

Abstract
Here we describe a method capable of identifying interactions between candidate trafficking proteins and a defined vesicle population in intact cells. The assay involves the expression of an FKBP12-rapamycin–binding domain (FRB)–tagged candidate vesicle-binding protein that can be inducibly linked to an FKBP-tagged molecular motor. If the FRB-tagged candidate protein binds the labeled vesicles, then linking the FRB and FKBP domains recruits motors to the vesicles and causes a predictable, highly distinctive change in vesicle trafficking. We describe two versions of the assay: a general protocol for use in cells with a typical microtubule-organizing center and a specialized protocol designed to detect protein-vesicle interactions in cultured neurons. We have successfully used this assay to identify kinesins and Rabs that bind to a variety of different vesicle populations. In principle, this assay could be used to investigate interactions between any category of vesicle trafficking proteins and any vesicle population that can be specifically labeled.
Keywords: Kinesin, vesicle, Rab, FKBP, FRB, membrane trafficking
INTRODUCTION
This unit describes an assay we developed to identify the trafficking proteins that bind to specific vesicle populations in intact cells. By governing the selectivity of vesicle budding, vesicle transport, and vesicle fusion, proteins on the cytosolic surface of intracellular organelles play an essential role in regulating membrane trafficking and in maintaining the organizational integrity of the endomembrane system. Examples of such trafficking proteins include clathrin adaptors, which regulate vesicle budding, Rab GTPases, which interact with and recruit other trafficking proteins, molecular motors, which provide the locomotive force for vesicle transport, and SNAREs, which mediate membrane fusion with the target compartment. Each of these families of trafficking proteins contains dozens of members, which fulfill similar functions on different vesicle populations (Vale, 2003; Spang, 2008; Wickner and Schekman, 2008). Identifying the particular complement of proteins associated with a given vesicle population is a necessary first step in understanding how its trafficking is regulated. Because of the large number of vesicle populations present in any cell, and the large size of the relevant protein families, this presents a challenging problem.
Several methods have been used to approach this problem. Yeast two-hybrid and immunoprecipitation strategies have identified many putative binding partners for trafficking proteins, but they cannot provide information about protein–vesicle interactions in vivo, which may be transient and highly regulated. Once candidate proteins have been identified, another strategy is needed to confirm their presence on vesicles in intact cells. Dominant-negative and RNAi approaches can disrupt the function of trafficking proteins, but require long expression times, which can lead to nonspecific secondary effects. Moreover, if vesicles carry multiple proteins that perform the same function, knockdown experiments require simultaneous targeting of multiple proteins, which is cumbersome. Another approach utilizes fluorescent colocalization, but such studies can be challenging to evaluate due to the density of intracellular organelles, differences in expression levels or staining efficiency of different proteins, and limitations in microscope resolution. Two-color live-cell imaging and immuno-EM are powerful alternative approaches, but these can be tedious to implement. Even when all of these strategies are applied in combination, it can be difficult to obtain a comprehensive picture of the trafficking proteins that associate with a given vesicle population and conflicting results are not unusual.
To definitively address such questions, we developed an assay that transduces the binding of a candidate protein into a distinctive change in vesicle position or vesicle dynamics (Jenkins et al., 2012; Bentley et al., 2015). As illustrated in Figure 1, the assay relies on three components: a candidate trafficking protein that has been modified to include an FKBP12-Rapamycin binding (FRB) domain, an FK506 binding protein (FKBP)-tagged motor protein capable of inducing a change in vesicle localization or movement, and a means of specifically labeling the vesicle population of interest. Adding a membrane permeant rapamycin analog links the FRB and FKBP domains (Belshaw et al., 1996). This results in the translocation of the FRB-FKBP protein complex by the motor; if the FRB-tagged protein binds the labeled vesicles, they will also undergo transport. This dimerization system has previously been used to recruit motors to peroxisomes (Kapitein et al., 2010a; 2010b) and to sequester cytosolic proteins on a membrane, thus inhibiting their activity in the cytoplasm (Robinson et al., 2010).
Figure 1. Components of the assay.
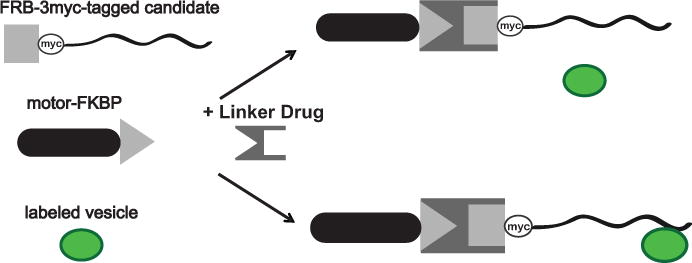
The assay relies on three components: an FRB-tagged candidate vesicle binding protein, an FKBP-tagged molecular motor, and a means to specifically label the vesicles of interest. In the presence of linker drug, the FKBP and FRB domains heterodimerize. The vesicle is unaffected if the FRB-tagged candidate protein does not bind it (upper right). If the vesicle and the FRB-tagged candidate protein interact, then the presence of linker drug results in the recruitment of motors to the vesicle (lower right). Adapted from Jenkins et al., 2012.
