

Summary
Phosphate is essential for life, being used in many core processes such as signal transduction and synthesis of nucleic acids. The waterborne agent of cholera, V ibrio cholerae, encounters phosphate limitation in both the aquatic environment and human intestinal tract. This bacterium can utilize extracellular DNA (eDNA) as a phosphate source, a phenotype dependent on secreted endo‐ and exonucleases. However, no transporter of nucleotides has been identified in V . cholerae, suggesting that in order for the organism to utilize the DNA as a phosphate source, it must first separate the phosphate and nucleoside groups before transporting phosphate into the cell. In this study, we investigated the factors required for assimilation of phosphate from eDNA. We identified PhoX, and the previously unknown proteins UshA and CpdB as the major phosphatases that allow phosphate acquisition from eDNA and nucleotides. We demonstrated separable but partially overlapping roles for the three phosphatases and showed that the activity of PhoX and CpdB is induced by phosphate limitation. Thus, this study provides mechanistic insight into how V . cholerae can acquire phosphate from extracellular DNA, which is likely to be an important phosphate source in the environment and during infection.
Introduction
As a waterborne, facultative pathogen, Vibrio cholerae transits between the aquatic environment and the human intestinal tract. During life in the environment, V. cholerae is often found in biofilms associated with phyto‐ and zooplankton but may also persist in a planktonic state (Lipp et al., 2002). The bacteria enter the human host via ingestion of contaminated water or food and subsequently colonize the small intestine (Peterson, 2002). Expression of cholera toxin, an Adenosine diphosphate (ADP)‐ribosylating enterotoxin, leads to excessive release of water from intestinal epithelial cells, resulting in massive secretory diarrhea and rapid dehydration of the host (Herrington et al., 1988; Peterson, 2002; Childers and Klose, 2007; Faruque et al., 2011). Expulsion from the host typically results in delivery of the bacteria back into aquatic reservoirs.
V. cholerae encounters phosphate limitation both in the aquatic environment and within the human host (Kamal et al., 2007; Schild et al., 2007; Nelson et al., 2008; McDonough et al., 2014). Due to the importance of phosphate, bacteria, including V. cholerae, have evolved several mechanisms to acquire it from the environment. Inorganic phosphate is the most readily available form of phosphate in the aquatic environment (White and Metcalf, 2007) and most bacteria encode two independent systems for its uptake into the cytoplasm: inorganic phosphate transport system (PitA or PitB) and phosphate‐specific transport system (Pst/PhoU) (Rosenberg et al., 1977; Willsky and Malamy 1980). V. cholerae harbors a functional Pst/PhoU system (Heidelberg et al., 2000; Pratt et al., 2009; McDonough et al., 2014), as well as an uncharacterized gene (VC2442) that encodes a protein with homology (37% identity) to the Escherichia coli PitA.
Some bacteria are able to utilize organophosphates, which are characterized by a phosphorous‐oxygen‐carbon ester bond (e.g. sugar phosphates), as sources of phosphate. Although many organophosphates can cross the outer membrane of Gram‐negative bacteria, they cannot be transported into the cytoplasm of cells with the exceptions of glycerol‐3‐phosphate and hexose‐6‐phosphates (van Veen 1997; Lamarche et al., 2008). Extra‐cytoplasmic phosphatases can facilitate the removal of phosphate groups from non‐transportable phosphate‐compounds. For example, V. cholerae alkaline phosphatase (PhoX), which is expressed in the periplasm, is able to remove the phosphate group from several organic phosphate compounds including glucose‐6‐phosphate, glucose‐1‐phosphate and β‐glycerophosphate (Roy et al., 1982).
Extracellular DNA (eDNA), which represents a major class of organophosphate compound, is present in picomolar to micromolar concentrations in aquatic environments, depending on the location tested (Lorenz and Wackernagel, 1994; Bjorkman and Karl, 2005). The source of the DNA in aquatic environments is unclear, although much of it may be released from decomposing microbes, zooplankton, fish or other aquatic‐dwelling organisms. Additionally, several marine‐ and fresh water‐dwelling bacterial species were found to secrete DNA under exponential growth conditions (Paul and David, 1989; Nielsen et al., 2007). The purpose of DNA secretion in microbes has been tied with biofilm formation, as eDNA can help provide structure to a biofilm matrix (Whitchurch et al., 2002; Qin et al., 2007). However, several organisms, including Ruegeria pomeroyi, Yersinia enterocolitica, Pseudomonas aeruginosa, Shewanella spp. and Corynebacterium glutamicum, have been shown to utilize DNA and/or nucleotides as sources of phosphate, carbon and nitrogen (Trulzsch et al., 2001; Rittmann et al., 2005; Pinchuk et al., 2008; Mulcahy et al., 2010; Sebastian and Ammerman, 2011).
Seper et al. (2011) recently demonstrated that V. cholerae is also able to utilize eDNA as a sole source of phosphate. During its life cycle, V. cholerae may encounter eDNA in both the aquatic environment or in the host. The aquatic environment provides ambient eDNA (as discussed above), but eDNA is also found within the matrix of V. cholerae biofilms (Seper et al., 2011). Within a host, V. cholerae can stimulate the release of neutrophil extracellular traps (NETs), which are web‐like structures comprised of neutrophil secreted‐eDNA and antimicrobial proteins (Kawasaki and Iwamuro, 2008; Branzk and Papayannopoulos, 2013; Zawrotniak and Rapala‐Kozik, 2013). Thus, DNA from these NETs could potentially provide a source of nutrients for the organism (Seper et al., 2013).
Utilization of eDNA as a source of phosphate requires the break down of eDNA into nucleotides. V. cholerae produces and secretes two nucleases into culture supernatant: Xds, with exonuclease activity; and Dns, with endonuclease activity (Focareta and Manning, 1987, 1991a, 1991b; Seper et al., 2011). These nucleases are both induced under low phosphate conditions, suggesting a role in phosphate scavenging during starvation conditions (Seper et al., 2011; McDonough et al., 2014). Although the main role of Xds and Dns may be to break down structural eDNA within a biofilm matrix or in NETs, it is not surprising that both are required for wild type growth on eDNA as a sole source of phosphate (Seper et al., 2011; 2013), since their activity presumably results in extracellular accumulation of nucleotides. In order to access the phosphate from these nucleotides, V. cholerae must separate the phosphate group from the nucleoside group through the action of one or more phosphatases.
In this work, we aimed to identify the phosphatases required for release of phosphate from nucleotides (i.e. nucleotidases). Nucleosides, which lack a phosphate group, are readily transported into the cytoplasm of V. cholerae via Nup transporters [Gumpenberger et al. (accompanying manuscript from separate group)]. However, no nucleotide transporter has been identified, suggesting that the phosphorylated nucleosides remain in the periplasm (Watanabe et al., 2011). Therefore, we hypothesized that expression of a periplasmic or extracellular phosphatase is required for growth on eDNA by releasing phosphate from the mononucleotides liberated by Xds and Dns. Presumably, once the phosphate is removed, it can be taken up into the cell by any of the phosphate transporters, i.e. Pst/PhoU or Pit. Here, we have presented evidence that three phosphatases, PhoX, UshA and CpdB, are the major phosphatases contributing to the ability of V. cholerae to assimilate phosphate from eDNA.
Results
PhoX is not required for utilization of eDNA as a phosphate source
Several organisms, including R. pomeroyi and Shewanella spp., use alkaline phosphatase to remove phosphate from nucleotides (Pinchuk et al., 2008; Sebastian and Ammerman, 2009). The V. cholerae alkaline phosphatase, PhoX, is expressed in the periplasm, and the gene is regulated by the major phosphate starvation response regulator, PhoB (von Kruger et al., 2006; Pratt et al., 2009). Therefore, we hypothesized that PhoX would provide the required phosphatase activity for growth on eDNA as a source of phosphate for V. cholerae.
To test our hypothesis, we assessed the growth of wild type and ΔphoX strains in MOPS‐glucose minimal medium that was either lacking phosphate or supplemented with sheared salmon sperm DNA as the sole source of phosphate (Figure S1). Prior to growth in the assay medium, the strains were pre‐grown in MOPS‐glucose minimal medium under phosphate replete conditions and washed thoroughly in the medium lacking phosphate. In this experiment, and in all other growth curves presented in this paper, strains were grown in no phosphate MOPS‐glucose medium as a negative control. Additionally, all strains were grown in MOPS‐glucose medium supplemented with 10 mM KH2PO4 (high phosphate), to ensure that the strains were able to grow equally well in phosphate replete conditions (data not shown). We included the ΔxdsΔdns strain, which is unable to utilize eDNA as a source of phosphate (Seper et al., 2011), as a negative control to ensure that there was no contaminating phosphate in the salmon sperm DNA. As had been reported earlier, we found that wild type V. cholerae was able to use eDNA as a source of phosphate; however, the rate of growth was severely decreased when compared with wild type growth even under phosphate limiting – 0.1 mM KH2PO4 – conditions. Growth of the phoX deletion strain in eDNA closely matched the wild type. Thus, we concluded that either PhoX does not contribute to utilization of eDNA as a source of phosphate, or its role is redundant with other phosphatases/nucleotidases.
Wild type, ΔphoX and ΔxdsΔdns all doubled a few times in medium lacking any phosphate source. This suggested that these strains accumulate internal phosphate stores (e.g. poly‐phosphate) during one of the pre‐growth conditions and resort to utilizing this phosphate store upon transition into phosphate limiting environments. In this experiment and in all other eDNA growth curves, we saw day‐to‐day variation in growth rate of the strains but very little variation between biological replicates within a single experiment. We believe this variation between experiments is connected to differences in phosphate storage of the bacteria prior to growth in the test media. Due to the growth rate variability, we did not calculate growth rates for the eDNA curves, and we have plotted only replicates from a single experiment.
Identification of additional putative phosphatases
We performed a genetic screen in order to identify additional phosphatases that may contribute to growth of V. cholerae on eDNA. A ΔphoX strain was mutagenized with a mTn10 transposon and plated on LB plates containing the colorimetric phosphatase substrate, 5‐bromo‐4‐chloro‐3‐indolyl phosphate (XP), which turns blue upon removal of the phosphate group and subsequent oxidation of the molecule. Colonies exhibiting phosphatase activity (e.g. wild type and ΔphoX) are blue on XP plates due to product accumulation in the periplasm, whereas colonies lacking phosphatase activity are white. Approximately 40 000 mTn10 mutants were screened on LB XP plates and seven white colonies were identified. Of these seven colonies, five represented unique insertion sites present throughout the VC2174 coding region and two unique insertions were in the 5′ end of VC2352 (Figure S2). An in frame deletion of VC2174 in the wild type background results in white colonies on LB XP plates, validating the transposon screen results. However, a clean deletion of VC2352 was still blue on LB XP plates, even when the VC2352 deletion was moved into the ΔphoX strain. VC2352, encoding a NupC‐homolog, appears to be the dominant nucleoside transporter in V. cholerae [Gumpenberger et al. [accompanying manuscript from separate group)]. Our work suggests that expression of a truncated VC2352 somehow inhibits transport and/or cleavage of XP; however, as we can see no obvious tie to phosphatase activity, we did not further characterize this protein.
