T5KO mice were given a high-fat diet for 8 weeks. (A) Fifteen-hour fasting blood glucose. (B) H&E stained pancreas (P, upper panel) and liver (L, lower panel). Arrows point to inflammatory infiltrates. (C) Mice on a regular diet were monitored for food intake. (D to G) Mice were subjected to food restriction and assayed for (D) body mass and (E) fat-pad mass. (F) Fifteen-hour fasting glucose. (G) Insulin sensitivity measured as described in Fig. 2E. Results in (A) and (B) are from a single experiment (n = 5 to 6) and representative of two independent experiments. Results in (C) are from a single experiment (n = 6) and representative of similar results with several groups of mice. (D) to (G) is from a single experiment (n = 10) and representative of two independent experiments. *, P < 0.05.
We next investigated whether changes in food intake contributed to the development of metabolic syndrome in T5KO mice. We found that T5KO mice consumed about 10% more food than WT littermates (Fig. 3C) and, concomitantly, had greater stool output (fig. S5). Bomb calorimetry and short-chain fatty acid analysis of T5KO and WT feces indicated that loss of TLR5 did not significantly impact the efficiency of dietary energy harvest (fig. S6). We examined serum levels of major orexigenic (ghrelin and neuropeptide Y) and anorexic (leptin and GLP-1) hormones, but these experiments did not provide definitive mechanistic insight into the hyperphagic phenotype of the T5KO mice (fig. S7). To explore the role of hyperphagia in the metabolic abnormalities exhibited by T5KO mice, we performed food restriction experiments. Beginning at 4 weeks of age, groups of WT and T5KO mice were no longer given unlimited access to food but rather were given only the amount of food consumed by a control group of ad libitum–fed WT mice. Twelve weeks of this food restriction regimen prevented many of the metabolic abnormalities normally seen in T5KO mice, including increased body mass and fat-pad mass and increased serum levels of glucose, lipids, and insulin (Fig. 3, D to F, and fig. S8). However, such lean T5KO mice still exhibited a decreased response to exogenous insulin (Fig. 3G), which suggests that their insulin resistance is not entirely dependent on increased food consumption or adiposity. Given that insulin suppresses food intake (12), these results raise the possibility that the low-grade proinflammatory signaling in T5KO mice (9) attenuates insulin signaling, resulting in increased food consumption that drives other manifestations of metabolic syndrome.
In light of observations that the lipopolysaccharide (LPS) receptor TLR4 promotes diet-induced metabolic syndrome (13, 14) and that TLR4 is required for T5KO colitis (9), we hypothesized that TLR4 might mediate the metabolic syndrome exhibited by T5KO mice. However, loss of TLR5 in mice already deficient in either TLR2 or TLR4 still produced features of metabolic syndrome (figs. S9 and S10), arguing against this possibility. Mice lacking MyD88 did not mimic the metabolic syndrome exhibited by T5KO mice (fig. S11), suggesting a potential role for another TLR [besides TLR2 or TLR4 (all TLRs except TLR3 use MyD88)] and/or a role for cytokines such as IL-1β and IL-18 whose levels are increased in noncolitic T5KO mice (9) and whose receptors signal in a MyD88-dependent manner. We also observed that loss of TLR5 in RAG1-deficient mice, which lack T and B lymphocytes, still resulted in impaired glucose regulation (fig. S12), which suggests that development of T5KO metabolic syndrome does not require adaptive immunity.