This unit explains the execution of two variations of this assay: Protocol 1 describes an assay designed for use in cells with a distinct microtubule-organizing center. In such cells, vesicles can be directed toward the cell center by linking them to dynein through the dynein adaptor Bicaudal D2 (BicD2), or toward the cell periphery by linking them to a kinesin motor domain (Figure 2). Positive interactions between the candidate protein and the labeled vesicles cause a profound, easily recognizable change in steady-state vesicle localization that is never observed in control cells. In neurons, a cell type of particular interest to our lab, linking vesicles to constitutively active motors produces a change in vesicle transport, but does not always lead to a change in steady-state vesicle localization. Thus Protocol 2 describes a version of this assay for use in nerve cells, in which binding of a candidate trafficking protein is transduced into an increase in vesicle transport into the axon, which is observable by live-cell imaging (Figure 3). The protocol we provide describes the use of this assay to identify kinesin motors that bind dendritic vesicle populations, but it is should be adaptable to a variety of trafficking proteins and to other cell types as well.
Figure 2. Application of the assay in cells with a microtubule-organizing center.
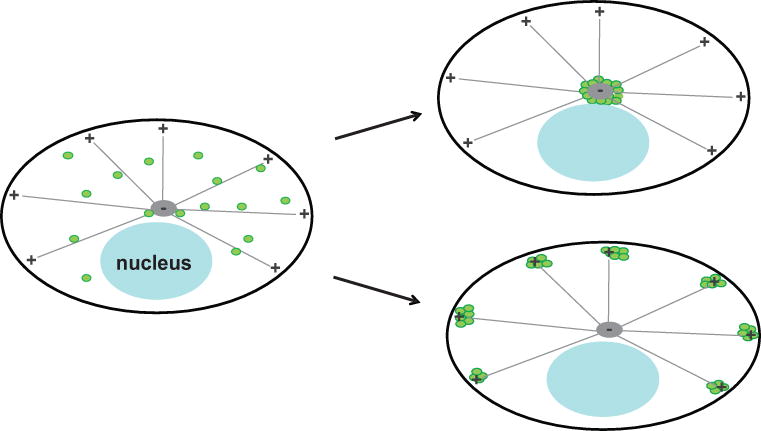
This schematic shows the predicted change in vesicle distribution resulting from an interaction between the candidate protein and labeled vesicles in the assay described in Protocol 1. Before adding linker drug, vesicles are distributed throughout the cell (left). If the candidate protein binds the vesicle, then in cells expressing FKBP-tagged Bicaudal D2, adding the linker drug results in dynein-mediated transport towards microtubule minus-ends, causing the accumulation of vesicles in the center of the cell (top right). In cells expressing FKBP-tagged KIF5C motor domain, addition of the linker drug causes accumulation of labeled vesicles in the periphery of the cell, near the plus-ends of microtubules (bottom right). If the candidate protein does not bind the labeled vesicles, then there will be no change in their distribution. Adapted from Bentley et al., 2015.
Figure 3. Applying the assay to identify proteins that bind to dendritic vesicles.
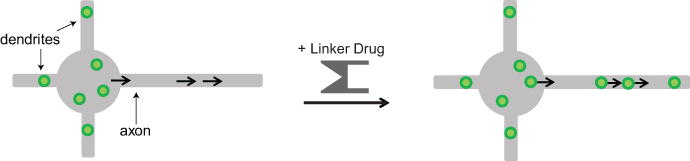
In the absence of linker drug, vesicles that carry dendritically polarized membrane proteins are confined to the somatodendritic region of the cell. If there is a positive interaction between the FRB-tagged candidate protein and the labeled vesicles, the addition of linker drug results in the recruitment of the KIF5C motor to the vesicles and their transport into the axon. The black arrows indicate movement of FKBP-tagged KIF5C motors, which translocate towards the tip of the axon. Adapted from Jenkins et al., 2012.
Protocol 1: Detecting protein-vesicle interactions based on an equilibrium change in vesicle distribution
Introduction
This protocol describes an assay in which inducible, motor-driven vesicle mislocalization serves as readout to indicate which of a series of candidate trafficking proteins associates with a given vesicle population. Vesicles can be misdirected toward the minus-ends of microtubules by expressing FKBP-tagged bicaudal, a dynein adaptor, or toward the plus-ends of microtubules by expressing an FKBP-tagged kinesin motor domain. The phenotype observed using Bicaudal D2 to recruit dynein and move vesicles toward the cell center is particularly distinctive since all the vesicles are directed to the same place, producing a particularly bright and highly distinctive readout (Figure 4 A, B). When kinesins are used to redirect vesicles, they accumulate at multiple locations around the cell periphery, which can be more difficult to detect, especially when screening a large number of cells (Figure 4 C, D). Since some vesicle populations may normally be distributed in the cell center or the cell periphery, the ability to misdirect vesicles in two different directions may be an advantage.
Figure 4. Detecting protein-vesicle interactions based on an equilibrium change in vesicle distribution.
