

Abstract
Macrophage accumulation is one of the hallmarks of progressive kidney disease. Tissue-type plasminogen activator (tPA) is known to promote macrophage infiltration and renal inflammation during chronic kidney injury. However, the underlying mechanism remains largely unknown. We examined the role of tPA in macrophage motility in vivo by tracking fluorescence-labeled bone marrow–derived macrophages, and found that tPA-deficient mice had markedly fewer infiltrating fluorescence-labeled macrophages than the wild-type (WT) mice. Experiments in bone marrow chimeric mice further demonstrated that myeloid cells are the main source of endogenous tPA that promotes macrophage migration. In vitro studies showed that tPA promoted macrophage motility through its CD11b-mediated protease-independent function; and focal adhesion kinase (FAK), Rac-1, and NF-κB were indispensable to tPA-induced macrophage migration as either infection of FAK dominant-negative adenovirus or treatment with a Rac-1–specific inhibitor or NF-κB inhibitor abolished the effect of tPA. Moreover, ectopic FAK mimicked tPA and induced macrophage motility. tPA also activated migratory signaling in vivo. The accumulation of phospho-FAK–positive CD11b macrophages in the obstructed kidneys from WT mice was clearly attenuated in tPA knockout mice, which also displayed lower Rac-1 activity than their WT counterparts. Therefore, our results indicate that myeloid-derived tPA promotes macrophage migration through a novel signaling cascade involving FAK, Rac-1, and NF-κB.
Regardless of the initial causes, most interstitial and glomerular renal diseases are characterized by macrophage accumulation.1, 2 Sustained macrophage infiltration in the diseased kidneys eventually becomes pathological, resulting in irreversible fibrosis, tissue destruction, and progressive chronic kidney disease (CKD).1 Although the expansion of local resident macrophages through proliferation contributes to the number of macrophages in the diseased kidneys,3 the recruitment of monocytes/macrophages from the circulation is generally considered to be the main pathway leading to the accumulation of these cells.1 In disease conditions, circulating monocytes derived from bone marrow myeloid progenitors migrate to the site of injury and form macrophages through multiple steps involving rolling, adhesion, crawling, and moving, in which proteases and members of the integrin family and Rho family GTPases play an important role.4, 5
Tissue-type plasminogen activator (tPA) is a member of the serine protease family and participates in the activation of various motility-related proteases and growth factors.6, 7, 8, 9 tPA also acts as a profibrotic cytokine to promote the progression of CKD by triggering profound intracellular signaling events that include integrin signaling pathways.6, 10, 11, 12, 13, 14, 15 Increasing evidence indicates that tPA modulates inflammatory infiltration in response to tissue injury in various disease models.15, 16, 17 tPA has been shown to mediate T-cell infiltration in a liver fibrosis model,16 and promote neutrophil infiltration in a model of acute kidney injury.18 Our recent data demonstrate that tPA activates NF-κB signaling and promotes renal inflammation by inducing macrophage accumulation and chemokine expression in a CKD model.15 Notably, tPA is also up-regulated in the obstructed kidneys with concomitant macrophage infiltration,11, 12 suggesting that tPA may be the endogenous factor that modulates macrophage motility in response to chronic kidney injury. However, the source of the endogenous tPA induction, as well as the underlying mechanisms whereby tPA modulates macrophage accumulation during injury, remain largely unknown.
Integrin signaling through focal adhesion kinase (FAK) is known to modulate cell motility.4, 19 FAK, the immediate downstream kinase of integrin, is a cytoplasmic protein tyrosine kinase that plays an essential role in the regulation of multiple cellular processes such as cell migration.20 FAK plays an integral role in the regulation of actin cytoskeleton and controls cell motility through several mechanisms including the interaction with members of Rho family of small GTPases.19, 20, 21 However, the role of FAK and its downstream signaling in tPA-mediated macrophage motility remains unknown. We hypothesized that tPA may promote macrophage motility by activating the downstream signaling of the integrin CD11b pathway.
Here, we investigated the role of tPA in macrophage motility and elucidated the underlying signaling mechanisms using both in vitro and in vivo approaches. Our data demonstrate that myeloid-derived tPA promotes macrophage migration through a protease-independent mechanism involving a novel signaling cascade of FAK, Rac1, and NF-κB.
Methods
Antibodies and Reagents
The anti–phospho-specific FAK (Tyr925) antibody and Alexa Fluor 488–conjugated anti-rabbit secondary antibody were purchased from Cell Signaling Technology (Beverly, MA). Mouse anti–α-tubulin and PKH26 Red Fluorescent Cell Linker kit were obtained from Sigma (St. Louis, MO). Monoclonal anti-mouse CD 11b PE-Cy5 and FITC antibodies and recombinant mouse macrophage colony-stimulating factor were purchased from eBioscience (San Diego, CA). The neutralizing CD11b antibody was provided by BD Biosciences (San Diego, CA). The monoclonal anti-F4/80 antibody was bought from AbD Serotec (Raleigh, NC). The Rac1 activation assay kit was supplied by Millipore (Temecula, CA). Recombinant human single-chain tPA was purchased from American Diagnostica (Stamford, CT). The nonenzymatic tPA was supplied by Molecular Innovations (Southfield, MI). The specific Rac1 inhibitor NSC23766 was ordered from Tocris Bioscience (Bristol, UK).
Cell Culture
Mouse macrophages J774.A1 were purchased from ATCC (Manassas, VA) and maintained as previously described.15 After 24 hours serum-free starvation, the macrophages were treated with vehicle and tPA for various periods of time as indicated and then collected for different assays.
