

Abstract
Amyloid-β (Aβ) and hyperphosphorylated tau (p-tau) aggregates form the two discrete pathologies of Alzheimer disease (AD), and oligomeric assemblies of each protein are localized to synapses. To determine the sequence by which pathology appears in synapses, Aβ and p-tau were quantified across AD disease stages in parietal cortex. Nondemented cases with high levels of AD-related pathology were included to determine factors that confer protection from clinical symptoms. Flow cytometric analysis of synaptosome preparations was used to quantify Aβ and p-tau in large populations of individual synaptic terminals. Soluble Aβ oligomers were assayed by a single antibody sandwich enzyme-linked immunosorbent assay. Total in situ Aβ was elevated in patients with early- and late-stage AD dementia, but not in high pathology nondemented controls compared with age-matched normal controls. However, soluble Aβ oligomers were highest in early AD synapses, and this assay distinguished early AD cases from high pathology controls. Overall, synapse-associated p-tau did not increase until late-stage disease in human and transgenic rat cortex, and p-tau was elevated in individual Aβ-positive synaptosomes in early AD. These results suggest that soluble oligomers in surviving neocortical synaptic terminals are associated with dementia onset and suggest an amyloid cascade hypothesis in which oligomeric Aβ drives phosphorylated tau accumulation and synaptic spread. These results indicate that antiamyloid therapies will be less effective once p-tau pathology is developed.
A large body of evidence indicates that soluble oligomers of amyloid-β (Aβ) are the primary toxic peptides that initiate downstream tau pathology in the amyloid cascade hypothesis of Alzheimer disease (AD).1, 2 However, the time course and severity of AD dementia have been generally found to correlate with neurofibrillary tangle development rather than plaque appearance,3, 4, 5, 6, 7, 8 although a few studies have linked plaques with early cognitive decline.9, 10, 11, 12 Soluble oligomeric Aβ has been highlighted as the primary toxin for loss of dendritic spines and synaptic function13 and has also been directly linked to downstream tau pathology. For example, suppression of a tau kinase pathway can prevent Aβ42 oligomer-induced dendritic spine loss,14 and injection of Aβ42 fibrils into mutant tau mice induces neurofibrillary tangles in cell bodies retrograde to the injections.15 In vivo, effects of Aβ oligomers versus fibrils are harder to separate; however, lowering soluble Aβ oligomers by halving β–site amyloid precursor protein (APP) cleaving enzyme reduces accumulation and phosphorylation of wild-type tau in a mouse model.16 Evidence for Aβ and tau association is particularly strong in the dendritic compartment, where tau was shown to mediate Aβ toxicity via linkage of fyn to downstream N-methyl-d-aspartate receptor toxicity.17
The earliest cognitive losses in AD have long been thought to correlate with synapse loss.8, 18, 19, 20, 21 In humans, electron microscopic studies have documented synapse-associated Aβ and tau,22, 23 and much work documents activity-dependent release of synaptic Aβ into interstitial fluid, which drives local Aβ deposition in human subjects and in rodents.4, 24, 25 Of importance, most synapse-associated Aβ in cortical synapses of AD patients consists of soluble oligomeric species,26 and synaptic tau pathology in AD also includes accumulations of SDS-stable tau oligomers.27, 28, 29, 30, 31 With the use of synaptosomes (resealed nerve terminals) from the cortex of postmortem human subjects and a transgenic rat model of AD, the present experiments were aimed at determining the sequence of appearance of Aβ and hyperphosphorylated tau (p-tau) pathology in synaptic terminals. In addition to early- and late-stage disease, the AD samples included nondemented high pathology controls (HPCs) with substantial AD-related pathology. Synaptic accumulation of Aβ occurred in the earliest plaque stages, before the appearance of synaptic p-tau, which did not appear until late-stage disease. Soluble Aβ oligomers in synaptic terminals were elevated in early AD cases compared with HPCs, indicating an association with the onset of a dementia diagnosis.
Materials and Methods
Materials
The monoclonal anti-Aβ antibody 10G4 is an N-terminal antibody with an epitope directed at residues 5 to 1732; reactivity against human and rodent Aβ was previously reported.33 The anti-tau Ser396/404 phosphospecific PHF-1 antibody was a generous gift from Peter Davies (Albert Einstein College of Medicine, Bronx, NY). The conformation-dependent anti-Aβ antibodies (prefibrillar oligomer-specific M55 and fibril-specific M116) were a generous gift from Charles G. Glabe (University of California Irvine, Irvine, CA). The following antibodies were purchased: anti–synaptosomal-associated protein, 25 kDa (SNAP-25; Sternberger Monoclonals, Inc., Lutherville, MD), 4G8 anti-Aβ (Senentek, Napa, CA), HT-7 anti-tau (Thermo Scientific, Rockford, IL), pS422 anti-tau Ser 422 phosphospecific (Invitrogen, Carlsbad, CA), and anti-synaptophysin (clone SY38) antibody (EMD Millipore, Billerica, MA). All isotype control mouse and rabbit IgG were purchased from BD Pharmingen (San Jose, CA). Recombinant Aβ-40 peptide was purchased from US PEPTIde (Rancho Cucamonga, CA). Zenon isotype-specific IgG labeling kits, Alexa 488-conjugated chicken anti-mouse and Alexa 594-conjugated chicken anti-rabbit IgG were purchased from Molecular Probes (Eugene, OR). Horseradish peroxidase-conjugated anti-mouse IgG was from Jackson ImmunoResearch (West Grove, PA). Pierce high-sensitivity streptavidin-horseradish peroxidase, Supersignal West Femto substrate, and bicinchoninic acid protein assay kit were from Thermo Scientific, (Rockford, IL). 3,3′,5,5′-Tetramethylbenzidine substrate was purchased from Sigma-Aldrich (St. Louis, MO).
Human Brain Specimens
Brain autopsy samples of parietal (A39 and A40), superior parietal (A7), and entorhinal cortex and hippocampus were obtained from the University of California Los Angeles (UCLA), University of Southern California, and University of California Irvine AD Research Centers. A total of 46 samples (25 women and 21 men) were used and were classified into groups on the basis of both clinical and pathologic criteria. Controls included four samples from cognitively normal elderly controls, two samples from spinocerebellar ataxia type II patients; 16 samples were from individuals without a clinical history of dementia but with histopathologic signs of AD-related pathology (HPCs); some of these subjects were cognitively impaired. Twenty-four samples were from individuals that were clinically demented and histopathologically diagnosed with AD; for the purpose of histologic staging, cases were classed as early versus late AD with the use of the Braak stage assignment. The mean age was 82 years for AD cases, and 90 years for normal and HPC cases. The mean postmortem interval was 6.9 hours for AD cases and 7 hours for normal and HPC cases. Detailed information about individual cases is presented in Table 1. For cryopreservation, samples were minced and slowly frozen in isotonic sucrose on the day of autopsy as previously described33; the same protocols were used at each of the three institutions that provided samples.
Table 1.