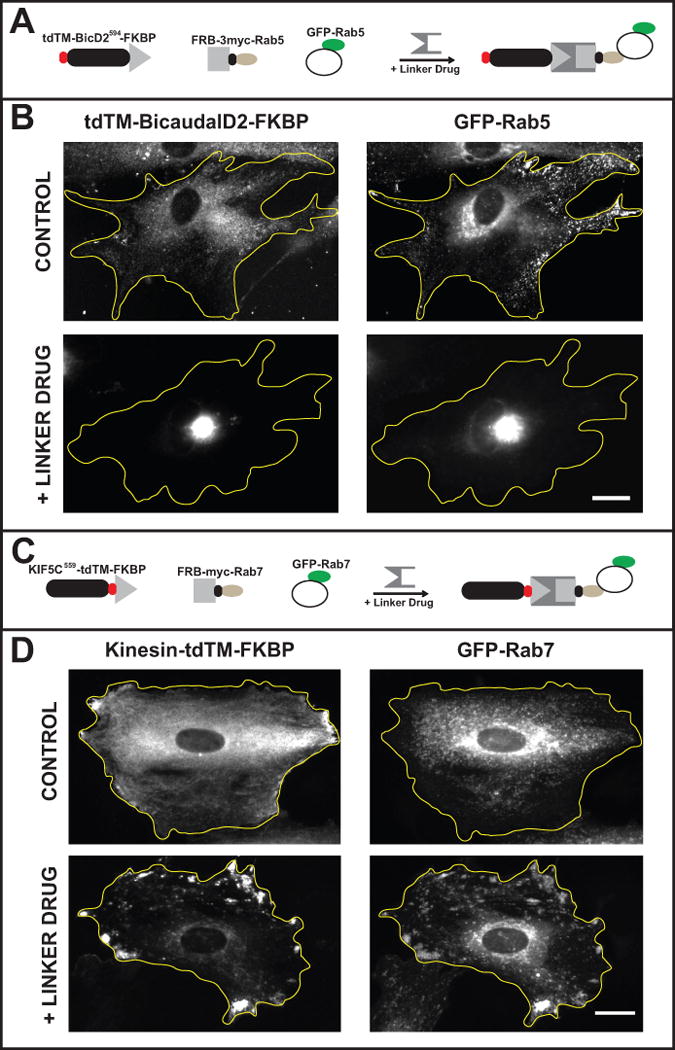
In this assay active motor proteins are targeted to labeled vesicles if, and only if, these vesicles bind the expressed FRB-tagged candidate protein. The examples show rat embryonic fibroblasts in which endosomes were mislocalized to the cell center by dynein or to the cell periphery by kinesin. (A & B) Expressing FRB-3myc-Rab5 and tdTM-BicD2594-FKBP resulted in a redistribution of early endosomes when linker drug was present. (A) A schematic showing the constructs expressed: tdTM-BicD2594-FKBP, FRB-3myc-Rab5, and GFP-Rab5. (B) Representative images show the distribution of tdTM-BicD2594-FKBP and GFP-Rab5 in control cells and in cells treated with linker drug. In control cells tdTM-BicD2594-FKBP was diffusely distributed with some vesicle association. GFP-Rab5 vesicles were distributed throughout the cell. In cells treated with linker drug, both GFP-Rab5 vesicles and tdTM-BicD2594-FKBP became concentrated in the center of the cell. (C & D) Expressing FRB-3myc-Rab7 and KIF5C559-tdTM-FKBP resulted in redistribution of late endosomes when linker drug was present. (C) A schematic showing the constructs expressed: KIF5C559-tdTM-FKBP, FRB-3myc-Rab7, and GFP-Rab7. (D) Representative images show the distribution of KIF5C559-tdTM-FKBP and GFP-Rab7 in control cells and in cells treated with linker drug. In control cells, the kinesin motor domain was diffusely distributed with small accumulations at a few points in the periphery of the cell. GFP-Rab7 vesicles had a mostly perinuclear distribution. In cells treated with linker drug, GFP-Rab7 vesicles accumulated in the periphery of the cell, together with the kinesin motor domain. The yellow lines outline cell boundaries. Bar, 30 μm. Adapted from Bentley et al., 2015.
Virtually any type of vesicle label is compatible with this assay. Fluorescently tagged proteins can be cotransfected with the FKBP- and FRB-tagged proteins (Step 2). It is also possible to use organelle dyes, such as LysoTracker, or fluorescently tagged proteins that are endocytosed, such as transferrin (Step 3). Alternatively, vesicles can be visualized by immunofluorescence after the assay is complete and cells have been fixed (Step 4).
Many of the details of this assay (e.g. the culture protocol, vesicle labeling method, optimal expression conditions, duration of drug treatment, etc.) depend on the cell type being investigated, the vesicle population to be labeled, and the candidate trafficking proteins of interest. Preliminary experiments are required to optimize these conditions and validate the assay, as discussed below (see “Critical Parameters”). As a starting point, we describe (in italics) the conditions we used for investigating the binding of kinesins with different endosomal populations in a rat embryonic fibroblast cell line.