Animal Model
Homozygous tPA knockout (KO) and wild-type (WT) mice on a C57BL/6 background were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained as previously described.10, 11, 13, 15 The animal protocol was approved by the Institutional Animal Care and Use Committee at the Penn State University College of Medicine. Unilateral ureteral obstruction (UUO) was performed in 20- to 22-g male mice (3 to 5 mice per group) using established procedures.10, 11, 13, 15
In Vivo Macrophage Motility Assay
Bone marrow–derived macrophages were prepared as previously described.22 Briefly, bone marrow isolated from the femurs of tPA KO mice were cultured in RPMI 1640 medium containing 20% fetal bovine serum, 2 mmol/L l-glutamine, and 10 ng/mL macrophage colony-stimulating factor for 7 days, followed by PKH26 red fluorescence labeling. Immune staining confirmed that >99% PKH26-labeled cells were CD11b-positive macrophages. The PKH26-labeled macrophages (4 × 106 cells per mouse) were then injected via tail vein into WT and tPA KO mice, followed by UUO for 7 days. Macrophages that migrated into the kidneys were quantitated by counting PKH26-positive cells under fluorescence microscopy (5 high power fields per mice, 5 mice per group) or by flow cytometry analysis (3 mice per group). PKH26-positive CD11b macrophages in the spleen were also quantitated by flow cytometry analysis (3 mice per group).
Generation of Chimeric Mice
Chimeric mice were generated by bone marrow transplantation between WT and tPA KO mice as previously described.23, 24 Briefly, donor bone marrow cells were transferred into lethally irradiated recipient mice using the following donor/recipient combinations: WT/WT, WT/KO, KO/WT, KO/KO. Eight to 10 weeks later, UUO was performed in these chimeric mice for 7 days. Chimerism of individual mouse was verified by genotyping of genomic DNA from peripheral blood as described elsewhere.23
Immunohistochemistry
Paraffin-embedded kidney tissue was sectioned at 4 μm and then subjected to staining with F4/80 antibody using the ABC method with NovaRED substrate and methyl green (Vector Laboratories) counterstaining.10, 15 A total of 500 interstitial cells were counted per slide, and the percentage of interstitial F4/80-positive cells in the total interstitial cell population was calculated, 5 mice per group.
Rac1 Activation Assay
Rac1 activation was measured by a Rac1 activation assay kit according to the instructions of the manufacturer (Millipore, Billerica, MA). Tissue homogeny or cell lysates were affinity precipitated by control beads or glutathione S-transferase–agarose beads bound with PAK1-PBD, which specifically recognized active Rac1, followed by Western blot against Rac1.
Flow Cytometry
Single-cell suspensions from the whole kidneys were prepared as previously described.15, 25 The cells were stained with anti–CD11b PE-Cy5 and/or anti–phospho-FAK antibodies, followed by flow cytometry analysis using a FACSCaliber machine (BD Biosciences, San Diego, CA) and FlowJo software version 7.6.1 (Tree Star, Ashland, OR).
Transwell Cell Motility Assay
J774 macrophages (1 × 105) were seeded in 8-μm pore size Transwell inserts (Corning-Costar, Tewksbury, MA). After overnight serum starvation and 10 nmol/L tPA treatment for 24 hours, cells were fixed with 3% paraformaldehyde and stained in Coomassie Blue solution (0.1% Coomassie Blue, 10% methanol, and 10% acetic acid) for 30 minutes. The migrated macrophages in the center of a filter (one field) were counted under a phase-contrast microscope with ×200 magnitude. The data were quantitated from five to six independent experiments.
Adenovirus Infection
The adenoviral vectors containing the WT FAK (Ad.FAK) or the dominant-negative FAK mutant FRNK (Ad.FRNK) were constructed as previously described,26, 27 and were provided by Dr. Christopher S. Chen (University of Pennsylvania) and Dr. Allen M. Samarel (Loyola University Medical Center). Transient adenovirus infection was performed as described elsewhere.10, 11, 12, 13
Western Blot Analysis
Samples were prepared and separated on 10% SDS polyacrylamide gels as previously described.10, 11, 12, 13, 15 The polyvinylidene difluoride membrane with transferred proteins was incubated overnight at 4°C with various primary antibodies, followed by incubation with horseradish peroxidase–conjugated secondary antibodies for 1 hour. After extensive washes, the signals on the membrane were visualized by a Chemiluminescent Substrate kit (Thermo Fisher Scientific, Waltham, MA).
Quantitative RT-PCR
Total RNA was extracted and reverse transcribed into cDNA and amplified using a SYBR Green PCR kit (Qiagen, Valencia, CA) as previously described.11, 15 The relative level of mRNAs was quantified and normalized to β-actin.
tPA ELISA
Plasma tPA was measured using an enzyme-linked immunosorbent assay (ELISA) kit from Oxford Biomedical Research (Oxford, MI) according to the manufacturer's instructions.
Measurement of Total Kidney Collagen Content
Total collagen content was evaluated using the Sirius Red/Fast Green Collagen detection kit (Chondrex, Redmond, WA) as previously described.15 Briefly, tissue sections were stained with Sirius Red and Fast Green, followed by the dye extraction and measurement of the optical density at 540 nm (collagen) and the optical density at 605 nm (noncollagen protein). The collagen content was expressed as micrograms per milligrams (collagen/total protein).
Statistical Analysis
All of the experimental data were presented as means ± SEM. Statistical analysis of the data were performed using SigmaStat software version 3.5 (Systat Software, San Jose, CA). Comparison between multiple groups was performed by using one-way analysis of variance followed by the Student-Newman-Keuls test or Student's t-test between two groups. A P value of <0.05 was considered statistically significant.
