

Abstract
BACKGROUND
Adenosine A2A receptors (A2AR) modulate dopamine and glutamate signaling and thereby may influence some of the psychomotor and cognitive processes associated with schizophrenia. Because astroglial A2AR regulate the availability of glutamate, we hypothesized that they might play an unprecedented role in some of the processes leading to the development of schizophrenia, which we investigated using a mouse line with a selective deletion of A2AR in astrocytes (Gfa2-A2AR knockout [KO] mice].
METHODS
We examined Gfa2-A2AR KO mice for behaviors thought to recapitulate some features of schizophrenia, namely enhanced MK-801 psychomotor response (positive symptoms) and decreased working memory (cognitive symptoms). In addition, we probed for neurochemical alterations in the glutamatergic circuitry, evaluating glutamate uptake and release and the levels of key proteins defining glutamatergic signaling (glutamate transporter-I [GLT-I], N-methyl-D-aspartate receptors [NMDA-R] and α-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors [AMPA-R]) to provide a mechanistic understanding of the phenotype encountered.
RESULTS
We show that Gfa2-A2AR KO mice exhibited enhanced MK-801 psychomotor response and decreased working memory; this was accompanied by a disruption of glutamate homeostasis characterized by aberrant GLT-I activity, increased presynaptic glutamate release, NMDA-R 2B subunit upregulation, and increased internalization of AMPA-R. Accordingly, selective GLT-I inhibition or blockade of GluR1/2 endocytosis prevented the psychomotor and cognitive phenotypes in Gfa2-A2AR KO mice, namely in the nucleus accumbens.
CONCLUSIONS
These results show that the dysfunction of astrocytic A2AR, by controlling GLT-I activity, triggers an astrocyte-to-neuron wave of communication resulting in disrupted glutamate homeostasis, thought to underlie several endophenotypes relevant to schizophrenia.
Keywords: Adenosine, Astrocytes, A2AR, GLT-I, NMDA-R, Schizophrenia
Schizophrenia is an intractable mental disorder characterized by a spectrum of positive, negative, and cognitive symptoms (1). Current pharmacotherapy, neurochemical, and neurohistological evidence emphasizes dopaminergic hyperfunction and N-methyl-D-aspartate receptor (NMDA-R) hypofunction as the possible basis of schizophrenia (2). The hypothesis of NMDA-R hypofunction is based on the observation that psychotomimetic agents such as phencyclidine and dizocilpine (MK-801) induce psychotic and cognitive disturbances in humans and animals similar to those observed in schizophrenia patients (3). Indeed, cortical glutamatergic dysfunction is well positioned to explain not only the positive and negative symptoms but also the cognitive decline that occurs in schizophrenia (4,5). This seems to result from the ability of NMDA-R to control the impaired connectivity between multiple brain regions, with a pronounced impact on the dysfunction of the striatal regulation of cortical glutamatergic activity (2,6). The current dopaminergic and glutamatergic models of schizophrenia are focused entirely on neurocentric processes. However, the modified function of astrocytes emerges as an attractive mechanism to bridge several findings related to schizophrenia: astrocytes are modified in the brains of schizophrenia patients (7), they affect the glutamatergic system through the uptake of glutamate (8), and they control integrated brain responses (9). Notably, an up-regulation of glutamate transporter 1 (GLT-I) messenger RNA (10,11) and its function (12,13) has been shown in the brains of schizophrenia patients. In addition, animal studies revealed that GLT-I expression and activity is decreased by antipsychotic drugs (14–17) and increased by psychotomimetics, such as phencyclidine (18), and the overexpression of GLT-I exacerbated deficits on prepulse inhibition (PPI) of the startle response induced by phencyclidine (19).
Adenosine, which exerts potent inhibitory or stimulatory influences on synaptic activity through the activation of adenosine A1 or A2A receptors (A2AR), respectively, is an endogenous modulator of dopamine and glutamate signaling (20). Therefore, adenosine may play an integrative role in controlling the expression of schizophrenia-related psychomotor and cognitive endophenotypes (21,22). A2AR are found mostly in striatal dopaminergic regions where they closely interact with dopamine type 2 receptors (D2R), decreasing their affinity for dopamine (23). However, in schizophrenia patients, a hypoadenosinergic state caused by a decreased production and transport in addition to an increased catabolism of adenosine (21,24–26) may diminish the essential activation of A2AR. Thus, several lines of evidence from animal studies suggest that the use of selective A2AR agonists as an indirect modulation of D2R might represent a novel approach to the treatment of positive symptoms associated with schizophrenia. Indeed, activation of A2AR has been shown to antagonize psychotomimetic-induced motor activity (27,28), whereas the A2AR antagonist caffeine has been reported to exacerbate psychosis in schizophrenic patients (29,30). Additionally, A2AR knockout mice display a reduced PPI and startle habituation (31), as well as blunted psychomotor responses to psychoactive drugs (32). In addition, an increase in the expression and levels of A2AR in schizophrenia has been supported with data from genetic (33) and postmortem (34,35) experiments. The upregulation of striatal A2AR could be a compensatory response triggered by reduced adenosinergic activity, which in turn could produce a hyperdopaminergic state (21,22).
Our recent findings showing that astroglial A2AR tightly regulate GLT-I activity (36,37) suggest a possible astroglial basis for some of the glutamatergic dysfunctions potentially associated with schizophrenia. Here, we report that the selective deletion of astrocytic A2AR triggered pathological hallmarks resembling schizophrenia, namely an enhanced psychomotor response to MK-801 and a decreased working memory.
METHODS AND MATERIALS
For detailed methods see Supplement 1.
CaMKIIα-A2AR KO and Gfa2-A2AR KO mice
The CaMKII-α gene promoter-driven forebrain A2AR knockout (KO) (CaMKIIα-A2AR KO, previously named Fb-A2AR KO) mice were previously characterized (39–42). The glial fibrillary acidic protein (GFAP)-Cre line was obtained from David Gutmann (Washington University School of Medicine, St. Louis, Missouri), using the gfa2 transgene construct (38). Both transgenic Gfa2-cre mice (30) and mice carrying the “floxed” A2AR gene (A2Aflox/flox) (39,40) were back-crossed for 10 to 12 generations to C57BL/6 mice (Charles River; Wilmington, Massachusetts). Gfa2-cre mice were then crossed with non-transgenic (no cre) A2Aflox/flox mice to generate Gfa2-A2AR KO mice and Gfa2-A2AR wild-type (WT) littermates at Michael Schwarzschild’s laboratory (Harvard Medical School) (39) and at the Boston University School of Medicine for this study. Mice were used at 90 days of age.
Fluorescence-activated Cell Sorting and Polymerase Chain Reaction Analysis of Gfa2-A2AR KO Mice
GFAP-positive astrocytes and β-tubulin III-positive neurons were separated by fluorescence-activated cell sorting, followed by polymerase chain reaction analysis for genomic detection of the floxed and recombinant DNA Cre-mediated A2AR deletion (40,41) (see Supplement 1 for details).
Glutamate Release and Uptake and Western Blot Analysis of Synaptosomes and Gliosomes
After cortical brain tissue was homogenized, purified synaptosomes and gliosomes were obtained using a discontinuous Percoll (Sigma-Aldrich, Munich, Germany) gradient (2%, 6%, 15%, and 23%, v/v, of Percoll in a medium containing .32 M sucrose and 1 mM EDTA, pH 7.4), as previously described (36,37). Western blot analysis (36,42), uptake of the non-metabolizable glutamate analogue D-[3H]aspartate (36,37), and release of L-[3H]glutamate (43) in gliosomes or synaptosomes were carried out as previously described.