Materials list*
18-mm circular coverslips (Fisher Scientific, catalog #12-545-84-1D)
12-well tissue culture plates
DMEM with 10 % fetal bovine serum or other appropriate cell culture medium
DNA construct encoding FKBP-tagged motor protein (one of the following: GFP-FLAG-BicD2594-FKBP, Addgene ID 63569; tdTM-FLAG-BicD2594-FKBP, Addgene ID 64205; KIF5C559-tdTM-FKBP, Addgene ID 64211)
DNA constructs encoding FRB-tagged candidate vesicle binding proteins
Vesicle label (DNA construct encoding fluorescently tagged protein, fluorescent dye, or immunofluorescence reagents)
Fugene 6 (Promega, catalogue number E2691) or other transfection reagent
Linker drug stock solution (AP21967, 0.5 mM in ethanol; Clontech A/C heterodimerizer, catalog number 635057)
Fixative (e.g., 4 % paraformaldehyde in PBS)
Microscope slides
Mounting medium: Elvanol (Banker and Goslin, 1998) or Prolong Gold (Life Technologies, Catalog number P10144)
Fluorescence microscope equipped with 40× and 60× objectives and a camera
*Here we describe the materials we routinely use in performing this assay in a rat embryonic fibroblast cell line. Different cell cultures and different imaging methods may require some modifications to this list of materials.
Steps of the procedure
Step 1: For each candidate protein you plan to test, prepare four coverslips, two to be drug-treated and two to serve as controls. For some cell types, it may be necessary to treat the coverslips with a suitable substrate for optimal cell attachment.
Step 2: Place each coverslip in a well of a 12-well dish and add 1 mL of culture medium to each well.
- Step 3: Plate cells into each well at a concentration that will yield the desired cell density on the day you plan to perform the experiment. Maintain the cultures in a CO2 incubator.For rat embyronic fibroblasts plate cells at a relatively low density (e.g., 2,500 rat embryonic fibroblasts per well) to enable imaging of isolated cells.
Step 4: Transfect the cultures with the appropriate constructs, using a transfection method suitable for the chosen cell type. For each combination of FRB-tagged candidate protein and FKBP-tagged motor (either BicD2 or KIF5C), transfect four cultures.
- OPTION: If vesicles will be labeled with a GFP-tagged protein, that construct should also be included in the transfection at this step. The colors of the fluorescent motor proteins can be adjusted to accommodate the desired vesicle labels.For rat embryonic fibroblasts, transfect 0.2 μg of each DNA construct per culture using Fugene 6 transfection reagent. For each set of constructs (FRB-trafficking protein and FKBP-motor) add 200 μL of DMEM to each of two microcentrifuge tubes. Add 12 μL of Fugene 6 to one tube and 0.8 μg of each DNA construct to the other. Vortex gently and centrifuge briefly to bring the contents to the bottom of the tube, then incubate at room temperature for 10 – 15 min. Add the medium containing the DNA to the tube with DMEM and Fugene 6. Vortex, centrifuge, and incubate again as before. Add 100 μL of this mixture to each well.
Step 5: Place cultures in a CO2 incubator to allow for expression of the exogenous proteins. For rat embryonic fibroblasts, incubate cultures for 1 – 2 days.
- Step 6: Add 0.2 μL of linker drug stock solution to each experimental well (yielding a final concentration of 100 nM) and return cultures to incubator for a sufficient period to allow for a change in vesicle distribution. Treat control wells with an equal volume of ethanol without drug. OPTION: If the vesicle marker is a dye or label that is taken up by the cell, add this reagent towards the end of the incubation period.For rat embryonic fibroblasts incubate with drug for 3 h.
- Step 7: Fix the cells according to an appropriate protocol and prepare the coverslips for fluorescence imaging. OPTION: If vesicles are to be antibody-labeled, conduct the immunostaining protocol in this step.For rat embryonic fibroblasts fix with 4 % paraformaldehyde in phosphate-buffered saline and mount on glass slides with Elvanol or Prolong Gold mounting medium (for details see Banker and Goslin, 1998).
Step 8: Scan each coverslip and record images of cells with labeled vesicles that express the fluorescent FKBP-tagged motor construct (BicD2 or KIF5C). Because only a fraction of the cells may be transfected, it may be helpful to first identify transfected cells based on their BicD2/KIF5C expression. Compare the vesicle distribution in control cells with that in cells treated with linker drug. If BicD2-FKBP was expressed, mislocalized vesicles will accumulate at a single place in the cell center; if KIF5C-FKBP was expressed, mislocalized vesicles will accumulate in several areas in the periphery of the cell.
Step 9: Determine the fraction of transfected cells that show a positive result.
Protocol 2: Detecting protein-vesicle interactions in living neurons based on directing vesicles to the axon
Introduction
This variation of the method was designed for a specific purpose: to identify trafficking proteins on dendritically polarized vesicles in cultured hippocampal neurons. Because neurons lack a microtubule-organizing center and because their neurites are long, it is not possible to induce a pronounced change in the equilibrium distribution of vesicles by linking them to kinesin or dynein, as in Protocol 1. Instead this assay is designed so that binding of the candidate protein to dendritic vesicles causes their movement into the axon. To accomplish this, the FRB-tagged candidate protein is linked to KIF5C559-tdTM-FKBP, an axon-selective motor protein (Huang and Banker, 2012). Since dendritically polarized vesicles do not normally enter the axon, an increase in their axonal trafficking is particularly apparent. This protocol can also be used to investigate protein binding to other vesicle populations, but if these vesicles normally undergo extensive axonal transport the drug-induced increase may be difficult to detect. Because the readout in this case is an increase in the flux of vesicles, this protocol requires live-cell imaging of the same neuron before and after the addition of linker drug. Thus each cell serves as its own control, enhancing the ability to detect drug-induced changes in vesicle trafficking.