We next considered the hypothesis that alterations in the gut microbiota resulting from loss of TLR5 promote the development of metabolic syndrome in these mice. To investigate this possibility, we placed T5KO mice on broad-spectrum antibiotics upon weaning for a period of 12 weeks. This treatment lowered total gut bacterial load by 90% and caused the enlargement of the ceca, as previously observed in germ-free mice (fig. S13). Analogous to observations made with mice exhibiting high-fat-diet–induced metabolic syndrome (13), we observed that decimation of the gut microbiota corrected T5KO metabolic syndrome relative to similarly treated WT mice (Fig. 4, A to C, and fig. S14). We next defined the extent to which loss of TLR5 altered the composition of the microbiota by pyrosequencing the 16S ribosomal RNA (rRNA) genes in the ceca. We generated a total of 22,712 partial 16S rRNA gene sequences (15). The relative abundance of bacterial phyla in the samples was similar to that reported in other studies (Firmicutes, 54%; Bacteroidetes, 39.8%; Proteobacteria, 1.1%; Tenericutes, Actinobacteria, TM7, and Verrucomicrobia, <0.2% of the sequences). In contrast to what has been seen with ob/ob mice, we found that the gut bacterial communities of T5KO and WT mice had similar relative abundances of Firmicutes and Bacteroidetes. However, UniFrac analysis, which compares bacterial communities based on the premise that similar communities are those that share a greater fraction of the overall phylogenetic tree (16), indicated that the gut microbiotas of T5KO and WT littermate mice were significantly different in their species composition (fig. S15). In addition, despite the marked interindividual differences in species diversity typical of mice (and humans), we observed 116 bacterial phylotypes from various phyla to be consistently enriched or reduced in T5KO mice relative to WT mice (figs. S16 and S17, and tables S1 and S2). Thus, in contrast to the ob/ob mouse model of obesity, where the alteration of the microbiota was characterized by a phylum-level shift without amplification or loss of particular species, here we have identified a specific suite of bacterial species whose abundance is altered by loss of TLR5. These phylotypes are typical of murine gut bacteria in general and related to a variety of gut bacteria identified by culture-independent analysis of human gut microbiota (table S3).
Fig. 4.
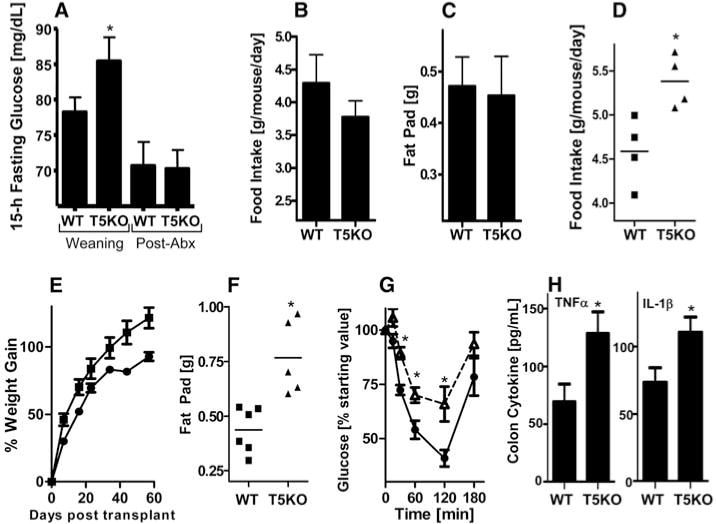
T5KO gut microbiota is necessary and sufficient to transfer metabolic syndrome phenotype to germ-free mice. (A to C) Four-week-old T5KO mice and WT littermates were placed on broad-spectrum antibiotics and monitored for 12 weeks. (A) Fifteen-hour fasting glucose. (B) Food intake. (C) Fat-pad mass. (D to H) Four-week-old WT germ-free mice were intragastrically administered cecal content from WT or T5KO mice and monitored for 7 weeks. (D) Food intake expressed as average consumption per mouse per day 10 to 13 days after transplant. (E) Body mass. (F) Fat-pad mass. (G) Insulin sensitivity. (H) Colonic tumor necrosis factor–α(TNF-α) and IL-1β. Results in (A) to (C) are from a single experiment (n = 5 to 6) and representative of two independent experiments (other shown in fig. S14). Results in (D) to (H) are from a single experiment (n = 5 to 6). *, P < 0.05.