Biotinylation Assay of Surface Receptors
The density of GluR1/GluR2 subunits of α-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPA-R) present at the plasma membrane surface of synaptosomes was estimated using a biotinylation assay, as previously described (44). A synaptosomal sample was stored for total protein quantification, and the remaining sample was incubated with 1 mg/mL EZ-Link sulfo-NHS-LC-biotin (ThermoFisher Scientific, Waltham, Massachusetts) for 30 minutes at 4°C. After being washed, the biotinylated proteins were extracted with a radioimmunoprecipitation assay buffer with protease inhibitors 20 μg/ml each of chymostatin, leupeptin, antipain and pepstatin (CLAPS) and analyzed by Western blotting.
Drug Administration, Locomotion, Working Memory
MK-801 (.5 mg/kg) was injected intraperitoneally (IP) and the motor activity recorded for the next 3 hours (32,40). Dihydrokainic acid (DHK; 10 mg/kg, IP) or vehicle were administrated 30 minutes before MK-801 injection or memory tests (45,46). SCH58261 was injected IP at an efficacious dose (.1 mg/kg) (47). CGS21680 (50 nM) was added to synaptosomes and gliosomes 30 minutes before L-[3H]glutamate release was probed, as previously described (36,37). The synthetic peptide (Tat-Glur23Y) or the scrambled control peptide (Tat-GluR23S) dissolved in .9% NaCl, were injected in mice (3 μmol/kg, IP) 60 minutes before memory tests and 90 minutes before MK-801 administration (48,49). Tat-GluR23Y or Tat-GluR23S (75 pmol/hemisphere, .5 μL) were bilaterally injected, via an implanted cannula, into the nucleus accumbens (NAc, anterior – posterior = 1.34 mm; medial – lateral = 1.0 mm; dorsal – ventral = 3.8 mm) (40,50). Horizontal locomotor activity was analyzed in polypropylene cages placed into adjustable frames equipped with seven infrared photocell beams (San Diego Instruments) (32). Working and reference memories were evaluated using the Y-maze spontaneous alternation test and the 8-baited-arms radial arm maze test, as previously described (32,42,51).
RESULTS
Selective A2AR Deletion in Astrocytes Triggers Psychomotor and Cognitive Endophenotypes Resembling Schizophrenia
To demonstrate cell type specificity of A2AR gene deletion in Gfa2-A2AR KO mice, we used flow cytometry to separate astrocytes (GFAP-positive cells) and neurons (β-tubulin III-positive cells) from whole-brain lysates of Cre-positive and -negative Gfa2-A2AR KO mice (32). Polymerase chain reaction analyses demonstrated the successful A2AR gene deletion in GFAP-expressing astrocytes sorted from Gfa2-A2AR KO mice (Cre+) but not in sorted neurons (β-tubulin III-positive cells) from these same mice or in either cell types from Cre− control mice (Figure 1A).
Figure 1.
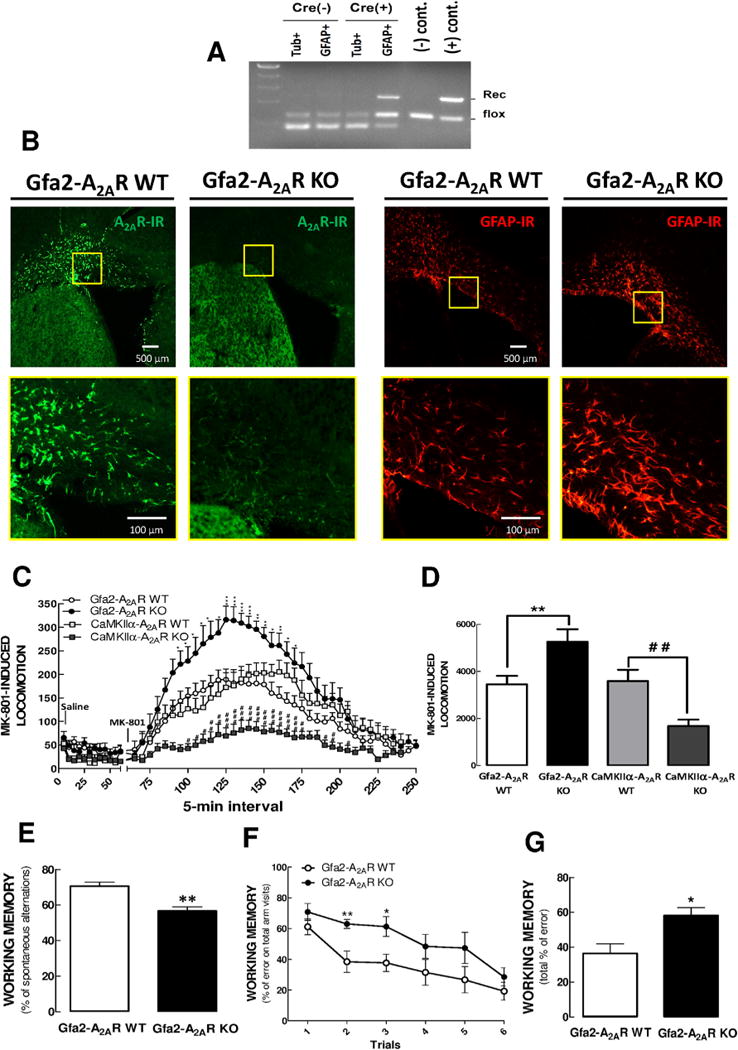
Selective deletion of A2AR in astrocytes triggered psychomotor and cognitive endophenotypes of schizophrenia. (A) Whole-brain astrocytes (GFAP+ cells) and neurons (β-tubulin III1 cells, Tub1) from Gfa2-A2AR KO mice [Cre(+)] and corresponding WT littermates [Cre(−)] were separated by flow cytometry, followed by PCR analysis of genomic DNA of the sorted cells. PCR analysis demonstrated the accuracy of the recombination, showing that the band corresponding to the recombined (“Rec”) allele was present only in astrocytes but not in neurons from Gfa2-A2AR KO mice. (B) Representative images show upregulation of A2AR density in astrocytic-like cells in the cerebral cortex of WT but not Gfa2-A2AR KO mice in response to MPTP treatment. Following MPTP treatment, both GFAP (red staining) and A2AR (green staining) levels were specifically upregulated in cortical astrocytes of WT mice (shown in the first panel from the right and second panel from the left, respectively). Conversely, the Gfa2-A2AR KO mice challenged with MPTP displayed no A2AR immunoreactivity in cortical astrocytes (second panel from the right), despite the increased GFAP immunoreactivity (first panel from the left). (B) Bottom row are 15× higher magnification sections of the boxes in the top row. These findings confirmed the selective deletion of A2AR in astrocytes of MPTP-intoxicated Gfa2-A2AR KO mice. Bar = 500 μm. (C) When mice were injected with MK-801 (.5 mg/kg, IP) and activity was recorded for another 3 hours, Gfa2-A2AR KO mice showed a 15× higher peak response than WT mice; in contrast, MK-801 caused a lower increase of locomotor activity in CaMKIIα-A2AR KO than in WT mice. (D) Graph shows the grouped analysis of the total locomotor activity in the MK-801 post-injection period (75–200 minutes), confirming that the selective deletion of astrocytic A2AR versus neuronal A2AR produced opposite effects on the MK-801-induced hyperlocomotor response. (E) Gfa2-A2AR KO mice displayed a spatial working memory deficit in a classical Y-maze paradigm. (F,G) Working memory performance was consistently impaired in Gfa2-A2AR KO mice compared to WT mice, in an 8-baited radial arms test, as evident by increased working memory errors over 6 consecutive trials (F) or by total number of errors for the entire 6 trials (G). Values are means 6 SEM, and statistical differences were assessed with two-way ANOVA and Bonferroni post-hoc analysis with n = 12 mice per group. *p < .05, **p < .01, GFAP-A2AR WT versus GFAP-A2A R KO mice. #p < .05, ## p < .01, CaMKIIα-A2AR- WT versus CaMKIIα-A2AR KO. DHK, dihydrokainate; KO, knockout; R, receptor; WT, wild type.