Here we provide a protocol for the application of this assay in cultured hippocampal neurons. With minor modifications this basic protocol can be adapted for any neuronal culture preparation.
Materials list
18 mm glass coverslips (Fisher Scientific, catalog number 12-545-84-1D)
DNA construct encoding FKBP-tagged Kinesin-1 motor domain (KIF5C559-tdTM-FKBP, Addgene ID 64211)
DNA constructs encoding FRB-tagged candidate vesicle binding proteins
Vesicle label (DNA construct encoding fluorescently tagged protein or fluorescent dye)
Lipofectamine 2000 (Life Technologies, catalogue number 11668027)
Imaging chamber: Chamlide CMB for 18 mm coverslips (Quorum Technologies, catalog number CM-B18-1)
Imaging medium (Hibernate E Low Fluorescence (BrainBits, SKU: HE-Lf)
Linker drug stock solution (AP21967, 0.5 mM in ethanol; Clontech A/C heterodimerizer, catalog number 635057)
Fluorescence microscope: we use a Nikon Ti-E microscope equipped with a Yokogawa CSU-W1 spinning disk attachment, hardware-based drift compensation to maintain focus, and an Andor Zyla sCMOS camera
Steps of the procedure
Step 1: Plate hippocampal neurons onto poly-L-lysine-treated 18 mm coverslips at a density of 150,000 – 250,000 cells per 6 cm dish (~7 - 12,000 cells/cm2) and maintain the cultures for 6 – 9 days (for details see Kaech and Banker, 2006).
Step 2: Transfect cultured neurons with DNA constructs encoding the KIF5C559-tdTM-FKBP motor domain, the FRB-tagged protein of interest, and a GFP-tagged vesicle label using Lipofectamine 2000 (for details see Kaech and Banker, 2006). Include 0.4 – 1 μg of DNA for each of the constructs.
Step 3. Return cultures to a CO2 incubator and allow time for expression of the exogenous proteins. Optimal expression times for imaging the transport of different vesicle labels may vary from 6 h to overnight.
Step 4: Prepare for live-cell imaging by equilibrating the environmental enclosure of the microscope and the imaging chamber to 37°C, and place imaging medium in a 37°C water bath.
Step 5: Prepare a working solution of linker drug by diluting 0.2 μL of stock solution in 30 μL of imaging medium.
Step 6: Mount a coverslip in the imaging chamber in 1 mL of imaging medium. Place the imaging chamber on the microscope, select a set of 3 to 8 transfected cells, and record their stage coordinates. Detailed information about optimal equipment and methods for imaging vesicle transport in cultured hippocampal neurons can be found elsewhere (Kaech et al., 2012a; 2012b).
Step 7: Ensure that the automated focus control is engaged and then record a 30 – 60 s movie of each cell. Focus the microscope on the proximal axon, but also include the cell body and some dendrites in the field of view when possible. These movies will serve as controls.
Step 8: Without touching any of the microscope hardware (chamber, stage, etc.), add the entire aliquot of linker drug working solution prepared in Step 4 to the imaging medium. Pipette up and down 4 – 5 times to mix the solution. This will yield a final linker drug concentration of 100 nM.
Step 9: Record movies of each cell about 10, 20, and 30 minutes after adding linker drug, using the same parameters as in step 7.
Step 10: Compare movies of each cell before and after adding linker drug to determine if there is an increase in vesicle transport in the axon. To quantify such changes, prepare kymographs illustrating vesicle transport in the axon (Kaech et al., 2012b). Count the number of anterograde events that occur in each axon before and after the addition of linker drug. We recommend doing this analysis blinded, with the analyst not knowing which FRB-tagged protein was expressed. Determine whether there is a statistically significant increase in axonal trafficking events that occurs after linker drug is added. This will require analysis of 10 – 20 cells.
COMMENTARY
Background
We have successfully used this assay to identify the kinesins that associate with different vesicle populations in both fibroblasts and hippocampal neurons. To compare the binding of Kinesin-3 family members with different endosomal populations in fibroblasts we used the approach described Protocol 1. Our results showed that two closely related Kinesin-3 family members, KIF13A and KIF13B, bind preferentially to early endosomes. Two other Kinesin-3 family members, KIF1A and KIF1Bbeta, bind preferentially to late endosomes and lysosomes (Bentley et al., 2015). We used the approach in Protocol 2 to identify the kinesins that may be responsible for the transport of dendritically polarized vesicles in cultured hippocampal neurons. Of the eight kinesins that we tested, three Kinesin-3 family members, KIF1A, KIF13A, and KIF13B, were found to interact with different dendritic vesicle populations. Taken together, our results demonstrate that kinesins bind vesicles with high specificity.
We believe that this assay will have broad application for investigating a variety of trafficking proteins and their interactions with different vesicle populations. From a practical perspective, the primary limitation of this assay is that it requires the generation of FRB-tagged constructs of the candidate proteins of interest. Once such constructs have been prepared, they can be screened against a large number of vesicle populations in a relatively short period of time. In particular, Protocol 1 could lend itself to large screens for detecting the “vesicle interactome” of trafficking proteins of interest.