Results
tPA Promotes the Migration and Accumulation of Macrophages in the Obstructive Kidneys
UUO was performed in WT and tPA KO mice for 7 days. Flow cytometry analyses showed that the number of CD11b-positive macrophages markedly increased in the WT kidneys after obstruction (Figure 1, A and B). However, the tPA KO mice showed fewer infiltrating CD11b macrophages, suggesting that tPA promotes macrophage accumulation in the fibrotic kidneys in vivo. This result is also consistent with our previous CD11b immunohistochemical data in these mice.15 Because cell motility is one of the key factors that determine the number of macrophages in diseased kidneys, we hypothesized that tPA may promote macrophage accumulation by increasing the motility of these cells. To this end, we performed an in vivo macrophage motility assay in tPA WT and KO mice. Bone marrow–derived macrophages, verified by CD11b staining (data not shown), were labeled with PKH26 red fluorescence. To minimize the interference of endogenous tPA, bone marrow cells were collected from tPA KO mice. The PKH26-positive macrophages were then intravenously injected into WT and tPA KO mice, followed by UUO for 7 days. The PKH26-positive macrophages that migrated into the obstructed kidneys were quantitated by cell counts or flow cytometry analysis. Obstructive renal injury induced the migration of PKH26-labeled macrophages (Figure 1, C–F). Notably, the number of these cells in the fibrotic kidneys from tPA KO mice was much lower than that in the WT mice (Figure 1, C–F). In addition, the redistribution of the PKH26-positive cells in the spleen, the major organ that stores the homing immune cells,28 was similar between WT and tPA KO mice (Figure 1G), suggesting that endogenous tPA in the diseased kidneys mediates the migration and accumulation of these macrophages. Thus, tPA promotes macrophage migration and accumulation in vivo.
Figure 1.

tPA promotes macrophage infiltration and migration in vivo. Unilateral ureteral obstruction (UUO) was performed in tPA wild-type (WT) and knockout (KO) mice (n = 4 mice per group) for 7 days. Single-cell suspensions were prepared from whole kidneys and were subjected to flow cytometry analysis using anti-CD11b antibody to identify macrophages. A: Representative flow cytometry analysis. Cells in the ovals were CD11-positive macrophages. Contralateral unobstructed kidneys served as controls. B: Quantitation of the ratio of CD11b macrophages in the gated cells. C: Fluorescence microscopy of PKH26-labeled macrophages in the fibrotic kidneys. Bone marrow–derived macrophages from tPA KO mice were labeled by PKH26 and intravenously transferred into tPA KO and WT mice, followed by UUO for 7 days. PKH-labeled macrophages in the obstructed kidneys were counterstained by DAPI and quantitated under the fluorescence microscope. D: Quantification of PKH26-positive macrophages. Units were expressed as number of PKH26-positive macrophages per ×400 field, 5 fields per mouse. E: Flow cytometry analysis of the PKH26-positive macrophages. Some WT and tPA KO mice receiving adoptive PKH26-positive macrophage transfer were subjected to flow cytometry analysis. Cells in the ovals are PKH26-labeled macrophages. Data are representative from one of the three animals analyzed. Contralateral unobstructed kidneys served as controls. F: Quantitation of flow cytometry analysis. G: Flow cytometry analysis of the ratio of PKH26-positive CD11b macrophages in the gated cells of the spleens from tPA KO and WT mice with UUO for 7 days. No statistical significance exists. ∗P < 0.05, ∗∗P < 0.01. n = 3 (F and G, mice per group); n = 4 (B, mice per group); n = 5 (C and D, mice per group). Original magnification, ×400 (C). CLK, contralateral unobstructed kidneys; SSC, side-scattered light.
Myeloid-Derived tPA Is the Endogenous Factor Inducing Macrophage Infiltration during Obstructive Renal Injury
The reduced ability of tPA-deficient macrophages to migrate in tPA KO, but not tPA WT mice, suggests that tPA from the WT animals is able to signal macrophages to migrate. To identify whether the source of tPA can be autocrine and/or juxtacrine, chimeric mice that lack tPA in either their myeloid cells or renal parenchyma were generated by bone marrow transplantation between tPA WT and KO mice (bone marrow donor/recipient: WT/WT, WT/KO, KO/WT, and KO/KO), and were subjected to UUO for 7 days. Chimerism of each mouse was verified as previously described.23 We found that WT/WT and WT/KO mice displayed similar and dramatically increased renal fibrosis and macrophage infiltration, as demonstrated by α-smooth muscle actin (α-SMA) induction (Figure 2, A–C), matrix (collagen and fibronectin) deposition (Figure 2, D–F), and the interstitial accumulation of F4/80-positive macrophages (Figure 2, G and H). However, both KO/WT and KO/KO mice showed similar, but significantly decreased, fibrosis (Figure 2, A–F) and remarkably fewer infiltrating macrophages (Figure 2, G and H). Notably, although KO/WT and KO/KO mice had similar lower levels of renal fibrosis (Figure 2, A–F) and macrophage accumulation (Figure 2, G and H), KO/WT mice showed a significantly higher plasma tPA level than KO/KO mice (Figure 2I). Similarly, WT/WT and KO/WT had a similar plasma level of tPA (Figure 2I), but displayed a significant difference in macrophage accumulation and fibrosis (Figure 2, A–H). Thus, it is likely that tPA in the circulation, mainly produced by the vascular endothelial cells, has little effect on the renal accumulation of macrophages and fibrosis. These results indicate that myeloid cells are the main source of endogenous tPA that induces macrophage accumulation and promotes renal fibrosis.
Figure 2.

Myeloid-derived tPA promotes macrophage infiltration in vivo. Chimeric mice were generated by bone-marrow transplantation between tPA WT and KO mice (donor/recipient: WT/WT, WT/KO, KO/WT, and KO/KO,). Unilateral ureteral obstruction (UUO) was performed in these mice for 7 days. A: Representative Western blot of α-SMA and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Number indicates individual mouse. Contralateral unobstructed kidneys (CLK) served as controls. B: Quantitation of renal α-SMA relative abundance. C: Immunofluorescence staining of αSMA in the obstructed kidneys. D: Immunostaining of collagen I in the obstructed kidneys. E: Quantitation of total collagen content in the obstructed kidneys by Sirius Red/Fast Green assay. F: Quantitative RT-PCR of fibronectin mRNA in the obstructed kidneys. G: Representative immunostaining of F4/80 in the obstructed kidneys. H: Quantitation of interstitial infiltration of F4/80-positive macrophages. I: Plasma concentration of tPA in the chimeric mice. ∗P < 0.05, ∗∗P < 0.01. n = 5 mice per group. Original magnification: ×200 (C); ×400 (D and G).
tPA Promotes Macrophage Migration in Vitro through a Protease-Independent Mechanism
J774 macrophages were seeded into the Transwell inserts in 24-well plates, followed by 10 nmol/L tPA treatment for 24 hours. tPA induced macrophage motility in vitro (Figure 3, A and B). It is known that tPA is a hybrid molecule of protease and cytokine.6, 10, 11, 12, 13 To determine whether tPA-induced macrophage motility depends on its protease activity or is mediated through its cytokine activity, J774 macrophages were treated with vehicle and a mutant, catalytically inactive tPA at the same concentration. The nonenzymatic tPA promoted the migration of macrophages comparable to the WT tPA, suggesting that tPA induces macrophage migration through a protease-independent mechanism (Figure 3C). Importantly, the interference from possible contamination by endotoxin in the tPA solution was excluded, because heat-inactivated nonenzymatic tPA had no effect on macrophage migration (Figure 3D). We also found that tPA still significantly induced macrophage motility in the presence of monocyte chemotactic protein-1 (MCP-1), although its additional effect on motility was relatively small (Figure 3E).