To further confirm the specificity of A2AR gene deletion, we treated Gfa2-A2AR KO and WT mice with MPTP, a toxin causing astrogliosis and Parkinsonism, to upregulate A2AR in astrocytes (41), because the levels of A2AR in astrocytes under physiological conditions are below immunohistochemistry detection limits. Seven days after MPTP treatment, A2AR were upregulated in the cerebral cortex of WT mice (where neuronal A2AR expression is low) (20) (Figure 1B, upper panel); on the other hand, the upregulation of A2AR immunoreactivity was largely abrogated in Gfa2-A2AR KO mice (Figure 1B, upper panel), whereas GFAP immunoreactivity was comparable in both genotypes (Figure 1B, lower panel). These results demonstrated the selective deletion of A2AR in GFAP-expressing astrocytes in Gfa2-A2AR KO mice. This allowed dissecting the role of astrocytic A2AR controlling psychomotor-like and cognitive-like endophenotypes associated with schizophrenia, which were modeled in mice by the MK-801-induced locomotor activity as a measure of psychomotor activity, as previously validated (2,52), and alterations of working memory performance as readout of cognitive alterations (53).
We report that the selective A2AR deletion in GFAP-expressing astrocytes exacerbated MK-801 psychomotor activity in Gfa2-A2AR KO compared to that in WT mice (bins [F59,1086 = 31.73, p < .0001]; bins × genotype interaction [F59,1086 = 2.53, p < .0001]. By contrast, A2AR deletion selectively in forebrain neurons in CaMKIIα-A2AR KO mice, decreased MK-801-induced hyperlocomotion compared to that in WT littermates (interaction with genotype: [F47,888 = 3.62, p < .0001]) (Figure 1C). One-way analysis of variance (ANOVA) of the total number of beam breaks in the MK-801 post-injection period (Figure 1D) corroborated this conclusion: Gfa2-A2AR KO mice showed an enhanced response to MK-801 (p < .01), whereas CaMKIIα-A2AR KO mice had a lower psychomotor response to MK-801 (p < .01) compared to their WT littermates.
Gfa2-A2AR KO mice also displayed a reduced working memory score than their WT littermates (n = 20/group, p < .01) when tested in a conventional one-trial Y-maze spontaneous alternation paradigm (Figure 1E). This memory deficit of Gfa2-A2AR-KO mice was selective for working memory because reference memory performance (i.e., both the number of entries and the time spent in the novel arm of a modified two-trial Y-maze test) scores were similar between Gfa2-A2AR KO and WT mice (data not shown). Working memory was also assessed using an 8-baited radial arm maze (Figure 1F,G), where the total working memory error score was significantly higher (p < .01; n = 10) in Gfa2-A2AR KO mice than WT mice. This indicates that astroglial A2AR deletion leads to a selective impairment of working memory, an effect opposite to the deletion of neuronal A2AR (39–40,54).
Deletion of Astrocytic A2AR Upregulates Glutamate Uptake in Gfa2-A2AR KO Mice
Because astroglial A2AR control both the activity and the expression of glutamate transporters (36,37), we next compared glutamate uptake in Gfa2-A2AR and CaMKIIα-A2AR KO mice. D-[3H]Aspartate uptake was significantly larger in gliosomes (astrocyte-enriched plasmalemma vesicles) from Gfa2-A2AR KO (175 ± 12% than from WT mice (p < .001, n = 6) but not from CaMKIIα-A2AR KO mice (Figure 2A,B) nor in synaptosomes (purified nerve terminals) from both transgenic lines (Figure 2C, D). The impact of astroglial A2AR on gliosomal glutamate uptake was selective for Gfa2-A2AR KO mice because the selective A2AR antagonist SCH58261 (100 nM) increased (167.7 ± 8.8%; p < .05, n = 6) glutamate uptake by gliosomes from CaMKIIα-A2AR KO mice (Figure 2B) but lacked any effects in gliosomes from Gfa2-A2AR KO mice (Figure 2A). In accordance with those functional observations, Western blot analysis showed that GLT-I density was increased (138.1 ± 4.4%; p < .001, n = 6) in the cortex of Gfa2-A2AR KO versus that in WT mice (Figure 2E), with no modifications of the densities of either glutamate-aspartate transporter (GLAST) (Figure 2F) or of the astrocytic marker GFAP (Figure 2G).
Figure 2.

Selective deletion of astrocytic A2AR upregulated glutamate uptake activity in Gfa2-A2AR KO mice. Gliosomes and synaptosomes from Gfa2-A2AR KO, CaMKIIα-A2AR KO mice, and corresponding WT mice were prepared after acute (30-minute) IP administration of vehicle or SCH58261 (SCH, .1 mg/kg, IP). Then, glutamate uptake was estimated by assaying D-[3H]aspartate (Asp) uptake for 10 minutes. Glutamate uptake was selectively enhanced in gliosomes (A) but not synaptosomes of Gfa2-A2AR KO mice (B) and was not modified in either gliosomes (B) or in synaptosomes (D) from CaMKIIα-A2AR KO mice. Furthermore, the A2AR antagonist SCH58261 enhanced glutamate uptake only in gliosomes from CaMKIIα-A2AR KO mice (B) but not from Gfa2-A2AR KO mice (B) and was devoid of effect in synaptosomes from both types of mice (C,D). Accordingly, GLT-I density (E) was also significantly increased in total membranes from the cerebral cortex of Gfa2-A2AR KO mice (black columns) compared to those in WT mice (white columns), with no differences in the densities of GLAST (F) or GFAP (G). (E–G) Bars represent the relative immunoreactivity obtained with each primary antibody compared to β-actin immunoreactivity (reference), expressed as percentages of the immunoreactivity found in WT littermates. Data are means 6 SEM from at least six independent mice or experiments. *p < .05, ***p < .001; one-way ANOVA Tukey’s post hoc test was used to compare results with those from naive WT mice. DHK, dihydrokainate; KO, knockout; R, receptor; WT, wild type.
GLT-I Inhibition Reverses MK-801-induced Psychomotor Activity and Working Memory Impairments in Gfa2-A2AR KO Mice
To causally link the hyperactive glutamate uptake and behavioral impairments in Gfa2-A2AR KO mice, we probed the effect of the selective GLT-I inhibitor DHK on psychomotor and working memory abnormalities in Gfa2-A2AR KO mice. DHK (10 mg/kg, IP), given 30 minutes prior to MK-801, selectively blunted the amplified psychomotor response to MK-801 in Gfa2-A2AR KO mice. DHK selectively affected Gfa2-A2AR KO mice but was devoid of effects in the MK-801 response of WT mice (Figure 3A,B) as probed by a two-way ANOVA analysis (DHK treatment × 5-minute bins) showing a significantly attenuated response to MK-801 in Gfa2-A2AR KO mice treated with DHK versus those treated with vehicle (genotype × DHK treatment interaction: [F45,683 = 2.47, p < .0001]). This was corroborated by the analysis of the total number of beam breaks (Figure 3B) showing that Gfa2-A2AR KO mice displayed a significantly amplified response to MK-801 (p < .05) compared to their WT littermates, and DHK-treated Gfa2-A2AR KO mice displayed a significant attenuation of the enhanced psychomotor response to MK-801 compared to vehicle-treated Gfa2-A2AR KO mice (p < .05). Finally, the working memory impairment observed in Gfa2-A2AR KO mice (p < .01) was prevented by DHK (p < .001), whereas DHK was devoid of effects in Gfa2-A2AR WT mice (p = .41) (Figure 3C).
