

Abstract
We previously identified transforming growth factor (TGF)-β signaling as a fibronectin-independent mechanism of type I collagen fibrillogenesis following adult liver injury. To address the contribution of TGF-β signaling during the development of liver fibrosis, we generated adult mice lacking TGF-β type II receptor (TGF-βIIR) from the liver. TGF-βIIR knockout livers indeed showed a dominant effect in reducing fibrosis, but fibrosis still remained approximately 45% compared with control and fibronectin knockout livers. Unexpectedly, this was accompanied by significant up-regulation of connective tissue growth factor mRNA levels. Organized type I collagen networks in TGF-βIIR knockout livers colocalized well with fibronectin. We provide evidence that elimination of TGF-βIIR is not sufficient to completely prevent liver fibrosis. Our results indicate a TGF-β–independent mechanism of type I collagen production and suggest connective tissue growth factor as its potent mediator. We advocate combined elimination of TGF-β signaling and connective tissue growth factor as a potential therapeutic target by which to attenuate liver fibrosis.
Liver fibrosis is defined as an abnormal response to persistent liver injury, characterized by the excessive accumulation of collagenous extracellular matrices (ECMs).1, 2 Liver fibrosis affects tens of millions of people worldwide and is of great clinical significance because normal liver architecture is disrupted and liver function is ultimately impaired. Because there is no effective treatment of liver fibrosis, many patients develop progressive liver cirrhosis, eventually requiring a liver transplant.3, 4
There is a long-standing concept that cells in culture cannot form a collagen fibril network without the ECM glycoprotein fibronectin.5 We recently established an adult mouse model lacking liver fibronectin and demonstrated that fibronectin-null livers, in fact, formed collagen fibril networks similarly to wild-type mice in response to carbon tetrachloride–induced chronic liver injury. The networks were found to be nucleated by type V collagen, induced by elevated local transforming growth factor (TGF)-β bioavailability.6 Therefore, we identified two mechanisms of collagen fibrillogenesis in response to liver injury: both fibronectin and TGF-β–signaling mediated.
Early in the fibrogenic process, inflammatory cytokines are important in initiating repair following injury. TGF-β acts as a fibrogenic master cytokine and plays a pivotal role in the progression of a variety of chronic fibrotic diseases by promoting myofibroblastic differentiation, stimulating synthesis of ECMs, and down-regulating ECM degradation.7 In TGF-β–mediated signaling, ligand TGF-βs bind to TGF-β type I and type II receptors that form heterotetrameric complexes. On ligand binding, downstream Smad signaling pathways are initiated. Activated (phosphorylated) Smads translocate to the nucleus where they are involved in the regulation of gene expression.8, 9 Currently, several monoclonal antibodies and small molecules targeting TGF-β are in the process of clinical application for chronic fibrotic diseases, including liver fibrosis.10 However, these studies were initiated without knowledge of how TGF-β signaling exerts its action in the development of liver fibrosis.
Elevated TGF-β activity in chronic fibrotic diseases is often accompanied by elevated expression of a matricellular protein, connective tissue growth factor (CTGF/CCN2).11 The manifestation of CTGF/CCN2 functions in vivo involves cooperative interactions with costimulatory factors in the microenvironment, such as TGF-β and fibronectin. One hypothesis arising from the in vivo models is that fibrosing liver injuries are exacerbated by the action of TGF-β–mediated CTGF/CCN2.12 Here, we addressed the extent to which fibrosis is dependent on the TGF-β/CTGF/CCN2 axis in chronic liver injury using an adult mouse model lacking liver TGF-β type II receptors (TGF-βIIR).
Materials and Methods
Mice
Because TGF-βIIR exclusively forms heterotetrameric complexes with the type I receptor in response to ligand binding of all TGF-βs,8, 9 we used TGF-βIIR floxed mice [a gift from Dr. Harold Moses (Vanderbilt University, Nashville, TN)].13 Liver TGF-IIR knockout mice were generated by i.p. injections of polyinosinic-polycytidic acid in 4- to 5-week-old TGF-βIIR(fl/fl)/Mx-Cre+ strain.14 Liver fibronectin knockout mice [fibronectin(fl/fl)/Mx-Cre+] were described previously.6, 15 All mice were maintained and bred at the animal facility in accordance with institutional guidelines. Mice were regularly monitored and had free access to standard mouse chow and water. Mice received humane care in accordance with institutional guidelines.
