

Abstract
Thrombospondin-1 (TSP1) is a multifunctional matricellular protein known to promote progression of chronic kidney disease. To gain insight into the underlying mechanisms through which TSP1 accelerates chronic kidney disease, we compared disease progression in Col4a3 knockout (KO) mice, which develop spontaneous kidney failure, with that of Col4a3;Tsp1 double-knockout (DKO) mice. Decline of excretory renal function was significantly delayed in the absence of TSP1. Although Col4a3;Tsp1 DKO mice did progress toward end-stage renal failure, their kidneys exhibited distinct histopathological lesions, compared with creatinine level–matched Col4a3 KO mice. Although kidneys of both Col4a3 KO and Col4a3;Tsp1 DKO mice exhibited a widened tubulointerstitium, predominant lesions in Col4a3 KO kidneys were collagen deposition and fibroblast accumulation, whereas in Col4a3;Tsp1 DKO kidney inflammation was predominant, with less collagen deposition. Altered disease progression correlated with impaired activation of transforming growth factor-β1 (TGF-β1) in vivo and in vitro in the absence of TSP1. In summary, our findings suggest that TSP1 contributes to progression of chronic kidney disease by catalyzing activation of latent TGF-β1, resulting in promotion of a fibroproliferative response over an inflammatory response. Furthermore, the findings suggest that fibroproliferative and inflammatory lesions are independent entities, both of which contribute to decline of renal function.
Progression of chronic kidney disease (CKD) toward end-stage renal failure (ESRF) is a prominent problem in clinical nephrology.1 The incidence of CKD is rising, but effective therapies to halt progression of disease remain elusive.2 Progression of CKD results from a complex interplay of pathologies that involve all constituents of the kidney, which makes it difficult to single out targets for effective therapeutic strategies.3
The extent of so-called tubulointerstitial fibrosis is often considered to be the rate-limiting step in progression of CKD.1 This idea is founded on histopathological analysis of large cohorts of kidney biopsies, which demonstrated that only tubulointerstitial fibrosis (which at the time was determined as the relative volume of the interstitium within a kidney biopsy section) correlates with and also predicts progression of CKD toward ESRF, irrespective of the underlying primary disease.4, 5, 6, 7 Widening of the tubulointerstitium, which is referred to as tubulointerstitial fibrosis, is caused by a composite of extracellular matrix (ECM) accumulation, sterile inflammation, accumulation of activated fibroblasts, and rarefaction of microvessels.1 Although the relevance of each of these events to progression of fibrosis and CKD is hotly debated, this knowledge led to the concept that tubulointerstitial fibrosis is a common pathway of all chronic progressive kidney diseases and that effective antifibrotic therapies could potentially halt progression of CKD irrespective of the underlying disease. However, such therapies are not yet available.1
Our aim was to gain insight into mechanisms that underlie the contribution of thrombospondin-1 (TSP1) to progression of CKD. TSP1 is the most-studied member of the thrombospondin family of matricellular proteins.8 Previous studies have demonstrated that pharmacological suppression or genetic depletion of TSP1 attenuates disease progression in animal models of CKD.9, 10, 11, 12, 13 TSP1 is a 450-kDa trimeric ECM protein, which does not fulfill primarily structural roles in the matrix, but instead functions as an extracellular modulator of cell function.8, 14 Most prominently, TSP1 is known to inhibit angiogenesis, inhibit inflammation, activate MMP-dependent ECM turnover, and facilitate fibroblast migration and activation, all of which are considered important contributors to progression of CKD.8, 10 To delineate through which of its known biological activities TSP1 impacts progression of CKD, we compared progression of kidney disease of Col4a3 knockout (KO) mice (deficient in type IV collagen α3 chain) with that of Col4a3;Tsp1 double-knockout (DKO) mutant mice.15
Here, we demonstrate that decrease of excretory renal function is delayed if TSP1 is absent. Furthermore, tissue analysis of plasma creatinine level–matched kidneys of Col4a3 KO and of Col4a3;Tsp1 DKO revealed that in Col4a3 KO mice disease progression is predominantly associated with fibrosis, whereas inflammation is the predominant interstitial pathology in Col4a3;Tsp1 DKO mice. We provide evidence that this altered disease progression is due to impaired activation of latent transforming growth factor-β1 (TGF-β1) in the absence of TSP1. Our findings provide evidence that both fibroproliferative injury and inflammation can independently cause expansion of the interstitium, leading to decline of excretory renal function.
Materials and Methods
Mice
Col4a3 KO mice and Tsp1 KO mice, both on a C57BL/6 background, have been described previously.16, 17, 18 To establish double-mutant mice, Col4a3 KO mice and Tsp1 KO mice were crossed, producing Col4a3+/−;Tsp1+/− mice in the F1 generation. Interbreeding of Col4a3+/−;Tsp1+/− mice resulted in Col4a3 KO, Tsp1 KO, and Col4a3;Tsp1 DKO mice in the expected Mendelian ratio. Mice were genotyped by PCR using established primers and protocols.16, 17 The TSP1-deficient mice were provided by Dr. Jack Lawler (Beth Israel Deaconess Medical Center, Boston, MA). All procedures were performed with the approval of the Institutional Animal Care and Use Committee of the Beth Israel Deaconess Medical Center.