VC2174 is annotated as a bifunctional UDP‐sugar hydrolase/5′nucleotidase (Heidelberg et al., 2000) called UshA in bacteria. The online program, PSORT (http://www.psort.org/psortb/index.html) categorizes the protein translation of VC2174 as periplasmic, consistent with it having a predicted secretion signal sequence according to SignalP (http://www.cbs.dtu.dk/services/SignalP‐4.1/) (Nakai and Kanehisa, 1991; Petersen et al., 2011). UDP‐sugar hydrolases are a broad class of enzymes with two catalytic activities: (i) UDP‐sugar to UMP and sugar‐phosphate, and (ii) UMP to uridine and phosphate (Neu, 1967a). In general, UDP‐sugar hydrolases are extra‐cytoplasmic and have broad substrate specificity for deoxy‐ and ribo‐nucleotides (Neu, 1967b; Rittmann et al., 2005). UshA is a highly conserved protein among bacterial species. The enzymatic function is most thoroughly studied in E. coli, but its physiological function remains unclear in this species. In C. glutamicum and Shewanella spp. UshA is essential for growth when eDNA and/or nucleotides is supplied as the sole source of phosphate (Rittmann et al., 2005; Pinchuk et al., 2008). BLAST analysis revealed that V. cholerae UshA has 79%, 41% and 72% sequence identity to UshA of E. coli, C. glutamicum and Shewanella oneidensis respectively (Altschul et al., 1997; 2005). Based on its identity to UshA in other organisms, as well as work presented in this manuscript, we have designated VC2174 as ushA.
Using the UshA sequence as a query for a BLAST analysis (Altschul et al., 1997; 2005) of the V. cholerae genome and by searching the V. cholerae genome annotation for ‘nucleotidase’ (Heidelberg et al., 2000), we identified three additional putative extra‐cytoplasmic nucleotidases: VCA0545, VCA0608 and VC2562 (Table S1). VCA0545 harbors a 5′nucleotidase domain and is 62% similar to UshA of S. oneidensis. This suggests that VCA0545 may represent an additional 5′nucleotidase present within the bacterium. VCA0608 does not share homology with traditional nucleotidase proteins. The gene encodes a protein carrying a haloacid dehalogenase‐like hydrolase domain (HAD) and is annotated as a provisional dUMP phosphatase. VC2562 is in the class of 2′3′cyclic phosphodiesterases. These enzymes have two independent active sites that catalyze the two‐step reaction: (i) 2′3′cyclic nucleotide to 3′ nucleotide, and (ii) 3′nucleotide to nucleoside and phosphate (Anraku, 1964a, 1964b). Although 2′3′cyclic phosphodiesterase and 3′nucleotidase activities have been described in several organisms including many of the Enterobacteriaceae, the physiological role of this enzyme is not well characterized (Neu, 1968). CpdB – the most commonly studied 2′3′cyclic phosphodiesterase in bacteria – of Y. pestis and Y. enterocolitica is essential for growth on 2′3′cAMP as the sole source of carbon. VC2562 shares 81% and at least 80% identity to CpdB of E. coli and Yersinia spp. respectively (Altschul et al., 1997; 2005). Based on this high level of identity to CpdB in other organisms, as well as work presented in this manuscript, we have designated VC2562 as cpdB.
In order to determine if UshA, CpdB, VCA0545 or VCA0608 contribute to growth of V. cholerae on eDNA, we made a quintuple knock‐out strain (Δ5 phosphatases) in which all four putative nucleotidases were deleted in conjunction with phoX. We tested this strain for its ability to utilize eDNA as a source of phosphate as described for the phoX single mutant. We found that a strain that was deleted for all five putative phosphates was unable to grow using eDNA as a source of phosphate (Fig. 1). Thus, we conclude that we have identified all major phosphatases involved in acquisition of phosphate from eDNA.
Figure 1.
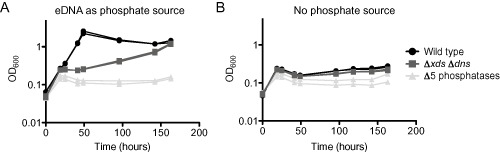
Deletion of five putative phosphatases hinders growth on eDNA as a source of phosphate.
Growth on eDNA was assessed using mid‐exponential phase bacteria, which were washed twice before putting in to the test conditions. The growth medium used was MOPS‐glucose supplemented with either (A) sheared salmon sperm DNA consisting of 0.5 mM phosphate or (B) no phosphate. The Δ5 phosphatases mutant is: ΔphoX ΔushA ΔcpdB ΔVCA0608 ΔVCA0545. Shown are two biological replicates assayed on the same day. The growth assay was performed two times, with a total of four biological replicates; each experiment exhibited the same results.
An unrelated finding in this experiment came from our using the ΔxdsΔdns as a negative control. Surprisingly, we found that this double mutant is able to reach wild type density but with a much delayed growth rate. The growth of the ΔxdsΔdns strain is not evident until at least 50 hours into the incubation, which explains why Seper et al. (2011) did not observe this phenotype. Presumably an additional, weak extracellular nuclease is present under this condition and contributes to the eDNA growth phenotype.
UshA is a non‐nucleotide‐specific 5′nucleotidase
The main product of eDNA degradation by Xds and Dns is thought to be monophosphorylated deoxynucleotides. We hypothesized that during growth on eDNA, the phosphate is removed from the mononucleotides, rather than the intact DNA strands. Therefore, in order to identify which of the five putative phosphatases are major contributors to the growth on eDNA, we performed growth curves in which 5′deoxy‐mononucleotides were supplied as the sole source of phosphate (Fig. 2 and Table 1). We identified that ushA is dominantly, and likely solely, responsible for supporting growth on all four 5′deoxy‐mononucleotides; the growth of ΔushA on all four nucleotides was comparable with that of the no phosphate control (Fig. 2A–D and Table 1). The deletion of ushA was complemented in trans by expression of ushA from an IPTG‐inducible promoter carried on a pMMB67EH vector (Fig. 2E).
Figure 2.
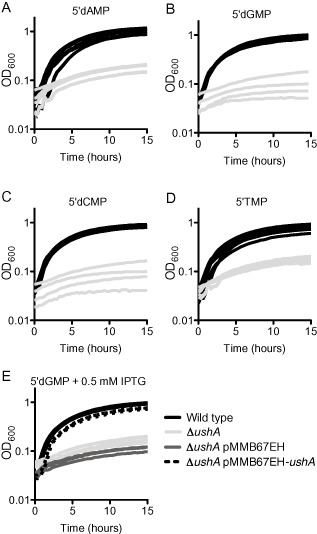
ushA is required for growth on all 5′ nucleotides when supplied as sources of phosphate.
A–D. Bacteria were pre‐grown to mid‐exponential phase in MOPS‐glucose minimal medium, supplemented with 10 mM KH2PO 4. Cultures were washed two times in MOPS medium containing no phosphate and inoculated into 200 μl MOPS‐glucose medium with either (A) 0.1 mM 5′dAMP, (B) 0.1 mM 5′dGMP, (C) 0.1 mM 5′dCMP, or (D) 0.1 mM 5′TMP. Strains were grown at 37°C with aeration in a 96 well plate. At least four biological replicates, assayed on at least two different days, are shown for all growth assays. Doubling times are reported in Table 1.
E. For complementation, ushA was expressed from the IPTG‐inducible Ptac promoter carried on the pMMB67EH plasmid. After pre‐growth and washing as described above, strains were inoculated into 200 μl MOPS‐glucose medium with 0.1 mM 5′dGMP + 0.5 mM IPTG. The strains harboring the expression vector were grown in the presence of Ap. Strains were grown at 37°C with aeration in a 96 well plate. Four biological replicates, assayed on at least two different days, are shown.
Table 1.
Nucleotide growth curve doubling times
| Strain | 0.1 mM KH2PO4 | 5′dAMP | 5′dGMP | 5′dCMP | 5′TMP | 3′AMP | 3′dGMP | 3′CMP | 3′TMP | No phosphate |
|---|---|---|---|---|---|---|---|---|---|---|
| Wild type | 56 (4) | 51 (7) | 58 (4) | 47 (4) | 47 (7) | 58 (5) | 54 (7) | 216 (3) | 144 (2) | 222 (12) |
| ΔphoX | 40 (4) | 56 (4) | 43 (2) | 41 (2) | 60 (4) | 60 (6) | 56 (6) | 228 (4) | 120 (2) | 222 (10) |
| ΔushA | 53 (4) | 174 (5) | 300 (4) | 300 (4) | 186 (5) | 60 (3) | 52 (5) | 348 (3) | 138 (2) | 396 (10) |
| ΔcpdB | 59 (5) | 42 (5) | 50 (2) | 52 (2) | 45 (5) | 181 (3) | 132 (6) | 222 (3) | 174 (2) | 186 (8) |
| ΔVCA0545 | 39 (4) | 32 (4) | 44 (4) | 45 (4) | 38 (4) | 49 (4) | 52 (4) | 216 (4) | ND | 198 (4) |
| ΔVCA0608 | 66 (3) | 53 (8) | 50 (2) | 49 (2) | 51 (8) | 59 (6) | 47 (6) | 180 (2) | 126 (2) | 204 (10) |
Doubling times are in minutes. Number of replicates shown in parentheses.
We performed nucleotidase assays, which use a mixture of ascorbic acid and molybdate to detect phosphate in solution upon release by nucleotidase activity, to continue assessment of UshA's 5′nucleotidase activity (Edwards et al., 1993). Using whole cell lysates as a source of UshA protein, we expected that the wild type bacterium would harbor 5′nucleotidase activity, whereas the ushA mutant would have undetectable activity. Indeed, V. cholerae cell lysate harbored 5′nucleotidase activity against all four 5′deoxy‐mononucleotides and that activity was drastically reduced in the ΔushA strain (Fig. 3 and Table S2). We saw no detectable accumulation of phosphate in either a no substrate control or a substrate only control (data not shown).
Figure 3.