Induction of Chronic Liver Injury by Carbon Tetrachloride
Chronic liver injury was induced by i.p. administration of the liver-damaging agent carbon tetrachloride solution (Fluka; Sigma-Aldrich, St. Louis, MO) in olive oil [0.5 mL/kg body weight as 50% (v/v), twice a week for up to 8 weeks] in sex-matched, 12- to 15-week-old mice.16, 17 Control and mutant mice were derived from the same litters.
Antibodies
The following antibodies were used for the analyses: rabbit polyclonal antibody (pAb; AB765P; Millipore, Billerica, MA) against type I collagen; rabbit monoclonal antibody (E184; Epitomics, Burlingame, CA) against alpha-smooth muscle actin (α-SMA); rabbit pAb (NB100-91994; Novus Biologicals, Littleton, CO) against TGF-βIIR; mouse monoclonal antibody against heat shock cognate protein 70 (HSC70; Santa Cruz Biotechnology, Santa Cruz, CA); chick pAb against fibronectin [a gift from Dr. Staffan Johansson (Uppsala University, Sweden)]; rabbit pAb against phospho-Smad2 [pSmad2; a gift from Dr. Koichi Matsuzaki (Kansai Medical University, Japan)]. Affinity-purified pAb against mouse CTGF/CCN2 was generated by immunization of rabbits to full-length recombinant proteins. Rabbit pAb (ab6992) against CTGF/CCN2 was from Abcam (Cambridge, UK). Fluorescein isothiocyanate– and Cy3-conjugated, and peroxidase-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories (West Grove, PA). DAPI was from Vector Laboratories (Burlingame, CA). Our and Abcam’s CTGF/CCN2 antibodies showed identical tissue and cellular distributions. No nonspecific binding of the anti-rabbit secondary antibody to mouse tissue Ig was confirmed by the immunohistochemical staining with normal rabbit IgG as a primary antibody (data not shown).
Histological Analysis, Immunohistochemistry, and Immunofluorescence
For histological analyses, liver samples were either directly frozen in optimal cutting temperature compound (Tissue-Tek; Sakura Finetek, Torrance, CA) or fixed overnight in 4% paraformaldehyde in phosphate-buffered saline (pH 7.2) and then dehydrated in a graded alcohol series before being embedded in paraffin. Sirius Red staining was performed according to standard protocols. Immunohistochemistry and immunofluorescence studies were performed as described previously.6, 14
For quantification of the fibrotic areas in response to chronic liver injury, images were captured with the same gain, offset, magnitude, and exposure time. A minimum of five different images were randomly selected and the intensities were quantified using Image-Pro Plus software version 6.1 (Media Cybernetics, Rockville, MD).
Real-Time PCR
Real-time PCR was performed as described previously.18 The following primers were used: ColIa1 forward, 5′-GGGCGAGTGCTGTGCTTT-3′; ColIa1 reverse, 5′-GGTCCCTCGACTCCTACATCTTC-3′; CTGF forward, 5′-ACTATGATGCGAGCCAACTG-3′; CTGF reverse, 5′-CTCCAGTCTGCAGAAGGTATTG-3′; fibronectin forward, 5′-CCAGACTTATGGTGGCAATTCA-3′; fibronectin reverse, 5′-TTCGTAATTGGAAGTTGTGCTACAC-3′; 18S rRNA forward, 5′-GGCGACGACCCATTCG-3′; 18S rRNA reverse, 5′-ACCCGTGGTCACCATGGTA-3′. All samples were analyzed in triplicate at a minimum. After the reactions, the specificity of amplifications in each sample was confirmed by dissociation analysis, showing that each sample gave a single melting peak. The relative mRNA levels were normalized to the level of 18S rRNA.
Western Blot Analysis
Western blot analyses were performed as described elsewhere.19 In some immunoblotting analyses, samples were transferred onto Immobilon-FL polyvinylidene fluoride membranes (Millipore) and probed with primary and IRDye 800CW- or IRDye 680-conjugated secondary antibodies (LI-COR Bioscience, Lincoln, NE). Immunoreactive bands were detected using the Odyssey Infrared Imaging System (LI-COR Bioscience).