Case Information for Human Samples
| Case number | Sex | Age, year | PMI, hour | 2012 NIA-Alz Assoc criteria | Diagnosis | MMSE score (months before death) |
|---|---|---|---|---|---|---|
| Control cases without AD-related pathology | ||||||
| U1 | F | 40 | 4 | A0, B0, C0 | SCA | NA |
| 824 | F | 86 | 12.5 | A0, B0, C0 | N | 30 (5) |
| 789 | F | ≥90 | 9 | A0, B0, C0 | N | 30 (29) |
| 726 | F | ≥90 | 8.5 | A0, B0, C0 | N | NA |
| 810 | M | 81 | 5 | A0, B0, C0 | SCA | 27 (>120) |
| 779 | F | 89 | 12 | A0, B0, C0∗ | N | 29 (1) |
| HPC cases | ||||||
| 2-11 | M | ≥90 | 4.1 | A1, B1, C1 | N | 28 (4) |
| 13-11 | F | ≥90 | 10.3 | A1, B1, C2 | CIND | 21 (3) |
| 4-12 | F | ≥90 | 5.25 | A2, B1, C2 | N | 28 (3) |
| 8-12 | M | ≥90 | 4 | A1, B1, C0 | N | 29 (5) |
| 11-12 | M | 86 | 5.25 | A1, B1, C0† | N | 27 (14) |
| 24-11 | M | 86 | 3.5 | A1, B2, C1 | CIND | 22 (18) |
| 27-10 | F | ≥90 | 4.3 | A0, B2, C0 | CIND | 29 (6) |
| 15-09 | M | 82 | 14.9 | A0, B2, C0 | Hippocampal sclerosis | 24 (58) |
| 33-10 | M | ≥90 | 6.05 | A3, B2, C2 | N | 29 (5) |
| 735 | M | 82 | 8.8 | A2, B2, C2 | N | NA |
| 34-11 | F | ≥90 | 4.25 | A1, B2, C1 | N | 30 (32) |
| 808 | M | ≥90 | 6 | A1, B3, C1 | N | 28 (22) |
| 22-12 | M | ≥90 | 6.5 | A2, B0, C1 | N | 25 (5) |
| 36-10 | M | ≥90 | 6 | A3, B2, C2 | CIND | 28 (4) |
| 38-10 | M | ≥90 | 9.4 | A2, B2, C2 | CIND | 24 (5) |
| 7-11 | F | ≥90 | 4.25 | A3, B2, C2 | CIND | 28 (5) |
| Early-stage AD cases (Braak stage IV or less) | ||||||
| 830 | F | 89 | 4.25 | A1, B1, C2 | AD | NA |
| 787 | F | 87 | 9 | A2, B2, C2 | AD/mild CAA | NA |
| 3-12 | M | 81 | 5 | A2, B2, C2 | AD | 19 (33) |
| 34-10 | F | ≥90 | 4.45 | A2, B2, C1 | AD | 23 (41) |
| 9-11 | F | ≥90 | 6.35 | A3, B2, C3 | AD | 17 (4) |
| 835 | F | 78 | 6.5 | A1, B2, C1 | AD/Parkinson's disease | 23 (12) |
| 737 | F | 76 | 10.8 | A2, B2, C1 | AD | NA |
| 788 | M | 82 | 9.5 | A1, B2, C1 | AD/mild CAA | 23 (13) |
| 825 | F | 68 | 9.75 | A1, B2, C2 | AD | 15 (9) |
| U2 | M | 75 | 13 | A1, B1, C2 | AD/Parkinson's disease | NA |
| Late-stage AD cases (Braak stage V to VI) | ||||||
| U3 | F | ≥90 | 6 | A2, B3, C2 | AD/Parkinson disease | NA |
| 811 | M | 59 | 5.5 | A3, B3, C3 | AD | NA |
| 11-09 | M | 77 | 5.5 | A3, B3, C3 | AD | NA |
| U4 | F | 80 | 11 | A3, B3, C3 | AD | NA |
| U5 | M | 82 | 5 | A3, B3, C3 | AD/Lewy body disease | NA |
| 10-09 | F | 82 | 4.5 | A3, B3, C3 | AD | 14 (>120) |
| 05-157 | F | 87 | 6 | A3, B3, C3 | AD | NA |
| 23-10 | M | 85 | 3.55 | A3, B3, C3 | AD | 5 (20) |
| 10-10 | F | 63 | 3.4 | A3, B3, C3 | AD | 2 (9) |
| 864 | M | 86 | 12 | A3, B3, C3 | AD | 2 (27) |
| 33-09 | M | ≥90 | 6.36 | A3, B3, C3 | AD | 19 (13) |
| 16-11 | F | 75 | 6.15 | A3, B3, C3 | AD | 13 (14) |
| 37-11 | F | ≥90 | 8 | A3, B3, C3 | AD/hippocampal sclerosis | 0 (12) |
| 819 | F | 80 | 5 | A2, B3, C2 | AD | NA |
Plaque stage determined by the method of Thal.48
F, female; M, male; AD, Alzheimer disease; Alz Assoc, Alzheimer’s Association; CAA, cerebral amyloid angiopathy; CIND, cognitive impairment no dementia; HPC, high pathology control; MMSE, mini-mental state examination; N, no signs of neuropathologic changes; NA, not available; NIA, National Institute on Aging; PMI, postmortem interval; SCA, spinocerebellar ataxia; WB, Western blot analysis.
Sparse-to-moderate tangles in hippocampus and entorhinal and primary visual cortexes.
Sparse plaques in frontal, parietal, temporal, and occipital cortexes.
Animals
We used a transgenic rat model of AD described previously.34, 35, 36 These animals are homozygous for the following three gene constructs: i) human APP 695 with the K670N/M671L mutation (rat synapsin-1 promoter); ii) human APP minigene with the K670N/M671L and V717F mutations (platelet-derived growth factor β promoter); and iii) human presenilin-1 with the M146V mutation (rat synapsin-1 promoter). This model exhibits age-associated Aβ pathology, with sparse hippocampal plaques seen by 9 months of age, and widespread cortical plaques seen by 18 to 20 months of age. Breeding pairs were obtained from Cephalon Inc. (West Chester, PA) and were bred and aged at the UCLA School of Medicine vivarium facility. Wild-type Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA) and kept at the UCLA School of Medicine vivarium facility before the experiments. All animals were housed under 12-hour light/dark cycle and had access to standard rat chow ad libitum. Animals were sacrificed at 3, 9, and 20 months of age, and their brains were dissected. Frontal cortex and association cortex (parietal, somatosensory, motor, and visual cortex, denoted as mixed cortex) were cryopreserved and processed to synaptosome-enriched fractions. All animal experiments were conducted in compliance with the guidelines of the UCLA Chancellor's Animal Research Committee.
Flow Cytometric Analysis of Synaptosome-Enriched P-2 Fraction
P-2 fractions (synaptosome-enriched fractions) were prepared from cryopreserved brain tissue as described previously,37 cryopreserved in 0.32 mol/L buffered sucrose solution and stored at −80°C as aliquots. On the day of the experiment, aliquots were quickly defrosted at 37°C, and P-2 pellets were collected by centrifugation. After fixation in 0.25% paraformaldehyde/phosphate-buffered saline (PBS; 1 hour at 4°C), and permeabilization in 0.2% Tween20/PBS solution (15 minutes at 37°C), the pellets were incubated with Aβ or tau/p-tau–specific antibodies, directly labeled with Alexa 488 or Alexa 647 fluorochromes with the use of Zenon isotype-specific labeling kits (30 minutes at room temperature). Immunolabeled P-2 pellets were washed with 0.2% Tween20/PBS and resuspended in 500 μL PBS for flow cytometric analysis. Data were acquired with the use of a BD-FACS Calibur analytical flow cytometer (Becton-Dickinson, San Jose, CA) equipped with argon 488 nm, helium-neon 635 nm, and helium-cadmium 325 nm lasers. Debris was excluded by establishing a size threshold set on forward light scatter. A total of 10,000 particles were collected and analyzed for each sample; an additional 5000 Aβ-positive particles were collected for experiments that investigated Aβ-positive synaptosomes. Alexa 488 and Alexa 647 were detected by FL-1 (fluorescein isothiocyanate) and FL-4 (allophycocyanin) channel photomultiplier tubes, respectively. Analysis was performed with the use of FCS Express software version 3 (DeNovo Software, ON, Canada).
Confocal Microscopy
Cryopreserved P-2 fractions were defrosted, fixed, and permeabilized as described in the section above. Fixed P-2 fractions were incubated with primary mouse anti-synaptophysin and conformation-dependent rabbit anti-Aβ prefibrillar oligomer-specific M55 or anti-Aβ fibril-specific M116 antibodies in 2% fetal bovine serum/PBS solution (2 hours at room temperature), followed by washing with 0.2%Tween20/PBS solution and incubation with Alexa 488-conjugated chicken anti-mouse and Alexa 594-conjugated chicken anti-rabbit IgG secondary (1 hour at room temperature). After washing, immunolabled P-2 fractions were dispersed with a pipette and spread on slides. Slides were dried, coverslipped with Vectashield mounting medium (Vector Laboratories Inc., Burlingame, CA), and stored at 4°C. Confocal fluorescence and differential interference contrast images of synaptosomes were performed with the use of a Leica TCS-SP2 confocal microscope (Heidelberg, Germany) with 63× oil immersion objective (numerical aperture 1.2). The Alexa Fluor 488 and 594 fluorescent signals were captured with the use of sequential line scanning with double excitation of 488-nm line from argon laser and 594-nm line from helium-neon laser. Confocal pinhole was set to 1 Airey unit.
Oligomeric and Total Aβ ELISA
To measure concentrations of the soluble Aβ oligomers in human and rat synaptosome-enriched P-2 fractions, we developed a sandwich enzyme-linked immunosorbent assay (ELISA) with the use of the same monoclonal anti-Aβ antibody (10G4) as both the capture and reporter antibodies. High-bind 96-well ELISA plates were coated with unlabeled 10G4 antibody in 0.1 mol/L carbonate buffer, pH 9.6 (overnight at 4°C), followed by blocking with 4% bovine serum albumin in PBS (4 hours at room temperature). A 7-point standard curve (range from 1.95 to 125 ng/mL) (Supplemental Figure S1A) was constructed with the use of photochemically cross-linked synthetic Aβ40 oligomers, which were described previously.38 The cross-linked oligomer standards remained stable in 0.1% 1,1,1,3,3,3-hexafluoro-2-proponol (Supplemental Figure S1A), and the assay did not detect monomeric Aβ (in 0.1% 1,1,1,3,3,3-hexafluoro-2-proponol) (Supplemental Figure S1B). Specificity of the assay for oligomeric Aβ species was also confirmed by measuring Aβ oligomer concentration in the presence of the excess amount of monomeric Aβ (Supplemental Figure S1B). The assay's limit of detection was calculated as 7.48 ng/mL; the limit of quantitation was 20.63 ng/mL. Soluble P-2 fractions were extracted by sonication in PBS with protease inhibitors and then spun at 100,000 × g for 30 minutes. The supernatant fluid was collected and used for the ELISA. Samples and standards were diluted in 1% bovine serum albumin in PBS and loaded into plates in duplicate (overnight at 4°C). After washing with PBS, biotinylated 10G4 antibody was applied (overnight at 4°C). The next day, plates were washed with PBS and incubated with streptavidin-horseradish peroxidase (1 hour at room temperature). The immunocomplex was reacted with 3,3′,5,5′-tetramethylbenzidine substrate and was detected with the use of a Versamax microplate reader (Molecular Devices, Sunnyvale, CA). Concentrations of Aβ oligomers in samples were calculated from the standard curve.