Figure 3.

Selective inhibition of the glutamate transporter GLT-I reverts psychomotor and cognitive abnormalities in Gfa2-A2AR KO mice. (A) Mice were habituated and then treated with dihydrokainate (DHK, 10 mg/kg, IP) 30 minutes before MK-801 (.5 mg/kg, IP), and activity was recorded for another 3 hours. DHK blunted the exacerbated response of Gfa2-A2AR KO mice to MK-801, whereas it was devoid of effects in WT mice. Statistical differences were assessed using Tukey’s post hoc test, applied after one-way ANOVA. (B) Grouped analysis of the total locomotor activity in the MK-801 post-injection period (75–200 minutes) shows DHK blockade of the enhanced psychomotor response to MK-801 in Gfa2-A2AR KO mice. (C) DHK treatment abrogated the impairment of working memory performance displayed by Gfa2-A2AR KO mice, assessed as the spontaneous alternation in a Y-maze test. Data are means ± SEM of the percentages of alternations of total possible alternations. Statistical differences, *p < .05 and **p < .01, comparing the indicated columns, were assessed using Tukey’s post-hoc test applied after one-way ANOVA with n = 8 to 12 mice per group. DHK, dihydrokainate; KO, knockout; MK, N-Methyl-D-aspartate receptor antagonist; R, receptor; WT, wild type.
Astrocytic A2AR Deletion Triggers an Astrocyte-to-Neuron Adaptation with Enhanced Glutamate Release and NR2B Density and Decreased Membrane Levels of GluR1/GluR2
Because the dynamics of the glutamatergic system depend on both the astrocytic clearance and the production of extracellular glutamate, we next investigated basal- and K+-stimulated L-[3H]glutamate release from synaptosomes (i.e., as a neurotransmitter) and from gliosomes (i.e., as a gliotransmitter). In Gfa2-A2AR KO mice, the depolarization of either gliosomes (Figure 4A) or synaptosomes (Figure 4B) triggered an increased outflow of glutamate in accordance with the known ability of synaptosomes, to sustain a vesicular release of glutamate, and gliosomes, to release glutamate through vesicular release and reversed glutamate transport activity (34). Notably, the evoked release of glutamate from synaptosomes (Figure 4B) but not from gliosomes (Figure 4A) was larger in Gfa2-A2AR KO than in WT littermates, as probed by a two-way ANOVA (genotype, [F1,46 = 47.05, p < .0001]). This became even more evident in synaptosomes in the presence of the A2AR agonist CGS21680 (50 nM; treatment, [F3,46 = 18.39, p < .0001]) (Figure 4B), with no significant interaction between genotype and treatment (p = .232). On the other hand, in gliosomes (Figure 4A), CGS21680 only enhanced the basal outflow of glutamate from WT mice (p < .05, n = 6) (Figure 4A) but not from Gfa2-A2AR KO mice, with no significant effect of genotype (p = .111) but with significant effect of CGS21680 or evoked treatment ([F3,46 = 6.33, p = .011], two-way ANOVA).
Figure 4.

Gfa2-A2AR KO mice displayed adaptive changes in glutamatergic synapses, with increased evoked release of glutamate from nerve terminals, enhanced density of NR2B, and decreased membrane surface levels of GluR1/GluR2. Release of basal or depolarization Ca21-dependent–evoked release of L-[3H]glutamate from cerebral cortical gliosomes (A) or synaptosomes (B) was evaluated in the absence or presence of the A2AR agonist CGS21680 (CGS, 50 nM). Depolarizing conditions triggered an evoked release from both the gliosomes (A) and the synaptosomes (B), which was larger in synaptosomes from Gfa2-A2AR KO mice, the only preparation where CGS21680 exacerbated the evoked release of glutamate. Comparison of Western blots of the densities of NMDA-R-NR1 (C), NMDA-R-NR2A (D), and NMDA-R-NR2B (E) in synaptosomal membranes and those of AMPA-R-GluR1 (F) and AMPA-R-GluR2 (G) in crude membranes and biotinylated extracts from synaptosomes from the cortex of Gfa2-A2AR KO and WT mice. Density ratios for AMPA-R subunits were determined by dividing the surface intensity by the total intensity. Data are means ± SEM from six independent mice or experiments performed in triplicate. *p < .05, **p < .01, compared with basal conditions. #p < .05, ##p < .01 compared with WT mice, one-way ANOVA, Tukey’s post-hoc test. DHK, dihydrokainate; KO, knockout; R, receptor; WT, wild type.
We next investigated the density of AMPA-R and NMDA-R, because NMDA-R levels are altered in some schizophrenia experimental models (2,3). Western blot analysis showed that, although there were no significant changes in the levels of NR1 (Figure 4C) and NR2A (Figure 4D) subunits, the density of NR2B subunits was significantly larger in cortical membranes of Gfa2-A2AR KO mice (p < .01) (Figure 4E). In accordance with the ability of NR2B-containing NMDA-R to control AMPA-R trafficking via endocytosis (48,49,55), biotinylation assays showed a significant decrease (p < .05, n = 3) of the surface density of AMPA GluR1/GluR2 proteins in crude cortical synaptosomes of Gfa2-A2AR KO mice (Figure 4F,G).
Thus, deletion of astroglial A2AR triggered a set of modifications of different glutamatergic components involving a neural adaptation, with a parallel increase of synaptic glutamate release from synaptic terminals, an upregulation of NR2B-containing NDMA-R and internalization of AMPA GluR1/GluR2.
Blocking the Regulated Endocytosis of AMPA-R with Tat-Glur23Y Blunts the Exacerbation of MK-801-induced Psychomotor Activity and Reverses the Working Memory Impairment in Gfa2-A2AR KO Mice
To establish a causal role of GluR2 endocytosis in mediating psychomotor dysfunction in Gfa2-A2AR KO mice, we tested the impact of Tat-GluR23Y, a peptide inhibiting GluR2 endocytosis (48,49,56), using the scrambled peptide Tat-GluR23S as control. We first confirmed that injection of Tat-GluR23Y, but not of Tat-GluR23S, reverted the decrease of biotin-labeled GluR1 (p < .001) (Figure 5A) and GluR2 (p < .05) (Figure 5B) levels in cortical synaptosomes from Gfa2-A2AR KO but not WT littermates, 60 minutes after peptide injection. These results showed the highly efficient ability of Tat-GluR23Y to control the regulated endocytosis of AMPA-R in cortical synapses. Tat-GluR23Y, but not Tat-GluR23S, selectively blunted the abnormally enhanced MK-801-induced psychomotor response in Gfa2-A2AR KO mice although lacking any effects in Gfa2-A2AR WT mice (Figure 5C). A two-way ANOVA of the number of beam breaks confirmed a significant reduction of MK-801 response in Gfa2-A2AR KO mice treated with Tat-GluR23Y versus control Tat-GluR23S (interaction with Tat-GluR treatment: [F71,1079 = 8.02, p < .001], n = 12) (Figure 5C). This was further supported by a one-way ANOVA of the total number of beam breaks in MK-801 post-injection (Figure 5D): the amplified response to MK-801 in Gfa2-A2AR KO mice was significantly attenuated by Tat-GluR23Y treatment in contrast with the lack of effect of the scrambled Tat-GluR23S peptide (p < .001). The intraperitoneal pretreatment of Gfa2-A2AR KO mice with Tat-GluR23Y, but not with the control Tat-GluR23S peptide, also prevented working memory impairment (Figure 5F) in the Y maze task (p < .001).