TGF-β Bioassay
A mink lung cell line (TMLC) stably transfected with a PAI-1 promoter fused to luciferase was used for the analysis.20 Active TGF-β levels were measured as described elsewhere.21
Data Presentation and Statistical Analysis
All experiments were performed in triplicate at a minimum, on separate occasions, and the data shown were chosen as representative of results consistently observed. Results are presented as means ± SD. Differences between groups were analyzed using the two-sided Student's t-test on raw data. A P value of <0.05 was considered significant.
Results
We previously demonstrated elevated local TGF-β bioavailability in fibronectin knockout livers in response to injury.6 When chronic liver injury was induced in fibronectin knockout mice with liver-damaging agent carbon tetrachloride at 0.5 mL/kg body weight, the expression of CTGF/CCN2 mRNA levels corresponded to locally activated TGF-β levels at 8 weeks of carbon tetrachloride treatment (Figure 1, A and B) (P < 0.05), suggesting an involvement of TGF-β/CTGF/CCN2 in liver fibrosis.
Figure 1.

Effect of fibronectin deficiency on active TGF-β and CTGF/CCN2 at 8 weeks of carbon tetrachloride treatment. A: Active TGF-β bioassay in control and fibronectin knockout livers (LivFn-KO). B: Real-time PCR analysis of CTGF/CCN2 mRNA level in control and fibronectin knockout livers. Relative expression levels are shown relative to a control value of 1. Error bars represent SD. n = 7 (A, control); n = 8 (A, LivFn-KO); n = 4 (B, control and fibronectin knockout livers). ∗P < 0.05.
Because both TGF-β and CTGF/CCN2 are identified as regulators of ECM protein transcription, including collagens and fibronectin,22 we hypothesized that TGF-β/CTGF/CCN2 pathway activation may be acting as an upstream modulator in the induction of liver fibrosis. To test this hypothesis, we used TGF-βIIR floxed mice and established liver TGF-βIIR knockout (mutant) mice [TGF-βIIR(fl/fl)/Mx-Cre+]. At 8 weeks after the final injection of polyinosinic-polycytidic acid to inactivate the TGF-βIIR gene, we confirmed by genomic Southern blotting that the TGF-βIIR gene was efficiently deleted from TGF-βIIR(fl/fl)/Mx-Cre+ livers (Figure 2A), indicating that Mx-Cre–mediated deletion of the floxed TGF-βIIR gene was complete in all cell types of the liver, both parenchymal and nonparenchymal cells. Because TGF-βIIR is the exclusive type II receptor for all TGF-β ligands and type I receptor complex formation,8, 9 lack of this receptor abolishes all TGF-β–mediated signaling in the liver. Chronic liver injury was then induced by carbon tetrachloride. We confirmed by immunohistochemistry that mutant livers did not show any positive staining for TGF-βIIR, whereas control livers showed positive staining in both hepatocytes and nonparenchymal cells (Figure 2B). Furthermore, mutant livers showed very weak and diffuse distribution of pSmad2, whereas control livers showed clear nuclear localizations in both some hepatocytes and nonparenchymal cells (Figure 2C).
Figure 2.

Generation of liver TGF-βIIR (TRII) knockout mice. A: Interferon- and polyinosinic-polycytidic acid–inducible Mx-Cre–mediated deletion of the floxed TGF-βIIR gene from the liver. Southern-blot analysis of liver genomic DNA (BglII digestion) from control [TGF-βIIR(fl/fl)] and TGF-βIIR(fl/fl)/Mx-Cre+ mice at 8 weeks after the final injection of polyinosinic-polycytidic acid. The sizes of the TGF-βIIR–floxed (floxed: 9.5 kb) and the TGF-βIIR–null (knockout: 6.0 kb) bands are indicated. Note that the floxed TGF-βIIR band is undetectable in the liver of TGF-βIIR(fl/fl)/Mx-Cre+ mice. B: Representative immunohistochemical staining for TGF-βIIR (TRII, in brown) in control and mutant livers at 8 weeks of carbon tetrachloride treatment. Sections were counterstained with hematoxylin. Note that nuclei of TGF-βIIR–positive hepatocytes (arrowheads) are approximately two- to threefold larger than those in nonparenchymal cells (arrows). C: Nuclear localization of pSmad2 in control and mutant livers at 8 weeks of carbon tetrachloride treatment. Representative immunofluorescence staining for pSmad2 (red), and double staining for pSmad2 (red) and DAPI for cell nuclei (blue) in the same image. Note that nuclei of pSmad2-positve hepatocytes (arrowheads) are approximately two- to threefold larger than those in nonparenchymal cells (arrows). Scale bars: 50 μm (B); 25 μm (C).