Total Aβ levels were measured with the use of a sandwich ELISA as described previously.39 Concentrations of Aβ oligomers and total Aβ in each sample were normalized against the total protein concentration measured with the use of Pierce BCA Protein Assay Kit according to the manufacturer's protocol.
Statistical Analysis
Statistical analyses were performed with SPSS 16.0 for Mac (SPSS Inc., Chicago, IL). Comparisons of Aβ and tau indices in human tissue between different neuropathologic stages of AD were analyzed by one-way analysis of variance. Additional post hoc comparisons were performed with Tukey's test, when applicable. Comparisons of Aβ and tau indices between transgenic and wild-type rat tissue of different ages were analyzed with unpaired t-tests. Associations between Aβ plaque density and total or oligomeric synaptic Aβ levels were analyzed with Spearman's rank correlation coefficient. Comparisons of synaptic p-tau levels between the overall pool of synaptosomes and the Aβ-positive subset were conducted with paired t-tests.
Results
Total in Situ Synaptic Aβ Is Associated with Plaque Level in Parietal Cortex
To study synaptic Aβ pathology across disease stage, synaptosomes (resealed nerve terminals) were prepared from human parietal cortex samples cryopreserved at the time of autopsy; tissue was finely minced in isotonic sucrose, followed by slow freezing. Synaptosomes are primarily presynaptic and contain mitochondria, endosomes, and exocytotic structures40; some postsynaptic elements are also present, with approximately 36% of particles positive for postsynaptic density protein 95.37 Comparison of synaptosomal proteins with proteins in undisturbed brain slices has shown that synaptic bouton protein composition is maintained in synaptosomes.41 Demographic and neuropathologic data for the normal control, HPC, and AD cases are shown in Table 1. HPCs indicated substantial Aβ and/or tau pathology but did not meet clinical criteria for dementia.
Using flow cytometry to focus the analysis on a large and pure population of synaptic terminals within the P-2 fraction, we first measured synaptic Aβ levels across disease stages in human samples. To label intracellular antigens, samples were first lightly fixed, then permeabilized before immunolabeling with the N-terminal antibody 10G4; therefore total synaptic terminal Aβ accumulation was measured in situ. Note that the protocol for fixation and immunolabeling includes multiple washes of the pellet in large volumes of PBS with 0.2% Tween, which would be expected to remove adherent artifactual Aβ. In addition, incubation of P-2 samples with a combination of trypsin and heparinase before fixation did not reduce synaptic Aβ labeling (Supplemental Figure S2, A and B). Previous work has shown that Aβ-positive synaptosomes are also SNAP-25 positive, and, therefore, not Aβ aggregates.42 In addition, preaggregated Aβ does not show up on flow dot plots,43 and synaptosomes physically sorted on size show a uniform population of spherical particles without visible artifacts.44
Representative dot plots from the flow cytometric analysis are shown in Figure 1; Aβ immunofluorescence is plotted against forward scatter, which is proportional to particle size. Two variables are reported for the fluorescence of each sample: the size of the positive fraction (% positive) and the brightness of fluorescence (relative fluorescence unit). For each experiment, background labeling was determined with an isotype control antibody (Figure 1A), and positive control samples were immunolabeled for the presynaptic docking protein SNAP-25 (Figure 1B). Figure 1B shows the purity of the synaptosomal population examined (approximately 95%) when the rectangular analysis gate, based on size standards, is drawn to include only particles between 0.75 and 1.5 μm. All flow cytometric data are routinely collected from this size-based (forward scatter) analysis gate to capture a relatively pure synaptosome population. We have extensively characterized 10G4 immunolabeling of synaptosomes and compared it with other Aβ and APP antibodies; in contrast to 10G4, the APP antibody 3E9 strongly labels most synaptosomes in both control and AD samples. The bright-specific flow cytometric signal of 10G4 also contrasts with the poorer flow cytometric results with the use of 6E10, which does not label synaptosomes in situ above background, despite mapping to a similar epitope. Like 10G4, MOAB2, a new monoclonal antibody specific for intraneuronal Aβ that does not label APP,45 shows a strong specific signal for synaptic Aβ with flow cytometric analysis (Supplemental Figure S2, C–E). Taken together with evidence from Western blot analysis that 10G4 strongly labels high molecular weight aggregates, SDS-stable oligomers, and monomer in a pattern that resembles labeling by an Aβ42-specific antibody,26 these results indicate that, for flow cytometry, 10G4 is selective for aggregated Aβ in situ. This interpretation is also supported by recent work showing that sequence-specific anti-Aβ antibodies are sensitive to conformation and differentially label aggregated Aβ preparations.46
Figure 1.

Flow cytometric analysis of Aβ immunolabeling across disease stage in Alzheimer disease parietal cortex synaptosomes. Representative dot plots show background in the presence of isotype-specific control antibody (A), SNAP-25 as a positive control and indicator of synaptosomal purity (B). Representative Aβ labeling in an aged cognitively normal control case (C), a case with Braak (synaptosomal-associated protein, 25 kDa NFT) stage IV and plaque stage 0 (D), and a late-stage case with extensive plaques and tangles (E). Ten thousand synaptosomes are plotted for each sample. The FSC variable is proportional to size. Percentages are positive fraction in analysis gate. RFU is mean relative fluorescence in analysis gate. Aβ, amyloid-β; FSC, forward scatter; NFT, neurofibrillary tangle; POS, positive; RFU, relative fluorescence unit; SNAP-25, synaptosomal-associated protein, 25 kDa.
As previously reported, synaptic terminals from control cases show little to no Aβ immunolabeling (Figure 1C)26, 33, 42, 47; representative plots from AD cases illustrate the rise in synaptic Aβ with increasing neuropathologic disease stage and the degree to which synaptic terminal Aβ increases between plaque stage 0 (no plaque deposition) and stage C (end-stage plaque deposition) (Figure 1, D and E).48
Figure 2A shows group differences in aggregate flow cytometric data [F(3,30) = 6.13, P = 0.002] in both early-stage (Braak II to IV) and late-stage (Braak V to VI) AD cases. Total in situ Aβ level was increased above the control and HPC groups (P< 0.05), which were not different from each other. When all cases in the present study are plotted by assigned neurofibrillary tangle and plaque stages (Figure 2B), the AD cases primarily cluster in the highest stage, and cognitively/neuropathologically normal control cases cluster in the lowest stage for both pathologies. However, the HPC cases are distributed across the four plaque stages, ranging from sparse to heavy plaque burden. The total in situ Aβ level measured by flow cytometry was highly correlated with the plaque score assigned by the pathologist for parietal cortex, which was taken from the neuropathology report. The correlation was strong when all cases were included in the analysis (rs = 0.67, P < 0.001) (Figure 2C) and when the HPC group was analyzed alone (rs = 0.68, P = 0.015) (Figure 2D). The progressive increase in total soluble synaptic Aβ across disease stages was confirmed biochemically by ELISA [F(3,18) = 4.91, P = 0.01] (Figure 2E) of the P-2 samples. These results indicate that in situ synaptic Aβ is associated with the local appearance of neuritic plaques, particularly in HPC samples.
Figure 2.

Synaptic Aβ pathology across disease stage. A: Group data show total in situ synaptic Aβ, measured by flow cytometry, plotted by Braak NFT stage. For Braak staging cases were divided into four groups: Con, HPC, AD ≤ IV, and AD ≥ V. B: The assigned NFT stage was plotted against plaque stage for each of the three groups' Cons, HPCs, and AD; HPCs are distributed across plaque stages. C and D: Total in situ Aβ level measured by flow cytometry significantly correlates with the local plaque level in the parietal cortex, taken from the neuropathology report, when all samples are included (C) and for the HPC group considered alone (D). E: ELISA measurements of total soluble Aβ in synaptosome-enriched P-2 samples across disease stage. F: Western blots with 6E10 antibody across disease stage in P-2 samples from parietal cortex. G: Soluble oligomeric Aβ levels in parietal cortex P-2 samples plotted across Braak stages. Data are expressed as means ± SEM. n = 6 Cons, n = 12 HPCs, n = 9 AD ≤ IV, and n = 7 AD ≥ V (A); n = 6 Cons, n = 12 HPCs, and n = 16 AD (B); n = 34 samples (C); n = 12 HPC samples (D); n = 5 Cons, n = 7 HPCs, n = 4 AD ≤ IV, and n = 6 AD ≥ V (E); controls n = 6 Cons, n = 12 HPCs, n = 7 AD ≤ IV, and n = 9 AD ≥ V (G). *P < 0.05, **P < 0.01. Aβ, amyloid-β; AD, Alzheimer disease; AD ≤ IV, Braak stage I to IV; AD ≥ V, Braak stage V to VI; B, Braak stage; Con, control; ELISA, enzyme-linked immunosorbent assay; HPC, high pathology control; NFT, neurofibrillary tangle.
