

Abstract
Inhibition of DNA repair and oxidative stress are 2 common mechanisms associated with arsenic-induced genotoxicity. The purpose of this study was to examine mechanisms of genotoxicity induced by environmentally relevant doses of arsenite (As+3) in mouse thymus cells. An increase in DNA damage and a decrease in poly(ADP-ribose) polymerase (PARP) activity were seen in vitro following exposure to 50 nM As+3 in primary mouse thymus cells and a murine thymus pre-T cell line, D1. 3,4-Dihydro-5[4-(1-piperindinyl) butoxyl]-1(2H)-isoquinoline, a well-characterized PARP inhibitor, also produced DNA damage in D1 cells, confirming the correlation between PARP inhibition and DNA damage increase. As+3 at 500 nM induced double strand breaks (DSBs) in DNA and oxidative stress at 4 h in D1 cells, which was reversed at 18 h. No apoptosis or decrease of viability was observed in these exposures. 4-Hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, a widely-used antioxidant, was utilized to confirm that oxidative stress is partially responsible for the increase of strand breaks in 500 nM As+3 exposure at 4 h. Expression of As+3 exporters, Mdr1 and Mrp1, were found to be induced by 500 nM As+3 in D1 cells, suggesting a possible mechanism for reversal of oxidative stress and DSBs at the 18-h timepoint. Finally, we showed that DNA damage and PARP inhibition by As+3 were reversed by zinc (Zn+2) at approximate equimolar doses. Collectively, these results demonstrate that As+3 at doses within the nanomolar range induce genotoxicity by inhibiting PARP, and produces oxidative stress at higher concentrations, which can be reversed by a Zn+2 treatment.
Keywords: arsenite-induced genotoxicity, mouse thymus cells, poly(ADP-ribose) polymerase (PARP) inhibition, oxidative stress, arsenic exporters, zinc
Two primary forms of inorganic arsenic (As), trivalent arsenite (As+3) and pentavalent arsenate (As+5), are found in air, food, and drinking water in many countries around the world. As+3 exposure is associated with diseases such as skin lesions, diabetes, cardiovascular diseases, and different types of cancers (Argos et al., 2010; Schuhmacher-Wolz et al., 2009; Vahter, 2008). Although growing evidence indicates that As+3 affects the immune system in multiple aspects, the exact mechanisms are still poorly understood (Dangleben et al., 2013). Oxidative stress due to formation of reactive oxygen species (ROS) and inhibition of DNA repair are the 2 most common mechanisms proposed for As-induced genotoxicity (Faita et al., 2013). An increase in ROS has been observed in multiple models after As+3 treatment such as keratinocytes, lymphoid cells, and many types of cancer cells (Cooper et al., 2009; Li et al., 2001; Zhang et al., 2011). ROS production leads to multiple effects in different types of cells, including apoptosis or necrosis, cell cycle arrest, generation of both single strand break (SSB) and double strand break (DSB), and gene mutation (Bauer et al., 2011; Shi et al., 2004; Wiseman and Halliwell, 1996).
Poly(ADP-ribose) polymerase (PARP), the ‘nick sensor’ in base excision repair (BER) for SSBs and direct participant in the repair of oxidative DNA damage, is directly inhibited by As+3 (Qin et al., 2012; Zhou et al., 2011). PARP is a protein with a DNA binding domain containing 2 C3H1 zinc (Zn) finger motifs, and replacement of Zn with As+3 is believed to be the mechanism that disrupts the coordination sphere in the Zn finger environment, impairing function of the protein (Zhou et al., 2014). There is also evidence that DNA damage caused by spontaneous base loss or genotoxic agents are repaired by BER (Zharkov, 2008), and inhibition of PARP may leave damaged DNA unrepaired, leading to the formation of DSBs in a replication-dependent manner (Carrozza et al., 2009). DSBs are repaired through non-homologous end joining, microhomology-mediated end joining, or homologous recombination, which are detrimental to cells and may lead to mutation or apoptosis (Kaina, 2003; Lieber, 2010).
Previous studies in our laboratory reported that in vivo drinking water exposure to As+3 suppresses mouse bone marrow and spleen cell function (Ezeh et al., 2014). Exposure of spleen cells in vitro suppressed T cell-dependent humoral immunity at concentrations as low as 500 nM (Li et al., 2010). Studies in human peripheral blood mononuclear cells showed a dose-dependent suppression of T cell proliferation at extremely low concentrations of As+3 (0.1–10 nM) in some individuals (Burchiel et al., 2014). These studies demonstrate that human and mouse lymphocytes are extremely sensitive to low concentrations of As+3 that likely reflect environmental exposures. Because our previous studies showed the bone marrow cells are sensitive to low doses of As+3 exposure (Ezeh et al., 2014), the purpose of the present studies was to examine whether thymus cells are also sensitive to As+3 and to determine potential mechanisms of genotoxicity.