Histological Analysis
Formalin-fixed, paraffin-embedded kidneys were sectioned at 3 μm and stained with hematoxylin and eosin (H&E), Masson’s trichrome stain (MTS), or periodic acid–Schiff’s stain (PAS) at the Beth Israel Deaconess Medical Center (Boston, MA) Histopathology Core facility. The relative interstitial volume was evaluated by morphometric analysis using a counting grid, as described previously,19 and 100 points per visual field (×60 magnification) were identified as tubule or interstitium. Results were summarized as interstitium among all scored areas (relative percentage of interstitial area). The fibrotic area was assessed using image processing software (Image-Pro Plus version 6.2; Media Cybernetics, Rockville, MD) as described previously.20 In brief, the blue (fibrotic) and nonblue areas were measured in photomicrographs at ×20 magnification. Glomerulosclerosis was similarly assessed with Image-Pro Plus software. Glomerulosclerosis was quantified as the PAS-positive area per glomerulus (×60 magnification). To assess cellularity, we counted all visible nuclei per visual field (×60 magnification) of H&E-stained sections. To assess the tubular atrophy index, tubules were evaluated for widened lumen and thickened basement membranes and the percentage of atrophic tubules was estimated.19
Staining Procedures
Immunolabeling was performed on formalin-fixed, paraffin-embedded kidney sections. Sections were deparaffinized and microwaved for 15 minutes in antigen unmasking solution (Vector Laboratories, Burlingame, CA). For immunohistochemistry, sections were blocked with avidin–biotin and 10% horse serum (both from Vector Laboratories) before incubation with the respective primary antibodies: αSMA and GPR44 (LifeSpan BioSciences); CD45 (BD Biosciences, San Jose, CA); CCR5, IL-13, ROR-γ, and FOXP3 (Abcam, Cambridge, MA); and TSP1 and p-SMAD2/3 (Santa Cruz Biotechnology, Dallas, TX). Immunolabeling was visualized using a Vectastain alkaline phosphatase kit with alkaline phosphatase substrate kit I (Vector Laboratories) according to the manufacturer’s recommendations. For immunofluorescence staining, kidney sections were incubated with primary antibodies to CD31 and collagen IV. Collagen IV antibodies were raised against the noncollagenous domain of type IV collagen and therefore detected all type IV collagen isoforms, including collagen IV α1 and/or α2 chains, which are abundantly present in the Col4a3 KO and Col4a3;Tsp1 KO mice. Nuclei were counterstained with DAPI. Images were captured with confocal microscopy.
Preparation of Primary Renal Fibroblasts
Primary renal fibroblasts were isolated from kidneys of C57BL/6 wild-type mice and of Tsp1 KO mice as described previously,21 with minor modifications. Kidney cortex from 6-month-old mice was minced and digested in 100 U/mL type II collagenase (Worthington Biochemical, Lakewood, NJ) for 30 minutes at 37°C. The tissue pieces (approximately 5 mm3) were washed to remove the collagenase and then were plated into T25 tissue-culture flasks in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and antibiotics (100 U/mL penicillin G, 100 g/mL streptomycin, and 0.25 μg/mL amphotericin B).
Proliferation Assay
Fibroblast proliferation in response to active and latent TGF-β1 was assayed as described previously,21 with minor modifications. Primary wild-type and Tsp1 KO fibroblasts were seeded into 96-well plates (4000 cells per well). On 24 hours of incubation in serum-free Dulbecco’s modified Eagle’s medium, the medium was exchanged for medium containing 5 ng/mL active or latent TGF-β1 (R&D Systems). Cell proliferation was measured using a colorimetric cell proliferation assay (WST-1–based, Roche) according to the manufacturer’s recommendations.
Quantification of Fibrillar Collagen Content
Fibrillar collagen was quantified in kidney protein lysates using a QuickZyme (Leiden, The Netherlands) fibrillar collagen assay kit according to the manufacturer’s recommendations. In brief, kidney tissues were homogenized in 0.5 mol/L acetic acid–pepsin. Total protein was quantified, and 100 μg per well of each sample was analyzed in the kit’s 96-well plates. A standard curve using the kit’s collagen standard was generated, and a best-fit linearized curve was generated to determine collagen content per well.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was isolated using Trizol reagent (Life Technologies, Carlsbad, CA) according to the manufacturer’s recommendation. Specific primers to collagen III α1 were designed using the Primer Express 1.5 program.21 TaqMan Gene Expression Assay (Life Technologies) were used for quantitative real-time PCR performed with an ABI 7000 sequence detection system (Life Technologies), and measurements were standardized to 18S rRNA (Life Technologies) using standard calculation methods as described previously.22
Statistical Analysis
Statistical analysis was performed using the Kruskal–Wallis method followed by Dunn’s test. If equal variance test failed, then U-test was used. All statistical analysis was performed using SigmaStat 2.0 software (Systat, San Jose, CA).
Results
Progression of Renal Fibrogenesis Is Delayed in Col4a3;Tsp1 DKO Mice
We compared progression of kidney disease of Col4a3 KO with that of Col4a3;Tsp1 DKO mutant mice.15 Col4a3 KO mice were originally generated as a mouse model for Alport syndrome; on a C57BL/6 genetic background, these mice exhibit robust and reproducible progression of spontaneous kidney disease toward ESRF due to a defined genetic defect.23 In Col4a3 KO mice, absence of the α3 chain of type IV collagen leads to glomerular basement membrane splitting and proteinuria starting at 6 weeks of age. Tubulointerstitial lesions (including ECM accumulation, fibroblast accumulation, sterile inflammation, and microvascular rarefaction) can be detected starting at 12 weeks of age.23 At the 22 weeks of age, kidneys of Col4a3 KO mice are severely fibrotic, and mice die of ESRF at the age of 24 to 26 weeks.23 Col4a3 KO mice on a C57BL/6 background allow for analysis of all aspects of chronic progressive kidney disease.23
We sacrificed predetermined cohorts of mice (Col4a3 KO, Col4a3;Tsp1 DKO, and C57BL/6 wild-type control mice) at 6, 12, 22, and 32 weeks of age. This study design was founded on our previous studies, which demonstrated that Col4a3;Tsp1 DKO mice survive to only 36 weeks of age, whereas Tsp1 KO mice with normal kidney function have a normal life span.15 MTS-stained sections of kidney tissue from Col4a3 KO mice indicated absence of interstitial fibrosis at the age of 6 weeks, but these mice had developed fibrosis by 12 weeks of age, with severe fibrosis at 22 weeks, confirming previous studies (Figure 1A).24 The progression of chronic kidney injury observed in MTS-stained sections correlated with progressive impairment of excretory renal function, as determined by plasma creatinine levels (Figure 1B). Compared with Col4a3 KO mice, decline of excretory renal function was delayed in Col4a3;Tsp1 DKO mice; plasma creatinine levels after 32 weeks were similar to those observed in the Col4a3 KO mice after 22 weeks (Figure 1B). Such delayed disease progression correlated with histopathological lesions noted in MTS-stained kidneys.