Next, fibrosis was assessed by Sirius Red staining, which directly marks the deposition of ECMs. TGF-βIIR knockout livers showed decreased fibrosis at 8 weeks of treatment, and levels were significantly less than control and fibronectin knockout livers (Figure 3A) (P < 0.01), indicating that TGF-β signaling is indeed a dominant pathway in the development of fibrosis. However, TGF-βIIR knockout livers did not completely resist fibrosis, which remained at approximately 46.4% and approximately 43.2% of controls and fibronectin knockout livers, respectively (Figure 3A). The expression levels of type I collagen mRNA were reduced but remained at approximately 59.1% and approximately 48.3% compared with control and fibronectin knockout livers, respectively (Figure 3B). Mutant livers showed a decreased number of α-SMA–positive cells but still remained at approximately 46.8% positive cells compared with controls (Figure 3C) [26.5 ± 3.7 cells/field in control (field = 0.036 mm2, n = 8) vs 12.4 ± 4.9 cells/field in mutant (n = 8; P < 0.01)]. α-SMA is a marker for myofibroblasts, including hepatic stellate cells, which play a central role in ECM production in response to liver injury. Type I collagen protein levels in mutant livers were also reduced (approximately 56.4% compared with controls), but substantial type I collagen network formations were confirmed by immunohistochemistry (Figure 3, D and E). Although the expression levels of fibronectin mRNA in mutant livers were down-regulated, organized type I collagen fibril networks colocalized well with fibronectin networks (Figure 3F), suggesting fibronectin-nucleated type I collagen network formation in TGF-βIIR knockout livers during the development of liver fibrosis.
Figure 3.

Characterization of TGF-βIIR knockout (TRII-KO) livers at 8 weeks of carbon tetrachloride treatment. A: Sirius Red staining of livers to detect the deposition of ECMs before (0 weeks) and after (8 weeks) chronic liver injury. Analysis of Sirius Red–positive areas. B: Real-time PCR analysis of type I collagen mRNA level. C: Expression of myofibroblast marker α-SMA (red) and DAPI (to visualize cell nuclei, blue) shown by immunofluorescence staining and quantification of α-SMA–positive areas. D: Western blot analysis of type I collagen protein expression and the analysis of type I collagen intensities. Band intensity was measured by densitometry and normalized to HSC70 (loading control). E: Deposition of type I collagen (in red) by immunofluorescence staining. F: Real-time PCR analysis of fibronectin mRNA level and triple immunofluorescence staining for type I collagen (red), fibronectin (green), and DAPI (blue) in TGF-βIIR–null liver. Expression levels are shown relative to a control value of 1. Error bars represent SD. n = 4 (D, mutant mice); n = 5 (A, each group; B and D, mutant mice; D, control); n = 8 (B and F, control; C, each group). ∗P < 0.05, ∗∗P < 0.01. Scale bars: 100 μm (A, B, C, and F).
Surprisingly, we found a 1.94-fold up-regulation of CTGF/CCN2 mRNA level in mutant livers (Figure 4A) (P < 0.01). Subsequent immunohistochemical analysis revealed that CTGF/CCN2 proteins in mutant livers localized mainly in the sinusoid (Figure 4B). CTGF/CCN2 can up-regulate ECM production, and increased expression and deposition of CTGF/CCN2 are observed in activated hepatic stellate cells, biliary epithelial cells, and hepatocytes in fibrotic animal liver models.22 Further immunofluorescence studies revealed that CTGF/CCN2-positive cells predominantly expressed hepatocyte marker albumin (Figure 4C). CTGF/CCN2 and myofibroblast marker α-SMA double-positive cells were detected as a minor proportion (data not shown). Our findings suggest that the participation of CTGF/CCN2 in type I collagen production is TGF-β independent during the development of liver fibrosis.
Figure 4.