In addition to identifying the trafficking proteins that bind to a given vesicle population, this assay can be adapted to explore other aspects of protein-vesicle interactions. For example, the assay can be used to ask whether two members of the same family of trafficking proteins bind to the same vesicles. This can be accomplished by tagging one family member with an FRB and another with GFP. If the GFP-tagged vesicles are misdirected, it implies that both proteins bind the same carriers (Bentley et al., 2015). The assay can also be used to determine the protein domains responsible for vesicle binding. For example, by generating truncation mutants of KIF13B, we showed that its interaction with early endosomes is mediated by the C-terminal portion of the protein (Bentley et al., 2015).
Critical parameters
Designing the components
This assay uses FKBP-tagged motor proteins to cause vesicle misdirection. To move vesicles towards the minus-ends of microtubules, we used the approach initially developed by the Hoogenraad laboratory (Hoogenraad et al., 2003; Kapitein et al., 2010a; 2010b), which employs the adaptor protein Bicaudal D2 to recruit dynein. To accomplish this, we express BicaudalD2594-FKBP, which consists of the N-terminal fragment of Bicaudal D2 (BicD2, amino acids 1-594) with an FKBP-tag on the C-terminus. The N-terminus is tagged with a FLAG tag, tandem-dimer Tomato (tdTM), or green fluorescent protein (GFP). To move vesicles toward the plus-ends of microtubules, we used a truncated version of Kinesin-1 fused to tdTM and FKBP (KIF5C559-tdTM-FKBP). KIF5C559, which lacks its autoinhibitory domain, is a constitutively active (Friedman and Vale, 1999), highly processive, plus-end directed motor that has previously been used to redirect peroxisomes (Kapitein et al., 2010b). In neurons, this protein preferentially walks on axonal microtubules (Jacobson et al., 2006; Huang and Banker, 2012). These FKBP-tagged proteins were all cloned into the pCAG expression vector (Niwa et al., 1991) and are available from Addgene.
Candidate vesicle-binding proteins, tagged with FRB, need to be carefully designed. It is important that placement of the FRB domain does not interfere with the protein’s ability to bind vesicles and that the FRB domain is accessible to interact with the FKBP-tagged proteins. For kinesins, we accomplished this by replacing the N-terminal motor domain with an FRB domain. This design is unlikely to interfere with kinesin-vesicle interactions, which are mediated by the C-terminal tail domain. We also inserted a glycine-serine linker to extend the FRB farther from the vesicle surface and to increase its flexibility, which may enhance its ability to dimerize with FKBP-tagged proteins. For Rab GTPases we added the FRB domain at the N-terminal, the position where Rabs are typically tagged with fluorescent proteins. This placement should not interfere with the vesicle-interacting domains. In addition to the FRB domain, candidate proteins should also include an epitope tag, so that their effective expression and steady state localization can be assessed. We used a 3myc tag for this purpose, but any convenient small epitope should be sufficient. We positioned the tag between the FRB and the candidate protein, so that it would not interfere with dimerization of the FRB- and FKBP-tagged proteins. These guidelines should also be useful for applying this strategy to other types of trafficking proteins.
Vesicles of interest can be labeled in several ways. One option is to co-express a fluorescently tagged vesicle protein along with the FKBP- and FRB-tagged proteins. Alternatively, vesicles can be labeled with a dye (e.g. LysoTracker or MitoTracker) or a fluorescently tagged marker that can be taken up by cells (e.g. fluorescently tagged transferrin). Finally, the assay described in Protocol 1 also allows for antibody staining to label particular vesicle components.
Setting up the assay
Before screening vesicle interactions with novel candidate proteins, it is important to confirm that the FRB- and FKBP-tagged proteins express at sufficient levels and that adding the linker drug results in their efficient dimerization. It is also important to verify that recruiting the FKBP-tagged motors to vesicles results in the expected change in their localization. This can best be accomplished by establishing conditions that give a strong positive response with proteins known to bind the labeled vesicles.
If a GFP-tagged protein can be used to label vesicles for use with Protocol 1, one straightforward approach to test the assay involves generating a second version of that protein tagged with an FRB domain (Figure 4). Since the GFP- and FRB-tagged proteins bind to the same vesicle, linking the FRB-tagged protein to BicD2594-FKBP or KIF5C559-FKBP should produce a profound redistribution of the GFP-tagged vesicles. If a clear change in vesicle distribution is not observed, it may be necessary to optimize some of the parameters of the assay, such as the concentration of each of the DNA constructs, the time of drug exposure, or construct design. Once conditions have been established that give a strong positive result, these can then be applied to assess the binding of other candidate proteins of interest.
Setting up a positive control for Protocol 2 could be done in a similar way, but if the construct chosen labels vesicles that are relatively abundant in the axon, it can be difficult to see an increase in axonal traffic when these vesicles are linked to a constitutively active kinesin. Instead, we recommend labeling vesicles with transferrin receptor-GFP (TfR-GFP) and co-expressing FRB-KIF13Btail and KIF5C559-tdTM-FKBP (Figure 5).