Figure 1.

Progression of CKD in Col4a3 KO and Col4a3;Tsp1 DKO mice. A: Representative photomicrographs of MTS-stained kidney sections of wild-type C57BL/6 control, Col4a3 KO, and Col4a3;Tsp1 DKO mice at 6, 12, 22, and 32 weeks. Col4a3 KO mice did not live to 32 weeks of age. B: Serum creatinine levels of wild-type C57BL/6, Tsp1 KO, Col4a3 KO mice, and Col4a3;Tsp1 DKO mice over time. Data are expressed as means ± SEM. Original magnification, ×10. CKD, chronic kidney disease; DKO, double knock-out; ESRF, end-stage renal failure; KO, knock-out; MTS, Masson’s trichrome stain.
Distinct Histopathologies in Fibrotic Kidneys of Col4a3 KO and Col4a3;Tsp1 DKO Mice
MTS-stained sections of end-stage diseased kidneys of Col4a3 KO mice (at 22 weeks) and of Col4a3;Tsp1 DKO mice (at 32 weeks) had markedly different pathologies (Figure 1), despite similar plasma creatinine levels. We therefore decided to systematically compare kidneys of creatinine-matched cohorts, as opposed to age-matched cohorts, to gain further insight into the role of TSP1 in renal fibrogenesis. We chose this approach of comparing kidneys with similar impairment of function over that of comparing age-matched kidneys of each group because we found that for kidneys at 22 weeks all parameters appeared to be improved in the Col4a3;Tsp1 DKO mice, providing little insight into why disease progression was delayed.
We first assessed the relative interstitial volume (ie, the cortical area not covered by glomeruli or tubules) in MTS-stained kidney sections of wild-type C57BL/6 (32 weeks) and of end-stage diseased Col4a3 KO and Col4a3;Tsp1 DKO mice (Figure 2). The interstitial volume was significantly increased in both Col4a3 KO and Col4a3;Tsp1 DKO kidneys, compared with the wild type, with no significant difference between the two mutant mouse groups (Figure 2). However, when we digitally quantified the relative blue area (reflective of collagen deposition) in MTS-stained kidney sections, we found that the relative blue area was significantly smaller in the Col4a3;Tsp1 DKO kidneys than in the creatinine-matched Col4a3 KO kidneys (Figure 2C). To confirm that reduced blue staining in the Col4a3;Tsp1 DKO kidneys is reflective of decreased collagen deposition, we analyzed fibrillar collagen content (per 100 μg total protein lysate) of kidney tissue. Collagen content in functionally impaired Col4a3;Tsp1 DKO kidneys was significantly reduced, compared with creatinine-matched Col4a3 KO kidneys (Figure 2D). In creatinine-matched kidneys, the Col4a3;Tsp1 DKO kidneys contained less collagen than Col4a3 KO kidneys, despite similar expansion of the interstitial space.
Figure 2.

Histopathology of fibrosis. A: Representative photomicrographs of MTS-stained kidney sections of wild-type control (32 weeks) and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. B: The degree of tubulointerstitial fibrosis (determined by relative interstitial volume) did not differ significantly between kidneys of Col4a3 KO and Col4a3;Tsp1 DKO mice. C: In MTS-stained kidney sections, the degree of collagen deposition (blue) was significantly higher in Col4a3 KO mice than in Col4a3;Tsp1 DKO mice. D: Fibrillar collagen content (determined as percent of total kidney protein) was higher in Col4a3 KO kidneys than in creatinine-matched Col4a3;Tsp1 DKO kidneys. Data are expressed as means ± SEM. *P < 0.05. Original magnification, ×20.
Identity of Interstitial Cells in Fibrotic Kidneys of Col4a3 KO and Col4a3;Tsp1 DKO Mice
We next compared interstitial cellularity (determined in H&E-stained sections as nuclei per visual field) between kidneys of creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. Cellularity was significantly increased in Col4a3;Tsp1 DKO kidneys, compared with creatinine-matched Col4a3 KO kidneys (Figure 3). To gain insight into the identity of the interstitial cells, we performed immunolabeling experiments using antibodies to the fibroblast marker α-smooth muscle actin (αSMA) in which bone marrow–derived inflammatory cells were immunolabeled with CD45. In kidneys of 22-week-old Col4a3 KO mice, the number of αSMA+ fibroblasts per visual field was substantially increased, compared with wild-type C57BL/6 control kidneys (Figure 4). The kidneys of 32-week-old creatinine-matched Col4a3;Tsp1 DKO mice exhibited significantly less αSMA+ interstitial fibroblast accumulation, compared with creatinine-matched Col4a3 KO kidneys (Figure 4). By contrast, CD45+ inflammatory cells were significantly increased in kidneys of 32-week-old Col4a3;Tsp1 DKO mice, compared with kidneys of creatinine-matched 22-week-old Col4a3 KO mice (Figure 5).
Figure 3.

Interstitial cellularity. A: Representative photomicrographs of H&E-stained kidney sections of wild-type control and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. B: The number of nuclei per visual field was significantly lower in kidneys of Col4a3 KO mice, compared with Col4a3;Tsp1 DKO mice. Data are expressed as means ± SEM. *P < 0.05. Original magnification, ×60. H&E, hematoxylin and eosin.
Figure 4.

Fibroblast accumulation during progression of CKD in Col4a3 KO and Col4a3;Tsp1 DKO mice. A: Representative photomicrographs of formalin-fixed, paraffin-embedded kidney sections of wild-type control, Col4a3 KO, and Col4a3;Tsp1 DKO mice at 12, 22, and 32 weeks of age. Sections were immunolabeled with antibodies against αSMA (red) and counterstained with methylene green. B: Fibroblast numbers were determined as αSMA+ cells per visual field. The fibroproliferative response over time was more robust in Col4a3 KO mice than in Col4a3;Tsp1 DKO mice and differences reached statistical significance at 22 weeks of age. Data are expressed as means ± SEM. ∗P < 0.05. Original magnification, ×20. αSMA, α-smooth muscle actin.