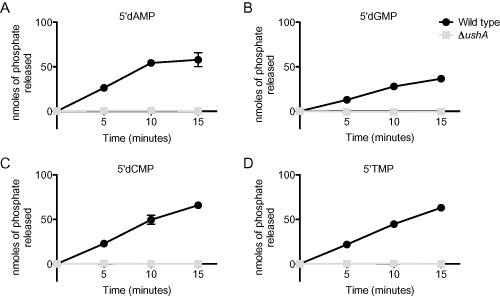
UshA is required for 5′ nucleotidase activity.
Wild type and ΔushA strains were grown to an OD 600 of ∼0.5 in 10 ml LB cultures. Cultures were washed once in 10 mM Tris pH 7.5 and lysed by sonication. Lysates were mixed with a final concentration of 1 mM (A) 5′dAMP, (B) 5′dGMP, (C) 5′dCMP or (D) 5′ TMP. At 0, 5, 10 and 15 min after addition of the substrate, aliquots of the reaction were removed and mixed with 0.1 N HCl to prevent further enzymatic activity. After all samples were collected, cellular debris was removed by centrifugation and the supernatants were incubated with the ammonium molybdate solution (1% ascorbic acid and 1 N H2SO 4) at 45°C for 20 min. Nanomoles of phosphate released by enzymatic activity was determined by measuring the OD at 820 nm and converting to nmole through use of a standard curve. The mean and standard error of at least three replicates, assayed on at least two different days, are shown for each assay.
As ushA appeared to be the only phosphatase required for growth on 5′deoxy‐mononucleotides, we hypothesized that it may account for the ability of V. cholerae to grow on eDNA as a source of phosphate. However, we found that the ΔushA strain grew at a similar rate to the wild type strain under this condition (Fig. 4). Thus, we concluded that while ushA accounts for all measurable 5′nucleotidase activity in the bacterium, at least one other phosphatase (phoX, cpdB, VCA0545 or VCA0608) also supports growth of V. cholerae on eDNA.
Figure 4.
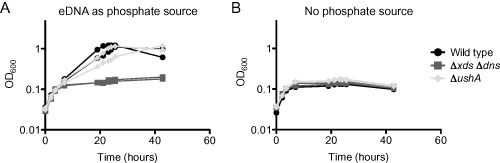
ushA is not required for growth on eDNA as a source of phosphate.
Growth on eDNA was assessed using mid‐exponential phase bacteria, which were washed twice before putting in to the test conditions. The growth medium used was MOPS‐glucose supplemented with either (A) 0.5 sheared salmon sperm DNA consisting of 0.5 mM phosphate or (B) no phosphate. Shown are two biological replicates assayed on the same day. The growth assay was performed four times, with a total of eight biological replicates; each experiment exhibited the same results.
CpdB is a purine‐specific 3′nucleotidase
Most DNases release 5′nucleotides, such that the phosphate group on the released nucleotide is attached to the 5′ carbon. However, the precise activities of the V. cholerae Xds and Dns extracellular nucleases have not been demonstrated, and thus it is unclear what nucleotide‐related substrates are produced by their activity on eDNA. BLAST analysis suggested that while Dns has strong identity to EndA‐type endonucleases that release 5′nucleotides, Xds harbors a YhcR domain (Altschul et al., 1997; 2005). The YhcR domain is named after the protein in which it was described, YhcR of Bacillus subtilis, an endonuclease that releases 3′monophosphorylated nucleotides (Oussenko et al., 2004; Seper et al., 2011). Therefore, it seems likely that V. cholerae has the capacity to release both 5′ and 3′phosphorylated mononucleotides. We hypothesized that the nucleotidase acting in concert with UshA during growth on eDNA is removing phosphate from 3′deoxy‐mononucleotides.
The single phosphatase deletion strains were tested in growth assays in which 3′mononucleotides were supplied as the sole source of phosphate (Fig. 5 and Table 1). Neither 3′dAMP or 3′dCMP were commercially available, so we used the ribo‐nucleotide form of these molecules. First, we found that V. cholerae is unable to utilize 3′CMP as a source of phosphate (Table 1). Second, the only gene contributing to growth on 3′AMP, 3′dGMP or 3′TMP appeared to be cpdB (Fig. 5A,B,D), consistent with its being annotated as a 3′nucleotidase. While 3′TMP did support growth of V. cholerae, it was only slightly greater than the no phosphate control. Thus, we suggest that CpdB has a preference for purine nucleotides. We were able to restore V. cholerae growth on 3′dGMP by expressing cpdB in trans (Fig. 5E).
Figure 5.
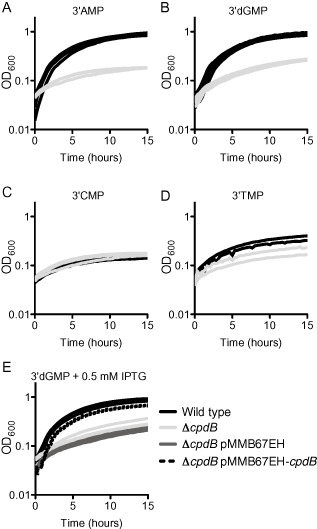
cpdB is required for growth on 3′AMP and 3′dGMP when supplied as sources of phosphate.
A–D. Bacteria were pre‐grown to mid‐exponential phase in MOPS‐glucose minimal medium, supplemented with 10 mM KH2PO 4. Cultures were washed twice in MOPS medium containing no phosphate and inoculated into 200 μl MOPS‐glucose medium with either (A) 0.1 mM 3′AMP, (B) 0.1 mM 3′dGMP, (C) 0.1 mM 3′CMP, or (D) 0.1 mM 3′TMP. Strains were grown at 37°C with aeration in a 96 well plate. At least two biological replicates are shown for each curve. Doubling times are reported in Table 1.
E. For complementation, cpdB was expressed from the IPTG‐inducible Ptac promoter carried on the pMMB67EH plasmid. After pre‐growth and washing as described above, strains were inoculated into 200 μl MOPS‐glucose medium with 0.1 mM 3′dGMP + 0.5 mM IPTG. The strains harboring the expression vector were grown in the presence of Ap. Strains were grown at 37°C with aeration in a 96 well plate. Four biological replicates, assayed on at least two different days, are shown.
In order to further support our finding that cpdB is a 3′nucleotidase, we performed nucleotidase assays using each of the four 3′mononucleotides as substrates. As expected from the growth assays, we found that wild type V. cholerae is able to remove the phosphate group from 3′AMP, 3′dGMP and 3′TMP, but not from 3′CMP (Fig. 6 and Table S3). However, although 3′dGMP supported full growth of wild type V. cholerae, the nucleotidase activity on this nucleotide is low, comparable with the activity on 3′TMP. This may be due to differences in how the bacteria were grown between the two assays (e.g. high phosphate LB for nucleotidase assays versus phosphate limiting MOPS‐glucose medium for the growth assays) and is addressed in Fig. 9. As expected, the ΔcpdB strain exhibited no detectable nucleotidase activity on any of the 3′mononucleotides, supporting its classification as a 3′nucleotidase.
Figure 6.
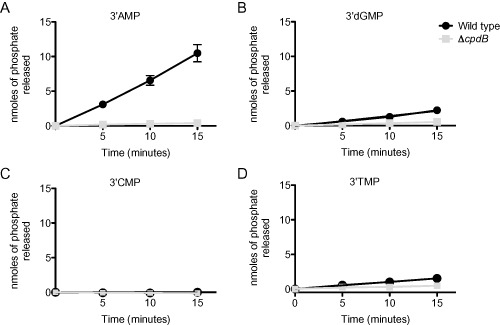
CpdB is required for 3′ nucleotidase activity on certain nucleotides.
Wild type and ΔcpdB strains were grown to an OD 600 of ∼0.5 in 10 ml LB cultures. Cultures were washed once in 10 mM Tris pH 7.5 and lysed by sonication. Lysates were mixed with a final concentration of 1 mM (A) 3′AMP, (B) 3′dGMP, (C) 3′CMP, or (D) 3′ TMP. At 0, 5, 10 and 15 min after addition of the substrate, aliquots of the reaction were removed and mixed with 0.1 N HCl to prevent further enzymatic activity. After all samples were collected, cellular debris was removed by centrifugation, and the supernatants were incubated with the ammonium molybdate solution (1% ascorbic acid and 1 N H2SO 4) at 45°C for 20 min. Nanomoles of phosphate released by enzymatic activity was determined by measuring the OD at 820 nm and converting to nmole through use of a standard curve. The mean and standard error of at least two replicates are shown for each assay.
Figure 9.
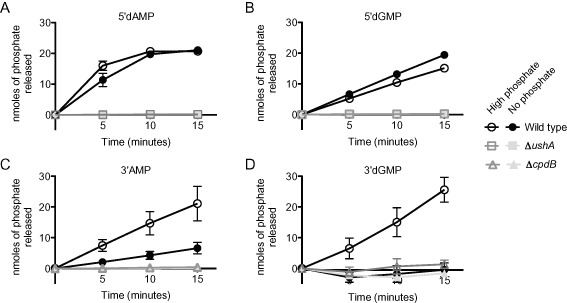
3′nucleotidase activity is induced by phosphate limitation.
Bacterial cultures were grown to mid‐exponential phase in MOPS‐glucose medium supplemented with 10 mM KH2PO 4, washed twice in no phosphate MOPS‐glucose, and resuspended into two test conditions: MOPS‐glucose medium supplemented with 10 mM KH2PO 4 or no phosphate. After 2 h of incubation at 37°C in the test conditions, the bacteria were washed once in 10 mM Tris pH 7.5 and resuspended in 100 μl of the same buffer. The cells were mixed with assay buffer and a final concentration of 1 mM (A) 5′dAMP, (B) 5′dGMP, (C) 3′AMP, or (D) 3′ dGMP. At 0, 5, 10 and 15 min after addition of the substrate, aliquots of the reaction were removed and mixed with 0.1 N HCl to prevent further enzymatic activity. After all samples were collected, cellular debris was removed by centrifugation, and the supernatants were incubated with the ammonium molybdate solution (1% ascorbic acid and 1 N H2SO 4) at 45°C for 20 min. Nanomoles of phosphate released by enzymatic activity was determined by measuring the OD at 820 nm and converting to nmole through use of a standard curve. The mean and standard error of four replicates (5′dGMP and 3′AMP) or two replicates (5′dAMP and 3′dGMP) are shown for each assay.