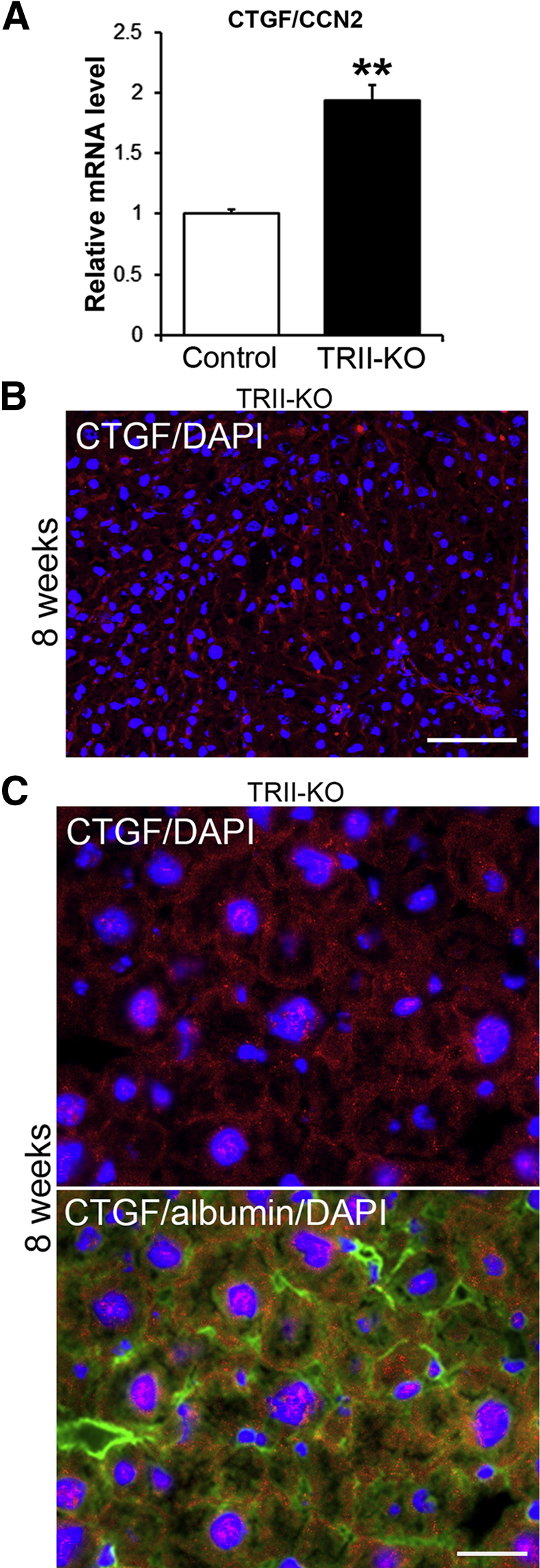
Up-regulated CTGF/CCN2 expression in TGF-βIIR knockout (TRII-KO) livers at 8 weeks of carbon tetrachloride treatment. A: Real-time PCR analysis of CTGF/CCN2 mRNA level. B: Tissue distribution of CTGF/CCN2 (CTGF, red) and DAPI for cell nuclei (blue) in the mutant liver by immunofluorescence staining. C: Cellular distribution of CTGF/CCN2 (CTGF) in the mutant liver. Double immunofluorescence staining for CTGF/CCN2 (red) and DAPI for cell nuclei (blue), and triple staining for CTGF/CCN2 (red), hepatocyte marker albumin (green), and DAPI (blue) in the same image. mRNA expression level is shown relative to the control value of 1. Error bars represent SD. n = 5 (mutant mice); n = 8 (control). ∗∗P < 0.01. Scale bars: 100 μm (B); 25 μm (C).