MATERIALS AND METHODS
Isolation of primary mouse thymus cells
C57BL/6 J male mice were purchased at 8–10 weeks of age from Jackson Laboratory (Bar Harbor, Maine). All animal experiments were performed following the protocols approved by the Institutional Animal Use and Care Committee at the University of New Mexico Health Sciences Center. Mouse thymuses were harvested and transferred to the laboratory in Hanks Balance Salt Solution (HBSS, Lonza, Walersville, Maryland). Cells from each thymus were prepared as single cell suspensions by placing the organ between the frosted ends of 2 microscope slides (Fisher Scientific, Pittsburgh, Pennsylvania) and squeezed into a dish containing 5 ml of the mouse medium (RPMI-1640 [Sigma-Aldrich, St. Louis, Missouri] with 10% FBS [HyClone Laboratories, Logan, Utah], 2 mM l-glutamine [Life Technologies, Grand Island, New York], 100 mg/ml streptomycin, 100 units/ml penicillin [Life Technologies]). Cells from 3 mice were pooled, centrifuged at 200 × g for 10 min and resuspended in the mouse medium. Cell viability was determined by acridine orange/propidium iodide (PI) staining and counting using the Nexcelom Cellometer 2000.
Culture and treatment of D1 cells
The D1 cell line is a CD3+CD4-CD8-, IL-7-dependent pre-T cell line established from p53−/− mouse thymocytes (Kim et al., 2003). It was a generous gift from Dr. Scott K. Durum (Center for Cancer Research, National Institute of Health, Frederick, Maryland). It is one of the few mouse pre-T cell lines that can be used to examine effects of xenobiotics on early thymic T cells. Cells were maintained in RPMI 1640 with 10% FBS (Atlanta Biologicals, Flowery Branch, Georgia), 2 mM l-glutamine, 100 mg/ml streptomycin, 100 units/ml penicillin, and 50 ng/ml recombinant mouse IL-7 (PeproTech, Rocky Hill, New Jersey). Cells were seeded at 5 × 104 cells/ml and subcultured every 3–4 days. Cell numbers and viabilities were determined by Trypan Blue and Nexcelom Cellometer 2000. For different experiments, cells were treated with sodium arsenite (Sigma-Aldrich), 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) (Sigma-Aldrich), or 3,4-dihydro-5[4-(1-piperindinyl) butoxyl]-1(2H)-isoquinoline (DPQ) (Santa Cruz Biotechnology, Dallas, Texas).
The single cell gel electrophoresis assay (Comet assay)
All reagents for the Comet assay were purchased from Trevigen (Gaithersburg, Maryland) unless otherwise noted. After treatments, cells were washed with Dulbecco’s phosphate-buffered saline without Ca+2 and Mg+2 (DPBS−, Mediatech, Manassas, Virginia) and immobilized in a bed of low melting point agarose on a Trevigen CometSlide. Cells were lysed with Lysis Solution + 10% DMSO (Sigma-Aldrich), and electrophoresed in basic buffer (pH >13) with 21 volts in 30 min. Slides were dried, stained with Sybr Green, and imaged by epifluorescence microscopy. From the image, 50 cells from each well on each of the slides were scored using CometScore (TriTek Corp., Sumerduck, Virginia). DNA damage was reported by percentage of DNA in tail (Collins, 2004).
PARP activity assay
A PARP/Apoptosis Kit from Trevigen was used to detect PARP activity in As+3-treated cells. Experiments were performed following the protocol described by Sun et al. (2012). After harvest, cells were lysed with Cell Extraction Buffer and total protein concentrations in cell extracts were determined by BCA Protein Assay (Thermo Scientific, Rockford, Illinois). 200 ng of total proteins was combined with activated DNA and nicotinamide adenine dinucleotide and loaded into a histone-coated strip well to form PAR and be fixed on the well bottom. After 30 min incubation at room temperature (RT), anti-PAR monoclonal antibody was added to the well to bind to PAR followed by an HRP conjugated secondary antibody. TACS-Sapphire was used to generate the chemiluminescence signal, then stopped by adding 0.2 M HCl. The signal was detected using SpectraMax 340PC microplate reader (Molecular Devices).