The inability to culture most gut bacteria makes assessment of their causal role in health and disease technically challenging. To investigate whether the changes in the gut microbiota were a cause or consequence of the metabolic syndrome in T5KO mice, we transplanted the T5KO microbiota into WT germ-free mice, which, like newborn mice, can be colonized by a diverse microbiota in a manner that preserves the complex composition of the transferred organisms (17). In principle, any gut microbial dysbiosis present in the host would be transferred to the recipient (7, 18). We found that the transplanted T5KO microbiota conferred many aspects of the T5KO phenotype to the WT germ-free hosts, including hyperphagia, obesity, hyperglycemia, insulin resistance, colomegaly, and elevated levels of proinflammatory cytokines (Fig. 4, D to H, and fig. S18). This suggests that the changes in the gut microbiota observed in the T5KO mice are likely to be a contributing factor in the development of metabolic syndrome in the mice.
In summary, we have shown that loss of TLR5 results in a phenotype reminiscent of human metabolic syndrome. The underlying molecular mechanisms remain to be defined, but we speculate that loss of TLR5 produces alterations in the gut microbiota that induce low-grade inflammatory signaling. This signaling may in turn cross-desensitize insulin receptor signaling, leading to hyperphagia, which then drives other aspects of metabolic syndrome (fig. S19). Our results suggest that the specific composition of microbiota to which individuals are first exposed may be an important means by which early environment exerts a lasting influence on metabolic phenotype. They also suggest that the excess caloric consumption driving the current epidemic of metabolic syndrome may be caused, at least in part, by alterations in host-microbiota interactions.
Supplementary Material
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/science.1179721/DC1
Materials and Methods
References
References and Notes
- 1.Wang Y, Beydoun MA, Liang L, Caballero B, Kumanyika SK. Obesity. 2008;16:2323. doi: 10.1038/oby.2008.351. [DOI] [PubMed] [Google Scholar]
- 2.Hotamisligil GS, Erbay E. Nat Rev Immunol. 2008;8:923. doi: 10.1038/nri2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bäckhed F, et al. Proc Natl Acad Sci USA. 2004;101:15718. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley RE, et al. Proc Natl Acad Sci USA. 2005;102:11070. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Nature. 2006;444:1022. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 6.Turnbaugh PJ, et al. Nature. 2009;457:480. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnbaugh PJ, et al. Nature. 2006;444:1027. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 8.Slack E, et al. Science. 2009;325:617. doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vijay-Kumar M, et al. J Clin Invest. 2007;117:3909. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Materials and methods are available as supporting material on Science Online.
- 11.Yan QW, et al. Diabetes. 2007;56:2533. doi: 10.2337/db07-0007. [DOI] [PubMed] [Google Scholar]
- 12.Woods SC, Lotter EC, McKay LD, Porte D., Jr Nature. 1979;282:503. doi: 10.1038/282503a0. [DOI] [PubMed] [Google Scholar]
- 13.Cani PD, et al. Diabetes. 2008;57:1470. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 14.Shi H, et al. J Clin Invest. 2006;116:3015. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The GenBank accession number for the 16S pyrosequencing data set is SRA009446.
- 16.Lozupone C, Knight R. Appl Environ Microbiol. 2005;71:8228. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turnbaugh PJ, et al. Sci Transl Med. 2009;1:6ra14. doi: 10.1126/scitranslmed.3000322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garrett WS, et al. Cell. 2007;131:33. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.We thank F. A. Anania, D. G. Harrison, and I. R. Williams for valuable discussions and comments, and N. Scalfone and M. Hamady for technical assistance. This work was supported by an NIH grant (DK061417 and an associated American Recovery and Reinvestment Act supplement) and a Senior Award from the Crohn’s and Colitis Foundation of America (CCFA) to A.T.G. M.V.-K. is a recipient of Career Development Awards from CCFA and K01 (DK083275) from NIH. This research used a Digestive Disease Research and Development Center (DDRDC) core facility that is supported by NIH grant DK06439.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.