Figure 3.

tPA promotes macrophage motility in vitro through its protease-independent activity. A: tPA induces macrophage migration in vitro. J774 macrophages were treated with 10 nmol/L tPA for 24 hours, followed by a Transwell motility assay. Data are representative of 5 independent experiments. B: Quantitation of tPA-induced macrophage motility. C: Nonenzymatic tPA (NE-tPA) also promotes macrophage motility. J774 cells were incubated with 10 nmol/L NE-tPA for 24 hours. Cell motility was assessed by Transwell assay. D: Heat-inactivated NE-tPA does not affect macrophage motility. J774 cells were treated with 10 nmol/L NE-tPA or heat-inactivated NE-tPA (Boil-tPA) for 24 hours, followed by motility assay. E: tPA induces macrophage motility in the presence of MCP-1. J774 cells were treated with control or 10 nmol/L tPA with or without 10 ng/mL MCP-1 in the lower chambers of a Transwell plate for 24 hours, followed by counting migrated cells. ∗P < 0.05, ∗∗P < 0.01. n = 5 (B–D); n = 6 (E). Original magnification, ×200 (A). Con, control.
Integrin CD11b Signaling Is Indispensable to tPA-Induced Macrophage Migration
Because tPA promoted macrophage migration through its protease-independent activity, it is plausible that tPA induces macrophage migration by activating intracellular signaling cascades that regulate cell motility. Integrins and Rho family GTPases play a central role in the regulation of cell motility. Our recent work demonstrated that tPA activates integrin CD11b signaling in macrophages.15 We examined the role of CD11b in tPA-mediated macrophage motility. CD11b neutralizing antibody suppressed the phosphorylation of its downstream mediator FAK (Figure 4, A and B) and inhibited tPA-induced macrophage migration (Figure 4C). Therefore, CD11b signaling is essential to tPA-mediated macrophage motility.
Figure 4.
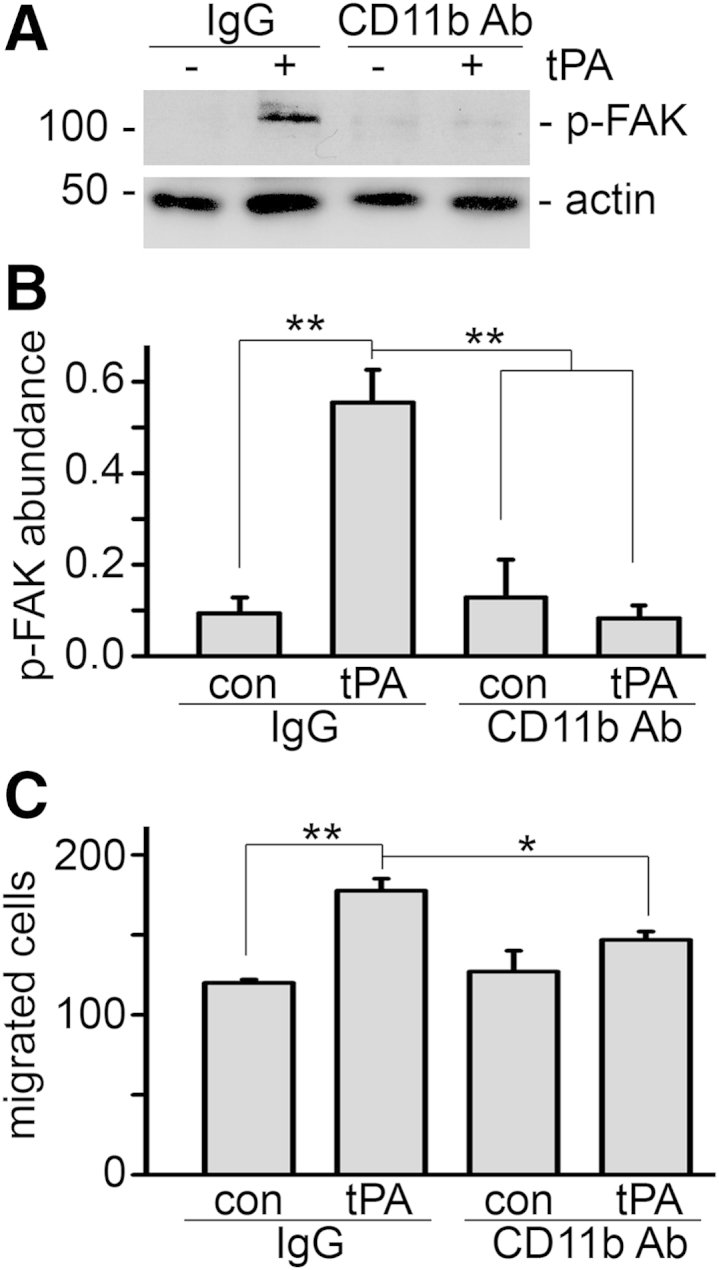
CD11b mediates tPA-induced macrophage migration. J774 macrophages were treated with 2.5 μg/mL control IgG or anti-CD11b neutralizing antibody (Ab) for 1 hour, followed by incubation with vehicle and 10 nmol/L tPA for 1 minute or 24 hours. A: Western blot for phospho-FAK (Tyr925) and α-tubulin. B: Quantitation of phospho-FAK abundance. C: Transwell motility assay. ∗P < 0.05, ∗∗P < 0.01. n = 3 (B); n = 5 (C).