Figure 5.

Intrarenal accumulation of inflammatory cells in the progression of CKD in Col4a3 KO and Col4a3;Tsp1 DKO mice. A: Representative photomicrographs of formalin-fixed, paraffin-embedded kidney sections of wild-type control, Col4a3 KO, and Col4a3;Tsp1 DKO mice at 12, 22, and 32 weeks of age. Sections were immunolabeled with antibodies against CD45 (red) and counterstained with methylene green. B: Inflammation was determined as numbers of CD45+ inflammatory cells per visual field. The inflammatory response over time was more robust in Col4a3;Tsp1 DKO mice than in Col4a3 KO mice and reached statistical significance at 22 weeks of age. Data are expressed as means ± SEM. ∗P < 0.05. Original magnification, ×20.
To further explore the contribution of different T-cell populations to the increase in inflammatory cells observed in Col4a3;Tsp1 DKO mice, we performed immunolabeling using antibodies to C-C chemokine receptor 5 (CCR5, a marker of Th1 cells), G-protein coupled receptor 44 (GPR44, a marker of Th2 cells), RAR-related orphan receptor-γ (ROR-γ, a marker of Th17 cells), or forkhead box protein P3 (FOXP3, a marker of T-regulatory cells). Fibroproliferative response in Col4a3 KO mice was associated with accumulation of GPR44+ Th2 cells (Figure 6, A and B); in creatinine-matched Col4a3;Tsp1 DKO mice, TSP1 deficiency caused a Th1 switch with accumulation of CCR5+ Th1 cells (Figure 6, C and D). This is consistent with recent findings describing a role of Th2 accumulation in progression of renal fibrogenesis and suggesting that inflammation not only changes quantitatively when TSP1 is absent, but also qualitatively, with a switch from a Th2 response to a Th1 response.25, 26, 27, 28 Furthermore, TSP1 deficiency in Col4a3;Tsp1 DKO mice was associated with loss of ROR-γ+ Th17 and FOXP3+ T-regulatory cells (Figure 7), supporting previous reports of a role of TSP1 in differentiation of Th17 and T-regulatory cells involved in regulating inflammatory response in the kidney.29, 30, 31, 32, 33 Because both TSP1 and the NC1 domain of the α3 chain of type IV collagen are inhibitors of tumor angiogenesis, and because rarefaction of microvessels is a consequence of tubulointerstitial fibrosis,1, 15 we next compared microvascular density in kidneys of creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. Analysis of CD31 immunostaining revealed that microvascular density was reduced in both Col4a3 KO and Col4a3;Tsp1 DKO mice, but did not differ in creatinine-matched kidneys (Figure 8).
Figure 6.

Interstitial accumulation of Th1 and Th2 cells. A and C: Representative microphotographs of formalin-fixed, paraffin-embedded kidney sections of wild-type control (32 weeks) and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. Sections were labeled with antibodies against C-C chemokine receptor type 5 (CCR5, a marker of Th1 cells) (A) or with G-protein coupled receptor 44 (GPR44, a marker of Th2 cells) (C) and counterstained with methylene green. B and D: Numbers of CCR5+ T cells (B) and GPR44+ T cells (D) per visual field. Col4a3 KO mice exhibited interstitial accumulation of GPR44+ Th2 cells; in creatinine-matched Col4a3;Tsp1 DKO mice, TSP1 deficiency was associated with a Th1 switch and accumulation of CCR5+ Th1 cells. Data are expressed as means ± SEM. *P < 0.05. Original magnification, ×40.
Figure 7.

Accumulation of Th17 and T-regulatory cells. A and C: Representative microphotographs of formalin-fixed, paraffin-embedded kidney sections of wild-type control (32 weeks) and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. Sections were labeled with antibodies against RAR-related orphan receptor-γ (ROR-γ, a marker of Th17 cells) (A) or forkhead box protein P3 (FOXP3, a marker of T-regulatory cells) (C) and counterstained with methylene green. B and D: Numbers of ROR-γ+ (B) and FOXP3+ (D) T cells per visual field. TSP1 deficiency in creatinine-matched Col4a3;Tsp1 DKO mice was associated with loss, relative to Col4a3 KO mice, of Th17 cells (ROR-γ+) and T-regulatory cells (FOXP3+). Data are expressed as means ± SEM. *P < 0.05. Original magnification, ×40.
Figure 8.

A: Representative photomicrographs of kidney sections of wild-type control (32 weeks) and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. Sections were immunolabeled with CD31 (red), a marker of vasculature; the basal membrane was stained with an antibody to collagen IV (green) that recognizes collagen IV α1/2 chains and thereby also detects collagen IV in Col4a3 KO and Col4a3;Tsp1 DKO mice. Nuclei were counterstained with DAPI. B: Numbers of microvascular intersections per visual field. TSP1 deficiency in Col4a3;Tsp1 DKO mice had no additional effect on microvascular density, compared with Col4a3 KO mice. Data are expressed as means ± SEM. Original magnification, ×60. Col4, collagen IV.
In summary, inflammation (due to accumulation of Th1 cells and loss of Th17 and T-regulatory cells) was more robust in Col4a3;Tsp1 DKO kidneys, compared with creatinine-matched Col4a3 KO mice, but fibroblast accumulation was reduced.