To confirm that flow cytometric increases are not related to APP, Western blot analysis of SDS-PAGE with the 6E10 antibody show large increases in Aβ monomer and lesser changes in small (<50 kDa) oligomers with neuropathologic disease stage in P-2 fractions from parietal cortex (Figure 2F). Some of these bands may also represent APP fragments that contain the 6E10 epitope. Some oligomers between 10 and 100 kDa did not change with disease stage, particularly a tetrameric peptide. This result is consistent with previous work showing that the Aβ42 tetramer structure is likely to be stable and long-lived and serves as a module for formation of larger Aβ assemblies.49 A prominent set of bands at approximately 55 kDa seems likely to include Aβ*56.50
Synapse-Associated Soluble Oligomers Are Associated with Onset of Dementia
In the same groups of staged parietal cortex samples, we also measured soluble Aβ oligomers (oAβ) in synaptosome-enriched P-2 fractions by ELISA. This assay, based on protocols developed recently by others,51, 52, 53, 54, 55 used the same antibody (10G4) for capture and detection and cross-linked oligomeric Aβ40 for the standards. Synaptic oAβ was markedly elevated in the early AD group relative to the neuropathologically normal (P = 0.01) and HPC (P = 0.05) groups [F(3,30) = 4.13, P = 0.015] (Figure 2G). oAβ was also numerically higher in early AD than in late AD (71.3 ± 29 ng/mg protein), but this difference did not reach significance after adjusting for multiple comparisons (P = 0.096). In contrast to total in situ Aβ measured by flow cytometry, parietal cortex oAβ did not correlate with Aβ plaque counts from neuropathology reports (data not shown). The sharp oAβ elevation in early AD cases suggests that the clinical syndrome of AD dementia may emerge once the level of synapse-associated soluble oligomers exceeds a certain threshold.
To confirm that synaptic Aβ is oligomeric, P-2 samples were immunolabeled with conformation-dependent monoclonal antibodies and spread on slides for confocal microscopy. These novel monoclonal antibodies were derived from the well-characterized polyclonal A11 (prefibrillar oligomers) and OC antibodies (fibrillar oligomers); only the fibril-specific antibody M116 labels Aβ deposits in the AD brain.56 Characterization of these antibodies has shown a number of distinct subtypes within the prefibrillar and fibrillar classes of Aβ oligomers, illustrating the diversity of Aβ oligomers in the AD brain.46, 57 A subset of spheroid AD synaptosomes showed diffuse labeling for both prefibrillar (antibody M55) (Figure 3, A–D) and fibrillar Aβ oligomers (antibody M116) (Figure 3, E–H), indicating structural heterogeneity in Aβ oligomers within terminals.
Figure 3.

Conformation-dependent antibodies label Aβ in synaptic terminals. A–D: Synaptosomes were dual-labeled with synaptotophysin and the monoclonal antibody M55 directed against pre-fibrillar oligomers: M55 (A); SYP (B); overlay image with arrows indicate colocalization (C); DIC image (D). E–H: Synaptosomes dual-labeled with SYP and the monoclonal antibody M116 directed against fibrillar oligomers: M116 (E); SYP (F); overlay image with arrows indicate co-localization (G); DIC image (H). Scale bar. Aβ, amyloid-β; DIC, differential interference contrast; SYP, synaptophysin. Scale bar = 5 μm.
Progression of Total in Situ Synaptic Aβ and oAβ in a Transgenic Rat Model of AD
Because animal models are widely used to examine pathology and to test therapies, particularly amyloid pathology, we next examined progression of synaptic Aβ pathology in a transgenic rat model of AD expressing two familial mutations and one familial presenilin-1 mutation. Representative plots from the flow cytometric analysis show little Aβ immunolabeling (approximately 10% to 18% positive) in mixed cortex from wild-type animals at all ages (not shown) and in 3- and 9-month-old transgenic animals (Figure 4, A and B). However, in 20-month-old transgenic animals, fluorescence for total synaptic Aβ increased approximately fourfold in a representative mixed cortex sample (Figure 4C).
Figure 4.

A–C: Flow cytometric analysis of Aβ in a transgenic rat model of Alzheimer disease (APP/PS1). Representative Aβ labeling in transgenic rat mixed cortex samples (Tg) is shown at 3 (A), 9 (B), and 20 (C) months; 10,000 synaptosomes are plotted for each sample; the FSC variable is proportional to size; percentage, positive fraction in analysis gate, RFU, mean relative fluorescence in analysis gate. D and E: Group data show total in situ synaptic Aβ at 3, 9, and 20 months measured by flow cytometry in WT and Tg rat mixed cortex (D) and frontal cortex (E). F: Soluble oligomeric Aβ level in mixed cortex P-2 samples is plotted for WT and Tg at 3, 9, and 20 months. Data are expressed as means ± SEM. n = 6 to 9 in each group. ***P < 0.001. Aβ, amyloid-β; APP, Aβ (A4) precursor protein; FSC, forward scatter; PS1, presenilin-1; RFU, relative fluorescence unit; Tg, transgenic; WT, wild-type.
The aggregate flow cytometric data for the animal experiments are shown in Figure 4, D and E, which confirm the huge synaptic amyloid burden at 20 months in both mixed cortex [t(15) = −8.89, P < 0.001] and frontal cortex [t(18) = −10.00, P < 0.001].
Corresponding to human results, the ELISA for soluble oAβ (Figure 4F) in transgenic rat P-2 samples also shows age and genotype effects with significant elevations in each age group, beginning at 3 months [age: F(2,53) = 44.10, P < 0.001; genotype: F(1,35) = 54.13, P < 0.001; age × genotype interaction: F(2,35) = 44.06, P < 0.001]. Importantly, subtle cognitive deficits are already detectable by 3 months of age in this model.
Synaptic p-Tau Is Not Elevated until Late-Stage AD in Parietal Cortex
Because tau and p-tau oligomers may contribute to synaptic dysfunction and loss,8, 27, 29, 58 we next examined timing of the appearance of synaptic p-tau for disease stage and amyloid pathology using the set of staged human samples, including nondemented HPCs. P-2 samples were immunolabeled for tau with the use of three antibodies: one directed against total tau (HT7) and two specific for p-tau (PHF-1 and pS422). The same immunolabeling protocols described above for flow cytometric analysis of synaptic Aβ were used; therefore, results likewise represent in situ tau associated with synaptic boutons. As noted above for Aβ in dual-labeled P-2 samples virtually all p-tau positives are also SNAP-25 positive, ruling out tangle remnants as artifacts (data not shown). Representative flow cytometric plots highlight the relatively low level of p-tau fluorescence in individual synaptosomes for PHF-1 until late-stage disease (Figure 5, A–C). Aggregate plots of the data across disease stages confirm that tau immunofluoresence is not increased until late-stage AD for all three antibodies examined [HT7 (total tau): F(3,30) = 9.67, P < 0.001) (Figure 5D), [PHF-1 (p-tau)]: F(3,30) = 8.08, P < 0.001] (Figure 5E), and [pS422 (p-tau): F(3,30) = 10.95, P < 0.001] (Figure 5F). HT7 antibody labels both phosphorylated and nonphosphorylated (total) tau; therefore, p-tau likely accounts for the increased HT7 fluorescence in late-stage AD (Figure 5D).
Figure 5.

Synaptic p-tau is not elevated until late-stage Alzheimer disease in parietal cortex. A, C, and E: Representative dot plots from the flow cytometric analysis are shown for p-tau labeling with the PHF-1 antibody; 10,000 synaptosomes are plotted for each sample; the FSC variable is proportional to size; percentage, positive fraction in analysis gate, RFU, mean relative fluorescence in analysis gate. B, D, and F: Aggregate plots of flow cytometric data from synaptosome-enriched P-2 samples from parietal cortex for total tau with HT-7 (B), and p-tau with PHF-1 (D), and pS422 (F) across disease stages. Cases were divided into four groups: Cons, HPCs, AD ≤ IV, and AD ≥ V. G: Representative dot plot from flow cytometric analysis for an early Braak stage case with elevated p-tau signal in parietal cortex P-2. Data are expressed as means ± SEM. n = 5 to 6 Cons, n = 11 to 12 HPCs, n = 9 AD ≤ IV, and n = 8 to 9 AD ≥ V (B, D, and F). ***P < 0.001. AD, Alzheimer disease; AD ≤ IV, Braak I to IV; AD ≥ V, Braak V to VI; Con, control; FSC, forward scatter; HPC, high pathology control; p-tau, hyperphosphorylated tau; RFU, relative fluorescence unit.
Although overall synaptic p-tau level in the parietal cortex is not elevated until late-stage AD, some cases found relatively high levels of synaptic p-tau in the parietal cortex in early-stage neurofibrillary disease. Figure 5G shows representative flow cytometric data for one such Braak III case with high p-tau fluorescence in parietal cortex. No apparent association was found between higher p-tau levels and mixed pathology in the present study. These data indicate that, in some circumstances, advanced neocortical synaptic tau pathology may be seen in early-stage disease, before the cortical deposition of NFTs.
Synaptic p-Tau Follows Amyloid Pathology in Aged Transgenic APP/PS1 Rats in the Absence of Tau Mutation or Overexpression
Tau hyperphosphorylation has been long observed in a number of animal models of AD that express only amyloid pathology, and several groups, including ours, have localized p-tau to synapses in such animal models.22, 33, 59 With the use of cryopreserved cortex samples from the same transgenic rat age groups as above (3, 9, and 20 months), we investigated the association of synaptic p-tau with disease stage and amyloid pathology. P-2 samples were immunolabeled for p-tau; flow cytometric analyses are illustrated in Figure 6, A–C and show a representative increase in synaptic PHF-1 fluorescence in transgenic relative to wild-type animals at 20 months. Aggregate group data are shown in Figure 6D. Synaptic PHF-1 labeling only found significant genotype differences at 20 months of age [t(13) = −3.01, P = 0.010]. However, no genotype effects were seen for synaptic pS422 labeling. These findings are consistent with human results above; however, as previously observed in mouse models with only Aβ mutations,60, 61 synaptic p-tau pathology in this rat model is less robust than that seen above in human disease.