Phospho-H2AX (Gamma H2AX) flow cytometry assays
Alexa Fluor 647 mouse anti-H2AX (pS139) was purchased from BD Biosciences (San Jose, California). Antibody was diluted with DPBS− 1:9 before use. Harvested cells were washed with cold DPBS−. 3.7% freshly prepared formaldehyde was added to fix the cells for 10 min at RT. After another wash, cells were permeablized with −20°C 90% methanol at RT for 5 min. Cells were washed again and then stained with 50 µl per sample of diluted antibody at RT for 1 h in dark. Stained cells were washed rinsed and resuspended with DPBS− and analyzed on BD Accuri C6 flow cytometer.
Gamma H2AX Western blot
Total protein lysate (10 μg) was resolved on a 10% Criterion Tris-HCl gels (Bio-Rad, Hercules, California) and transferred onto a nitrocellulose membrane (Bio-Rad). After blocking for 1 h at RT in TBST [50 mM Tris + 150 mM NaCl + 0.05% Tween 20] containing 5% blotting-grade blocker (Bio-Rad), the membrane was then incubated with a phospho-H2AX (Ser139) antibody (Rabbit, 1:1000, Cell Signaling Technology, Danvers, Massachusetts) overnight at 4°C. After washing with TBST, the membrane was incubated for 1 h at RT with anti-rabbit IgG, HRP-linked antibody (1:1000, Cell Signaling Technology). Following incubation, the membrane was washed and the resulting signal was detected with the SuperSignal West Femto Chemiluminescent Substrate (Thermo Scientific).
Dihydroethidium (DHE) staining
DHE was purchased from Life Technologies, resuspended with 158 µl DMSO and diluted with 20 ml DPBS− to a final concentration of 5 µM. D1 cells were treated with 0, 50, or 500 nM As+3 for 2, 4, or 18 h. After treatment, cells were washed twice with cold wash buffer (DPBS− + 1% FBS + 0.9% sodium azide [Sigma-Aldrich]). Each sample was stained using 5 µM DHE in DPBS− with 37°C incubation for 30 min. Cells were then washed with cold wash buffer, resuspended in 500 µl DPBS− and analyzed on BD Accuri C6 flow cytometer.
Annexin V/PI staining
FITC Annexin V Apoptosis Detection Kit II (Catalog No. 556570) was purchased from BD Biosciences. D1 cells exposed to 50 and 500 nM As+3 in vitro were washed twice with DPBS−. 1 × 105 cells were resuspended in 100 μl 1 × Annexin V Binding Buffer, 5 μl of FITC Annexin V, and 5 µl of PI Staining Solution were added to each sample for 15 min at RT in dark. 400 µl of 1 × Annexin V Binding Buffer was added and the samples were analyzed by BD Accuri C6 flow cytometer. D1 cells treated with 10 μM Etoposide for 4 h were used as positive control. D1 cells blocked with 5 μg of purified recombinant Annexin V and unstained D1 cells were used for gating.
RNA isolation and HO-1 qPCR
RNeasy Mini Kit and QIAshredder (Qiagen, Valencia, California) were used according to the manufacturer’s instructions to isolate RNA from As+3-treated D1 cells. RNA was then quantified on an Agilent Nanodrop spectrophotometer. All samples were stored at −20°C until assayed. For each reverse transcriptase (RT) reaction, 60 µl containing a minimum of 1080 ng RNA was run on PTC-200 Thermal Cycler (MJ Research, Reno, Nevada) using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, California) under the following conditions: 25°C for 10 min, 37°C for 2 h. Samples were then diluted to ∼9 ng/µl with RNase, DNase-free water and stored at −20°C. Real-time PCR (qPCR) reactions utilizing Mm_Hmox1_1_SG QuantiTect Primer Assay and QuantiTect SYBR Green PCR Kit (Qiagen) were carried out on Applied Biosystem’s 7900HT system in a 384 well plate at 10 µl per reaction and 1 µL cDNA (∼9 ng total RNA) was added to each well. qPCR thermal cycling parameters used were polymerase activation 15 min 95°C, denature 15 s at 94°C, anneal 30 s at 55°C, and extend 30 s at 72°C (denature-anneal-extend for 40 cycles). Comparative CT (first amplification cycle exceeding threshold) method was applied for relative quantification, using GADPH gene as the endogenous reference. To calculate the comparative CT, briefly, the difference in CT (ΔCT) values between target and endogenous control were determined. Then the fold differences in gene expression (ΔΔCT) and the ΔCT of the control was subtracted from the ΔCT of the target (ΔCT target − ΔCT calibrator). This calculation is described in detail in the manual from Applied Biosystems.