Glomerular Sclerosis and Tubular Atrophy in Fibrotic Kidneys of Col4a3 KO and Col4a3;Tsp1 DKO Mice
In PAS-stained kidney sections, we digitally quantified deposition of PAS-positive material within glomeruli as an indicator of glomerulosclerosis. We found that glomerulosclerosis did not differ between Col4a3;Tsp1 DKO mice and creatinine-matched Col4a3 KO mice (Figure 9), suggesting that delayed fibrogenesis in Col4a3;Tsp1 DKO mice is not due to attenuated glomerular injury. This observation correlated with absence of glomerular TSP1 immunolabeling, even in sclerotic glomeruli of Col4a3 KO mice (Figure 10A), suggesting that TSP1 is not involved in progression of glomerular disease in Col4a3 KO mice. Instead, TSP1 was robustly up-regulated in tubules and interstitium within fibrotic kidneys of Col4a3 KO mice (Figure 10A). Tubular atrophy, determined by masked scoring of the percentage of atrophic tubules among all tubules, was more pronounced in the Col4a3 KO mice (Figure 10B). The growth factor TGF-β1 is a principal mediator of progression of CKD.1, 34 To gain insight into the role of TSP1 on TGF-β1 activation and progression of CKD in Col4a3 KO mice, we performed immunostaining with antibodies to phosphorylated SMADs 2 and 3 (p-SMAD2/3), which detects active TGF-β1 signaling.35 Consistent with the robust up-regulation of TSP1 observed in tubules and interstitium of fibrotic kidneys of Col4a3 KO mice, tubular and interstitial p-SMAD2/3 was markedly increased (Figure 10, C and D). Compared with Col4a3 KO mice (increased TSP1), tubular and interstitial immunostaining of p-SMAD2/3 was attenuated in creatinine-matched Col4a3;Tsp1 DKO mice (Figure 10, C and D).
Figure 9.

A: Representative photomicrographs of PAS-stained kidney sections of wild-type control (32 weeks) and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. B: Relative sclerotic glomerular area (PAS-positive area per glomerulus) did not differ significantly between kidneys of Col4a3 KO mice and Col4a3;Tsp1 DKO mice. Data are expressed as means ± SEM. Original magnification, ×60. PAS, periodic acid–Schiff.
Figure 10.

TSP1 deficiency attenuates TGF-β1 signaling. A and C: Representative photomicrographs of formalin-fixed, paraffin-embedded kidney sections of wild-type control (32 weeks) and creatinine-matched Col4a3 KO and Col4a3;Tsp1 DKO mice. A: Sections were labeled with antibodies to TSP1 (red) and counterstained with methylene green. In the fibrotic Col4a3 KO kidneys, TSP1 was localized predominantly in tubular epithelial cells, blood vessels, and interstitial cells. B: The percentage of atrophic tubules was significantly greater in kidneys of Col4a3 KO mice than in Col4a3;Tsp1 DKO mice. C: Sections were labeled with antibodies to phosphorylated SMADs 2 and 3 (p-SMAD2/3), indicative of active signaling in response to TGF-β1 (red) and counterstained with methylene green. D: Numbers of p-SMAD2/3+ nuclei per visual field. SMAD2/3 signaling was significantly enhanced in tubular and interstitial compartments of kidneys of Col4a3 KO mice, compared with creatinine-matched Col4a3;Tsp1 DKO kidneys. Data are expressed as means ± SEM. *P < 0.05. Original magnification, ×40.
Proliferation and Collagen Expression in Response to Latent TGF-β1 Is Blunted in TSP1-Deficient Fibroblasts
Previous studies have demonstrated that TSP1 activates latent TGF-β1.36 To determine whether altered disease progression in the absence of TSP1 is due to failure to activate latent TGF-β1, we next explored the capacity of TSP1-deficient cells to respond to latent TGF-β1 in vitro. Because fibroblast accumulation and ECM deposition were blunted in kidneys of end-stage diseased Col4a3;Tsp1 DKO mice (at 32 weeks), compared with the Col4a3 KO counterparts (at 22 weeks), and because TGF-β1 is known to induce both proliferation and ECM synthesis in fibroblasts, we focused on renal fibroblasts. We isolated primary fibroblasts from kidneys of Tsp1 KO mice and C57BL/6 control mice and compared their proliferation and procollagen III expression in response to either latent or active TGF-β1. Although the proliferative response of Tsp1 KO fibroblasts to latent TGF-β1 was significantly decreased, compared with wild-type C57BL/6 fibroblasts, their response to active TGF-β1 was equal (Figure 11A). Similarly, collagen III expression was blunted in response to latent TGF-β1 but not to active TGF-β1 (Figure 11B). In summary, these findings suggest that ineffective activation of latent TGF-β1 is likely the underlying cause for altered kidney disease progression in Col4a3;Tsp1 DKO mice.
Figure 11.
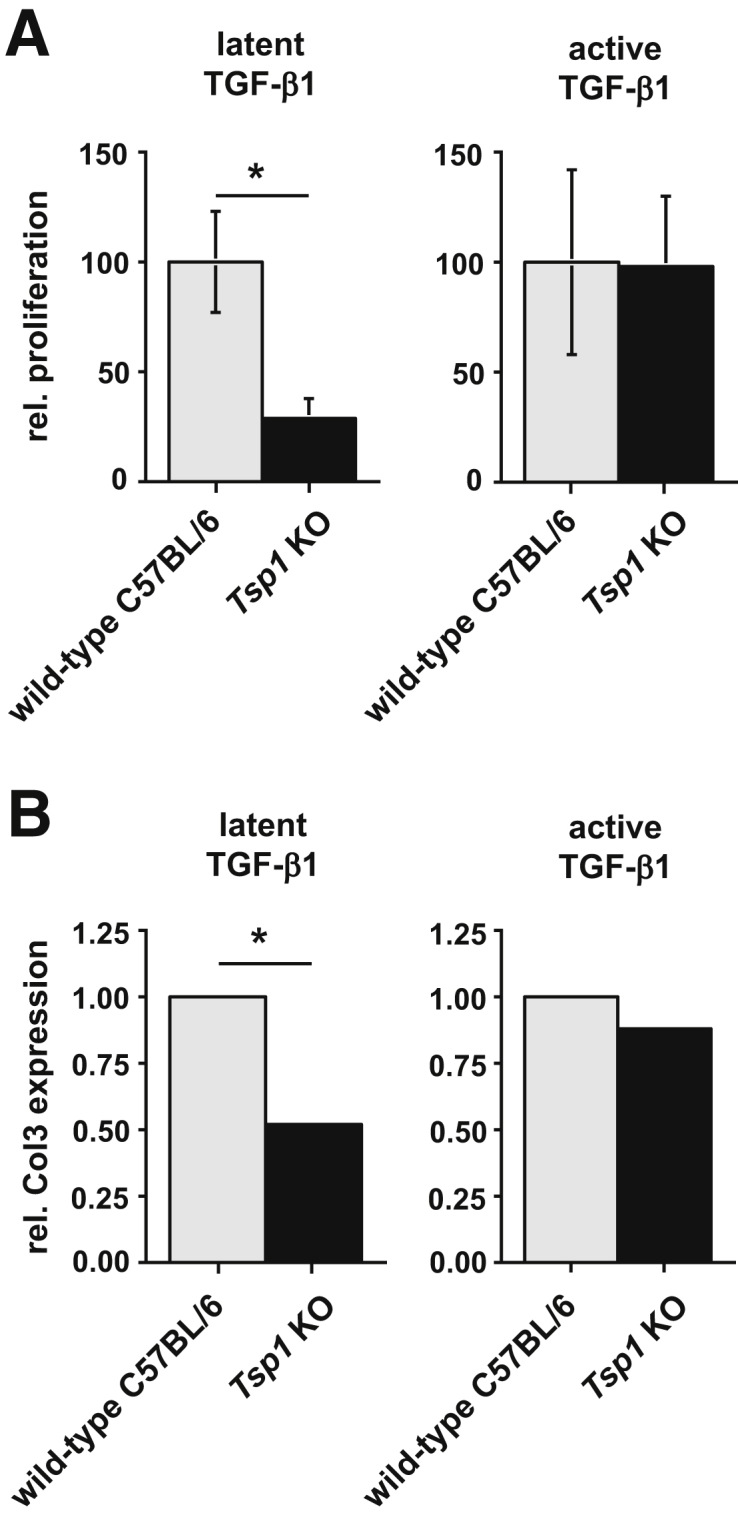