UshA, CpdB and PhoX account for the majority of phosphatase activity required for growth on eDNA as a source of phosphate
Our work demonstrated that UshA and CpdB are required for growth on 5′ and 3′nucleotides as sole sources of phosphate respectively. Therefore, we hypothesized that deletion of both genes would result in the loss of V. cholerae growth when eDNA is supplied as the sole source of phosphate. To test this, we performed a growth assay with wild type, ΔushAΔcpdB and ΔxdsΔdns. While the double phosphatase mutant displayed a modest decrease in the growth rate, ΔushAΔcpdB reached the same final optical density as the wild type control after approximately 70 hours of growth (Fig. 7). Therefore, we concluded that one (or more) of the remaining three putative nucleotidases (phoX, VCA0545 or VCA0608) was responsible for the ability the bacterium to grow in the absence of ushA and cpdB. To address this hypothesis, we tested the triple mutants (ΔushAΔcpdB combined with an additional phosphatase mutant) for growth on eDNA as the sole source of phosphate. Indeed, we found that the additional deletion of phoX, but not VCA0545 or VCA0608, resulted in a growth phenotype nearly identical to the Δ5 phosphatase mutant strain (Fig. 8). By day six, the ΔushAΔcpdBΔphoX mutant exhibited slight growth that was not apparent in the Δ5 phosphatase mutant. The amount of growth varied between replicates and experiments and is not immediately obvious in the curve presented in Fig. 8. We did not determine if the triple mutant could continue to grow and reach wild type turbidity. We hypothesized that this slight growth is due to activity of either VCA0545 and/or VCA0608. These data support that UshA, CpdB and PhoX are the major phosphatases that allow V. cholerae to utilize eDNA as a source of phosphate.
Figure 7.
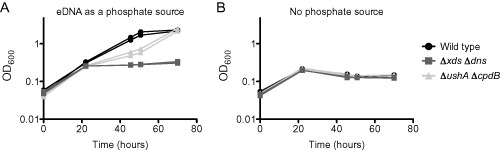
Deletion of cpdB, together with ushA, does not abolish growth on eDNA as a sole source of phosphate.
This growth assay was performed using mid‐exponential phase bacteria, which were washed twice before putting into the test conditions. The growth medium used was MOPS‐glucose supplemented with either (A) sheared salmon sperm DNA consisting of 0.5 mM phosphate or (B) no phosphate source. The experiment was performed three times with a total of five biological replicates. Two biological replicates assayed in the same experiment are shown.
Figure 8.
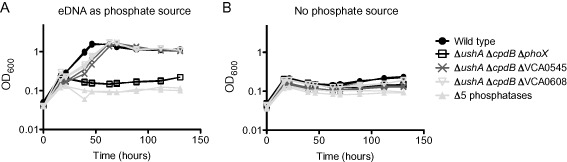
Deletion of ush A, cpd B and pho X mimics the delta 5 phosphatase mutant.
This growth assay was performed using mid‐exponential phase bacteria, which were washed twice before putting into the test conditions. The growth medium used was MOPS‐glucose supplemented with either (A) sheared salmon sperm DNA consisting of 0.5 mM phosphate or (B) no phosphate source. The experiment was performed twice with two biological replicates each time. Two replicates from the same experiment are shown.
CpdB, but not UshA, is activated by phosphate limitation
We predicted that the nucleotide phosphatases would be induced under phosphate limitation, when they would be required for phosphate scavenging by V. cholerae. Indeed, phoX is a low phosphate induced gene (von Kruger et al., 2006). We performed quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) experiments to determine if ushA and cpdB are also expressed upon transition to phosphate limiting conditions. Although we were able to induce expression of phoX after incubation of bacteria in a no‐phosphate medium, we did not detect increased transcription of ushA or cpdB when compared with the high phosphate condition (data not shown and Supplementary experimental procedures).
To determine if UshA or CpdB nucleotidase activity is regulated by phosphate conditions, we performed nucleotidase assays using bacteria that were incubated in either high phosphate or no phosphate MOPS‐glucose media. The wild type rate of phosphate removal from 5′dAMP and 5′dGMP was very similar under high phosphate and no phosphate conditions (Fig. 9A and B). Conversely, 3′nucleotidase activity was induced in the wild type by phosphate limitation (Fig. 9C and D). Incubation of the bacteria in high phosphate rendered 3′nucleotidase activity against 3′dGMP undetectable, but slight activity still remained against 3′AMP. CpdB accounts for all 3′nucleotidase activity under both high phosphate and no phosphate environments, as the deletion strain did not exhibited any detectable phosphate release under either condition. As we did not test activity against 3′dAMP or 3′GMP, we cannot say whether CpdB 3′nucleotidase activity under high phosphate is specific to the adenosine nucleotide or ribo‐nucleotides in general. These results explain why the 3′dGMP nucleotidase activity presented in Fig. 6 is unexpectedly low based on the growth assays in Fig. 5; the medium used in that experiment, LB, is a high phosphate environment. From these experiments, we conclude that CpdB activity is phosphate regulated due to some post‐transcriptional process.
Discussion
Being comprised of phosphate, sugars and nucleic acids, DNA is a rich source of phosphate, carbon and nitrogen. Several bacterial species, including V. cholerae, are able to utilize eDNA as a source of nutrients (Trulzsch et al., 2001; Rittmann et al., 2005; Pinchuk et al., 2008; Mulcahy et al., 2010; Sebastian and Ammerman, 2011; Seper et al., 2011). The utilization of eDNA as a source of phosphate requires break down of the DNA strands into nucleotides, removal of the phosphate from the nucleoside and uptake of the phosphate into the cell via dedicated transporters (e.g. Pit and Pst/PhoU). Under phosphate‐limiting conditions, V. cholerae expresses and secretes an exo‐ and endonuclease, Xds and Dns respectively (Seper et al., 2011; McDonough et al., 2014). A mutant deleted for both of these nuclease genes exhibits a severe growth defect when eDNA is supplied as the sole source of phosphate (Seper et al., 2011). This suggests that if there is an additional secreted nuclease produced by V. cholerae, it does not significantly contribute to the acquisition of phosphate from eDNA under in vitro conditions. Although the cytoplasmic‐uptake machinery for phosphate is well characterized in V. cholerae, no phosphatases involved in utilization of DNA/nucleotides as sources of phosphate had been described in V. cholerae. Therefore, the goal of this work was to identify the phosphatases involved in this phenotype. A model of our findings is presented in Fig. 10. In summary, we identify UshA, CpdB and PhoX as the major phosphatases that allow V. cholerae to utilize eDNA as a source of phosphate.
Figure 10.
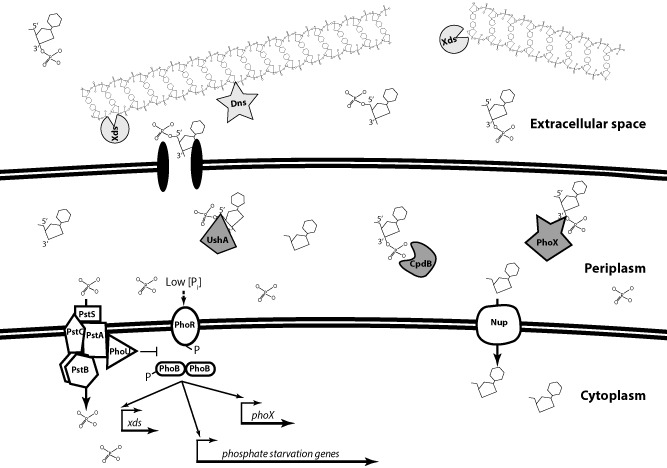
Model for the utilization of eDNA as a source of phosphate in V . cholerae.
Extracellular DNA is broken down by Xds and Dns in the extracellular space. Dns is an endonuclease in the EndA family of nuclease and presumably cleaves at the 3′ carbon, leaving a 5′ phosphate attached to the DNA strand. Alternatively, Xds is an exonuclease and may cleave at the 5′ carbon, leaving a 3′ phosphate. Once produced, nucleotides can pass across the outer membrane into the periplasm through porins. UshA and CpdB, which we hypothesize as located in the periplasm, remove phosphate groups from 5′ and 3′nucleotides respectively. Additionally, we hypothesize that PhoX contributes to removal of phosphate from 3′dAMP and 3′dCMP. Released phosphate can traverse the inner membrane via the Pst/PhoU system, whereas nucleosides can pass through the nucleoside transporters (e.g. NupC). Low phosphate conditions induce transcription of phosphate starvation genes such as xds and pho X, in a PhoB‐dependent manner. Additionally, CpdB activity is induced under phosphate limiting conditions.
Although we have focused on describing V. cholerae UshA and CpdB as 5′ and 3′nucleotidases, respectively, we have not determined if these proteins carry the bifunctional activities (UDP‐sugar hydrolase and 2′3′phosphodiesterase, respectively) as described in other organisms. Our bioinformatic searches for phosphatases identified two additional genes: VCA0545 and VCA0608. VCA0545 is homologous to UshA in V. cholerae and in other species. VCA0608 is annotated as a dUMP phosphatase, and thus may be specific for this ribo‐nucleotide intermediate. We were unable to verify the phosphatase activities of these two proteins, however, as described below, our phenotypic data implicate one or both of these proteins in residual phosphatase activity.
A quintuple mutant (all five putative phosphatases deleted) demonstrated an inability to use eDNA as a phosphate source after 7 days of incubation in the growth medium. Similarly, a triple mutant, ΔushAΔcpdBΔphoX, was a near phenocopy of the Δ5 phosphatase mutant. However, slight growth of the triple mutant was observed by day six, suggesting that another phosphatase, likely VCA0545 or VCA0608, provides minimal phosphatase activity against eDNA or nucleotides. While we can easily attribute the role of UshA and CpdB in eDNA growth to their nucleotidase activities, the role of PhoX is less clear. V. cholerae PhoX was previously demonstrated to be inactive against 5′AMP or 5′ATP, and thus presumed inactive against all nucleotides (Roy et al., 1982). Although these authors did not test PhoX for phosphatase activity against other nucleotides, it seems unlikely that the generally non‐specific phosphatase would act on some nucleotides but not others. However, our data clearly suggest that PhoX must be active as a phosphatase against either a subset of nucleotides that we were unable to test (3′dAMP or 3′dCMP) or DNA. Further biochemical analysis of PhoX could elucidate the precise role that the protein plays in eDNA growth.
While expression of phoX is induced by phosphate limitation (von Kruger et al., 2006), we did not detect increased transcription of ushA or cpdB. However, we did observe an increase in CpdB nucleotidase activity after incubation of the bacteria in media lacking phosphate. Thus, we conclude that CpdB is likely post‐transcriptionally regulated by phosphate. For example, either the protein level or activity of the enzyme is induced by phosphate limitation.