Discussion
Whether CTGF/CCN2 drives tissue/organ fibrosis has remained a fundamental question for many years. We have recently shown that constitutive overexpression of CTGF/CCN2 in fibroblasts under the collagen α2(I) fibroblast enhancer results in significant up-regulation of collagen type I mRNA and protein levels in vitro and that CTGF/CCN2 alone is sufficient to cause spontaneous multiorgan fibrotic phenotypes in vivo.23 Importantly, these CTGF/CCN2-mediated pathways do not involve canonical TGF-β signaling but promote several other signaling pathways.23 However, the liver did not show significant fibrosis compared to wild type. Here, we demonstrated significant up-regulation of CTGF/CCN2 expression in both TGF-βIIR and fibronectin-null livers in chronic injury. Our results, therefore, indicate a TGF-β–independent mechanism of type I collagen production, and CTGF/CCN2 as a potent mediator in liver fibrosis. Hence, the progression of liver fibrosis requires two hits: the presence or activation of the matricellular protein CTGF/CCN2 and the induction of adult tissue injury. Furthermore, up-regulated expression of CTGF/CCN2 by TGF-β–independent signaling such as IL-1324 and/or by down-regulation of CTGF/CCN2 scavenger low-density lipoprotein-receptor-related protein 125 would be critical in fibronectin/TGF-βIIR double-knockout mice to induce liver fibrosis in chronic injury. This latter speculation for the requirement of adult tissue injury is based on evidence that overexpression of CTGF/CCN2 in concert with signaling pathways associated with chronic fibrosing injuries can lead to the initiation or exacerbation of fibrosis.12 These findings support the hypothesis that CTGF/CCN2 plays a role in modifying cellular function during the development of liver fibrosis by sensing changes in the microenvironment through the engagement of molecular binding partners. We propose CTGF/CCN2 as a key element in TGF-β–independent type I collagen production during the development of liver fibrosis. Thus, it remains to be elucidated how the expression of CTGF is regulated in fibronectin/TGF-βIIR double-knockout livers in the development of liver fibrosis.
To our knowledge, this is the first study to extensively examine the effects of TGF-β signaling on the development of liver fibrosis. Currently, several antifibrotic therapies targeting TGF-β are in the process of clinical application for chronic fibrotic diseases, including liver fibrosis.10 However, our present study provides compelling evidence that elimination of TGF-βIIR is not sufficient to completely prevent liver fibrosis. Clearly, it is vital to generate the most appropriate therapeutic strategy based on a precise molecular framework of targeting pathways. We propose the combined elimination of TGF-β signaling and CTGF/CCN2 could be a potential therapeutic target to attenuate liver fibrosis. This finding is of critical importance to the field of TGF-β research and chronic fibrotic diseases, where the focus is on accelerating the generation of the most effective treatment for patients, and on an optimum drug development, ie, drugs that target the entire, not partial, mechanisms of disease development.
Acknowledgments
We thank Dr. Harold Moses for TGF-β type II receptor floxed mice, Drs. Staffan Johansson and Koichi Matsuzaki for antibodies, Peter Metcalfe for technical assistance, and Drs. Dusko Ilic and Emma Stephenson for editorial assistance.
T.Sak. conceived ideas, designed experiments, and supervised the project; K.S. and S.J. performed experiments; T.Sas. generated anti–type I collagen and CTGF/CCN2 antibodies; K.S., S.J., G.B.-G., and T.Sak. analyzed the data; and T.Sak. wrote and edited the manuscript.
Footnotes
Supported in part by NIH grant DK074538 (T. Sak.) and the Institute of Translational Medicine, University of Liverpool (T. Sak.).
K.S. and S.J. contributed equally to this work.
Disclosures: None declared.