Figure 5.
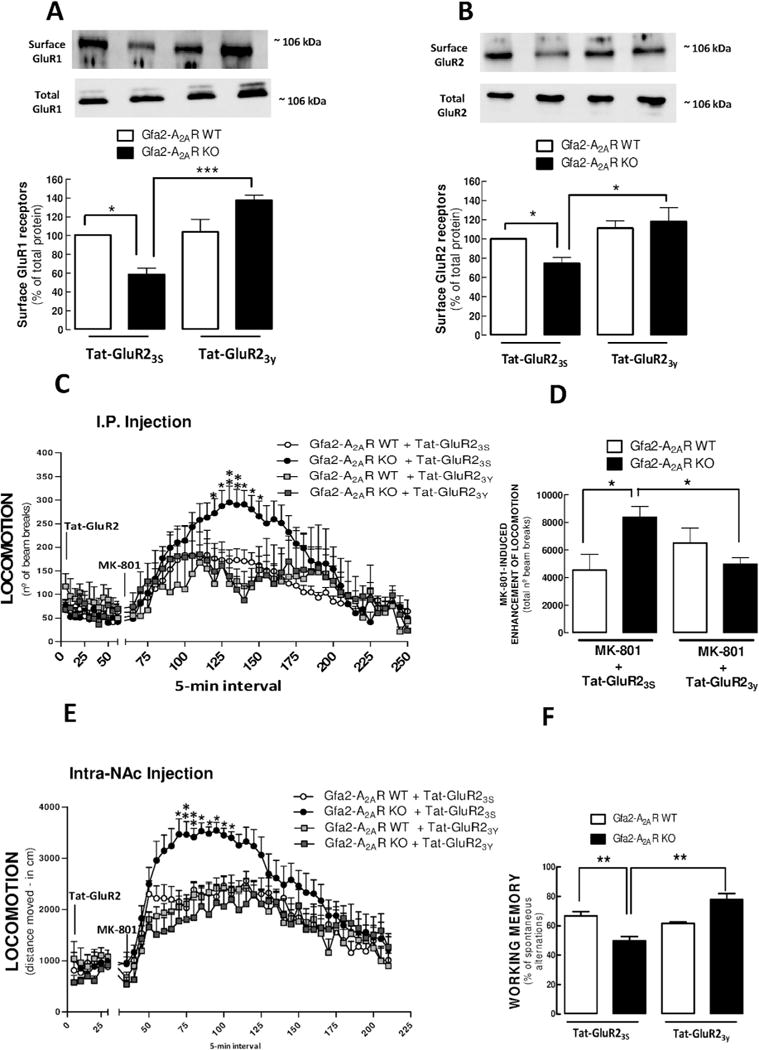
Blockade of the regulated endocytosis of AMPA-R GluR2 subunits with Tat-Glur23Y reversed the working memory impairment and blunted the exacerbation of MK-801-induced psychomotor activity in Gfa2-A2AR KO mice, namely in the nucleus accumbens (NAc). (A,B) Tat-GluR23Y and the scrambled Tat-GluR23S control peptide were injected IP in Gfa2-A2AR KO and WT mice 60 minutes before they were sacrificed for preparation of cerebral cortical synaptosomes, followed by biotinylation and Western blot analysis. Decrease of the plasma membrane levels of GluR1 (A) and GluR2 (B) in Gfa2-A2AR KO mice was reverted by Tat-Glur23Y but unaffected by Tat-GluR23S. (C–F) Tat-GluR23Y and Tat-GluR23S were IP injected in Gfa2-A2AR KO and WT mice 90 minutes before MK-801 administration or 60 minutes before Y-maze testing. (C) Gfa2-A2AR KO mice treated with Tat-GluR23Y did not display the exacerbated response to MK-801, observed in the presence of Tat-GluR23S. (D) Grouped analysis of the total locomotion in the MK-801 post-injection period confirmed that Tat-Glur23Y prevented the enhanced MK-801 response in Gfa2-A2AR KO mice. (E) Gfa2-A2AR KO mice bilaterally injected in the NAc with Tat-GluR23Y did not display the significantly increased response to MK-801 observed with intra-NAc injection of Tat-GluR23S. (F) Data show that the impairment of working memory performance observed in Gfa2-A2AR KO mice subject to a conventional Y-maze test was reverted by Tat-Glur23Y but not by Tat-GluR23S. Values are the means ± SEM of n = 8 to 12 mice per group. *p < .05, **p < .01, ***p < .001, comparing the indicated columns in the bar graphs (A,B,D,F) and *p < .05, **p < .01 comparing Gfa2-A2AR KO versus WT mice in the time course graphs (C,E), using two-way ANOVA followed by Tukey’s post hoc test. DHK, dihydrokainate; KO, knockout; MK, N-Methyl-D-aspartate receptor antagonist; R, receptor; WT, wild type.
Previous studies have identified the NAc as the key structure where AMPA-R internalization critically affects psychostimulant activity (48) and regulates cortical glutamatergic activity in schizophrenia (6,57). Accordingly, we found that the intra-accumbens bilateral injection of Tat-GluR23Y, but not Tat-GluR23S inactive peptide, prevented the exacerbated MK-801-induced psychomotor response in Gfa2-A2AR KO mice (Figure 5E) to a level similar to that of WT mice pretreated with either Tat-GluR23Y or Tat-GluR23S peptide (Figure 5E).
Results suggest that modification of the regulated endocytosis of GluR1/2-containing AMPA-R may be a key process responsible for psychomotor and working memory dysfunctions found in Gfa2-A2AR KO mice and imply the NAc as a significant brain locus for this process.
DISCUSSION
The present study shows that a mouse transgenic line with a selective deletion of A2AR in astrocytes exhibits motor and memory dysfunctions relevant to schizophrenia, namely the exacerbation of MK-801-induced psychomotor response and a decrease of working memory. This was paralleled by neurochemical alterations in glutamatergic synapses typified by an enhanced astrocytic uptake of glutamate, an enhanced release of glutamate from nerve terminals, an upregulation of NR2B-containing NMDA receptors, and an internalization of AMPA receptors. The causal relationship between these modifications of the glutamatergic network and the cognitive and psychomotor dysfunctions upon genetic deletion of astrocytic A2AR is heralded by the ability of inhibitors of glutamate uptake and of AMPA receptor internalization to revert the behavioral alterations emerging upon deletion of astrocyte A2AR. Although it still remains to be directly defined if the adenosine system is altered in schizophrenia or if A2AR polymorphisms are associated with this disease, it is worth noting that our present findings are in line with our adenosine hypofunction hypothesis of schizophrenia (21,22), which is based on findings that the enhanced production of adenosine ameliorates positive and cognitive endophenotypes of schizophrenia (26). Therefore, a possible impact of A2AR on schizophrenia-like behavioral abnormalities is unlikely to be limited to the ability of A2AR to interfere with dopamine D2 receptors, as previously noted (27,28); instead, astroglial A2AR may directly affect glutamate transport activity as a primary target to drive an astrocyte-to-neuron communication, leading ultimately to the deregulated glutamate neurotransmission implicated in schizophrenia.