Despite its universal role in phosphate assimilation, expression of ushA in relation to phosphate concentrations is not consistent between other bacterial species. For example, ushA is expressed under low phosphate conditions in C. glutamicum and this expression is regulated through the phosphate starvation regulator, PhoS (Rittmann et al., 2005; Kocan et al., 2006). Alternatively, E. coli ushA is not regulated by phosphate conditions or PhoB, a homolog of PhoS (Burns and Beacham, 1986). The V. cholerae ushA, which encodes a protein more similar to E. coli than to C. glutamicum, is likely not regulated by phosphate. However, it is important to note that there may be small changes in transcription of ushA that we are unable to detect with the qRT‐PCR assay.
Regulation of cpdB in other organisms has not been thoroughly studied; however, the gene is not induced by phosphate limitation in Salmonella Typhimurium (Kier et al., 1977). Alternatively, cpdB of both Salmonella Typhimurium and Yersinia spp. are under control of carbon catabolite repression (Kier et al., 1977; Trulzsch et al., 2001). A cAMP‐CRP binding site has been identified in the cpdB promoter of Y. enterocolitica, and CpdB allows growth of this species on cAMP as the sole source of carbon (Trulzsch et al., 2001). Using the online promoter prediction tool, Softberry BRPOM (Solovyev and Salamov, 2011), we identified two putative CRP binding sites in the V. cholerae cpdB promoter. Thus, it will be interesting to see if cpdB from this organism is carbon catabolite repressed while phosphate levels control the enzymatic activity.
The physiological relevance of phosphate acquisition from eDNA by V. cholerae is unclear. Within a human host, V. cholerae may encounter eDNA in the form of NETs secreted by attacking neutrophils. Indeed, the secreted nucleases, Xds and Dns, were shown to be important in defending V. cholerae from NET attack by breaking down the structural DNA of the NET (Seper et al., 2013). As we have shown that the small intestine of an infant mouse is a phosphate limiting environment, we hypothesize that DNA from NETs can also serve as a source of phosphate for the pathogen (McDonough et al., 2014).
DNA is a known structural component of V. cholerae biofilms, which form in the aquatic environment where nutrients are often limiting. In the context of a biofilm, Xds and Dns were demonstrated as important for allowing individual bacteria to degrade the DNA structure and escape (Seper et al., 2011). Because DNA is a rich source of phosphate, it seems likely that the organism would take advantage of the released nucleotides and consume them as nutrients. Therefore, we hypothesize that the genes involved in acquiring nutrients from eDNA (e.g. ushA, cpdB and phoX) would be important for survival of V. cholerae in a biofilm.
Vibrio cholerae biofilms in the aquatic environment may develop on chitinous surfaces, e.g. exoskeletons of copepods (Meibom et al., 2005). Under these conditions, V. cholerae is naturally transformable; chitin induces expression of the DNA uptake machinery, such as the competence pilus (Meibom et al., 2004; 2005). The process of DNA uptake via natural transformation has been suggested as a mechanism of nutrient acquisition in other organisms (Sinha et al., 2013), although this hypothesis is highly debated (Johnston et al., 2014). Considering that upon transport into the cytoplasm by competence machinery, single‐stranded DNA is very quickly coated with single‐stranded binding protein (SSB), it seems unlikely that this DNA could be accessed by degradation enzymes that would allow its use as a source of nutrients (Dubnau, 1999). The strand of DNA not taken up by the cell is degraded into nucleotides, providing a source of energy for the transport of the intact strand across the inner membrane. Thus, it is possible that this broken down strand is further degraded by UshA, CpdB and/or PhoX into nucleoside and phosphate and used as nutrients. Until further work has been complete, it will be unclear if V. cholerae natural competence is tied with nutrients acquisition.
The source of eDNA within the V. cholerae biofilm is unknown. Neisseria spp. are known to secrete chromosomal DNA, a phenotype that has been hypothesized as important in both natural competence as well as biofilm formation (Hamilton et al., 2005; Lappann et al., 2010). Like Neisseria spp., V. cholerae might actively secrete DNA for use in the biofilm matrix. Alternatively, the eDNA isolated from the biofilm matrix of V. cholerae may be released upon cell lysis. Aside from the eDNA present in V. cholerae biofilms, the aquatic environment contains a great deal of eDNA. Reported concentrations of eDNA in the aquatic environment range from picomolar to micromolar amounts (Lorenz and Wackernagel, 1994; Bjorkman and Karl, 2005). Although we have used a concentration of eDNA approximately 10‐fold less than Seper et al. (2011), we did not attempt to titrate the concentration of eDNA to determine whether picomolar or micromolar amounts of eDNA are enough to support survival of V. cholerae. However, the DNA found in biofilms or in NETS is likely more concentrated and may be similar to the concentration of DNA that we have used.
In summary, we have described results that demonstrate the requirement of a 5′ and 3′nucleotidase (UshA and CpdB, respectively), for the ability of V. cholerae to grow using nucleotides as a phosphate source. To our knowledge, we are the first to show that V. cholerae can utilize both 5′ and 3′nucleotides as sources of phosphate, as well as being the first to describe the ability of any bacterium to survive using 3′nucleotides as a source of phosphate. Interestingly, we have also shown that the organism is unable to use 3′CMP. We demonstrated that UshA and CpdB, work together with alkaline phosphatase, PhoX, to release phosphate from eDNA. Although phoX is induced by phosphate limitation, ushA and cpdB do not appear to be. However, CpdB enzymatic activity is increased in phosphate‐depleted medium. Prior to our work, UshA and CpdB had not been characterized in V. cholerae or any of the Vibrionaceae, and PhoX was considered to be inactive against all nucleotides (Roy et al., 1982) – a result that this work throws into question. Additionally, this is the first demonstration of a 2′3′cyclic phosphodiesterase/3′nucleotidase, CpdB, being involved in phosphate acquisition from DNA.
Experimental procedures
Media and bacterial strains
Bacterial strains were propagated in LB broth with aeration or on LB agar at 37°C, unless otherwise noted. When indicated bacteria were grown in MOPS‐glucose minimal media [1× MOPS salts (40 mM MOPS pH 7.4 (3 –(N‐morpholino)propanesulfonic acid) (Sigma Aldrich), 4 mM tricine, 0.1 mM FeSO4●7H2O, 9.5 mM NH4Cl, 0.28 mM KCl, 0.53 mM MgCl2●6H2O and 50 mM NaCl); 1× NRES (25 mM of each of the amino acids N, R, E and S); 1× trace metals (0.005% MgSO4, 0.0005% MnCl2●4H2O, 0.0005% FeCl3 and 0.0004% nitrilotriacetic acid); and 0.5% glucose], supplemented with various sources of phosphate. Unless otherwise noted, antibiotics were used at the following concentrations: 100 μg ml−1 streptomycin (Sm), 50 μg ml−1 ampicillin (Ap), 2 μg ml−1 chloramphenicol (Cm), 50 μg ml−1 spectinomycin (Sp) and 50 μg ml−1 kanamycin (Kn). Addition of 0.5 mM IPTG to broth was used to induce transcription from the Ptac promoter.
Strain construction
Bacterial strains and plasmids used are listed in Table 2. PCR primers used are listed in Table 3. All V. cholerae strains were constructed using standard molecular techniques in an Sm resistant derivative of the clinical O1 El Tor isolate E7946 (Mekalanos, 1983). Unless stated otherwise, all mutations generated in this study were confirmed by Sanger sequencing by the Tufts University Core Facility or by Eton Bioscience (Charlestown, MA). Plasmids were maintained in E. coli DH5αλpir. The donor strain, E. coli MFDpir, was used for conjugative transfer of plasmids (Ferrieres et al., 2010).
Table 2.
List of strains used in this study
| Strain or plasmid | Genotype or phenotype | Reference |
|---|---|---|
| V. cholerae | ||
| Wild type | E7946 El tor Ogawa, HapR+, ApR | Laboratory strain |
| Δxds Δdns | In frame deletion of xds and dns | Laboratory strain |
| ΔphoX | In frame deletion of phoX | Laboratory strain |
| ΔushA | In frame deletion of ushA, FRT scar | This study |
| ΔnupC | In frame deletion of nupC, FRT scar | This study |
| ΔcpdB | In frame deletion of cpdB, clean deletion | This study |
| ΔVCA0545 | In frame deletion of VCA0545, FRT scar | This study |
| ΔVCA0608 | In frame deletion of VCA0608, FRT scar | This study |
| ΔushA ΔcpdB | In frame deletion of ushA (FRT scar) and cpdB (FRT scar) | This study |
| ΔushA ΔcpdB ΔphoX | In frame deletion of ushA (FRT‐Spec‐FRT cassette), cpdB (FRT scar) and phoX (clean deletion) | This study |
| ΔushA ΔcpdB ΔVCA0545 | In frame deletion of ushA (FRT scar), cpdB (FRT scar) and VCA0545 (FRT scar) | This study |
| ΔushA ΔcpdB ΔVCA0608 | In frame deletion of ushA (FRT scar), cpdB (FRT scar) and VCA0608 (FRT scar) | This study |
| Δ5 phosphatases | In frame deletion of ushA (FRT‐Spec‐FRT cassette), cpdB (FRT scar), phoX (clean deletion), VCA0545 (FRT scar) and VCA0608 (FRT scar) | This study |
| ΔushA pMMB67EH | In frame deletion of ushA carrying pMMB67EH, ApR | This study |
| ΔushA pMMB67EH‐ushA | In frame deletion of ushA carrying pMMB67EH‐ushA, ApR | This study |
| ΔcpdB pMMB67EH | In frame deletion of cpdB carrying pMMB67EH, ApR | This study |
| ΔcpdB pMMB67EH‐cpdB | In frame deletion of cpdB carrying pMMB67EH‐cpdB, ApR | This study |
| E. coli | ||
| DH5αλpir | F‐ Δ(lacZYA‐argF)U169 recA1 endA1 hsdR17 supE44 thi‐1 gyrA96 relA1 λ::pir | Laboratory strain |
| TG1 pBAD33kan | F′ [traD36 proAB+ lacIq lacZΔM15] supE thi‐1 Δ(lac‐proAB) Δ(mcrB‐hsdSM) 5, (rk‐, mk‐), carrying pBAD33kan | Laboratory strain |
| MFDpir | MG1655 RP4‐2‐Tc::[ΔMu1::aac(3)IV‐ΔaphA‐Δnic35‐ΔMu2::zeo] ΔdapA::(erm‐pir) ΔrecA | Ferrieres et al. (2010) |
| Plasmids | ||
| pDL1098‐flp | Temperature‐sensitive Flp recombinase delivery vector, Fig. S2, CmR | This study; McDonough et al. (2014) |
| pBAD33kan | Laboratory plasmid | |
| pDL1098 | Temperature‐sensitive mTn10 delivery vector, Fig. S1, CmR, SpR | This study; McDonough et al. (2014) |
| pMMB67EH | pMMB67EH IncQ lacI q bla (ApR) Ptac rrnB | Furste et al. (1986) |
| pMMB67EH‐ushA | pMMB67EH with the ushA ORF cloned into SacI and PstI restriction sites | This study; McDonough et al. (2014) |
| pMMB67EH‐cpdB | pMMB67EH with the cpdB ORF cloned into SacI and PstI restriction sites | This study; McDonough et al. (2014) |
Table 3.