Figure 6.

Synaptic p-tau follows amyloid pathology in a transgenic rat model of Alzheimer disease. A–C: Representative dot plots from the flow cytometric analysis are shown for background staining in the presence of isotype-specific control antibody (A), and p-tau immunolabeling with the PHF-1 antibody in a 20-month-old WT (B)] and Tg (C) P-2 samples. Ten thousand synaptosomes are plotted for each sample; the FSC variable is proportional to size; percentage, positive fraction in analysis gate, RFU, mean relative fluorescence in analysis gate. D: Group data for 3-, 9-, and 20-month-old animals; p-tau immunolabeling is shown for the PHF-1 in frontal cortex P-2 samples. Data are expressed as means ± SEM. n = 8 animals per group. **P = 0.01. FSC, forward scatter; p-tau, hyperphosphorylated tau; RFU, relative fluorescence unit; Tg, transgenic; WT, wild-type.
P-Tau Immunolabeling Is Elevated in Individual Aβ-Positive Synaptosomes in Early AD
If early synaptic Aβ accumulation drives tau phosphorylation within terminals, synaptic p-tau should be higher in the population of terminals positive for Aβ. To test this hypothesis, we dual-labeled human parietal cortex P-2 samples for Aβ (10G4) and p-tau (PHF-1), and used flow cytometry to collect data from Aβ-positive synaptosomes (5000 per sample). This experiment used cases with early-stage disease (Braak II to III), in which the mean synaptic p-tau level was not different from controls (Figure 5); we also included hippocampus and entorhinal cortex samples as regions that are affected earlier by tau pathology. Figure 7, A–C, illustrates the analysis for a single dual-labeled sample; Figure 7A shows the total PHF-1-positive fraction (10,000 events), and Figure 7B shows the analysis gate for collection of Aβ-positive terminals in the same sample (5000 events); note that for this analysis strategy only data from the Aβ-positives were saved. P-tau immunofluorescence increased from 8.9% in the total sample (Figures 7A) to 21.5% when only Aβ-positives were included (Figure 7C). The aggregate data (Figure 7D) found that p-tau immunofluorescence was elevated in the Aβ-positive fraction relative to the total sample in all three regions examined, increasing more than twofold in each region [t(11) = −6.15, P < 0.001], hippocampal [t(6) = −5.99, P = 0.001], and entorhinal cortex [t(5) = −9.05, P < 0.001].
Figure 7.

Elevation of p-tau immunolabeling in Aβ-positive synaptosomes. A–C: Flow cytometric analysis of a single AD parietal cortex synaptosome-enriched P-2 sample dual-labeled for Aβ (10G4 antibody) and p-tau (PHF-1 antibody). Representative plots of the same dual-labeled sample show the size of the total p-tau positive fraction (8.82% (A); 10,000 particles], the collection of Aβ-positive synaptosomes inside the analysis gate (B), and increased p-tau in the Aβ-positive synaptosomes (21.5%; C, upper right quadrant). Five thousand particles were collected for each sample; the FSC variable is proportional to size; percentage, positive fraction in analysis gate, RFU, mean relative fluorescence in analysis gate. D: Group data for dual-labeling analysis in PAR, HIPP, and EC. E and F: Correlation analysis of Aβ and p-tau immunolabeling in early-stage AD (F) and Aβ and p-tau correlation in early-stage AD HIPP and EC cortex. Data are expressed as means ± SEM. n = 28 PAR, n = 7 HIPP, and n = 6 EC (D); n = 20 ≤Braak IV (E); n = 20 ≤Braak IV (F). ***P < 0.001. Ab, antibody; Aβ, amyloid-β; AD, Alzheimer disease; EC, entorhinal cortex; FSC, forward scatter; HIPP, hippocampus; PAR, parietal cortex; p-tau, hyperphosphorylated tau; RFU, relative fluorescence unit.
To further examine the relation, correlation analyses were performed; despite the increased PHF-1 labeling in Aβ-positives, the PHF-1 level in each sample was not strongly correlated with the Aβ level for parietal cortex across all disease stages (r = 0.20, n = 30; not shown). Reasoning that terminal accumulation of Aβ in mid- and late-stage disease might obscure a relation, we next included only parietal cortex samples with early-stage AD (≤Braak IV), which likewise showed no significant association (r = 0.33, n = 20) (Figure 6E). However, early-stage samples from hippocampus and entorhinal cortex, affected earlier in disease, found a strong correlation (r = 0.92, n = 10; P < 0.0001) (Figure 7F). On the basis of the data presented earlier, which establishes the early appearance of Aβ compared with p-tau, this association is consistent with Aβ-driven induction of tau phosphorylation in synaptic terminals relatively early in the disease process.
Discussion
Multiple lines of evidence document that substantial proportions of cognitively intact elderly show abundant AD pathology on autopsy or substantial amyloid deposition by neuroimaging.62, 63, 64, 65 However, robust biochemical and neuropathologic differences have not been observed between cognitively intact and demented individuals with similar levels of AD pathology. On the basis of a working hypothesis that synaptic pathology may drive clinical symptoms, the present work used synaptosome preparations made from cryopreserved samples to focus on changes within synapses and shows that high levels of soluble amyloid oligomers within synaptic terminals are present in early dementia but not in HPCs, suggesting that clinical dementia emerges when the Aβ oligomers within terminals reach a certain level. Aβ accumulation occurs in the early stages of AD in the parietal cortex, but tau is not substantially elevated until late-stage disease. However, even in early-stage disease, p-tau may be elevated in individual Aβ-positive terminals.
The total synaptic Aβ measured by flow cytometry in the present experiments includes all forms of Aβ in synaptic terminals detectable by an N-terminal antibody in situ. In contrast, the oAb assay measures only oligomeric Aβ extracted from synaptosome-enriched P-2 fractions into buffer. Because Western blot analysis of P-2 samples shows a strong signal for postsynaptic density protein 95,26 it is possible that some of the Aβ signal in P-2 samples arises from extracellular Aβ bound to postsynaptic surface receptors, particularly heparan sulfate proteoglycan.66 However, prior electron microscopic studies reported by our group and others document the spherical and primarily presynaptic structure of synaptosomes,33, 40 and incubation of AD synaptosomes with a combination of trypsin and heparinase does not reduce Aβ labeling. Confirmation of presynaptic localization of oligomers by electron microscope would be optimal; however, detection of intraneuronal, particularly oligomeric, Aβ is limited by the technical difficulty of detecting Aβ antigens, which are hydrophobic and difficult to detect by conventional immunocytochemical methods, especially in postmortem tissue.67 On the basis of previous and current results, we posit that most synaptosomal Aβ represents an intraterminal pool largely localized within endosomal and autophagic vesicles, consistent with a strong literature showing synaptic release of Aβ25, 68, 69 and disruption of autophagy in AD.69, 70, 71, 72
Relatively few examples in the literature have found cellular or pathologic factors associated with cognitive protection in subjects with AD pathology.73 A theory that altered synaptic structure or function may be a key factor for protection against AD-related cognitive decline is attractive for a number of reasons, in part because of the strong association of cognitive decline with synapse loss, and because established AD pathology localizes to either extracellular (plaques) or somatic (tangles) regions. Along this line, two groups have recently showed the relative preservation of synaptic markers in cortex from nondemented high AD pathology subjects.74, 75 One of these studies included a discovery microarray approach that identified a number of proteins that are differentially expressed in such subjects, including APP binding protein and regulators of apoptosis, trafficking, cytoskeletal structure, and cell-cycle proteins.74 Work by Perez-Nievas et al75 showed more fibrillar and oligomer-positive Aβ plaques, increased p-tau oligomers, and glial activation in demented compared with nondemented subjects with underlying AD pathology. In another study, low molecular weight SDS-stable Aβ oligomers were associated with the postsynaptic density in patients with Alzheimer dementia but not in nondemented HPCs.76 Results are mixed for soluble Aβ oligomers in cortical homogenates, but increased oligomers were reported in demented compared with HPC cases.51 Our data come from synaptosome-enriched P-2 fractions with membrane cryopreservation, which would be expected to retain soluble oligomers; this is in contrast to typical homogenate samples, which are more dilute and represent multiple cell types and cellular compartments. In the present experiments, the concentration of synaptic oligomers was the only Aβ measure to distinguish HPCs from early dementia cases. Although noting that this study is based on a relatively small set of cases, this observation signals an association between synaptic Aβ oligomers and clinical diagnoses of dementia.
The correlation between total in situ synaptic Aβ and local plaque levels in the parietal cortex could be explained by accumulation of insoluble Aβ deposits in two sinks: an extracellular one in plaques and an intraterminal one within endosomes/autophagic vacuoles. After eventual loss and degradation of the synaptic terminal, the insoluble synaptic vacuole pool would then serve as a seed for a new extracellular deposit. This explanation would be consistent with our observation of a (nonsignificant) reduction of oAβ in late-stage compared with early-stage disease and with the view that amyloid plaques are predominately a sink rather than a source for soluble Aβ.2 However, plaques could still serve as local reservoirs for small oligomers, operating with a dynamic equilibrium.77 For the oAβ assay, note that the sandwich ELISA may detect multiple Aβ molecules bound to an unrelated carrier protein such as a proteoglycan; this possibility should be somewhat mitigated by the use of an aqueous soluble fraction without membranes or membrane fragments.