tPA Sequentially Activates FAK and Rac1 Signaling Cascade
We further found that tPA rapidly induced Tyr925 phosphorylation of FAK, an immediate downstream effector of integrin signaling, as early as 1 minute after tPA treatment (Figure 5A), followed by Rac1 activation from 10 minutes to 2 hours (Figure 5B). The temporal order of Rac1 activation after phosphorylation of FAK suggested that FAK may be the upstream mediator of Rac1 in the tPA signaling cascade. To further clarify the role of FAK in tPA-induced Rac1 activation, J774 macrophages were infected with adenoviral vectors containing the dominant-negative FAK noncatalytic mutant FRNK (Ad.FRNK) or Ad.LacZ, followed by tPA treatment. The FRNK is the C-terminal region of FAK, which lacks the N-terminal and the central catalytic domains.26, 27, 29 Infection of Ad.FRNK inhibited tPA-induced FAK phosphorylation (Figure 5C) and subsequently abolished Rac1 activation (Figure 5D). However, NSC23766, a specific Rac1 inhibitor, had no effect on tPA-induced phosphorylation of FAK (data not shown). Thus, FAK mediates Rac1 activation in the tPA signaling cascade.
Figure 5.

tPA promotes macrophage migration through the FAK/Rac1-mediated pathway. A: J774 macrophages were treated with 10 nmol/L tPA for 1, 2, 5, or 10 minutes, followed by Western blot for phospho-FAK (Tyr925) and α-tubulin. B: Representative Western blot of activated Rac1. Cells were treated with 10 nmol/L nonenzymatic tPA for 10, 30, and 120 minutes. Cell lysates were subjected to a Rac1 activity assay. C: Dominant-negative FAK mutant (FRNK) blocks tPA-induced phosphorylation of FAK (Tyr925). Cells were infected with Ad.LacZ and Ad.FRNK, followed by treatment with 10 nmol/L tPA for 1 minute and Western blot for phospho-FAK (Tyr925) and α-tubulin. D: Ad.FRNK infection abolishes nonenzymatic tPA-induced Rac1 activation. E: Transwell motility assay shows that FRNK inhibits tPA-induced macrophage migration. F: Ectopic overexpression of WT FAK mimics tPA and promotes macrophage migration. J774 macrophages were infected with Ad.LacZ and Ad.FAK for 48 hours, followed by a Transwell motility assay. G: NSC23766, a specific Rac1 inhibitor, suppresses tPA-induced migration of macrophages. H: NF-κB–specific inhibitor SN50 (0.1 mg/mL) abrogates tPA-induced macrophage motility. ∗∗P < 0.01. n = 5.
FAK Mediates tPA-Induced Macrophage Motility
We also examined the role of FAK in tPA-induced macrophage motility. J774 macrophages were seeded in the Transwell inserts and subjected to infection with Ad.LacZ and Ad.FRNK, followed by tPA treatment for 24 hours. Ad.FRNK, the dominant-negative FAK mutant, dramatically suppressed tPA-induced macrophage migration (Figure 5E). The efficiency of FAK inhibition by Ad.FRNK infection was verified by Western blot for FAK phosphorylation (Figure 5C). Conversely, ectopic overexpression of WT FAK by Ad.FAK infection mimics tPA and promoted the migration of J774 macrophages (Figure 5F). Therefore, FAK phosphorylation is both necessary and sufficient for tPA-induced macrophage migration.
Rac1 Is Essential to tPA-Promoted Macrophage Migration
To investigate the functional role of Rac1 in macrophage migration, macrophages were pretreated with NSC23766, the specific chemical inhibitor of Rac1, for 1 hour, followed by tPA treatment for 24 hours and subsequently, a Transwell migration assay. NSC23766 abolished tPA-induced macrophage migration, indicating that Rac1 is indispensable to tPA-modulated macrophage motility (Figure 5G).
NF-κB Is Indispensable to tPA-Induced Macrophage Motility
We have shown that CD11b mediates tPA-induced NF-κB signaling in macrophages,15 and we further examined the role of NF-κB in tPA-induced macrophage motility. We found that the NF-κB–specific inhibitor SN50 eliminated the effect of tPA on macrophage motility (Figure 5H).
tPA Modulates Cell Motility Signaling in Vivo
The in vitro mechanistic studies suggest that tPA-induced FAK/Rac1 migratory signaling pathway is also operative in vivo. To this end, we investigated the FAK/Rac1 signaling in the obstruction-induced fibrotic kidneys from tPA WT and KO mice. We found that phosphorylation of FAK (Tyr925) was markedly induced in the CD11b-positive macrophages, as indicated by flow cytometry analysis (Figure 6, A and B), in the fibrotic kidneys from the WT mice with UUO for 7 days. However, obstruction-induced accumulation of phospho-FAK–positive CD11b macrophages was clearly attenuated in tPA KO mice (Figure 6, A and B). In addition, double immunofluorescence staining of phospho-FAK (red, intracellular focal adhesions) and CD11b (green, cell surface) confirmed that most of the CD11b macrophages were also phospho-FAK positive in diseased kidneys (Figure 6, C and D). We also discovered that Rac1 was strongly activated in the obstructed kidneys from tPA WT mice, but its activation was almost completely alleviated in tPA KO mice (Figure 6, E and F). These results strongly support the view that tPA modulates macrophage FAK/Rac1 migratory signaling in vivo, leading to the accumulation of macrophages in the fibrotic kidneys.
Figure 6.