List of primers used in this study
| Primer use | Primer name | Sequence (5′ to 3′ orientation) |
|---|---|---|
| Arbitrary primed PCR | ||
| Arb1 | Hava and Camilli (2002) | |
| olj363 | McDonough et al. (2014) | |
| Arb2 | Hava and Camilli (2002) | |
| olj386 | McDonough et al. (2014) | |
| qPCR primers | ||
| phoX | VCA0033 qPCR F | CGGTGTCACCATTGTTGAAG |
| VCA0033 qPCR R | TGATCCGACGATTACGTTCA | |
| phoB | PhoBqF | AGGGCTATCAGGCGGTTGAG |
| PhoBqR | TACCACCAGGCAACATCCAG | |
| cpdB | cpdB qRT F | AGATAAAGCCTCCGATCAAAT |
| cpdB qRT R | GATCAAATCACCGTTATCGAC | |
| ushA | ushA qPCR F | GTACCAGAATCAGACCTACAAGA |
| ushA qPCR R | GGATTATCAAATTCGTGGTTAC | |
| Strain construction | ||
| ΔushA | ushA FRT F1 | TCACATCGAGTTAGCACGTCTG |
| ushA FRT R1 | GTCGACGGATCCCCGGAATCATTGTCATACCTTTGAACTGATG | |
| ushA FRT F2 | GAAGCAGCTCCAGCCTACATAATAAGGTTTGACTCGCAAAGTTG | |
| ushA FRT R2 | AGAGGTTACAGGAGTGCGTCAG | |
| ushA R0 | CTTTGCGCACTTTGATGAAT | |
| ΔnupC | nupC FRT F1 | TACACTGAGCTGCAACGCATTG |
| nupC FRT R1 | GTCGACGGATCCCCGGAATCAAATTGTGAGTAGAACAGGAAAGG | |
| nupC FRT F2 | GAAGCAGCTCCAGCCTACATGATCACAGATTGATGGATTGAG | |
| nupC FRT R2 | GTTAAGGGTAATAGTGCCTTCAGC | |
| nupC R0 | CCGACTAAAAACTCCACCTGA | |
| ΔcpdB | cpdB F1 | TCTCGGTCTCTCCCTGTAAATG |
| cpdB R1 | TGTAGGCTGGAGCTGCTTCTTCACTCATAACCAAATTGTGATGTG | |
| cpdB F2 | GAAGCAGCTCCAGCCTACATAAGCACCGATAATGCCCCTATTG | |
| cpdB R2 | CCTCATAGAAAAGAAAACAGCC | |
| cpdB R0 | CCTCATAGAAAAGAAAACAGCC | |
| ΔVCA0545 | VCA0545 F1 | GGTGTGAAAAGTACCAAGGGA |
| VCA0545 R1 | TGTAGGCTGGAGCTGCTTCGGCATACGTCTTCTCTCTTTTC | |
| VCA0545 F2 | GAAGCAGCTCCAGCCTACATAAAACGGATATCTCTTTGCCTT | |
| VCA0545 R2 | CAGTTCCAAAGCTCACTCC | |
| VCA05454 R0 | CGTTCGGCTTACCATTTTTCT | |
| ΔVCA0608 | VCA0608 F1 | TTTCGTTGGATGTTGACACTG |
| VCA0608 R1 | TGTAGGCTGGAGCTGCTTCTTCATGATGATCTCCTTAAAATCAG | |
| VCA0608 F2 | GAAGCAGCTCCAGCCTACATAATGCCATGAATAAGCGAGG | |
| VCA0608 R2 | TCATCACCTCTTTCTATTCACC | |
| VCA0608 R0 | AGAGCTAGAGAAACTGGAAGAA | |
| FRT scar screening | ABD725 | GAAGCAGCTCCAGCCTACA |
| pMMB67EH‐ushA | pMMB‐ushA F | ATCGGAGCTCATGAAACAAGGCCTCATTCTA |
| pMMB‐ushA R | ATCGCTGCAGTTAACGATAAACAATCTCGCCCG | |
| pMMB67EH‐cpdB | pMMB‐cpdB F | ATCGGAGCTCGTGAAACCTTTGTTTCATCGA |
| pMMB‐cpdB R | ATCGCTGCAGTTATTTTTGTAAGTCGATGCGAT | |
The single mutants – ΔushA and ΔnupC – were constructed using the Trans‐FLP method, which utilizes the natural transformability of chitin‐grown V. cholerae and Flp‐recombination, as described previously. Briefly, a gene of interest is replaced with a selectable FRT cassette, which is flipped out of the genome by the Flp recombinase carried on pBR‐flp (De Souza Silva and Blokesch, 2010; Blokesch, 2012; McDonough et al., 2014).
Natural co‐transformation as previously described was used to make the single mutants: ΔcpdB, ΔVCA0545, VCA0608 (Dalia et al., 2014). The selectable marker used in this construction was pBAD33kan isolated from E. coli TG1 cells. The PCR constructs used for transformation resulted in the exchange of the desired open reading frame for a FRT scar (GAAGCAGCTCCAGCCTACA), leaving only the start and stop codon of the deleted gene. Tranformants were selected on LB plates with 75 μg ml−1 Kn and subsequently screen by MASC PCR in order to confirm the genotype of each transformant at all loci of interest (Wang and Church, 2011a). The ΔushA ΔcpdB mutant was constructed using natural co‐transformation and ΔushA as the parental strain. A second round of natural co‐transformation on this strain was used to make the ΔushA ΔcpdB ΔVCA0545 and ΔushA ΔcpdB ΔVCA0608 mutants. The triple mutant ΔushA ΔcpdB ΔphoX was constructed by first using Trans‐Flp to delete ushA in the phoX deletion background. We were unable to obtain a mutant with the FRT cassette flipped out, so ushA is replaced by the FRT‐Spec‐FRT cassette in this strain. Second, natural co‐transformation was used to replace cpdB with a FRT scar. The Δ5 phosphatase mutant was constructed by using the ΔushA (FRT‐spec‐FRT) ΔphoX parental strain and two rounds of natural co‐transformation to additionally delete cpdB, VCA0545 and VCA0608.
The complementation plasmids pMMB67EH‐ushA and pMMB67EH‐cpdB were constructed in the pMMB67EH vector. V. cholerae ushA was amplified from E7946 genomic DNA using the primers pMMB‐ushA F and pMMB ushA R. Likewise, cpdB was amplified from E7946 genomic DNA using the primers pMMB‐cpdB F and pMMB‐cpdB R. The PCR fragments were digested with SacI and PstI restriction enzymes and then ligated into pMMB67EH that had been similarly digested. pMMB67EH‐ushA, pMMB67EH‐cpdB and pMMB67EH were cloned in E. coli DH5αλpir and transferred into the conjugation donor strain E. coli MFDpir (Ferrieres et al., 2010). The plasmids were moved into V. cholerae using filter mating, and the exconjugates were selected by plating on Sm and Ap. The primers pMMB‐F and pMMB‐R were used to screen for isolates carrying the expression vector and insert.
Phosphatase screen
The mTn10 library was constructed in ΔphoX using pDL1098 as described previously (McDonough et al., 2014). Aliquots of the library were thawed and diluted to 10−5 in LB, and 225 μl was plated on 150 mm LB plates supplemented with 100 μg ml−1 Sp and 40 μg ml−1 XP. Plates were incubated overnight at 37°C and white colonies were identified (7/∼40 000) and colony purified on the same medium. Genomic DNA was prepped from each white colony and arbitrary primed PCR followed by sequencing of the PCR product was used to determine the location of each mTn10 insertion (Hava and Camilli, 2002). Briefly, two rounds of PCR were performed using the primers (i) Arb1/olj363 and (ii) Arb2/olj386. For PCR 1, the following program was used: 95°C for 5 min, followed by six rounds of 95°C for 30 s, 30°C for 30 s and 72°C for 1 min; followed by 30 rounds of 95°C for 30 s, 45°C for 30 s and 72°C for 1 min. For PCR 2, the following program was used: 95°C for 5 min, followed by 35 rounds of 95°C for 30 s, 55°C for 30 s and 72°C for 1 min. PCR products were cleaned and sent for sequencing using the primer olj386.
Phosphate growth curves
Strains were struck on LB Sm plates and grown overnight at 37°C. Three single colonies per culture were used to inoculate 2 ml cultures of LB, and grown for 4 h at 37°C with aeration. Next, strains were back‐diluted to an OD600 ∼0.05 and grown to mid‐exponential phase (OD600 ∼0.5) in 3 ml MOPS‐glucose medium supplemented with 10 mM KH2PO4. Bacteria were washed twice in MOPS‐glucose medium (no phosphate) and inoculated into the growth curve test cultures.
For testing growth in minimal medium plus eDNA, strains were inoculated into 2 ml cultures of MOPS‐glucose medium supplemented with the desired source of phosphate (either KH2PO4, DNA or no phosphate). Culture tubes were grown at 37°C with aeration, and OD600 readings were taken through the glass tube after blanking the spectrophotometer with the appropriate medium. New glass tubes were always used to avoid misreading of the optical density due to scratches in the glass, as well as to avoid phosphate contamination from the phosphoric acid used to wash the dishes.
The source of eDNA used in these growth experiments was sheared salmon sperm DNA (Life Technologies). According to agarose gel electrophoresis analysis, the DNA strands range from ∼50 base pairs to 500 base pairs in length. The concentration of 0.5 mM used in these experiments refers to the total amount of phosphate molecules in the DNA, and not the molarity of the DNA. The stock of 10 mg ml−1 sheared salmon sperm DNA was calculated to be 25.5 mM phosphate.