Because tau aggregates can transfer from one cell to another, some p-tau in synapses may enter from the extracellular compartment.78, 79 However, considered together with the early appearance of Aβ relative to p-tau, the marked increase in p-tau in individual Aβ-positive synapses is consistent with a hypothesis that p-tau induction is directly driven by synaptic oAβ; synaptic p-tau may then lead to anterograde and retrograde trans-synaptic spread of tau pathology. Such a hypothesis is supported by the following multiple lines of evidence: i) the exclusive initial appearance of Aβ in the neocortex before dementia onset, which has extensive reciprocal projections with hippocampus and entorhinal cortex48, 80; ii) the well-established regional propagation of tau pathology in human disease and animal models79, 81, 82, 83; iii) the observation that low levels of dimeric oAβ from the AD cortex are sufficient for induction of tau phosphorylation and beaded dystrophic neurites84; and iv) evidence to indicate tau intracellular protein misfolding can be transmitted by direct protein-protein contact.78 Indeed, such a prion-like mechanism of endogenous templated protein corruption was suggested as a common mechanism for multiple neurodegenerative diseases.85 Additional Aβ-mediated mechanisms may also contribute to propagation of AD pathology; for example, oligomer binding to membrane lipids or to α7 nicotinic cholinergic receptors might modulate the downstream signaling.84, 86, 87
Our results suggest that effective therapies will need to target synaptic Aβ oligomers and that antiamyloid therapies will be much less effective once synaptic p-tau pathology has developed, thus providing a potential explanation for the failure of amyloid-based trials. An additional important clinical implication is that therapies slowing Aβ oligomer accumulation in the synaptic compartment will delay onset of dementia. Clarifying the precise mechanisms by which soluble Aβ oligomers induce p-tau induction and contribute to synaptic dysfunction in early dementia is an important next step; the correspondence between our human and animal data suggest that this and related animal models will be useful for understanding the progress of synaptic pathology and for developing therapies to protect synaptic terminals.
Acknowledgments
Confocal laser scanning microscopy was performed at the CNSI Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA. PHF-1 and the anti-Aβ antibodies were generous gifts from Drs. Peter Davies (Albert Einstein College of Medicine, Bronx, NY) and Charles G. Glabe (University of California Irvine, Irvine, CA), respectively.
Footnotes
Supported by NIH grants AG27465 (K.H.G.), NS038328 (D.B.T.), and AG041295 (D.B.T.); National Institute on Aging (NIA) grants AG18879 (C.A.M.) and AG34628 (jointly sponsored by the NIA, American Federation for Aging Research, the John A. Hartford Foundation, the Atlantic Philanthropies, the Starr Foundation, and an anonymous donor; to E.T.); the Jim Easton Consortium for Alzheimer's Drug Discovery and Biomarkers (D.B.T.); University of California Los Angeles Clinical and Translational Science Institute (UCLA CTSI) grant UL1TR000124 (E.Y.H.); UCLA Older Americans Independence Center grant P30 AG028748 (E.Y.H.); and the Daljit S. and Elaine Sarkaria Chair in Diagnostic Medicine (H.V.V.). Tissue was obtained from the AD Research Center Neuropathology Cores NIA grants P50 AG05142 (USC), P50 AG16970 (UCLA), and P50 AG16573 (UC Irvine). Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility, supported by NIH grants CA16042 and AI 28697 and by the JCCC, the UCLA AIDS Institute, the David Geffen School of Medicine, and the Chancellor's Office at UCLA. Diagnosis, characterization, and follow-up of >90 study subjects was supported by NIA grant R01AG21055.
Disclosures: None declared.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2015.09.018.
Supplemental Data
Supplemental Figure S1.
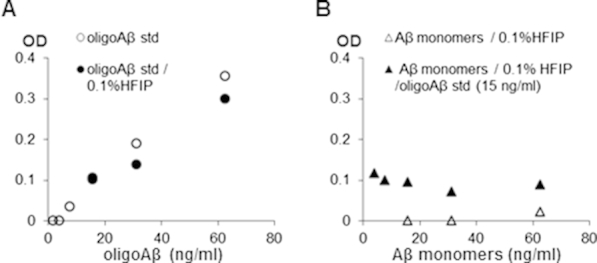
Controls for oAβ ELISA for soluble synaptic Aβ oligomers. A: Standard curve for cross-linked oligomeric Aβ 40 standards and stability of cross-linked oligomeric Aβ standards in 0.1% HFIP. B: Cross-linked oAβ standard (15 ng/mL) measured by ELISA in the presence of excess monomeric Aβ40. Aβ, amyloid-β; ELSIA, enzyme-linked immunosorbent assay; HFIP, 1,1,1,3,3,3-hexafluoro-2-proponol; oAβ, soluble Aβ oligomers.
Supplemental Figure S2.
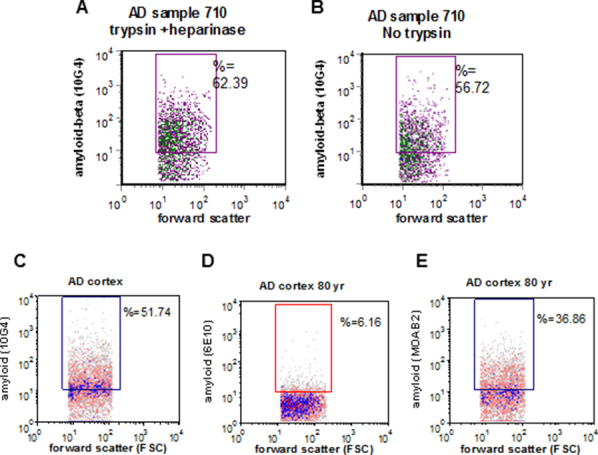
Total in situ synaptic Aβ is not removed by incubation with trypsin and heparinase. Aβ immunolabeling with the 10G4 antibody was quantified by flow cytometry in an AD sample with (A) and without (B) incubation with 0.01% trypsin (2 minutes room temperature) and 10 u/mL heparinase (5 minutes, 32°C) before the fixation step. C–E: Comparison of flow cytometric immunolabeling of synaptosomes with 10G4 (C), 6E10 (D), and MOAB2 (E) antibodies. Aβ, amyloid-β; AD, Alzheimer disease.