Figure 5. Detecting protein-vesicle interactions in living neurons based on directing vesicles to the axon.
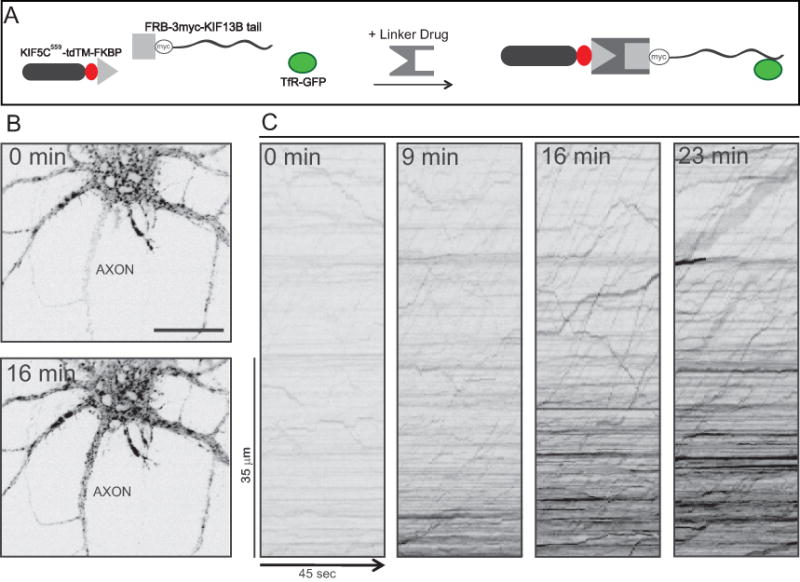
In the example shown, the assay was used to identify one of the kinesins that bind to dendritic vesicles labeled with transferrin receptor-GFP. (A) A schematic showing the three constructs expressed in this assay before and after assembly of the split kinesin. (B) Images showing the cell body and proximal axon of a neuron imaged immediately before (0 min) and 16 min after addition of the linker drug. Note the increase in intensity of TfR-GFP in the axon after 16 min. Bar, 20 μm. (C) Kymographs showing the transport of transferrin receptor vesicles in the axon before and at varying times after adding the linker drug. Time is shown on the x axis and position along the axon on the y axis. Diagonal lines with positive slope represent movements away from the cell body. Before adding the linker drug there was nearly no anterograde transport of vesicles in the axon. Addition of the linker drug resulted in a pronounced increase in long-range anterograde vesicle transport in the axon. In these images the contrast was reversed so that bright vesicles appear black. Adapted from Jenkins et al., 2012.
Anticipated results
Positive results in this assay are clear and powerful. For Protocol 1, binding of the candidate protein results in a pronounced change in vesicle localization; vesicles are concentrated either at the center or the periphery of the cell, depending on which motor protein is coexpressed. The localization of the FKBP-tagged motor protein is also altered. In the absence of linker drug, BicD2 and the KIF5C motor domain have a mostly diffuse distribution. In the case of positive interaction between the candidate protein and the vesicle, the FKBP-tagged proteins also accumulate in the same location as the vesicles. Because the FKBP and FRB domains bind tightly when linker drug is present, the FKBP-tagged motor proteins appear to remain associated with the labeled vesicles they bind. As a result, the soluble pool of FKBP-tagged motor proteins is vastly reduced. In some cases the motor protein accumulates strongly, but the labeled vesicles do not. We interpret this as indicating an interaction between the FRB-tagged candidate protein and an unlabeled vesicle population, which accumulates together with the motor. When using Protocol 2, a positive result is indicated by a pronounced increase in the stream of vesicles moving into the axon. This change in vesicle trafficking occurs relatively quickly, about 10 – 20 minutes after adding linker drug.
For both protocols, even in conditions that yield a positive interaction between a candidate protein and a vesicle population, not every transfected cell will exhibit a positive result. Since this assay depends on the co-expression of multiple proteins at levels appropriate to induce vesicle movement, this observation is not surprising. Because the profound changes caused by a positive interaction are never observed in control cells, we interpret vesicle accumulation and misdirection in a significant number of cells as strong evidence that the FRB-tagged candidate protein binds the labeled vesicles.
As with any experiment, negative results are not as straightforward to interpret. The expression levels of the FKBP- and FRB-tagged proteins can affect the performance of the assay. If too few copies of these molecules are expressed, then an insufficient number of motor proteins may be recruited to misdirect vesicles. The balance of FKBP- to FRB-tagged proteins may also be important: a low expression level or a large soluble fraction of the FRB-tagged protein could also prevent enough motors from being targeted to vesicles. Another problem could arise with proteins that quickly cycle on and off the vesicle membrane. In such a case the motors would also only spend a short period of time associated with the vesicle, which may not be long enough to cause misdirection.
Another limitation of the assay is that it does not provide quantitative information about the interaction under investigation. These experiments provide largely binary results: either the FRB-tagged candidate protein binds the labeled vesicle population or it does not. The assay does not indicate what percentage of labeled vesicles interacts with the FRB-tagged candidate protein. It may be possible to leverage careful quantification to generate an estimate, but this remains an inherent limitation of these experiments.