For growth in minimal medium plus nucleotides, strains were pre‐grown and washed as described for eDNA growth curves. Following this, strains were inoculated into 200 μl MOPS‐glucose medium supplemented with the desired source of phosphate (either KH2PO4, nucleotides or nothing). Nucleotides were added at a concentration of 0.1 mM total phosphate. The cultures were grown in 96 well plates at 37°C with aeration using the BioTek Synergy Plate Reader (BioTek Instruments, Inc., Winooski, VT, USA). Optical density readings were measured and recorded every 15 min using the Gen5 Data Analysis software (BioTek Instruments, Inc., Winooski, VT, USA). All nucleotides were obtained from Santa Cruz Biotechnology, except 5′dAMP, 5′dCMP and 5′dGMP, which were obtained from Sigma‐Aldrich. The nucleotides were obtained in powder form and resuspended in pure H2O, except 5′dAMP, which was resuspended in 200 mM NaOH.
Nucleotidase assays
Single colonies were used to inoculate 10 ml LB cultures, which were grown at 37°C with aeration to an optical density of ∼0.5. For each replicate, the equivalent of 10 ml of OD600 = 0.5 culture was centrifuged in a 15 ml conical tube at 4500 × g for 20 min at room temperature. The supernatants were removed, and the cultures were resuspended in 1 ml 10 mM Tris HCl pH 7.5. The cultures were spun again, and the pellets were resuspended in 0.5 ml 10 mM Tris HCl pH 7.5. At this point, cells were lysed by transferring the cells to 2 ml eppendorf tubes and sonicating for 1 min with 50% amplitude, ½ s on and ½ s off using a high‐intensity cuphorn sonifier (Branson). Cell lysates were clarified by spinning the tubes at 8000 × g for 10 min at 4°C.
For nucleotidase assays performed under defined phosphate conditions, bacteria were pre‐grown in LB and back diluted to OD600 = 0.05 in 10 mM phosphate MOPS‐glucose medium. After reaching OD600 = 0.4–0.6, the bacteria were washed twice in MOPS‐glucose medium lacking phosphate and resuspended in either no phosphate or 10 mM phosphate MOPS‐glucose medium. Bacteria were incubated for 2 h at 37°C. The equivalent of 1 ml at OD600 = 0.6 bacteria were washed once in 10 mM Tris pH 7.5, resuspended in 100 μl of the same buffer and used directly in the nucleotidase assay.
Nucleotidase assays were performed as described previously with a few modifications (Edwards et al., 1993). One hundred microlitres of the cells lysates or washed cells was mixed with 890 μl of assay buffer (150 μl 0.5 M Sodium Acetate pH 6.0, 30 μl 150 mM CoCl2, 30 μl 480 mM CaCl2 and water to a final volume of 890 μl) and equilibrated to 37°C for 5 min. The nucleotidase reaction was started by the addition of 10 μl 100 mM nucleotide substrate and samples were immediately mixed by vortexing and placed at 37°C. At times 0, 5, 10 and 15 min after addition of the substrate, 150 μl samples of each reaction were removed and transferred to eppendorf tubes with 100 μl 0.1 N HCl and placed on ice to stop the reaction. Once all samples were acquired, the tubes were centrifuged at 16 000 × g for 5 min at 4°C to pellet cell debris. Subsequently, 60 μl of the supernatant was mixed with 140 μl of the development reagent (one part 10% ascorbic acid, six parts 0.42% Ammonium molybdate in 1 N H2SO4). Samples were incubated at 45°C for 20 min, after which 150 μl of each sample was transferred to a 96 well plate, and the absorbance at 820 nm was measured using a BioTek Synergy Plate Reader.
To convert the absorbance readings to nmoles of phosphate released, a standard curve was performed. Fivefold serial dilutions of KH2PO4 corresponding to 1000, 200, 40, 8, 1.6 or 0 pmoles of phosphate were mixed with the assay buffer and then mixed with the development reagent and incubated as described above. After the absorbance was measured, the readings were plotted against starting concentration and the slope corresponded to the conversion factor (i.e. absorbance readings in subsequent assays were converted to pmoles released by the slope). The standard curve was performed twice, in duplicate each time.
Supporting information
Supporting information
Acknowledgements
H.K. performed several of the nucleotidase assays presented in this manuscript. E.M. designed and completed all other experiments. A.C. provided financial and intellectual support. The manuscript was written by E.M. and edited by H.K. and A.C. The authors would like to thank Eleanor Fleming for her careful reading of the manuscript and Ankur Dalia for many helpful discussions about the data. This work was supported by US National Institutes of Health grants AI055058 (A.C.). A.C. is a Howard Hughes Medical Institute investigator.
References
- Altschul, S.F. , Madden, T.L. , Schaffer, A.A. , Zhang, J. , Zhang, Z. , Miller, W. , and Lipman, D.J. (1997) Gapped blast and psi‐blast: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F. , Wootton, J.C. , Gertz, E.M. , Agarwala, R. , Morgulis, A. , Schaffer, A.A. , and Yu, Y.K. (2005) Protein database searches using compositionally adjusted substitution matrices. FEBS J 272: 5101–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anraku, Y. (1964a) A new cyclic phosphodiesterase having a 3′‐nucleotidase activity from Escherichia coli b. I. Purification and some properties of the enzyme. J Biol Chem 239: 3412–3419. [PubMed] [Google Scholar]
- Anraku, Y. (1964b) A new cyclic phosphodiesterase having a 3′‐nucleotidase activity from Escherichia coli b. II. Further studies on substrate specificity and mode of action of the enzyme. J Biol Chem 239: 3420–3424. [PubMed] [Google Scholar]
- Bjorkman, K.M. , and Karl, D.M. (2005) Presence of dissolved nucleotides in the north pacific subtropical gyre and their role in cycling of dissolved organic phosphorus. Aquat Microb Ecol 39: 193–203. [Google Scholar]
- Blokesch, M. (2012) TransFLP – a method to genetically modify Vibrio cholerae based on natural transformation and FLP‐recombination. J Vis Exp 68: e3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzk, N. , and Papayannopoulos, V. (2013) Molecular mechanisms regulating netosis in infection and disease. Semin Immunopathol 35: 513–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns, D.M. , and Beacham, I.R. (1986) Nucleotide sequence and transcriptional analysis of the E. coli ushA gene, encoding periplasmic UDP‐sugar hydrolase (5′‐nucleotidase): regulation of the ushA gene, and the signal sequence of its encoded protein product. Nucleic Acids Res 14: 4325–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childers, B.M. , and Klose, K.E. (2007) Regulation of virulence in Vibrio cholerae: the ToxR regulon. Future Microbiol 2: 335–344. [DOI] [PubMed] [Google Scholar]
- Dalia, A.B. , McDonough, E. , and Camilli, A. (2014) Multiplex genome editing by natural transformation. Proc Natl Acad Sci USA 111: 8937–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza Silva, O. , and Blokesch, M. (2010) Genetic manipulation of Vibrio cholerae by combining natural transformation with FLP recombination. Plasmid 64: 186–195. [DOI] [PubMed] [Google Scholar]
- Dubnau, D. (1999) DNA uptake in bacteria. Annu Rev Microbiol 53: 217–244. [DOI] [PubMed] [Google Scholar]
- Edwards, C.J. , Innes, D.J. , Burns, D.M. , and Beacham, I.R. (1993) UDP‐sugar hydrolase isozymes in Salmonella enterica and Escherichia coli: silent alleles of ushA in related strains of group I Salmonella isolates, and of ushB in wild‐type and K12 strains of E. coli, indicate recent and early silencing events, respectively. FEMS Microbiol Lett 114: 293–298. [DOI] [PubMed] [Google Scholar]
- Faruque, S.M. , Nair, G.B. , and Takeda, Y. (2011) Molecular epidemiology of toxigenic Vibrio cholerae . Epidemiological and molecular aspects on cholera. T. R. a. S. K. Bhattacharya, Springer Science+Business Media, LLC.