References
- 1.Larson M.E., Lesne S.E. Soluble Abeta oligomer production and toxicity. J Neurochem. 2012;120 Suppl 1:125–139. doi: 10.1111/j.1471-4159.2011.07478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hefti F., Goure W.F., Jerecic J., Iverson K.S., Walicke P.A., Krafft G.A. The case for soluble Abeta oligomers as a drug target in Alzheimer's disease. Trends Pharmacol Sci. 2013;34:261–266. doi: 10.1016/j.tips.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 4.Bierer L.M., Hof P.R., Purohit D.P., Carlin L., Schmeidler J., Davis K.L., Perl D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 5.Giannakopoulos P., Herrmann F.R., Bussiere T., Bouras C., Kovari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 6.Gomez-Isla T., Price J.L., McKeel D.W., Jr., Morris J.C., Growdon J.H., Hyman B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–4500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyman B.T., Marzloff K., Arriagada P.V. The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol. 1993;52:594–600. doi: 10.1097/00005072-199311000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Ingelsson M., Fukumoto H., Newell K.L., Growdon J.H., Hedley-Whyte E.T., Frosch M.P., Albert M.S., Hyman B.T., Irizarry M.C. Early Abeta accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–931. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 9.Cummings B.J., Cotman C.W. Image analysis of beta-amyloid load in Alzheimer's disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 10.Defigueiredo R.J., Cummings B.J., Mundkur P.Y., Cotman C.W. Color image analysis in neuroanatomical research: application to senile plaque subtype quantification in Alzheimer's disease. Neurobiol Aging. 1995;16:211–223. doi: 10.1016/0197-4580(94)00151-0. [DOI] [PubMed] [Google Scholar]
- 11.Kanne S.M., Balota D.A., Storandt M., McKeel D.W., Jr., Morris J.C. Relating anatomy to function in Alzheimer's disease: neuropsychological profiles predict regional neuropathology 5 years later. Neurology. 1998;50:979–985. doi: 10.1212/wnl.50.4.979. [DOI] [PubMed] [Google Scholar]
- 12.Haroutunian V., Perl D.P., Purohit D.P., Marin D., Khan K., Lantz M., Davis K.L., Mohs R.C. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–1191. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- 13.Selkoe D.J. Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–113. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mairet-Coello G., Courchet J., Pieraut S., Courchet V., Maximov A., Polleux F. The CAMKK2-AMPK kinase pathway mediates the synaptotoxic effects of Abeta oligomers through Tau phosphorylation. Neuron. 2013;78:94–108. doi: 10.1016/j.neuron.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gotz J., Chen F., van Dorpe J., Nitsch R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 16.Chabrier M.A., Blurton-Jones M., Agazaryan A.A., Nerhus J.L., Martinez-Coria H., LaFerla F.M. Soluble abeta promotes wild-type tau pathology in vivo. J Neurosci. 2012;32:17345–17350. doi: 10.1523/JNEUROSCI.0172-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ittner L.M., Ke Y.D., Delerue F., Bi M., Gladbach A., van Eersel J., Wolfing H., Chieng B.C., Christie M.J., Napier I.A., Eckert A., Staufenbiel M., Hardeman E., Gotz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 18.DeKosky S.T., Scheff S.W., Styren S.D. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5:417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- 19.DeKosky S.T., Scheff S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457–464. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 20.Terry R.D., Masliah E., Salmon D.P., Butters N., DeTeresa R., Hill R., Hansen L.A., Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30:572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 21.Masliah E., Mallory M., Hansen L., DeTeresa R., Alford M., Terry R. Synaptic and neuritic alterations during the progression of Alzheimer's disease. Neurosci Lett. 1994;174:67–72. doi: 10.1016/0304-3940(94)90121-x. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi R.H., Capetillo-Zarate E., Lin M.T., Milner T.A., Gouras G.K. Co-occurrence of Alzheimer's disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging. 2010;31:1145–1152. doi: 10.1016/j.neurobiolaging.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi R.H., Milner T.A., Li F., Nam E.E., Edgar M.A., Yamaguchi H., Beal M.F., Xu H., Greengard P., Gouras G.K. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma T., Klann E. Amyloid beta: linking synaptic plasticity failure to memory disruption in Alzheimer's disease. J Neurochem. 2012;120 Suppl 1:140–148. doi: 10.1111/j.1471-4159.2011.07506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cirrito J.R., Yamada K.A., Finn M.B., Sloviter R.S., Bales K.R., May P.C., Schoepp D.D., Paul S.M., Mennerick S., Holtzman D.M. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–922. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 26.Sokolow S., Henkins K.M., Bilousova T., Miller C.A., Vinters H.V., Poon W., Cole G.M., Gylys K.H. AD synapses contain abundant Abeta monomer and multiple soluble oligomers, including a 56-kDa assembly. Neurobiol Aging. 2012;33:1545–1555. doi: 10.1016/j.neurobiolaging.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Henkins K.M., Sokolow S., Miller C.A., Vinters H.V., Poon W., Cornwell L.B., Saing T., Gylys K.H. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer's cortical synapses. Brain Pathol. 2012;22:826–833. doi: 10.1111/j.1750-3639.2012.00598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tai H.C., Serrano-Pozo A., Hashimoto T., Frosch M.P., Spires-Jones T.L., Hyman B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am J Pathol. 2012;181:1426–1435. doi: 10.1016/j.ajpath.2012.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasagna-Reeves C.A., Castillo-Carranza D.L., Sengupta U., Clos A.L., Jackson G.R., Kayed R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol Neurodegener. 2011;6:39. doi: 10.1186/1750-1326-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maeda S., Sahara N., Saito Y., Murayama S., Ikai A., Takashima A. Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer's disease. Neurosci Res. 2006;54:197–201. doi: 10.1016/j.neures.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 31.Patterson K.R., Remmers C., Fu Y., Brooker S., Kanaan N.M., Vana L., Ward S., Reyes J.F., Philibert K., Glucksman M.J., Binder L.I. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011;286:23063–23076. doi: 10.1074/jbc.M111.237974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang F., Mak K., Vinters H.V., Frautschy S.A., Cole G.M. Monoclonal antibody to the C-terminus of beta-amyloid. Neuroreport. 1994;5:2117–2120. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]
- 33.Fein J.A., Sokolow S., Miller C.A., Vinters H.V., Yang F., Cole G.M., Gylys K.H. Co-localization of amyloid beta and tau pathology in Alzheimer's disease synaptosomes. Am J Pathol. 2008;172:1683–1692. doi: 10.2353/ajpath.2008.070829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flood D.G., Lin Y.G., Lang D.M., Trusko S.P., Hirsch J.D., Savage M.J., Scott R.W., Howland D.S. A transgenic rat model of Alzheimer's disease with extracellular Abeta deposition. Neurobiol Aging. 2009;30:1078–1090. doi: 10.1016/j.neurobiolaging.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 35.Teng E., Kepe V., Frautschy S.A., Liu J., Satyamurthy N., Yang F., Chen P.P., Cole G.B., Jones M.R., Huang S.C., Flood D.G., Trusko S.P., Small G.W., Cole G.M., Barrio J.R. [F-18]FDDNP microPET imaging correlates with brain Abeta burden in a transgenic rat model of Alzheimer disease: effects of aging, in vivo blockade, and anti-Abeta antibody treatment. Neurobiol Dis. 2011;43:565–575. doi: 10.1016/j.nbd.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L., Orozco I.J., Planel E., Wen Y., Bretteville A., Krishnamurthy P., Wang L., Herman M., Figueroa H., Yu W.H., Arancio O., Duff K. A transgenic rat that develops Alzheimer's disease-like amyloid pathology, deficits in synaptic plasticity and cognitive impairment. Neurobiol Dis. 2008;31:46–57. doi: 10.1016/j.nbd.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gylys K.H., Fein J.A., Yang F., Cole G.M. Enrichment of presynaptic and postsynaptic markers by size-based gating analysis of synaptosome preparations from rat and human cortex. Cytometry A. 2004;60:90–96. doi: 10.1002/cyto.a.20031. [DOI] [PubMed] [Google Scholar]
- 38.Rosensweig C., Ono K., Murakami K., Lowenstein D.K., Bitan G., Teplow D.B. Preparation of stable amyloid beta-protein oligomers of defined assembly order. Methods Mol Biol. 2012;849:23–31. doi: 10.1007/978-1-61779-551-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lim G.P., Yang F., Chu T., Chen P., Beech W., Teter B., Tran T., Ubeda O., Ashe K.H., Frautschy S.A., Cole G.M. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunkley P.R., Jarvie P.E., Robinson P.J. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- 41.Wilhelm B.G., Mandad S., Truckenbrodt S., Krohnert K., Schafer C., Rammner B., Koo S.J., Classen G.A., Krauss M., Haucke V., Urlaub H., Rizzoli S.O. Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science. 2014;344:1023–1028. doi: 10.1126/science.1252884. [DOI] [PubMed] [Google Scholar]
- 42.Gylys K.H., Fein J.A., Yang F., Miller C.A., Cole G.M. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer's disease cortex. Neurobiol Aging. 2007;28:8–17. doi: 10.1016/j.neurobiolaging.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 43.Gylys K.H., Fein J.A., Tan A.M., Cole G.M. Apolipoprotein E enhances uptake of soluble but not aggregated amyloid-beta protein into synaptic terminals. J Neurochem. 2003;84:1442–1451. doi: 10.1046/j.1471-4159.2003.01643.x. [DOI] [PubMed] [Google Scholar]