As+3 exporter assays
RNA samples of 500 nM As+3-treated D1 cells harvested at 2, 4, and 18 h were prepared as above. The concentration of total RNA was quantified by UV spectrophotometry at 260/280 nm using a Nanodrop spectrophotometer 2000 (Thermo Scientific). Complementary DNA was generated from 1 µg RNA using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). The mRNA expression of mouse transporters was quantified by qPCR using SYBR Green to detect amplified products in the Applied Biosystem’s 7900HT PCR system. Primers used in the analysis are listed below. Ct values were converted to ΔΔCt values by comparing to a reference gene, ribosomal protein 13 a (Rpl13a). For Mrp1 and Mdr1 protein expressions, Cell lysates (10–13 µg protein/well) were separated by SDS-PAGE electrophoresis and transferred to a nitrocellulose membrane at 4°C overnight. After blocking with 5% nonfat dry milk in 0.5% phosphate buffered saline with 0.5% of Tween 20 (PBS/T), membranes were incubated with primary antibodies against Mdr1 (C219) (Novus Biologicals, Littleton, Colorado) and Mrp1 (Alexis, Farmingdale, New York) followed by incubations with species-appropriate secondary antibodies for 1–2 h. The SuperSignal West Dura Chemiluminescent Substrate (Thermo Scientific) was applied to the membranes prior to detection of luminescence using a FluorChem Imager (Alpha Innotech, San Leandro, California). Target protein band intensities were semi-quantified and normalized to β-actin levels (ab8227 antibody, Abcam).
Primers used for As+3 exporter qPCR analysis
| Transporters | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| Mrp1 | GCTGTGGTGGGCGCTGTCTA | CCCAGGCTCAGCCACAGGAA |
| Mrp2 | AGCAGGTGTTCGTTGTGTGT | AGCCAAGTGCATAGGTAGAGAAT |
| Mdr1a | TGCCCCACCAATTTGACACCCT | ATCCAGTGCGGCCTGAACCA |
| Mdr1b | GTGTTAAAGGGGCGATGGGCG | AGGCTTGGCCAGACAACAGCTT |
Statistics
Data were analyzed using Excel 2010 and Sigma Plot 12.5 software. One-way analysis of variance (ANOVA) and Dunnett’s t test were used to determine differences between control and treatment groups.
RESULTS
As+3 Exposure Increased DNA Damage and Inhibited PARP in Primary Mouse Thymus Cells In Vitro
Primary thymus cells from C57BL/6 J male mice were isolated and treated with 5 and 50 nM As+3 and harvested at 4 and 18 h. An increase in DNA damage and a decrease in PARP activity were seen in cells treated with 50 nM As+3 at both time points (Figs. 1A and 1B), indicating the genotoxic effects of As+3 on mouse thymus cells in vitro. These results not only demonstrate that environmentally relevant doses of As+3 induce genotoxic events in mouse thymus cells, but also suggest that inhibition of PARP activity by As+3 is involved in, and may be the mechanism responsible for the increase in DNA damage.
FIG. 1.
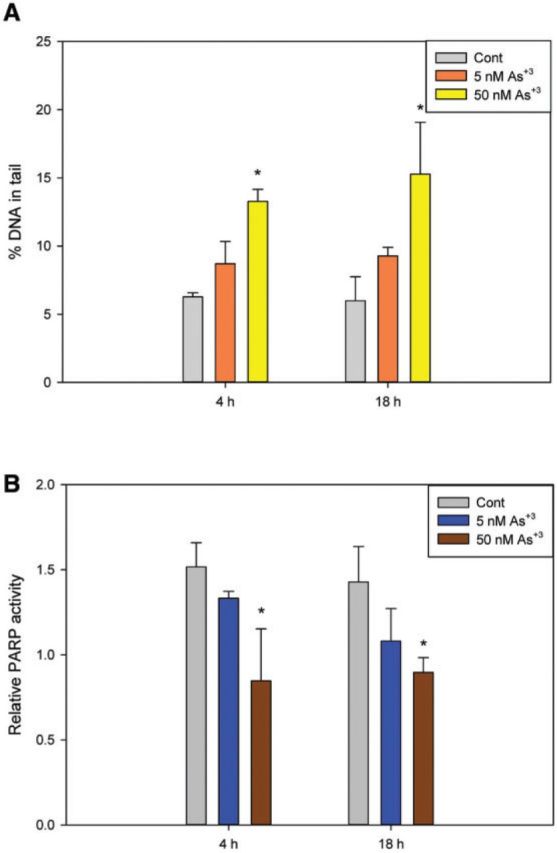
Genotoxicity in primary mouse thymus cells exposed to As+3 in vitro. Naïve primary thymus cells from C57BL/6 J male mice were exposed to 5 and 50 nM for As+3 for 4 and 18 h in vitro. A, DNA damage was measured by percentage of DNA in tail using the alkaline Comet assay. B, PARP activity was measured with Trevigen ELISA kits represented by absorbance at 450 nm. *Significantly different compared with control (P < .05, n = 5). Results are means ± SD.