- Ferrieres, L. , Hemery, G. , Nham, T. , Guerout, A.M. , Mazel, D. , Beloin, C. , and Ghigo, J.M. (2010) Silent mischief: bacteriophage mu insertions contaminate products of Escherichia coli random mutagenesis performed using suicidal transposon delivery plasmids mobilized by broad‐host‐range RP4 conjugative machinery. J Bacteriol 192: 6418–6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focareta, T. , and Manning, P.A. (1987) Extracellular proteins of Vibrio cholerae: molecular cloning, nucleotide sequence and characterization of the deoxyribonuclease (DNase) together with its periplasmic localization in Escherichia coli K‐12. Gene 53: 31–40. [DOI] [PubMed] [Google Scholar]
- Focareta, T. , and Manning, P.A. (1991a) Distinguishing between the extracellular dnases of Vibrio cholerae and development of a transformation system. Mol Microbiol 5: 2547–2555. [DOI] [PubMed] [Google Scholar]
- Focareta, T. , and Manning, P.A. (1991b) Genetic analysis of the export of an extracellular DNase of Vibrio cholerae using DNase‐beta‐lactamase fusions. Gene 108: 31–37. [DOI] [PubMed] [Google Scholar]
- Furste, J.P. , Pansegrau, W. , Frank, R. , Blocker, H. , Scholz, P. , Bagdasarian, M. , and Lanka, E. (1986). Molecular cloning of the plasmid rp4 primase region in a multi‐host‐range tacp expression vector. Gene 48: 119–131. [DOI] [PubMed] [Google Scholar]
- Hamilton, H.L. , Dominguez, N.M. , Schwartz, K.J. , Hackett, K.T. , and Dillard, J.P. (2005) Neisseria gonorrhoeae secretes chromosomal DNA via a novel type IV secretion system. Mol Microbiol 55: 1704–1721. [DOI] [PubMed] [Google Scholar]
- Hava, D.L. , and Camilli, A. (2002) Large‐scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol Microbiol 45: 1389–1406. [PMC free article] [PubMed] [Google Scholar]
- Heidelberg, J.F. , Eisen, J.A. , Nelson, W.C. , Clayton, R.A. , Gwinn, M.L. , Dodson, R.J. , et al (2000) DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae . Nature 406: 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington, D.A. , Hall, R.H. , Losonsky, G. , Mekalanos, J.J. , Taylor, R.K. , and Levine, M.M. (1988) Toxin, toxin‐coregulated pili, and the ToxR regulon are essential for Vibrio cholerae pathogenesis in humans. J Exp Med 168: 1487–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, C. , Martin, B. , Fichant, G. , Polard, P. , and Claverys, J.P. (2014) Bacterial transformation: distribution, shared mechanisms and divergent control. Nat Rev Microbiol 12: 181–196. [DOI] [PubMed] [Google Scholar]
- Kamal, D. , Khan, A.N. , Rahman, M.A. , and Ahamed, F. (2007) Study on the physico chemical properties of water of Mouri River, Khulna, Bangladesh. Pak J Biol Sci 10: 710–717. [DOI] [PubMed] [Google Scholar]
- Kawasaki, H. , and Iwamuro, S. (2008) Potential roles of histones in host defense as antimicrobial agents. Infect Disord Drug Targets 8: 195–205. [DOI] [PubMed] [Google Scholar]
- Kier, L.D. , Weppelman, R. , and Ames, B.N. (1977) Regulation of two phosphatases and a cyclic phosphodiesterase of Salmonella Typhimurium. J Bacteriol 130: 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocan, M. , Schaffer, S. , Ishige, T. , Sorger‐Herrmann, U. , Wendisch, V.F. , and Bott, M. (2006) Two‐component systems of Corynebacterium glutamicum: deletion analysis and involvement of the PhoS‐PhoR system in the phosphate starvation response. J Bacteriol 188: 724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kruger, W.M. , Lery, L.M. , Soares, M.R. , de Neves‐Manta, F.S. , Batista e Silva, C.M. , Neves‐Ferreira, A.G. , et al (2006) The phosphate‐starvation response in Vibrio cholerae O1 and phoB mutant under proteomic analysis: disclosing functions involved in adaptation, survival and virulence. Proteomics 6: 1495–1511. [DOI] [PubMed] [Google Scholar]
- Lamarche, M.G. , Wanner, B.L. , Crepin, S. , and Harel, J. (2008) The phosphate regulon and bacterial virulence: a regulatory network connecting phosphate homeostasis and pathogenesis. FEMS Microbiol Rev 32: 461–473. [DOI] [PubMed] [Google Scholar]
- Lappann, M. , Claus, H. , van Alen, T. , Harmsen, M. , Elias, J. , Molin, S. , and Vogel, U. (2010) A dual role of extracellular DNA during biofilm formation of Neisseria meningitidis . Mol Microbiol 75: 1355–1371. [DOI] [PubMed] [Google Scholar]
- Lipp, E.K. , Huq, A. , and Colwell, R.R. (2002). Effects of global climate on infectious disease: the cholera model. Clin Microbiol Rev 15: 757–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, M.G. , and Wackernagel, W. (1994) Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev 58: 563–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough, E. , Lazinski, D.W. , and Camilli, A. (2014) Identification of in vivo regulators of the Vibrio cholerae xds gene using a high‐throughput genetic selection. Mol Microbiol 92: 302–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meibom, K.L. , Li, X.B. , Nielsen, A.T. , Wu, C.Y. , Roseman, S. , and Schoolnik, G.K. (2004) The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci USA 101: 2524–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meibom, K.L. , Blokesch, M. , Dolganov, N.A. , Wu, C.Y. , and Schoolnik, G.K. (2005) Chitin induces natural competence in Vibrio cholerae . Science 310: 1824–1827. [DOI] [PubMed] [Google Scholar]
- Mekalanos, J.J. (1983) Duplication and amplification of toxin genes in Vibrio cholerae . Cell 35: 253–263. [DOI] [PubMed] [Google Scholar]
- Mulcahy, H. , Charron‐Mazenod, L. , and Lewenza, S. (2010) Pseudomonas aeruginosa produces an extracellular deoxyribonuclease that is required for utilization of DNA as a nutrient source. Environ Microbiol 12: 1621–1629. [DOI] [PubMed] [Google Scholar]
- Nakai, K. , and Kanehisa, M. (1991) Expert system for predicting protein localization sites in Gram‐negative bacteria. Proteins 11: 95–110. [DOI] [PubMed] [Google Scholar]
- Nelson, E.J. , Chowdhury, A. , Flynn, J. , Schild, S. , Bourassa, L. , Shao, Y. , et al (2008) Transmission of Vibrio cholerae is antagonized by lytic phage and entry into the aquatic environment. PLoS Pathog 4: e1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu, H.C. (1967a) The 5′‐nucleotidase of Escherichia coli. I. Purification and properties. J Biol Chem 242: 3896–3904. [PubMed] [Google Scholar]
- Neu, H.C. (1967b) The 5′‐nucleotidase of Escherichia coli. II. Surface localization and purification of the Escherichia coli 5′‐nucleotidase inhibitor. J Biol Chem 242: 3905–3911. [PubMed] [Google Scholar]
- Neu, H.C. (1968) The 5′‐nucleotidases and cyclic phosphodiesterases (3′‐nucleotidases) of the Enterobacteriaceae. J Bacteriol 95: 1732–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, K.M. , Johnsen, P.J. , Bensasson, D. , and Daffonchio, D. (2007) Release and persistence of extracellular DNA in the environment. Environ Biosafety Res 6: 37–53. [DOI] [PubMed] [Google Scholar]
- Oussenko, I.A. , Sanchez, R. , and Bechhofer, D.H. (2004) Bacillus subtilis YhcR, a high‐molecular‐weight, nonspecific endonuclease with a unique domain structure. J Bacteriol 186: 5376–5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, J.H. , and David, A.W. (1989) Production of extracellular nucleic acids by genetically altered bacteria in aquatic‐environment microcosms. Appl Environ Microbiol 55: 1865–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, T.N. , Brunak, S. , von Heijne, G. , and Nielsen, H. (2011) Signalp 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- Peterson, K.M. (2002) Expression of Vibrio cholerae virulence genes in response to environmental signals. Curr Issues Intest Microbiol 3: 29–38. [PubMed] [Google Scholar]
- Pinchuk, G.E. , Ammons, C. , Culley, D.E. , Li, S.M. , McLean, J.S. , Romine, M.F. , et al (2008) Utilization of DNA as a sole source of phosphorus, carbon, and energy by Shewanella spp.: ecological and physiological implications for dissimilatory metal reduction. Appl Environ Microbiol 74: 1198–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt, J.T. , McDonough, E. , and Camilli, A. (2009) PhoB regulates motility, biofilms, and cyclic di‐GMP in Vibrio cholerae . J Bacteriol 191: 6632–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, Z. , Ou, Y. , Yang, L. , Zhu, Y. , Tolker‐Nielsen, T. , Molin, S. , and Qu, D. (2007) Role of autolysin‐mediated DNA release in biofilm formation of Staphylococcus epidermidis . Microbiology 153: 2083–2092. [DOI] [PubMed] [Google Scholar]
- Rittmann, D. , Sorger‐Herrmann, U. , and Wendisch, V.F. (2005) Phosphate starvation‐inducible gene ushA encodes a 5′ nucleotidase required for growth of Corynebacterium glutamicum on media with nucleotides as the phosphorus source. Appl Environ Microbiol 71: 4339–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, H. , Gerdes, R.G. , and Chegwidden, K. (1977) Two systems for the uptake of phosphate in Escherichia coli . J Bacteriol 131: 505–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, N.K. , Ghosh, R.K. , and Das, J. (1982) Monomeric alkaline phosphatase of Vibrio cholerae . J Bacteriol 150: 1033–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schild, S. , Tamayo, R. , Nelson, E.J. , Qadri, F. , Calderwood, S.B. , and Camilli, A. (2007) Genes induced late in infection increase fitness of Vibrio cholerae after release into the environment. Cell Host Microbe 2: 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastian, M. , and Ammerman, J.W. (2009) The alkaline phosphatase PhoX is more widely distributed in marine bacteria than the classical PhoA. ISME J 3: 563–572. [DOI] [PubMed] [Google Scholar]
- Sebastian, M. , and Ammerman, J.W. (2011) Role of the phosphatase PhoX in the phosphorus metabolism of the marine bacterium Ruegeria pomeroyi DSS‐3. Environ Microbiol Rep 3: 535–542. [DOI] [PubMed] [Google Scholar]
- Seper, A. , Fengler, V.H. , Roier, S. , Wolinski, H. , Kohlwein, S.D. , Bishop, A.L. , et al (2011) Extracellular nucleases and extracellular DNA play important roles in Vibrio cholerae biofilm formation. Mol Microbiol 82: 1015–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seper, A. , Hosseinzadeh, A. , Gorkiewicz, G. , Lichtenegger, S. , Roier, S. , Leitner, D.R. , et al (2013) Vibrio cholerae evades neutrophil extracellular traps by the activity of two extracellular nucleases. PLoS Pathog 9: e1003614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha, S. , Mell, J. , and Redfield, R. (2013) The availability of purine nucleotides regulates natural competence by controlling translation of the competence activator Sxy. Mol Microbiol 88: 1106–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovyev, V. , and Salamov, A. (2011) Automatic annotation of microbial genomes and metagenomic sequences In Metagenomics and Its Applications in Agriculture, Biomedicine and Environmental Studies. Li R.W. (ed.). Hauppauge, NY: Nova Science Publishers, pp. 61–78. [Google Scholar]
- Trulzsch, K. , Roggenkamp, A. , Pelludat, C. , Rakin, A. , Jacobi, C. , and Heesemann, J. (2001) Cloning and characterization of the gene encoding periplasmic 2′,3′‐cyclic phosphodiesterase of Yersinia enterocolitica O:8. Microbiology 147: 203–213. [DOI] [PubMed] [Google Scholar]
- van Veen, H.W. (1997). Phosphate transport in prokaryotes: molecules, mediators and mechanisms. Antonie Van Leeuwenhoek 72: 299–315. [DOI] [PubMed] [Google Scholar]
- Wang, H.H. , and Church, G.M. (2011a) Multiplexed genome engineering and genotyping methods applications for synthetic biology and metabolic engineering. Methods Enzymol 498: 409–426. [DOI] [PubMed] [Google Scholar]
- Watanabe, K. , Tomioka, S. , Tanimura, K. , Oku, H. , and Isoi, K. (2011) Uptake of AMP, ADP, and ATP in Escherichia coli W. Biosci Biotechnol Biochem 75: 7–12. [DOI] [PubMed] [Google Scholar]
- Whitchurch, C.B. , Tolker‐Nielsen, T. , Ragas, P.C. , and Mattick, J.S. (2002) Extracellular DNA required for bacterial biofilm formation. Science 295: 1487. [DOI] [PubMed] [Google Scholar]
- White, A.K. , and Metcalf, W.W. (2007) Microbial metabolism of reduced phosphorus compounds. Annu Rev Microbiol 61: 379–400. [DOI] [PubMed] [Google Scholar]
- Willsky, G.R. , and Malamy, M.H. (1980) Characterization of two genetically separable inorganic phosphate transport systems in Escherichia coli . J Bacteriol 144: 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawrotniak, M. , and Rapala‐Kozik, M. (2013) Neutrophil extracellular traps (NETs) – formation and implications. Acta Biochim Pol 60: 277–284. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information