References
- 1.Bataller R., Brenner D.A. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iredale J.P. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forbes S.J., Parola M. Liver fibrogenic cells. Best Pract Res Clin Gastroenterol. 2011;25:207–217. doi: 10.1016/j.bpg.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Friedman S.L. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 5.Velling T., Risteli J., Wennerberg K., Mosher D.F., Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins alpha 11beta 1 and alpha 2beta 1. J Biol Chem. 2002;277:37377–37381. doi: 10.1074/jbc.M206286200. [DOI] [PubMed] [Google Scholar]
- 6.Moriya K., Bae E., Honda K., Sakai K., Sakaguchi T., Tsujimoto I., Kamisoyama H., Keene D.R., Sasaki T., Sakai T. A fibronectin-independent mechanism of collagen fibrillogenesis in adult liver remodeling. Gastroenterology. 2011;140:1653–1663. doi: 10.1053/j.gastro.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gressner O.A., Weiskirchen R., Gressner A.M. Biomarkers of hepatic fibrosis, fibrogenesis and genetic pre-disposition pending between fiction and reality. J Cell Mol Med. 2007;11:1031–1051. doi: 10.1111/j.1582-4934.2007.00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moustakas A., Heldin C.H. The regulation of TGFbeta signal transduction. Development. 2009;136:3699–3714. doi: 10.1242/dev.030338. [DOI] [PubMed] [Google Scholar]
- 9.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wynn T.A., Ramalingam T.R. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mason R.M. Fell-Muir lecture: connective tissue growth factor (CCN2)—a pernicious and pleiotropic player in the development of kidney fibrosis. Int J Exp Pathol. 2013;94:1–16. doi: 10.1111/j.1365-2613.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brigstock D.R. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: lessons from transgenic animals. J Cell Commun Signal. 2010;4:1–4. doi: 10.1007/s12079-009-0071-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chytil A., Magnuson M.A., Wright C.V., Moses H.L. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–75. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- 14.Sakai T., Johnson K.J., Murozono M., Sakai K., Magnuson M.A., Wieloch T., Cronberg T., Isshiki A., Erickson H.P., Fassler R. Plasma fibronectin supports neuronal survival and reduces brain injury following transient focal cerebral ischemia but is not essential for skin-wound healing and hemostasis. Nat Med. 2001;7:324–330. doi: 10.1038/85471. [DOI] [PubMed] [Google Scholar]
- 15.Moriya K., Sakai K., Yan M.H., Sakai T. Fibronectin is essential for survival but is dispensable for proliferation of hepatocytes in acute liver injury in mice. Hepatology. 2012;56:311–321. doi: 10.1002/hep.25624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Constandinou C., Henderson N., Iredale J.P. Modeling liver fibrosis in rodents. Methods Mol Med. 2005;117:237–250. doi: 10.1385/1-59259-940-0:237. [DOI] [PubMed] [Google Scholar]
- 17.Hayashi H., Sakai T. Animal models for the study of liver fibrosis: new insights from knockout mouse models. Am J Physiol Gastrointest Liver Physiol. 2011;300:G729–G738. doi: 10.1152/ajpgi.00013.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Honda K., Sakaguchi T., Sakai K., Schmedt C., Ramirez A., Jorcano J.L., Tarakhovsky A., Kamisoyama H., Sakai T. Epidermal hyperplasia and papillomatosis in mice with a keratinocyte-restricted deletion of csk. Carcinogenesis. 2007;28:2074–2081. doi: 10.1093/carcin/bgm112. [DOI] [PubMed] [Google Scholar]
- 19.Sakai T., Li S., Docheva D., Grashoff C., Sakai K., Kostka G., Braun A., Pfeifer A., Yurchenco P.D., Fassler R. Integrin-linked kinase (ILK) is required for polarizing the epiblast, cell adhesion, and controlling actin accumulation. Genes Dev. 2003;17:926–940. doi: 10.1101/gad.255603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abe M., Harpel J.G., Metz C.N., Nunes I., Loskutoff D.J., Rifkin D.B. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 21.Maeda T., Sakabe T., Sunaga A., Sakai K., Rivera A.L., Keene D.R., Sasaki T., Stavnezer E., Iannotti J., Schweitzer R., Ilic D., Baskaran H., Sakai T. Conversion of mechanical force into TGF-beta-mediated biochemical signals. Curr Biol. 2011;21:933–941. doi: 10.1016/j.cub.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gressner O.A., Gressner A.M. Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int. 2008;28:1065–1079. doi: 10.1111/j.1478-3231.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 23.Sonnylal S., Shi-Wen X., Leoni P., Naff K., Van Pelt C.S., Nakamura H., Leask A., Abraham D., Bou-Gharios G., de Crombrugghe B. Selective expression of connective tissue growth factor in fibroblasts in vivo promotes systemic tissue fibrosis. Arthritis Rheum. 2010;62:1523–1532. doi: 10.1002/art.27382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y., Meyer C., Muller A., Herweck F., Li Q., Mullenbach R., Mertens P.R., Dooley S., Weng H.L. IL-13 induces connective tissue growth factor in rat hepatic stellate cells via TGF-beta-independent Smad signaling. J Immunol. 2011;187:2814–2823. doi: 10.4049/jimmunol.1003260. [DOI] [PubMed] [Google Scholar]
- 25.Huang G., Brigstock D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front Biosci (Landmark Ed) 2012;17:2495–2507. doi: 10.2741/4067. [DOI] [PubMed] [Google Scholar]