Systematically testing the binding of a series of related candidate proteins can be helpful in evaluating and enhancing confidence in negative results. If a particular candidate protein can misdirect one type of vesicle, its inability to move another type is likely not due to a problem with the assay, but accurately reflects its physiological binding specificity. Noting changes in the distribution of the FKBP-tagged BicD2 or the FKBP-tagged kinesin motor domain can also help evaluating negative results. If the expressed FRB-tagged protein binds vesicles efficiently, then the FKBP-tagged motor will become linked to these vesicles and accumulate in a predictable manner. If the motor accumulates but the labeled vesicles do not, this can be interpreted as a lack of interaction between the FRB-tagged protein and the vesicles of interest.
In spite of such caveats, in our hands it has not been difficult to achieve conditions that result in a positive result. Because the pattern of vesicle mislocalization induced by association with active motors is never observed in control cells and because this mislocalization requires that the FRB-tagged protein binds to the labeled vesicles, we believe a positive result in the assay provides strong evidence of vesicle binding.
Time considerations
Protocol 1 can be completed in about 3 – 4 days. With our cell line and expression system we prefer transfecting on day 1 and conducting the experiment (i.e. adding linker drug, fixing, and mounting coverslips) on day 3. We then image the coverslips on day 3 or 4.
Protocol 2 requires the culturing of primary rat hippocampal neurons. This is an involved procedure described elsewhere (Kaech and Banker, 2006). Once the cells have reached the appropriate stage of maturity, the experiments described in Protocol 2 take 10 – 18 h to complete. Some cargoes require expression times of about 6 – 8 h for optimal vesicle labeling, while others label efficiently after overnight expression. In either case, imaging time on the microscope will be about 45 – 50 minutes per coverslip.
Acknowledgments
We thank Helena Decker, Brian Jenkins, and Julie Luisi who played an integral role in the development of these assays. We also thank Zoe Bostick for her comments on the manuscript. This work was supported by NIH grant R01MH066179.
Footnotes
The authors declare no conflict of interest.
References
- Banker G, Goslin K. Culturing Nerve Cells. MIT Press; 1998. [Google Scholar]
- Belshaw PJ, Ho SN, Crabtree GR, Schreiber SL. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc Natl Acad Sci U S A. 1996;93:4604–4607. doi: 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley M, Decker H, Luisi J, Banker G. A novel assay reveals preferential binding between Rabs, kinesins, and specific endosomal subpopulations. The Journal of Cell Biology. 2015;93:4604. doi: 10.1083/jcb.201408056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman DS, Vale RD. Single-molecule analysis of kinesin motility reveals regulation by the cargo-binding tail domain. Nat Cell Biol. 1999;1:293–297. doi: 10.1038/13008. Available at: http://www.nature.com/doifinder/10.1038/13008. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, Wulf P, Schiefermeier N, Stepanova T, Galjart N, Small JV, Grosveld F, De Zeeuw CI, Akhmanova A. Bicaudal D induces selective dynein-mediated microtubule minus end-directed transport. The EMBO Journal. 2003;22:6004–6015. doi: 10.1093/emboj/cdg592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CF, Banker G. The Translocation Selectivity of the Kinesins that Mediate Neuronal Organelle Transport. Traffic. 2012;13:549–564. doi: 10.1111/j.1600-0854.2011.01325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson C, Schnapp B, Banker GA. A change in the selective translocation of the Kinesin-1 motor domain marks the initial specification of the axon. Neuron. 2006;49:797–804. doi: 10.1016/j.neuron.2006.02.005. Available at: http://linkinghub.elsevier.com/retrieve/pii/S089662730600095X. [DOI] [PubMed] [Google Scholar]
- Jenkins B, Decker H, Bentley M, Luisi J, Banker G. A novel split kinesin assay identifies motor proteins that interact with distinct vesicle populations. The Journal of Cell Biology. 2012;198:749–761. doi: 10.1083/jcb.201205070. Available at: http://www.jcb.org/cgi/doi/10.1083/jcb.201205070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaech S, Banker G. Culturing hippocampal neurons. Nature protocols. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, Kuijpers M, Wulf PS, van Spronsen M, MacKintosh FC, Hoogenraad CC. Mixed microtubules steer dynein-driven cargo transport into dendrites. Curr Biol. 2010a;20:290–299. doi: 10.1016/j.cub.2009.12.052. [DOI] [PubMed] [Google Scholar]
- Kapitein LC, Schlager MA, van der Zwan WA, Wulf PS, Keijzer N, Hoogenraad CC. Probing intracellular motor protein activity using an inducible cargo trafficking assay. Biophys J. 2010b;99:2143–2152. doi: 10.1016/j.bpj.2010.07.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Robinson MS, Sahlender DA, Foster SD. Rapid inactivation of proteins by rapamycin-induced rerouting to mitochondria. Developmental Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A. Membrane traffic in the secretory pathway: The life cycle of a transport vesicle. Cellular and Molecular Life Sciences CMLS. 2008 doi: 10.1007/s00018-008-8349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD. The molecular motor toolbox for intracellular transport. CELL. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Wickner W, Schekman R. Membrane fusion. Nature structural & molecular biology. 2008;15:658–664. doi: 10.1038/nsmb.1451. [DOI] [PMC free article] [PubMed] [Google Scholar]