TSP1 deficiency causes an altered response to latent TGF-β1. A: Fibroblast proliferation in response to active and latent TGF-β1. Primary fibroblasts isolated from kidneys of wild-type control and TSP1-deficient mice (Tsp1 KO) were exposed for 24 hours to 5 ng/mL of either latent TGF-β1 or active TGF-β1. Relative proliferation was assessed by a WST assay. TSP1 deficiency significantly impaired the proliferative response to latent TGF-β1, but not to active TGF-β1. B: Type III collagen expression in response to active and latent TGF-β1. Wild-type control and Tsp1 KO renal fibroblasts were exposed for 24 hours to 5 ng/mL of either latent TGF-β1 or active TGF-β1. Type III procollagen expression was assessed by quantitative real-time PCR. TSP1 deficiency significantly decreased collagen III expression in response to latent TGF-β1. Data are expressed as means ± SEM. *P < 0.05. Col3, collagen III; rel., relative.
Discussion
We conducted the present study to gain mechanistic insight into the role of TSP1 in the progression of CKD. For this purpose, we compared kidney disease progression in Col4a3 KO mice with that of Col4a3;Tsp1 DKO mice. Decline of excretory renal function was delayed by the absence of TSP1, confirming previous reports.9, 10, 11, 12, 13 Unexpectedly, comparison of creatinine-matched kidneys of Col4a3 KO and Col4a3;Tsp1 DKO mice revealed that disease progression was not simply delayed, but that kidney disease followed different paths. Despite similar interstitial volumes at matched creatinine levels, absence of TSP1 in Col4a3;Tsp1 DKO mice was associated with lesser collagen deposition, lesser fibroblast accumulation, and lesser tubular atrophy, compared with Col4a3 KO mice, but increased Th1-mediated inflammation. The extent of glomerulosclerosis and the density of microvessels were not altered by the absence of TSP1. Our findings suggest that the altered course of disease progression in Col4a3;Tsp1 DKO mice is caused by an inability of kidney cells to activate latent TGF-β1 in the absence of TSP1. Our findings are consistent with the literature on the role of TSP1 in TGF-β1 activation. TGF-β1 is known to induce fibroblast proliferation and collagen deposition, and to contribute to formation of tubular atrophy, all of which are reduced in the absence of TGF-β1–activating TSP1.1, 37 TGF-β1–deficient mice exhibit severe inflammatory lesions, and TGF-β1 in general is known to be anti-inflammatory.38, 39 In the present study, inability to activate latent TGF-β1 was associated with an increased inflammatory response in chronic progressive kidney disease. With regard to progression of CKD, it is an unexpected finding that decline of kidney function is caused by different pathways, depending on the presence or absence of TSP1. Our findings suggest that fibroproliferative and inflammatory responses are distinct entities, both of which (and possibly independent of each another) can determine progression of CKD. Our present findings confirm earlier findings indicating that relative interstitial volume is the most accurate correlate of chronic progression of kidney disease, but challenge the concept of one common uniform pathway leading to ESRF.
Additionally, our findings suggest that a fibrosis-dominated pathology is more detrimental for kidney function than is inflammation-dominated response, but that either pathological entity can ultimately cause decline of kidney function. Although the common denominator appears to be increased interstitial volume, it remains unclear how such expansion of the interstitium causes decline in kidney function. We speculate that both fibrosis and inflammation can cause vascular and/or tubular occlusion through volume expansion. The present findings give reason for caution, and suggest that future therapeutic strategies exclusively targeting fibrosis could be less effective than combined inhibition of fibrosis and inflammation. The concept of a uniform pathway of chronic progressive kidney disease may require further consideration, and fibroproliferative and inflammatory components of interstitial disease may need to be viewed as separate causal entities.
Acknowledgments
We thank Sai Ram Reddy, Scott McGoohan, Lauren Orefice, and Margot Martino for technical assistance.
Footnotes
Supported by NIH grants R01DK55001-14, DK81576, CA125550, CA155370, CA151925, and CA163191 (R.K.); Metastasis Research Center; by German Research Foundation (DFG) grants ZE523/2-1 and ZE523/3-1 (M.Z.) and SFB1002/TPC01 (E.Z.); the Cancer Prevention and Research Institute of Texas (R.K.); and the seed funding research program of the Faculty of Medicine, Georg August University Göttingen (B.T., D.T.).