We suggest that a primary outcome of astroglial A2AR deletion involves the upregulation of glutamate transport, namely GLT-I, in GFAP-expressing astrocytes. This hypothesis is supported by the upregulation of D-[3H]aspartate uptake selectively in gliosomes of Gfa2-A2AR-KO mice, which extends our recent demonstration of a physical and functional interaction between A2AR and the Na+-K+-ATPase-GLT-I protein complex as a mechanistic basis for the control of astroglial GLT-I activity by A2AR (37). Interestingly, studies with schizophrenic patients showed an increase of glutamate uptake sites (10–13). In addition, GLT-I activity and expression is increased by psychotomimetics (18) and decreased by antipsychotic drugs both in vivo (14–16) and in vitro (17). Finally, GLT-I overexpression exacerbates deficits on PPI of the startle response induced by phencyclidine (19). Therefore, although not without its share of disagreement (58), there are a fair amount of data linking GLT-I dysfunction to schizophrenia (59). The identification of astrocytic A2AR as a critical trigger and astrocytic GLT-I as primary downstream target underlying motor and memory dysfunctions illustrates the critical role that astrocytes may operate in the development of some of the brain modifications observed in schizophrenia (7). This shift toward modified astrocytic function as primers of neurological dysfunction is in line with the increased awareness of the importance of astrocytes to control higher brain functions (9), typified by the observations that altering astrocytic functions such as ATP or glutamate release (56,60), glycogenolysis or lactate transport (61), or the density of aquaporin-4 channels (62) impair cognitive processes.
The present results also indicate that both neuronal and astrocytic A2AR, despite their low abundance in cortical areas (20), have evident but opposite neuromodulatory effects on memory: thus, the suppression of neuronal A2AR activity is procognitive (40,42,47,54), whereas the deletion of astrocyte A2AR potentiated behavioral impairments. This suggests a reverse but coordinated function of different populations of A2AR to fine-tune information processing in neuronal networks. This joins previous reports showing that A2AR can modulate behavior in different ways depending on the brain area, cell type and cell compartment where they are expressed (40,41), which is probably related with different sources of adenosine fulfilling different roles as modulators and homeostatic regulators of synaptic and brain function (63). In this respect, it is worth noting that a recent study also found that the selective interference with another modulation system typically associated with neuronal adaptive control, cannabinoid CB1R, also impacts working memory in opposite manners according to whether neuronal or astrocytic CB1R are manipulated (56), regardless of the fact that astrocyte CB1R levels are considerably lower than neuronal, as with A2AR. This parallel analogy suggests that both A2AR and CB1R are powerful regulators of astrocyte function, normally kept at low levels owing to their remarkable impact on glutamatergic circuitry, thus prompting the hypothesis that homeostatic mechanisms sensed by astrocytes are designed to decisively control the functional integration between brain circuits.
In fact, we found that the astrocytic A2AR-induced modification of GLT-I function was associated with several striking neuronal adaptations at the glutamatergic circuitry, typified by an increase of synaptic glutamate release, an enhanced density of NR2B subunits of NMDA-R, and an increased internalization of AMPA-R. Importantly we found a selective upregulation of NR2B-containing NMDA-R rather than of other NMDA-R isoforms, which may fit in the global hypothesis of glutamatergic dysfunction of schizophrenia because NR2B-containing NMDA-R often trigger responses qualitatively opposite to other glutamate receptors (64). In fact, NR2B-containing NMDA-R have been associated with the control of both working memory (65) and the response to drugs of abuse (66), as well as with AMPA-R internalization (49,56), which underlies the deterioration of working memory performance found in schizophrenia patients (57). Arguing for a causal relationship between these neurochemical alterations and the abnormal behavior of Gfa2-A2AR KO mice were our observations that the inhibition of either the activity of astrocytic glutamate transporters or of the regulated AMPA-R internalization in neurons blunted the abnormal working memory and the amplified psychomotor response to MK-801 in Gfa2-A2AR KO mice. Our findings also propose that an altered rate of AMPA-R internalization in a defined brain locus (NAc) may be a critical process underlying psychomotor and cognitive dysfunction underlying schizophrenia-associated processes (6,48,57). Thus, alterations of AMPA-R trafficking restricted to the NAc were sufficient to account for the enhanced psychomotor responses to MK-801 administration, an animal pharmacological model for cognitive symptoms of schizophrenia (52,53). In spite of this evidence linking alterations of the homeostasis of glutamatergic synapses to behavioral alterations upon disruption of astrocytic A2AR, several aspects still remain to be characterized in greater detail. In fact, we did not determine an accurate casual and temporal sequence between the neurochemical and behavioral modifications. Furthermore, glutamate was not directly measured but only indirectly estimated. Additionally, there are still critical aspects related to the specificity of the effect of astrocytic A2AR on the relative activity GLT-I and of other glutamate transporters that may shed light on this striking ability of DHK to selective restore behavioral deficits in Gfa2-A2AR KO mice. Finally, it is important to recognize that we focused this study on alterations of glutamatergic synapses, and we cannot rule out the eventual involvement of the gamma-aminobutyric acid (GABA) ergic system (GABA release and transport mechanisms) on the endophenotypes observed in the Gfa2-A2AR KO mice, because alterations of the GABAergic system are also associated with schizophrenia (67), particularly when considering recent evidence showing a role of A1 and A2A receptors in the control of GABA transport in astrocytes (68). Finally, our tentative association of the cognitive and psychomotor alterations of Gfa2-A2AR KO mice with schizophrenia also need to be tempered by the heterogeneous causes underlying schizophrenia, which still cannot be faithfully recapitulated by animal models, each with its own limitations (53). The relatively early (between E18 and P5) induction of the Gfa2-induced Cre recombination (see Supplementary data) may suggest that Gfa2-A2AR KO mice could fit into a developmental model of cognitive dysfunction present in prodromal stages of schizophrenia (4,5). Further support of Gfa2-A2AR KO mice as a possible model of schizophrenia will require determining the specificity of the alterations of psychomotor activity by testing other NMDA-R antagonists and psychostimulants applied both acutely and chronically.
Supplementary Material
Acknowledgments
The work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke and National Institute of Mental Health grants NS041083-11, NS073947, and MH083973; National Alliance for Research on Schizophrenia and Depression, Defense Advanced Research Projects Agency grants 09-68-ESR-FP-010 and W911NF-10-1-0059; the Portuguese Foundation for Science and Technology (FCT, PTDC/SAU-NSC/122254/2010) and Quadro de Referência Estratégico Nacional (CENTRO-07-ST24-FEDER-002006).
Drs. Matos and Augusto acknowledge FCT/Fundo Social Europeu fellowship awards SFRH/BD/36289/2007 and SFRH/BD/47824/2008 and support from Boston University Clinical and Translational Science Institute.
Footnotes
DISCLOSURES
All authors declare no biomedical financial interests or potential conflicts of interest.
Supplementary material cited in this article is available online at http://dx.doi.org/10.1016/j.biopsych.2015.02.026.