- 44.Sokolow S., Henkins K.M., Williams I.A., Vinters H.V., Schmid I., Cole G.M., Gylys K.H. Isolation of synaptic terminals from Alzheimer's disease cortex. Cytometry A. 2012;81:248–254. doi: 10.1002/cyto.a.22009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Youmans K.L., Tai L.M., Kanekiyo T., Stine W.B., Jr., Michon S.C., Nwabuisi-Heath E., Manelli A.M., Fu Y., Riordan S., Eimer W.A., Binder L., Bu G., Yu C., Hartley D.M., LaDu M.J. Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol Neurodegener. 2012;7:8. doi: 10.1186/1750-1326-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hatami A., Albay R., 3rd, Monjazeb S., Milton S., Glabe C. Monoclonal antibodies against Abeta42 fibrils distinguish multiple aggregation state polymorphisms in vitro and in Alzheimer disease brain. J Biol Chem. 2014;289:32131–32143. doi: 10.1074/jbc.M114.594846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gylys K.H., Fein J.A., Yang F., Wiley D.J., Miller C.A., Cole G.M. Synaptic changes in Alzheimer's disease: increased amyloid-beta and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. Am J Pathol. 2004;165:1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thal D.R., Rub U., Orantes M., Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–1800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 49.Bernstein S.L., Dupuis N.F., Lazo N.D., Wyttenbach T., Condron M.M., Bitan G., Teplow D.B., Shea J.E., Ruotolo B.T., Robinson C.V., Bowers M.T. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nat Chem. 2009;1:326–331. doi: 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lesne S.E., Sherman M.A., Grant M., Kuskowski M., Schneider J.A., Bennett D.A., Ashe K.H. Brain amyloid-beta oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Esparza T.J., Zhao H., Cirrito J.R., Cairns N.J., Bateman R.J., Holtzman D.M., Brody D.L. Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls. Ann Neurol. 2013;73:104–119. doi: 10.1002/ana.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Youmans K.L., Tai L.M., Nwabuisi-Heath E., Jungbauer L., Kanekiyo T., Gan M., Kim J., Eimer W.A., Estus S., Rebeck G.W., Weeber E.J., Bu G., Yu C., Ladu M.J. APOE4-specific changes in Abeta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012;287:41774–41786. doi: 10.1074/jbc.M112.407957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bruggink K.A., Jongbloed W., Biemans E.A., Veerhuis R., Claassen J.A., Kuiperij H.B., Verbeek M.M. Amyloid-beta oligomer detection by ELISA in cerebrospinal fluid and brain tissue. Anal Biochem. 2013;433:112–120. doi: 10.1016/j.ab.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 54.Kasai T., Tokuda T., Taylor M., Kondo M., Mann D.M., Foulds P.G., Nakagawa M., Allsop D. Correlation of Abeta oligomer levels in matched cerebrospinal fluid and serum samples. Neurosci Lett. 2013;551:17–22. doi: 10.1016/j.neulet.2013.06.029. [DOI] [PubMed] [Google Scholar]
- 55.Yang T., Hong S., O'Malley T., Sperling R.A., Walsh D.M., Selkoe D.J. New ELISAs with high specificity for soluble oligomers of amyloid beta-protein detect natural Abeta oligomers in human brain but not CSF. Alzheimers Dement. 2013;9:99–112. doi: 10.1016/j.jalz.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McLean D., Cooke M.J., Albay R., 3rd, Glabe C., Shoichet M.S. Positron emission tomography imaging of fibrillar parenchymal and vascular amyloid-beta in TgCRND8 mice. ACS Chem Neurosci. 2013;4:613–623. doi: 10.1021/cn300226q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kayed R., Canto I., Breydo L., Rasool S., Lukacsovich T., Wu J., Albay R., 3rd, Pensalfini A., Yeung S., Head E., Marsh J.L., Glabe C. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Abeta oligomers. Mol Neurodegener. 2010;5:57. doi: 10.1186/1750-1326-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kopeikina K.J., Polydoro M., Tai H.C., Yaeger E., Carlson G.A., Pitstick R., Hyman B.T., Spires-Jones T.L. Synaptic alterations in the rTg4510 mouse model of tauopathy. J Comp Neurol. 2013;521:1334–1353. doi: 10.1002/cne.23234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pooler A.M., Polydoro M., Wegmann S.K., Pitstick R., Kay K.R., Sanchez L., Carlson G.A., Gomez-Isla T., Albers M.W., Spires-Jones T.L., Hyman B.T. Tau-amyloid interactions in the rTgTauEC model of early Alzheimer's disease suggest amyloid-induced disruption of axonal projections and exacerbated axonal pathology. J Comp Neurol. 2013;521:4236–4248. doi: 10.1002/cne.23411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sturchler-Pierrat C., Abramowski D., Duke M., Wiederhold K.H., Mistl C., Rothacher S., Ledermann B., Burki K., Frey P., Paganetti P.A., Waridel C., Calhoun M.E., Jucker M., Probst A., Staufenbiel M., Sommer B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997;94:13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moechars D., Dewachter I., Lorent K., Reverse D., Baekelandt V., Naidu A., Tesseur I., Spittaels K., Haute C.V., Checler F., Godaux E., Cordell B., Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- 62.Aizenstein H.J., Nebes R.D., Saxton J.A., Price J.C., Mathis C.A., Tsopelas N.D., Ziolko S.K., James J.A., Snitz B.E., Houck P.R., Bi W., Cohen A.D., Lopresti B.J., DeKosky S.T., Halligan E.M., Klunk W.E. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mintun M.A., Larossa G.N., Sheline Y.I., Dence C.S., Lee S.Y., Mach R.H., Klunk W.E., Mathis C.A., DeKosky S.T., Morris J.C. [11C]PIB in a nondemented population: potential antecedent marker of Alzheimer disease. Neurology. 2006;67:446–452. doi: 10.1212/01.wnl.0000228230.26044.a4. [DOI] [PubMed] [Google Scholar]
- 64.Rowe C.C., Ng S., Ackermann U., Gong S.J., Pike K., Savage G., Cowie T.F., Dickinson K.L., Maruff P., Darby D., Smith C., Woodward M., Merory J., Tochon-Danguy H., O'Keefe G., Klunk W.E., Mathis C.A., Price J.C., Masters C.L., Villemagne V.L. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 65.Riley K.P., Snowdon D.A., Desrosiers M.F., Markesbery W.R. Early life linguistic ability, late life cognitive function, and neuropathology: findings from the Nun Study. Neurobiol Aging. 2005;26:341–347. doi: 10.1016/j.neurobiolaging.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 66.Kanekiyo T., Zhang J., Liu Q., Liu C.C., Zhang L., Bu G. Heparan sulphate proteoglycan and the low-density lipoprotein receptor-related protein 1 constitute major pathways for neuronal amyloid-beta uptake. J Neurosci. 2011;31:1644–1651. doi: 10.1523/JNEUROSCI.5491-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gouras G.K., Willen K., Tampellini D. Critical role of intraneuronal Abeta in Alzheimer's disease: technical challenges in studying intracellular Abeta. Life Sci. 2012;91:1153–1158. doi: 10.1016/j.lfs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cirrito J.R., Kang J.E., Lee J., Stewart F.R., Verges D.K., Silverio L.M., Bu G., Mennerick S., Holtzman D.M. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008;58:42–51. doi: 10.1016/j.neuron.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bero A.W., Yan P., Roh J.H., Cirrito J.R., Stewart F.R., Raichle M.E., Lee J.M., Holtzman D.M. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang D.S., Stavrides P., Mohan P.S., Kaushik S., Kumar A., Ohno M., Schmidt S.D., Wesson D., Bandyopadhyay U., Jiang Y., Pawlik M., Peterhoff C.M., Yang A.J., Wilson D.A., St George-Hyslop P., Westaway D., Mathews P.M., Levy E., Cuervo A.M., Nixon R.A. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cataldo A.M., Barnett J.L., Berman S.A., Li J., Quarless S., Bursztajn S., Lippa C., Nixon R.A. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 72.Cataldo A.M., Hamilton D.J., Barnett J.L., Paskevich P.A., Nixon R.A. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci. 1996;16:186–199. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bennett D.A., Arnold S.E., Valenzuela M.J., Brayne C., Schneider J.A. Cognitive and social lifestyle: links with neuropathology and cognition in late life. Acta Neuropathol. 2014;127:137–150. doi: 10.1007/s00401-013-1226-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yu J.T., Tan L., Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci. 2014;37:79–100. doi: 10.1146/annurev-neuro-071013-014300. [DOI] [PubMed] [Google Scholar]
- 75.Perez-Nievas B.G., Stein T.D., Tai H.C., Dols-Icardo O., Scotton T.C., Barroeta-Espar I., Fernandez-Carballo L., de Munain E.L., Perez J., Marquie M., Serrano-Pozo A., Frosch M.P., Lowe V., Parisi J.E., Petersen R.C., Ikonomovic M.D., Lopez O.L., Klunk W., Hyman B.T., Gomez-Isla T. Dissecting phenotypic traits linked to human resilience to Alzheimer's pathology. Brain. 2013;136:2510–2526. doi: 10.1093/brain/awt171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bjorklund N.L., Reese L.C., Sadagoparamanujam V.M., Ghirardi V., Woltjer R.L., Taglialatela G. Absence of amyloid beta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer's disease neuropathology. Mol Neurodegener. 2012;7:23. doi: 10.1186/1750-1326-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koffie R.M., Meyer-Luehmann M., Hashimoto T., Adams K.W., Mielke M.L., Garcia-Alloza M., Micheva K.D., Smith S.J., Kim M.L., Lee V.M., Hyman B.T., Spires-Jones T.L. Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–4017. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kfoury N., Holmes B.B., Jiang H., Holtzman D.M., Diamond M.I. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287:19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.de Calignon A., Polydoro M., Suarez-Calvet M., William C., Adamowicz D.H., Kopeikina K.J., Pitstick R., Sahara N., Ashe K.H., Carlson G.A., Spires-Jones T.L., Hyman B.T. Propagation of tau pathology in a model of early Alzheimer's disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naslund J., Haroutunian V., Mohs R., Davis K.L., Davies P., Greengard P., Buxbaum J.D. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 81.Braak H., Braak E. Staging of Alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271–278. doi: 10.1016/0197-4580(95)00021-6. discussion 278–84. [DOI] [PubMed] [Google Scholar]
- 82.Clavaguera F., Bolmont T., Crowther R.A., Abramowski D., Frank S., Probst A., Fraser G., Stalder A.K., Beibel M., Staufenbiel M., Jucker M., Goedert M., Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dujardin S., Lecolle K., Caillierez R., Begard S., Zommer N., Lachaud C., Carrier S., Dufour N., Auregan G., Winderickx J., Hantraye P., Deglon N., Colin M., Buee L. Neuron-to-neuron wild-type Tau protein transfer through a trans-synaptic mechanism: relevance to sporadic tauopathies. Acta Neuropathol Commun. 2014;2:14. doi: 10.1186/2051-5960-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Jin M., Shepardson N., Yang T., Chen G., Walsh D., Selkoe D.J. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Walker L.C., Diamond M.I., Duff K.E., Hyman B.T. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013;70:304–310. doi: 10.1001/jamaneurol.2013.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ittner L.M., Gotz J. Amyloid-beta and tau–a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 87.Hernandez C.M., Cortez I., Gu Z., Colon-Saez J.O., Lamb P.W., Wakamiya M., Yakel J.L., Dineley K.T. Research tool: validation of floxed alpha7 nicotinic acetylcholine receptor conditional knockout mice using in vitro and in vivo approaches. J Physiol. 2014;592:3201–3214. doi: 10.1113/jphysiol.2014.272054. [DOI] [PMC free article] [PubMed] [Google Scholar]