Increased DNA Damage After As+3 Exposure Is Directly Related to PARP Inhibition in Pre-T Cells
We utilized an IL-7 dependent, CD3+CD4-CD8- pre-T cell line, D1, to further examine the genotoxicity induced by As+3 in mouse thymus cells. D1 cells were treated with As+3 at 5, 50, and 500 nM in vitro for 4 and 18 h. A significant increase of DNA damage was observed in 50 and 500 nM As+3-treated cells for 4 and 18 h (Figure 2A). A significant decrease in PARP activity was also seen at 4 and 18 h in 50 and 500 nM As+3-treated cells (Figure 2B). Therefore, As+3 induced similar genotoxic events in D1 cells as compared with primary thymus cells.
FIG. 2.

Genotoxicity in D1 cells exposed to As+3 or PARP inhibitor (DPQ). D1 cells were treated with 5, 50, or 500 nM As+3 for 4 and 18 h. A, DNA damage was measured by percentage of DNA in tail using alkaline Comet assay. B, PARP activities were measured with Trevigen ELISA kit represented by absorbance at 450 nm. C and D, DNA damage were measured by alkaline Comet assay in D1 cells treated with 5, 50, and 500 nM As+3 and 0.1, 1, and 10 µM PARP inhibitor, DPQ for 4 h (C) and 18 h (D). *Significantly different compared to control (P < .05). Results are means ± SD.
To determine if DNA damage increased by As+3 is related to PARP inhibition, we treated the D1 cells with a potent PARP inhibitor, DPQ. A significant increase in DNA damage was observed with 1 µM DPQ at 4 h (Figure 2C) and 0.1 µM DPQ at 18 h (Figure 2D), indicating that the DNA damage observed with As+3 treatments was likely to be the result of PARP inhibition, which leaves damaged DNA unrepaired. However, because the DNA damage in the DPQ treatments seemed to have a limitation at around 15% of DNA in tail, while As+3-induced DNA damage easily exceeded this amount, it was thought that As+3 may act through additional mechanisms than inhibiting PARP alone, such as oxidative stress at higher concentrations (Figs. 2C and 2D).
As+3 Exposure at 50 and 500 nM Induces DSBs in D1 Cells at Early Time Points
The DNA damage observed in alkaline Comet assay represents the total strand breaks including both SSB and DSB (Collins, 2004). We have already shown that 500 nM As+3 caused significant genotoxicity which exceeded the effects of PARP inhibitor, so it was hypothesized that besides the inhibition of DNA repair, oxidative stress may also be involved higher levels of As+3 exposure (eg, 500 nM). When DSBs occur, H2AX is phosphorylated on serine 139, also known as gamma H2AX and is often used as a marker for DSBs (Mah et al., 2010). Therefore, we treated D1 cells with 0, 50, and 500 nM of As+3 and harvested cells at 2, 4, and 18 h. Phospho-H2AX antibody was used to stain the cells in order to measure the phospho-H2AX level using flow cytometry. An increase in phospho-H2AX mean channel fluorescence was observed in cells exposed to 500 nM As+3 at both 2 and 4 h time points, but not at 18 h (Figure 3A), indicating that DSBs were generated by As+3 treatment in D1 cells in early stages at high concentration (500 nM). We also confirmed the observation by Western blot (Figure 3B).
FIG. 3.

Phospho-H2AX (γ-H2AX) expression and oxidative stress in D1 cells exposed to As+3. D1 cells were treated with 50 or 500 nM As+3. Phospho-H2AX expression levels were measured by flow cytometry (A), represented by Mean Channel Fluorescence or Western blot (B). C, Mean Channel Fluorescence of DHE. D, HO-1 RNA levels were measured by qPCR on cDNA samples from As+3-treated D1 cells. *Significantly different compared with control (P < .05). Results are means ± SD.
Time-Dependent Oxidative Stress in D1 Cells Exposed to As+3
Oxidative stress is commonly associated with the induction of DSBs. We used DHE staining to examine the superoxide level and Heme Oxygenase-1 (HO-1) expression by qPCR to see if oxidative stress is involved in the genotoxicity induced by As+3. As determined by DHE fluorescence, a significant increase in superoxide level was observed at 500 nM following 2–4 h of treatment (Figure 3C), but not at the 18 h time point, indicating that it is an early stage event occurring at high exposure levels. HO-1 is known to be induced by oxidative stress and considered to be protective to cells (Vile et al., 1994). An ∼8-fold increase in HO-1 expression was observed at 2 h and a more significant 20-fold increase was seen at 4 h in 500 nM As+3 treatment, but dropped to control levels at 18 h (Figure 3D). These results suggested a time-dependent oxidative stress and DSBs occurrence in D1 cells treated with a high dose of As+3 (500 nM).