tPA modulates cell motility signaling in vivo. Unilateral ureteral obstruction (UUO) was performed in tPA WT and KO mice for 7 days. A: Representative flow cytometry analysis using probes for CD11b and phospho-FAK (Tyr925). Upper right quarter indicates CD11b-positive macrophages with activated FAK signaling. Contralateral unobstructed kidneys served as controls. B: Quantitation of relative amount of CD11b macrophages with activated FAK phosphorylation. Units are expressed as percentage of both (Tyr925)-phospho-FAK–positive and CD11b-positive cells in gated cells. C: Immunostaining for CD11b (green) and phospho FAK (red). Arrows indicate cells with positive double immunostaining of phospho-FAK (intracellular focal adhesions) and CD11b (cell surface) in diseased kidneys after UUO for 7 days. Nuclei were indicated by DAPI. Insets show enlarged images (×3.8) of the area bordered by white lines. Contralateral unobstructed kidneys served as controls. D: Quantitation of the CD11b macrophages with FAK phosphorylation in the obstructed kidneys. Units were expressed as number of CD11b and phospho-FAK double-positive cells per ×400 field. E: Representative Western blot of activated Rac1 in the diseased kidney homogenized from tPA WT and KO mice. Number indicates individual mouse. F: Quantitation of relative Rac1 abundance. ∗∗P < 0.01. n = 5 mice per group. Original magnification, ×400 (C). CLK, contralateral unobstructed kidneys.
Discussion
Infiltration of inflammatory cells is one of the histological hallmarks of both acute kidney injury and CKD. In response to persistent injury cues, inflammatory cells, including monocytes/macrophages and other leukocytes, migrate to the sites of injury and produce a broad spectrum of proinflammatory and profibrotic factors that not only promote disease progression, but also form a cycle of deterioration.1, 2, 15 Proteases such as plasminogen activators and matrix metalloproteinases (MMPs) and their inhibitors, as well as members of the integrin family and Rho GTPases, play a fundamental role in the process of inflammatory infiltration.4, 5, 19 tPA has been shown to modulate inflammatory infiltration in numerous disease models.15, 16, 17, 30 In an acute brain injury model, tPA was shown to mediate F4/80 macrophage accumulation and activation in the ischemic brain.30 In a carbon tetrachloride–induced liver fibrosis model, tPA KO mice demonstrated decreased infiltration of T cells in comparison with their WT counterparts.16 Moreover, it was found that tPA promoted infiltration of macrophages or other leukocytes in both models of acute17 and chronic kidney injury.15 Thus, tPA appears to have broad implication in the modulation of infiltration and inflammation in diverse organs. Intriguingly, the increased inflammatory infiltration in most of those models is accompanied by the concomitant induction of tPA,11, 12, 31, 32 suggesting that tPA may be a common endogenous factor that modulates inflammatory infiltration and response in multiple organ systems.
Here, we showed that tPA promoted the accumulation of CD11b macrophages in response to obstructive injury (Figure 1, A and B). We hypothesized that tPA may promote macrophage accumulation by enhancing the motility of these cells. We designed a new approach to assess macrophage motility in vivo by tracking PKH26 red fluorescence–labeled bone marrow–derived macrophages in WT and tPA KO mice. Obstruction-induced migration of PKH26-labeled macrophages was attenuated in tPA KO mice (Figure 1, C–F). However, the fact that little difference of the splenic redistribution of these PKH26-positive cells was found between WT and tPA KO mice (Figure 1G) suggests a fundamental role of renal endogenous tPA in mediating the obstruction-induced macrophage accumulation. Experiments in bone marrow chimeric mice demonstrated that myeloid-derived tPA is the primary endogenous factor that modulates macrophage migration (Figure 2). This notion is further supported by the finding in the chimeric mice that the concentration of tPA (Figure 2I) in the circulation has little effect on renal fibrosis and interstitial macrophage accumulation (Figure 2, A–H). In addition, exogenous tPA also promoted J774 macrophage migration in vitro (Figure 3). Thus, it is likely that myeloid-derived tPA modulates inflammatory infiltration by affecting cell motility. Intriguingly, tPA-induced macrophage motility is likely through a mechanism independent of the MCP-1 and CCR2 axis, because the levels of MCP-1 and CCR2 are similar in tPA KO and WT mice after UUO for 7 days (data not shown) and MCP-1 has little effect on tPA-induced macrophage motility (Figure 3E).
Although tPA, as a potent serine protease, activates several proteases (plasminogen and MMPs)6 and growth factors (TGF-β1 and platelet-derived growth factor-CC)7, 8, 9 that participate in the regulation of cell motility, its protease activity is dispensable to tPA-mediated macrophage migration (Figure 3C). Instead, tPA activated a series of intracellular signaling events that control cell motility. tPA promoted FAK Tyr925 phosphorylation and Rac1 activation in a temporal order (Figure 5, A and B). We further confirmed that FAK is the upstream kinase that activates Rac1, because FRNK, a dominant-negative FAK mutant,21, 27 inhibited the phosphorylation of FAK Tyr925, eliminated the activation of Rac1, and subsequently abolished tPA-induced macrophage migration (Figure 5, C–E). By contrast, ectopic expression of WT FAK mimicked tPA and promoted macrophage migration (Figure 5F). tPA induces phosphorylation of FAK at Tyr925 through activation of integrin CD11b signaling (Figure 4). These results are consistent with our recent finding that tPA activates CD11b signaling in J774 macrophages,15 and that CD11b has been shown to mediate tPA-initiated peritoneal macrophage efflux in a lipopolysaccharide-induced peritonitis model.33 On activation, integrin induces FAK autophosphorylation at Tyr397 and its exposure to Src family kinases, which phosphorylates additional tyrosine residues on FAK, leading to its full activation.19 In contrast to a previous report34 that phosphorylation of FAK at Src-dependent Tyr576, Tyr577, and Tyr881 are responsible for FAK-mediated Rac1 activation in smooth muscle cells, our results indicate that FAK Tyr925 is a new tyrosine site that mediates tPA-induced Rac1 activation and subsequent macrophage migration (Figure 5). Recent studies suggest that FAK might regulate cytoskeletal events through modulation of the paxillin kinase linker (PKL/Git2) and β-pix complex.20 β-pix, as an exchange factor for Cdc42, is connected to focal adhesions through binding of PKL/Git2 to paxillin,35 and also serves as a scaffold to activate Rac and PAK signaling.36 FAK induces the tyrosine phosphorylation of β-pix, leading to the recruitment and activation of Rac1 and, subsequently, the rearrangement of actin cytoskeleton and cell migration.37 Of note, urokinase-type plasminogen activator receptor (uPAR), the receptor of tPA's close cousin, uPA, also modulates Rac1 activity and cell motility through interaction with integrins.38, 39 Moreover, CD11b-mediated NF-κB is also indispensable to tPA-induced macrophage motility (Figure 5H).