Disclosures: None declared.
Contributor Information
Michael Zeisberg, Email: mzeisberg@med.uni-goettingen.de.
Raghu Kalluri, Email: rkalluri@mdanderson.org.
References
- 1.Zeisberg M., Neilson E.G. Mechanisms of tubulointerstitial fibrosis. J Am Soc Nephrol. 2010;21:1819–1834. doi: 10.1681/ASN.2010080793. [DOI] [PubMed] [Google Scholar]
- 2.McClellan A.C., Plantinga L., McClellan W.M. Epidemiology, geography and chronic kidney disease. Curr Opin Nephrol Hypertens. 2012;21:323–328. doi: 10.1097/MNH.0b013e3283521dae. [DOI] [PubMed] [Google Scholar]
- 3.Zeisberg M., Kalluri R. Experimental strategies to reverse chronic renal disease. Blood Purif. 2004;22:440–445. doi: 10.1159/000080790. [DOI] [PubMed] [Google Scholar]
- 4.Risdon R.A., Sloper J.C., De Wardener H.E. Relationship between renal function and histological changes found in renal-biopsy specimens from patients with persistent glomerular nephritis. Lancet. 1968;2:363–366. doi: 10.1016/s0140-6736(68)90589-8. [DOI] [PubMed] [Google Scholar]
- 5.Bohle A., Bader R., Grund K.E., Mackensen S., Neunhoeffer Serum creatinine concentration and renal interstitial volume. Analysis of correlations in endocapillary (acute) glomerulonephritis and in moderately severe mesangioproliferative glomerulonephritis. Virchows Arch A Pathol Anat Histol. 1977;375:87–96. doi: 10.1007/BF00428097. [DOI] [PubMed] [Google Scholar]
- 6.Bohle A., Glomb D., Grund K.E., Mackensen S. Correlation between relative interstitial volume of the renal cortex and serum creatinine concentration in minimal changes with nephrotic syndrome and in focal sclerosing glomerulonephritis. Virchows Arch A Pathol Anat Histol. 1977;376:221–232. doi: 10.1007/BF00432398. [DOI] [PubMed] [Google Scholar]
- 7.Bohle A., Wehrmann M., Bogenschutz O., Batz C., Müller C.A., Müller G.A. The pathogenesis of chronic renal failure in diabetic nephropathy. Investigation of 488 cases of diabetic glomerulosclerosis. Pathol Res Pract. 1991;187:251–259. doi: 10.1016/s0344-0338(11)80780-6. [DOI] [PubMed] [Google Scholar]
- 8.Armstrong L.C., Bornstein P. Thrombospondins 1 and 2 function as inhibitors of angiogenesis. Matrix Biol. 2003;22:63–71. doi: 10.1016/s0945-053x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 9.Daniel C., Wiede J., Krutzsch H.C., Ribeiro S.M., Roberts D.D., Murphy-Ullrich J.E., Hugo C. Thrombospondin-1 is a major activator of TGF-beta in fibrotic renal disease in the rat in vivo. Kidney Int. 2004;65:459–468. doi: 10.1111/j.1523-1755.2004.00395.x. [DOI] [PubMed] [Google Scholar]
- 10.Lu A., Miao M., Schoeb T.R., Agarwal A., Murphy-Ullrich J.E. Blockade of TSP1-dependent TGF-β activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol. 2011;178:2573–2586. doi: 10.1016/j.ajpath.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bige N., Shweke N., Benhassine S., Jouanneau C., Vandermeersch S., Dussaule J.C., Chatziantoniou C., Ronco P., Boffa J.J. Thrombospondin-1 plays a profibrotic and pro-inflammatory role during ureteric obstruction. Kidney Int. 2012;81:1226–1238. doi: 10.1038/ki.2012.21. [DOI] [PubMed] [Google Scholar]
- 12.Daniel C., Takabatake Y., Mizui M., Isaka Y., Kawashi H., Rupprecht H., Imai E., Hugo C. Antisense oligonucleotides against thrombospondin-1 inhibit activation of TGF-beta in fibrotic renal disease in the rat in vivo. Am J Pathol. 2003;163:1185–1192. doi: 10.1016/s0002-9440(10)63478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hochegger K., Knight S., Hugo C., Mayer G., Lawler J., Mayadas T.N., Rosenkranz A.R. Role of thrombospondin-1 in the autologous phase of an accelerated model of anti-glomerular basement membrane glomerulonephritis. Nephron Exp Nephrol. 2004;96:e31–e38. doi: 10.1159/000076402. [DOI] [PubMed] [Google Scholar]
- 14.Chen H., Herndon M.E., Lawler J. The cell biology of thrombospondin-1. Matrix Biol. 2000;19:597–614. doi: 10.1016/s0945-053x(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 15.Xie L., Duncan M.B., Pahler J., Sugimoto H., Martino M., Lively J., Mundel T., Soubasakos M., Rubin K., Takeda T., Inoue M., Lawler J., Hynes R.O., Hanahan D., Kalluri R. Counterbalancing angiogenic regulatory factors control the rate of cancer progression and survival in a stage-specific manner. Proc Natl Acad Sci USA. 2011;108:9939–9944. doi: 10.1073/pnas.1105041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sugimoto H., Mundel T.M., Sund M., Xie L., Cosgrove D., Kalluri R. Bone-marrow-derived stem cells repair basement membrane collagen defects and reverse genetic kidney disease. Proc Natl Acad Sci USA. 2006;103:7321–7326. doi: 10.1073/pnas.0601436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamano Y., Sugimoto H., Soubasakos M.A., Kieran M., Olsen B.R., Lawler J., Sudhakar A., Kalluri R. Thrombospondin-1 associated with tumor microenvironment contributes to low-dose cyclophosphamide-mediated endothelial cell apoptosis and tumor growth suppression. Cancer Res. 2004;64:1570–1574. doi: 10.1158/0008-5472.can-03-3126. [DOI] [PubMed] [Google Scholar]