References
- 1.Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;5:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 2.Moghaddam B, Javitt D. From revolution to evolution: The glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Field JR, Walker AG, Conn PJ. Targeting glutamate synapses in schizophrenia. Trends Mol Med. 2011;3:689–698. doi: 10.1016/j.molmed.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Snellenberg JX. Working memory and long-term memory deficits in schizophrenia: Is there a common substrate? Psychiatry Res. 2009;174:89–96. doi: 10.1016/j.pscychresns.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Kristiansen LV, Huerta I, Beneyto M, Meador-Woodruff JH. NMDA receptors and schizophrenia. Curr Opin Pharmacol. 2007;7:48–55. doi: 10.1016/j.coph.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 6.Mikell CB, McKhann GM, Segal S, McGovern RA, Wallenstein MB, Moore H. The hippocampus and nucleus accumbens as potential therapeutic targets for neurosurgical intervention in schizophrenia. Stereotact Funct Neurosurg. 2009;87:256–265. doi: 10.1159/000225979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takahashi N, Sakurai T. Roles of glial cells in schizophrenia: Possible targets for therapeutic approaches. Neurobiol Dis. 2013;53:49–60. doi: 10.1016/j.nbd.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 8.Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- 9.Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH. Expression of excitatory amino acid transporter transcripts in the thalamus of subjects with schizophrenia. Am J Psychiatry. 2001;158:1393–1399. doi: 10.1176/appi.ajp.158.9.1393. [DOI] [PubMed] [Google Scholar]
- 11.Matute C, Melone M, Vallejo-Illarramendi A, Conti F. Increased expression of the astrocytic glutamate transporter GLT-I in the prefrontal cortex of schizophrenics. Glia. 2005;49:451–500. doi: 10.1002/glia.20119. [DOI] [PubMed] [Google Scholar]
- 12.Deakin JF, Slater P, Simpson MD, Gilchrist AC, Skan WJ, Royston MC, et al. Frontal cortical and left temporal glutamatergic dysfunction in schizophrenia. J Neurochem. 1989;52:1781–1786. doi: 10.1111/j.1471-4159.1989.tb07257.x. [DOI] [PubMed] [Google Scholar]
- 13.Simpson MD, Slater P, Deakin JF. Comparison of glutamate and gamma-aminobutyric acid uptake binding sites in frontal and temporal lobes in schizophrenia. Biol Psychiatry. 1998;15:423–427. doi: 10.1016/s0006-3223(98)00077-8. [DOI] [PubMed] [Google Scholar]
- 14.Schneider JS, Wade T, Lidsky TI. Chronic neuroleptic treatment alters expression of glial glutamate transporter GLT-1 mRNA in the striatum. Neuroreport. 1998;9:133–136. doi: 10.1097/00001756-199801050-00026. [DOI] [PubMed] [Google Scholar]
- 15.Melone M, Vitellaro-Zuccarello L, Vallejo-Illarramendi A, Perez-Samartin A, Matute C, Cozzi A, et al. The expression of glutamate transporter GLT-1 in the rat cerebral cortex is down-regulated by the antipsychotic drug clozapine. Mol Psychiatry. 2001;6:380–386. doi: 10.1038/sj.mp.4000880. [DOI] [PubMed] [Google Scholar]
- 16.Bragina L, Melone M, Fattorini G, Torres-Ramos M, Vallejo-Illarramendi A, Matute C, et al. GLT-1 down-regulation induced by clozapine in rat frontal cortex is associated with synaptophysin upregulation. J Neurochem. 2006;99:134–141. doi: 10.1111/j.1471-4159.2006.04030.x. [DOI] [PubMed] [Google Scholar]
- 17.Vallejo-Illarramendi A, Torres-Ramos M, Melone M, Conti F, Matute C. Clozapine reduces GLT-1 expression and glutamate uptake in astrocyte cultures. Glia. 2005;50:276–279. doi: 10.1002/glia.20172. [DOI] [PubMed] [Google Scholar]
- 18.Fattorini G, Melone M, Bragina L, Candiracci C, Cozzi A, Pellegrini-Giampietro DE, et al. GLT-I expression and Glu uptake in rat cerebral cortex are increased by phencyclidine. Glia. 2008;56:1320–1327. doi: 10.1002/glia.20700. [DOI] [PubMed] [Google Scholar]
- 19.Bellesi M, Melone M, Gubbini A, Battistacci S, Conti F. GLT-I upregulation impairs prepulse inhibition of the startle reflex in adult rats. Glia. 2009;57:703–713. doi: 10.1002/glia.20798. [DOI] [PubMed] [Google Scholar]
- 20.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. Int Rev Neurobiol. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 21.Boison D, Singer P, Shen HY, Feldon J, Yee BK. Adenosine hypothesis of schizophrenia—Opportunities for pharmacotherapy. Neuropharmacology. 2012;62:1527–1543. doi: 10.1016/j.neuropharm.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rial D, Lara DR, Cunha RA. The adenosine neuromodulation system in schizophrenia. Int Rev Neurobiol. 2014;119:395–449. doi: 10.1016/B978-0-12-801022-8.00016-7. [DOI] [PubMed] [Google Scholar]
- 23.Ferré S, Quiroz C, Woods AS, Cunha RA, Popoli P, Ciruela F, et al. An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr Pharm Des. 2008;14:1468–1474. doi: 10.2174/138161208784480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shan D, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE. Expression of equilibrative nucleoside transporter type 1 protein in elderly patients with schizophrenia. Neuroreport. 2012;23:224–227. doi: 10.1097/WNR.0b013e3283500987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aliagas E, Villar-Menéndez I, Sévigny J, Roca M, Romeu M, Ferrer I, et al. Reduced striatal ecto-nucleotidase activity in schizophrenia patients supports the “adenosine hypothesis”. Purinergic Signal. 2014;9:599–608. doi: 10.1007/s11302-013-9370-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen HY, Singer P, Lytle N, Wei CJ, Lan JQ, Williams-Karnesky RL, et al. Adenosine augmentation ameliorates psychotic and cognitive endophenotypes of schizophrenia. J Clin Invest. 2012;122:2567–2577. doi: 10.1172/JCI62378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferré S. Adenosine-dopamine interactions in the ventral striatum. Implications for the treatment of schizophrenia. Psychopharmacology (Berl) 1997;133:107–120. doi: 10.1007/s002130050380. [DOI] [PubMed] [Google Scholar]
- 28.Rimondini R, Ferré S, Ogren SO, Fuxe K. Adenosine A2A agonists: A potential new type of atypical antipsychotic. Neuropsychopharmacology. 1997;17:82–91. doi: 10.1016/S0893-133X(97)00033-X. [DOI] [PubMed] [Google Scholar]
- 29.Hughes JR, McHugh P, Holtzman S. Caffeine and schizophrenia. Psychiatr Serv. 1998;49:1415–1417. doi: 10.1176/ps.49.11.1415. [DOI] [PubMed] [Google Scholar]
- 30.Peng PJ, Chiang KT, Liang CS. Low-dose caffeine may exacerbate psychotic symptoms in people with schizophrenia. J Neuropsychiatry Clin Neurosci. 2014;26:E41. doi: 10.1176/appi.neuropsych.13040098. [DOI] [PubMed] [Google Scholar]
- 31.Wang JH, Short J, Ledent C, Lawrence AJ, van den Buuse M. Reduced startle habituation and prepulse inhibition in mice lacking the adenosine A2A receptor. Behav Brain Res. 2003;143:201–207. doi: 10.1016/s0166-4328(03)00036-6. [DOI] [PubMed] [Google Scholar]
- 32.Chen JF, Beilstein M, Xu YH, Turner TJ, Moratalla R, Standaert DG, et al. Selective attenuation of psychostimulant-induced behavioral responses in mice lacking A2A adenosine receptors. Neuroscience. 2000;97:195–204. doi: 10.1016/s0306-4522(99)00604-1. [DOI] [PubMed] [Google Scholar]
- 33.Deckert J, Nothen MM, Bryant SP, Schuffenhauer S, Schofield PR, Spurr NK, et al. Mapping of the human adenosine A2a receptor gene: relationship to potential schizophrenia loci on chromosome 22q and exclusion from the CATCH 22 region. Hum Genet. 1997;99:326–328. doi: 10.1007/s004390050366. [DOI] [PubMed] [Google Scholar]
- 34.Kurumaji A, Toru M. An increase in [3H]CGS21680 binding in the striatum of postmortem brains of chronic schizophrenics. Brain Res. 1998;808:320–323. doi: 10.1016/s0006-8993(98)00840-3. [DOI] [PubMed] [Google Scholar]
- 35.Deckert J, Brenner M, Durany N, Zöchling R, Paulus W, Ransmayr G, et al. Up-regulation of striatal adenosine A2A receptors in schizophrenia. Neuroreport. 2003;14:313–316. doi: 10.1097/00001756-200303030-00003. [DOI] [PubMed] [Google Scholar]