Exposure to As+3 Did Not Induce Apoptosis in D1 Cells
Since DNA strand breaks and oxidative stress apoptosis may lead to cell death and the loss of PARP activity through caspase catalyzed PARP cleavage (Los et al., 2002), Annexin V and PI were used to check if D1 cells were apoptotic after As+3 exposure. No significant changes were found in both viability (% of Annexin V-, PI- cells, Figs. 4A and 4B) and % of apoptotic cells (% of Annexin V+ cells, Figs. 4A and 4C) in D1 cells exposed to 50 and 500 nM As+3 at 2, 4, and 18 h. The results excluded the possibility that decrease of PARP activity observed in low concentrations of As+3 exposure was induced by increase of apoptosis.
FIG. 4.

Annexin V and PI staining in D1 cells exposed to As+3. D1 cells were treated with 50 or 500 nM As+3 for 2, 4, and 18 h, or 10 µM Etoposide as positive control. A, Flow cytometry results showing D1 cells which are Annexin V-PI- (LL), Annexin V+PI- (LR), Annexin V-PI+ (UL), or Annexin V+PI+ (UR). B, Viability (% of Annexin V-PI- cells). C, % of apoptotic cells (% of Annexin V+ cells).
Oxidative Stress Was Partially Responsible for the Increase of DNA Damage in 500 nM As+3 Exposure at 4 h
In order to confirm that oxidative stress and superoxide production is directly related to the increase of DNA damage at 500 nM As+3, we utilized a superoxide scavenger, TEMPOL, to perform co-exposure studies with As+3 for 4 h, at which time point oxidative stress was observed (Figs. 3C and 3D). A significant decrease of superoxide levels was observed in 500 nM As+3 and 100 µM TEMPOL co-exposure comparing to 500 nM As+3 only (Figure 5A), indicating that TEMPOL inhibited the production of superoxide induced by 500 nM As+3. At the same time, DNA damage was also decreased by a small amount (∼2%) in As+3 and TEMPOL co-exposure (Figure 5B), suggesting that oxidative stress and superoxide production was directly related to the DNA damage in 500 nM As+3 exposure at 4 h, but that oxidative stress and DSBs represent only a small percentage of total DNA strand breaks. PARP activity was not affected by adding TEMPOL in As+3 exposure (Figure 5C). Collectively, these results indicate that oxidative stress contributes to DNA damage in D1 cells exposed to 500 nM As+3 at 4 h.
FIG. 5.
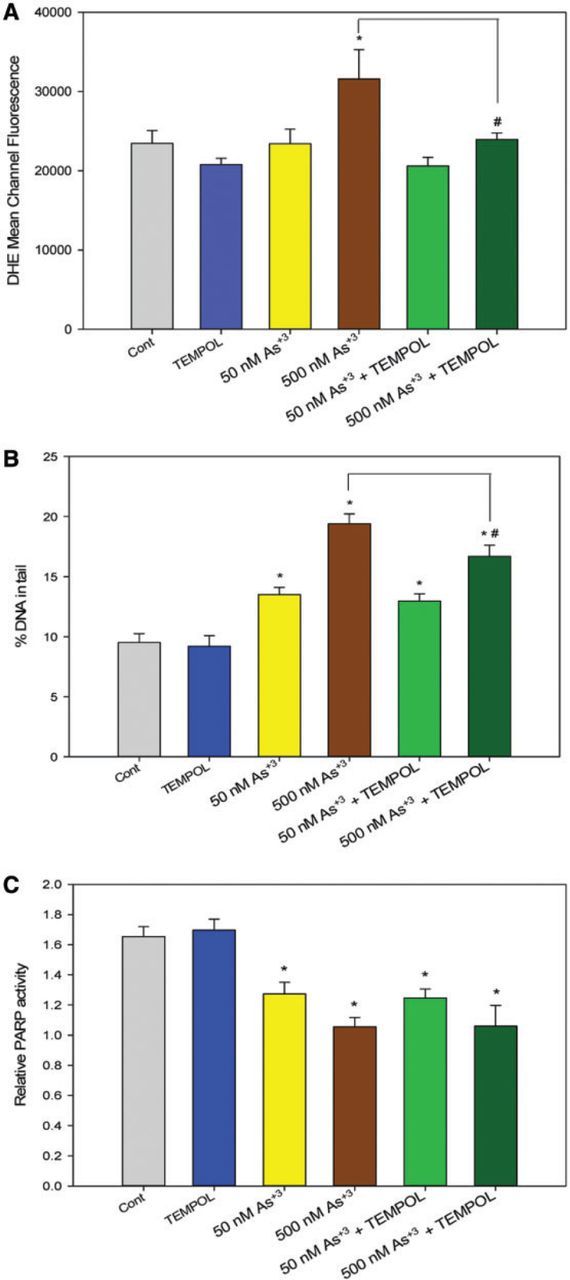
Superoxide level and genotoxicity in D1 cells exposed to As+3 and TEMPOL for 4 h. D1 cells were exposed with 50 or 500 nM As+3, with or without co-exposure to 100 µM TEMPOL. A, Mean Channel Fluorescence of DHE represents superoxide level. B, DNA damage was measured by percentage of DNA in tail using alkaline Comet assay. C, PARP activity was measured with Trevigen ELISA kits represented by absorbance at 450 nm. *Significantly different compared with control (P < .05). #Significantly different compared with 500 nM As+3 (P < .05). Results are means ± SD.