Myeloid-derived tPA promotes macrophage migration through a novel signaling cascade involving FAK, Rac1, and NF-κB. This migratory signaling pathway is also operative in vivo because tPA promoted FAK phosphorylation in macrophages during the course of obstructive renal injury, as indicated by flow cytometry and double immunofluorescence staining (Figure 6, A–D); and obstruction-induced Rac1 activation in the WT mice was mostly alleviated in tPA-deficient mice (Figure 6, E and F). tPA also promotes NF-κB activation in macrophages, which is related to the interstitial accumulation of the macrophages.15 Therapeutic strategy targeting this migratory signaling, not only suppresses the inflammatory infiltration, but also reduces the cells that generate the pathogenic myeloid-derived tPA, making these findings clinically significant.
Of note, it is known that macrophages, in response to injury, differentiate into two distinct subsets through classical activation (M1) or alternative activation (M2).1 Generally, M1 macrophages promote inflammation and exaggerate damage, whereas M2 macrophages help to resolve inflammation and promote tissue remodeling.1, 2 It is not clear whether tPA differentially promotes the infiltration and accumulation of one subset of macrophages over the other. However, we found that obstruction-induced infiltration of CD11b macrophages, fibroblast activation, and matrix deposition are attenuated in tPA-deficient mice.13, 15 Together with our previous finding that exogenous tPA induces the expression of M1-like chemokines in J774 macrophages,15 it is reasonable to speculate that tPA might preferably promote the infiltration of M1 macrophages. However, in disease conditions, differentiation between M1 and M2 macrophages is dynamic and macrophages can switch their phenotype at different disease stages.40 Future investigation is required to address these questions.
In summary, we have established that myeloid-derived tPA promotes macrophage accumulation in the diseased kidneys by enhancing the motility of these cells. We have also defined a novel tPA-mediated migratory signaling pathway involving FAK phosphorylation, Rac1 activation, and the resultant increase of macrophage motility.
Acknowledgments
We thank Drs. Christopher S. Chen and Allen M. Samarel for providing the necessary adenoviral vectors as well as Terrell Smith and Jessica Carswell for their assistance in cell counting.
Footnotes
Supported by NIH grants DK102624 (K.H.) and DK081876 (W.B.R.), American Heart Association grant 14GRNT20380289 (K.H.), Barsumian Trust grant 157904 (K.H.), and by the Kidney Foundation of Central Pennsylvania (K.H.).
Disclosures: None declared.
References
- 1.Ricardo S.D., van Goor H., Eddy A.A. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y., Harris D.C. Macrophages in renal disease. J Am Soc Nephrol. 2011;22:21–27. doi: 10.1681/ASN.2010030269. [DOI] [PubMed] [Google Scholar]
- 3.Zhang M.Z., Yao B., Yang S., Jiang L., Wang S., Fan X., Yin H., Wong K., Miyazawa T., Chen J., Chang I., Singh A., Harris R.C. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 2012;122:4519–4532. doi: 10.1172/JCI60363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huttenlocher A., Horwitz A.R. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ley K., Laudanna C., Cybulsky M.I., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 6.Hu K., Mars W.M., Liu Y. Novel actions of tissue-type plasminogen activator in chronic kidney disease. Front Biosci. 2008;13:5174–5186. doi: 10.2741/3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mars W.M., Zarnegar R., Michalopoulos G.K. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 8.Yee J.A., Yan L., Dominguez J.C., Allan E.H., Martin T.J. Plasminogen-dependent activation of latent transforming growth factor beta (TGF beta) by growing cultures of osteoblast-like cells. J Cell Physiol. 1993;157:528–534. doi: 10.1002/jcp.1041570312. [DOI] [PubMed] [Google Scholar]
- 9.Fredriksson L., Li H., Fieber C., Li X., Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu K., Lin L., Tan X., Yang J., Bu G., Mars W.M., Liu Y. tPA protects renal interstitial fibroblasts and myofibroblasts from apoptosis. J Am Soc Nephrol. 2008;19:503–514. doi: 10.1681/ASN.2007030300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu K., Wu C., Mars W.M., Liu Y. Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J Clin Invest. 2007;117:3821–3832. doi: 10.1172/JCI32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu K., Yang J., Tanaka S., Gonias S.L., Mars W.M., Liu Y. Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J Biol Chem. 2006;281:2120–2127. doi: 10.1074/jbc.M504988200. [DOI] [PubMed] [Google Scholar]
- 13.Lin L., Bu G., Mars W.M., Reeves W.B., Tanaka S., Hu K. tPA activates LDL receptor-related protein 1-mediated mitogenic signaling involving the p90RSK and GSK3beta pathway. Am J Pathol. 2010;177:1687–1696. doi: 10.2353/ajpath.2010.100213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi Y., Mantuano E., Inoue G., Campana W.M., Gonias S.L. Ligand binding to LRP1 transactivates Trk receptors by a Src family kinase-dependent pathway. Sci Signal. 2009;2:ra18. doi: 10.1126/scisignal.2000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin L., Wu C., Hu K. Tissue plasminogen activator activates NF-kappaB through a pathway involving annexin A2/CD11b and integrin-linked kinase. J Am Soc Nephrol. 2012;23:1329–1338. doi: 10.1681/ASN.2011111123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Higazi A.A., El-Haj M., Melhem A., Horani A., Pappo O., Alvarez C.E., Muhanna N., Friedman S.L., Safadi R. Immunomodulatory effects of plasminogen activators on hepatic fibrogenesis. Clin Exp Immunol. 2008;152:163–173. doi: 10.1111/j.1365-2249.2008.03606.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roelofs J.J., Rouschop K.M., Leemans J.C., Claessen N., de Boer A.M., Frederiks W.M., Lijnen H.R., Weening J.J., Florquin S. Tissue-type plasminogen activator modulates inflammatory responses and renal function in ischemia reperfusion injury. J Am Soc Nephrol. 2006;17:131–140. doi: 10.1681/ASN.2005010089. [DOI] [PubMed] [Google Scholar]