- 36.Matos M, Augusto E, Santos-Rodrigues A, Schwarzschild MA, Chen JF, Cunha RA, et al. Adenosine A2A receptors modulate glutamate uptake in cultured astrocytes and gliosomes. Glia. 2012;60:702–716. doi: 10.1002/glia.22290. [DOI] [PubMed] [Google Scholar]
- 37.Matos M, Augusto E, Agostinho P, Cunha RA, Chen JF. Antagonistic interaction between adenosine A2A receptors and Na+/K+-ATPase-α2 controlling glutamate uptake in astrocytes. J Neurosci. 2013;33:18492–18502. doi: 10.1523/JNEUROSCI.1828-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bastia E, Xu YH, Scibelli AC, Day YJ, Linden J, Chen JF, et al. A crucial role for forebrain adenosine A2A receptors in amphetamine sensitization. Neuropsychopharmacology. 2005;30:891–900. doi: 10.1038/sj.npp.1300630. [DOI] [PubMed] [Google Scholar]
- 40.Shen HY, Coelho JE, Ohtsuka N, Canas PM, Day YJ, Huang QY, et al. A critical role of the adenosine A2A receptor in extrastriatal neurons in modulating psychomotor activity as revealed by opposite phenotypes of striatum and forebrain A2A receptor knock-outs. J Neurosci. 2008;19:2970–2975. doi: 10.1523/JNEUROSCI.5255-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu L, Shen HY, Coelho JE, Araújo IM, Huang QY, Day YJ, et al. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Ann Neurol. 2008;63:338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]
- 42.Augusto E, Matos M, Sévigny J, El-Tayeb A, Bynoe MS, Müller CE, et al. Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J Neurosci. 2013;33:11390–11399. doi: 10.1523/JNEUROSCI.5817-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Milanese M, Bonifacino T, Zappettini S, Usai C, Tacchetti C, Nobile M, et al. Glutamate release from astrocytic gliosomes under physiological and pathological conditions. Int Rev Neurobiol. 2009;85:295–318. doi: 10.1016/S0074-7742(09)85021-6. [DOI] [PubMed] [Google Scholar]
- 44.Schenk U, Verderio C, Benfenati F, Matteoli M. Regulated delivery of AMPA receptor subunits to the presynaptic membrane. EMBO J. 2003;22:558–568. doi: 10.1093/emboj/cdg059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Namura S, Maeno H, Takami S, Jiang XF, Kamichi S, Wada K, et al. Inhibition of glial glutamate transporter GLT-I augments brain edema after transient focal cerebral ischemia in mice. Neurosci Lett. 2002;324:117–120. doi: 10.1016/s0304-3940(02)00193-3. [DOI] [PubMed] [Google Scholar]
- 46.John CS, Smith KL, Van’t Veer A, Gompf HS, Carlezon WA, Cohen BM, et al. Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology. 2012;37:2467–2475. doi: 10.1038/npp.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Canas PM, Porciúncula LO, Cunha GMA, Silva CG, Machado NJ, Oliveira JMA, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β-amyloid peptides via p38 mitogen-activated protein kinase pathway. J Neurosci. 2009;29:14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brebner K, Wong TP, Liu L, Liu Y, Campsall P, Gray S, et al. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science. 2005;310:1340–1343. doi: 10.1126/science.1116894. [DOI] [PubMed] [Google Scholar]
- 49.Ge Y, Dong Z, Bagot RC, Howland JG, Phillips AG, Wong TP, et al. Hippocampal long-term depression is required for the consolidation of spatial memory. Proc Natl Acad Sci U S A. 2010;107:16697–16702. doi: 10.1073/pnas.1008200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yee BK, Singer P, Chen JF, Feldon J, Boison D. Transgenic overexpression of adenosine kinase in brain leads to multiple learning impairments and altered sensitivity to psychomimetic drugs. Eur J Neurosci. 2007;26:3237–3252. doi: 10.1111/j.1460-9568.2007.05897.x. [DOI] [PubMed] [Google Scholar]
- 51.Singer P, McGarrity S, Shen HY, Boison D, Yee BK. Working memory and the homeostatic control of brain adenosine by adenosine kinase. Neuroscience. 2012;213:81–92. doi: 10.1016/j.neuroscience.2012.03.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blot K, Bai J, Otani S. The effect of non-competitive NMDA receptor antagonist MK-801 on neuronal activity in rodent prefrontal cortex: An animal model for cognitive symptoms of schizophrenia. J Physiol Paris. 2013;107:448–451. doi: 10.1016/j.jphysparis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 53.Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol. 2011;164:1162–1194. doi: 10.1111/j.1476-5381.2011.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wei CJ, Singer P, Coelho J, Boison D, Feldon J, Yee BK, et al. Selective inactivation of adenosine A2A receptors in striatal neurons enhances working memory and reversal learning. Learn Mem. 2011;18:459–474. doi: 10.1101/lm.2136011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tigaret CM, Thalhammer A, Rast GF, Specht CG, Auberson YP, Stewart MG, et al. Subunit dependencies of N-methyl-D-aspartate (NMDA) receptor-induced alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor internalization. Mol Pharmacol. 2006;69:1251–1259. doi: 10.1124/mol.105.018580. [DOI] [PubMed] [Google Scholar]
- 56.Han J, Kesner P, Metna-Laurent M, Duan T, Xu L, Georges F, et al. Acute cannabinoids impair working memory through astroglial CB1 receptor modulation of hippocampal LTD. Cell. 2012;48:1039–1050. doi: 10.1016/j.cell.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 57.Hammond JC, McCullumsmith RE, Funk AJ, Haroutunian V, Meador-Woodruff JH. Evidence for abnormal forward trafficking of AMPA receptors in frontal cortex of elderly patients with schizophrenia. Neuropsychopharmacology. 2010;35:2110–2119. doi: 10.1038/npp.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shan D, Yates S, Roberts RC, McCullumsmith RE. Update on the neurobiology of schizophrenia: A role for extracellular microdomains. Minerva Psychiatr. 2012;53:233–249. [PMC free article] [PubMed] [Google Scholar]
- 59.Nanitsos Moghaddam B, Javitt EK, Nguyen KT, St’astný F, Balcar VJ. Glutamatergic hypothesis of schizophrenia: Involvement of Na+/K+-dependent glutamate transport. J Biomed Sci. 2005;12:975–984. doi: 10.1007/s11373-005-9015-0. [DOI] [PubMed] [Google Scholar]
- 60.Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, et al. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, et al. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144:810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li YK, Wang F, Wang W, Luo Y, Wu PF, Xiao JL, et al. Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology. 2012;37:1867–1878. doi: 10.1038/npp.2012.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: Different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- 64.Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA, Roche KW. Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep. 2013;3:607–614. doi: 10.1016/j.celrep.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang M, Yang Y, Wang CJ, Gamo NJ, Jin LE, Mazer JA, et al. NMDA receptors subserve persistent neuronal firing during working memory in dorsolateral prefrontal cortex. Neuron. 2013;77:736–749. doi: 10.1016/j.neuron.2012.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, et al. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12:602–610. doi: 10.1038/nn.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lewis DA. Inhibitory neurons in human cortical circuits: Substrate for cognitive dysfunction in schizophrenia. Curr Op Neurobiol. 2013;26C:22–26. doi: 10.1016/j.conb.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cristóvão-Ferreira S, Navarro G, Brugarolas M, Pérez-Capote K, Vaz SH, Fattorini G, et al. A1R-A2AR heteromers coupled to Gs and Gi/0 proteins modulate GABA transport into astrocytes. Purinergic Signal. 2013;9:433–449. doi: 10.1007/s11302-013-9364-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.