Induction of As+3 Exporters by 500 nM As+3
Because 500 nM As+3 induced DSBs and oxidative stress in D1 cells at early time points (2 and 4 h) but not at 18 h, we examined potential mechanisms responsible for this reversal. One possible mechanism is that 500 nM As+3 induces the expression of As+3 exporters such as the multidrug resistance-associated proteins 1 and 2 (Mrp1, Mrp2) or the multidrug resistance protein 1 (Mdr1) (Liu et al., 2001, 2002). Enhancement of efflux transporter expression could reduce intracellular levels of As+3 thereby reducing the toxic effects. Therefore, we examined the mRNA expression of Mrp1, Mrp2, Mdr1a, and Mdr1b in D1 cells treated with 500 nM As+3 at 2, 4, and 18 h. An increase of Mrp2 (and to some extent Mrp1) expression was observed in D1 cells treated with 500 nM As+3 at 18 h (Figure 6B). Interestingly, Mdr1a and Mdr1b were up-regulated at 2 h and down-regulated by 18 h (Figs. 6C and 6D), which was different from the expression of Mrp1 and 2 (Figs. 6A and 6B). Western blots confirmed that the protein level of Mrp1 was up-regulated at 18 h (Figs. 7A and 7B). However, Mdr1 protein levels were not decreased at 18 h, which was different from the mRNA expression (Figs. 7C and 7D). Mrp2 protein was not detectable in D1 cell lysates. Collectively, these results suggest that the induction of As+3 exporters may play a role in regulating As+3-induced genotoxicity at high concentrations (500 nM).
FIG. 6.

mRNA expression of As+3 exporters in D1 cells treated with 500 nM As+3. D1 cells were treated 500 nM As+3, expression of Mrp1, Mrp2, Mdr1a, and Mdr1b at 2, 4, and 18 h was examined by RT-PCR. A, Mrp1. B, Mrp2. C, Mdr1a. D, Mdr1b. *Significantly different compared with control (P < .05). Results are means ± SD.
FIG. 7.

Protein levels of Mrp1 and Mdr1 in D1 cells treated with 500 nM As+3. D1 cells were treated 500 nM As+3, expression of Mrp1 and Mdr1 at 2, 4, and 18 h was examined by Western blot. A, Mrp1 Western blot. B, Mrp1 band intensity normalized to Actin. C, Mdr1 Western blot. D, Mdr1 band intensity normalized to Actin. *Significantly different compared with control (P < .05). Results are means ± SD.
One Micomolar Zn+2 Reversed the DNA Damage and PARP Inhibition Induced by As+3
Since PARP is a zinc finger protein and Zn+2 was found to reverse the PARP inhibition induced by As+3 in HaCat cells (Sun et al., 2012), we wanted to know if Zn+2 would reverse the DNA damage induced by As+3 in mouse pre-T cells. D1 cells were treated with 50 or 500 nM As+3, 1 µM Zn+2 and the combinations as Zn+2 + As+3. 1 µM Zn+2 reversed the DNA damage induced by 50 and 500 nM As+3 at both 4 and 18 h (Figure 8A). In addition, PARP inhibition induced by 50 and 500 nM As+3 at both 4 and 18 h was also reversed by 1 µM Zn+2 (Figure 8B). Therefore, the genotoxic effects induced by As+3 in thymus cells can be reversed by Zn+2, suggesting that Zn may be involved in the mechanism of action of As+3.