- 18.Liot G., Roussel B.D., Lebeurrier N., Benchenane K., Lopez-Atalaya J.P., Vivien D., Ali C. Tissue-type plasminogen activator rescues neurones from serum deprivation-induced apoptosis through a mechanism independent of its proteolytic activity. J Neurochem. 2006;98:1458–1464. doi: 10.1111/j.1471-4159.2006.03982.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhao X., Guan J.L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv Drug Deliv Rev. 2011;63:610–615. doi: 10.1016/j.addr.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schaller M.D. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J Cell Sci. 2010;123:1007–1013. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 21.Hall J.E., Fu W., Schaller M.D. Focal adhesion kinase: exploring Fak structure to gain insight into function. Int Rev Cell Mol Biol. 2011;288:185–225. doi: 10.1016/B978-0-12-386041-5.00005-4. [DOI] [PubMed] [Google Scholar]
- 22.Marim F.M., Silveira T.N., Lima D.S., Jr., Zamboni D.S. A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One. 2010;5:e15263. doi: 10.1371/journal.pone.0015263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu H., Chen G., Wyburn K.R., Yin J., Bertolino P., Eris J.M., Alexander S.I., Sharland A.F., Chadban S.J. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang B., Ramesh G., Uematsu S., Akira S., Reeves W.B. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol. 2008;19:923–932. doi: 10.1681/ASN.2007090982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tadagavadi R.K., Reeves W.B. Renal dendritic cells ameliorate nephrotoxic acute kidney injury. J Am Soc Nephrol. 2010;21:53–63. doi: 10.1681/ASN.2009040407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pirone D.M., Liu W.F., Ruiz S.A., Gao L., Raghavan S., Lemmon C.A., Romer L.H., Chen C.S. An inhibitory role for FAK in regulating proliferation: a link between limited adhesion and RhoA-ROCK signaling. J Cell Biol. 2006;174:277–288. doi: 10.1083/jcb.200510062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koshman Y.E., Kim T., Chu M., Engman S.J., Iyengar R., Robia S.L., Samarel A.M. FRNK inhibition of focal adhesion kinase-dependent signaling and migration in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2010;30:2226–2233. doi: 10.1161/ATVBAHA.110.212761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swirski F.K., Nahrendorf M., Etzrodt M., Wildgruber M., Cortez-Retamozo V., Panizzi P., Figueiredo J.L., Kohler R.H., Chudnovskiy A., Waterman P., Aikawa E., Mempel T.R., Libby P., Weissleder R., Pittet M.J. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612–616. doi: 10.1126/science.1175202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hayasaka H., Martin K.H., Hershey E.D., Parsons J.T. Disruption of FRNK expression by gene targeting of the intronic promoter within the focal adhesion kinase gene. J Cell Biochem. 2007;102:947–954. doi: 10.1002/jcb.21329. [DOI] [PubMed] [Google Scholar]
- 30.Zhang C., An J., Strickland D.K., Yepes M. The low-density lipoprotein receptor-related protein 1 mediates tissue-type plasminogen activator-induced microglial activation in the ischemic brain. Am J Pathol. 2009;174:586–594. doi: 10.2353/ajpath.2009.080661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yepes M., Sandkvist M., Moore E.G., Bugge T.H., Strickland D.K., Lawrence D.A. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. doi: 10.1172/JCI19212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang Y.F., Tsirka S.E., Strickland S., Stieg P.E., Soriano S.G., Lipton S.A. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 33.Cao C., Lawrence D.A., Li Y., Von Arnim C.A., Herz J., Su E.J., Makarova A., Hyman B.T., Strickland D.K., Zhang L. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 2006;25:1860–1870. doi: 10.1038/sj.emboj.7601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sundberg L.J., Galante L.M., Bill H.M., Mack C.P., Taylor J.M. An endogenous inhibitor of focal adhesion kinase blocks Rac1/JNK but not Ras/ERK-dependent signaling in vascular smooth muscle cells. J Biol Chem. 2003;278:29783–29791. doi: 10.1074/jbc.M303771200. [DOI] [PubMed] [Google Scholar]
- 35.Turner C.E., Brown M.C., Perrotta J.A., Riedy M.C., Nikolopoulos S.N., McDonald A.R., Bagrodia S., Thomas S., Leventhal P.S. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–863. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frank S.R., Hansen S.H. The PIX-GIT complex: a G protein signaling cassette in control of cell shape. Semin Cell Dev Biol. 2008;19:234–244. doi: 10.1016/j.semcdb.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chang F., Lemmon C.A., Park D., Romer L.H. FAK potentiates Rac1 activation and localization to matrix adhesion sites: a role for betaPIX. Mol Biol Cell. 2007;18:253–264. doi: 10.1091/mbc.E06-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kjoller L., Hall A. Rac mediates cytoskeletal rearrangements and increased cell motility induced by urokinase-type plasminogen activator receptor binding to vitronectin. J Cell Biol. 2001;152:1145–1157. doi: 10.1083/jcb.152.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma Z., Thomas K.S., Webb D.J., Moravec R., Salicioni A.M., Mars W.M., Gonias S.L. Regulation of Rac1 activation by the low density lipoprotein receptor-related protein. J Cell Biol. 2002;159:1061–1070. doi: 10.1083/jcb.200207070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee S., Huen S., Nishio H., Nishio S., Lee H.K., Choi B.S., Ruhrberg C., Cantley L.G. Distinct macrophage phenotypes contribute to kidney injury and repair. J Am Soc Nephrol. 2011;22:317–326. doi: 10.1681/ASN.2009060615. [DOI] [PMC free article] [PubMed] [Google Scholar]