- 18.Miner J.H., Sanes J.R. Molecular and functional defects in kidneys of mice lacking collagen alpha 3(IV): implications for Alport syndrome. J Cell Biol. 1996;135:1403–1413. doi: 10.1083/jcb.135.5.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeisberg M., Bottiglio C., Kumar N., Maeshima Y., Strutz F., Müller G.A., Kalluri R. Bone morphogenic protein-7 inhibits progression of chronic renal fibrosis associated with two genetic mouse models. Am J Physiol Renal Physiol. 2003;285:F1060–F1067. doi: 10.1152/ajprenal.00191.2002. [DOI] [PubMed] [Google Scholar]
- 20.Zeisberg E.M., Potenta S.E., Sugimoto H., Zeisberg M., Kalluri R. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J Am Soc Nephrol. 2008;19:2282–2287. doi: 10.1681/ASN.2008050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E., Chandraker A., Yuan X., Pu W.T., Roberts A.B., Neilson E.G., Sayegh M.H., Izumo S., Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- 22.Okuno Y., Huettner C.S., Radomska H.S., Petkova V., Iwasaki H., Akashi K., Tenen D.G. Distal elements are critical for human CD34 expression in vivo. Blood. 2002;100:4420–4426. doi: 10.1182/blood-2002-03-0788. [DOI] [PubMed] [Google Scholar]
- 23.Zeisberg M., Khurana M., Rao V.H., Cosgrove D., Rougier J.P., Werner M.C., Shield C.F., 3rd, Werb Z., Kalluri R. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 2006;3:e100. doi: 10.1371/journal.pmed.0030100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimoto H., LeBleu V.S., Bosukonda D., Keck P., Taduri G., Bechtel W., Okada H., Carlson W., Jr., Bey P., Rusckowski M., Tampe B., Tampe D., Kanasaki K., Zeisberg M., Kalluri R. Activin-like kinase 3 is important for kidney regeneration and reversal of fibrosis. Nat Med. 2012;18:396–404. doi: 10.1038/nm.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann K.F., McCarty T.C., Segal D.H., Chiaramonte M., Hesse M., Davis E.M., Cheever A.W., Meltzer P.S., Morse H.C., 3rd, Wynn T.A. Disease fingerprinting with cDNA microarrays reveals distinct gene expression profiles in lethal type 1 and type 2 cytokine-mediated inflammatory reactions. FASEB J. 2001;15:2545–2547. doi: 10.1096/fj.01-0306fje. [DOI] [PubMed] [Google Scholar]
- 26.Liu L., Kou P., Zeng Q., Pei G., Li Y., Liang H., Xu G., Chen S. CD4+ T lymphocytes, especially Th2 cells, contribute to the progress of renal fibrosis. Am J Nephrol. 2012;36:386–396. doi: 10.1159/000343283. [DOI] [PubMed] [Google Scholar]
- 27.Sandler N.G., Mentink-Kane M.M., Cheever A.W., Wynn T.A. Global gene expression profiles during acute pathogen-induced pulmonary inflammation reveal divergent roles for Th1 and Th2 responses in tissue repair. J Immunol. 2003;171:3655–3667. doi: 10.4049/jimmunol.171.7.3655. [DOI] [PubMed] [Google Scholar]
- 28.Wynn T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang H.P., Wu Y., Liu J., Jiang J., Geng X.R., Yang G., Mo L., Liu Z.Q., Liu Z.G., Yang P.C. TSP1-producing B cells show immune regulatory property and suppress allergy-related mucosal inflammation. Sci Rep. 2013;3:3345. doi: 10.1038/srep03345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang K., Vega J.L., Hadzipasic M., Schatzmann Peron J.P., Zhu B., Carrier Y., Masli S., Rizzo L.V., Weiner H.L. Deficiency of thrombospondin-1 reduces Th17 differentiation and attenuates experimental autoimmune encephalomyelitis. J Autoimmun. 2009;32:94–103. doi: 10.1016/j.jaut.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu S., Zhang N., Yopp A.C., Chen D., Mao M., Zhang H., Ding Y., Bromberg J.S. TGF-beta induces Foxp3+ T-regulatory cells from CD4+ CD25− precursors. Am J Transplant. 2004;4:1614–1627. doi: 10.1111/j.1600-6143.2004.00566.x. [DOI] [PubMed] [Google Scholar]
- 32.Gandolfo M.T., Jang H.R., Bagnasco S.M., Ko G.J., Agreda P., Satpute S.R., Crow M.T., King L.S., Rabb H. Foxp3+ regulatory T cells participate in repair of ischemic acute kidney injury. Kidney Int. 2009;76:717–729. doi: 10.1038/ki.2009.259. [DOI] [PubMed] [Google Scholar]
- 33.Kinsey G.R., Huang L., Vergis A.L., Li L., Okusa M.D. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. 2010;77:771–780. doi: 10.1038/ki.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wynn T.A. Fibrosis under arrest. Nat Med. 2010;16:523–525. doi: 10.1038/nm0510-523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeisberg M., Hanai J., Sugimoto H., Mammoto T., Charytan D., Strutz F., Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 36.Schultz-Cherry S., Lawler J., Murphy-Ullrich J.E. The type 1 repeats of thrombospondin 1 activate latent transforming growth factor-beta. J Biol Chem. 1994;269:26783–26788. [PubMed] [Google Scholar]
- 37.Bechtel W., McGoohan S., Zeisberg E.M., Müller G.A., Kalbacher H., Salant D.J., Müller C.A., Kalluri R., Zeisberg M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christ M., McCartney-Francis N.L., Kulkarni A.B., Ward J.M., Mizel D.E., Mackall C.L., Gress R.E., Hines K.L., Tian H., Karlsson S., Wahl S.M. Immune dysregulation in TGF-beta 1-deficient mice. J Immunol. 1994;153:1936–1946. [PubMed] [Google Scholar]
- 39.Kulkarni A.B., Karlsson S. Inflammation and TGF beta 1: lessons from the TGF beta 1 null mouse. Res Immunol. 1997;148:453–456. doi: 10.1016/s0923-2494(97)82669-7. [DOI] [PubMed] [Google Scholar]