FIG. 8.
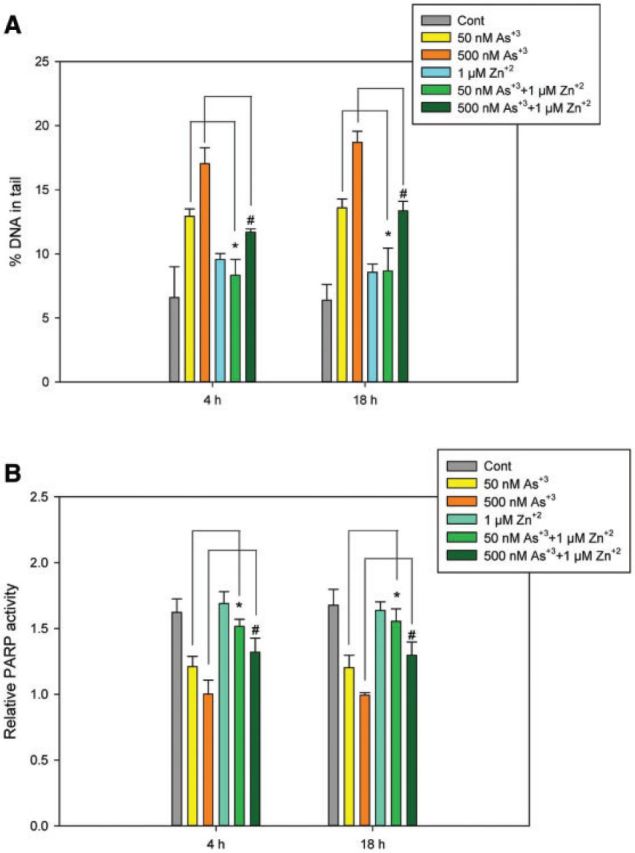
Protective effect of Zn+2 supplement in As+3-induced DNA damage and PARP activity in D1 cells. D1 cells were treated with 50 and 500 nM As+3, 10 µM Zn+2, or the combinations. A, DNA damages were measured by percentage of DNA in tail using alkaline Comet assay. B, PARP activities were measured with Trevigen ELISA kit represented by absorbance at 450 nm. *Significantly different compared with 50 nM As+3 (P < .05), #Significantly different compared with 500 nM As+3 (P < .05). Results are means ± SD.
DISCUSSION
Suppression of immune function by As has been observed in vivo and in vitro by several groups (Ahmed et al., 2014; Biswas et al., 2008; Burchiel et al., 2014; Ezeh et al., 2014; Gonsebatt et al., 1994; Nadeau et al., 2014; Soto-Peña et al., 2006; Vahter, 2008), and may potentially contribute to unregulated cancer cell growth. As+3 is considered to be a co-carcinogen in keratinocytes, as it does not induce detectable DNA damage at low concentrations without the co-existence of other carcinogens such as UV light (Qin et al., 2012). As+3 inhibits DNA repair enzymes such as PARP and XPA (Zhou et al., 2011), which may decrease the ability of cells to repair spontaneous endogenous strand breaks (Swenberg et al., 2011), leading to an increase in DNA damage. Our results showed that As+3 alone induced a significant increase in DNA damage in mouse thymus cells (Figs. 1A, 1B, and 2A) by inhibiting PARP. Also, thymus cells required less As+3 to induce DNA damage than keratinocytes, indicating there is a difference between their sensitivities to As+3. The difference may be due to the exposure relating to passive and active transporters of As+3 (Jiang et al., 2006), the ability to metabolize As+3, or differences in other intra/extracellular pathways affected by As+3. It is important to further identify the mechanisms underlying these differences in sensitivity.
PARP is the initiator of BER, which has a critical role in DNA repair. Recent studies have also revealed the relationship between PARP inhibition and immunosuppression. PARP-1 was found to regulate TGF-β receptor expression in immune cells, and inhibition of PARP-1 up-regulated Foxp3, a primary marker for Tregs, and an increase in Th17 cell number (Zhang et al., 2013). In our experiment, there was a significant decrease in PARP activity and increased DNA damage in both primary thymus cells and the D1 cell line treated with very low concentrations of As+3 (50 nM). PARP inhibition induced by As+3 may not only affect T cell development in the thymus, but may also cause immunosuppression in peripheral T cells.
PARP is also known to affect other DNA repair pathways, including non-homologous end-joining and homologous recombination (Yelamos et al., 2011). We found that As+3 at 500 nM induced DSBs at 4 h, which may be the result of ROS production (Qin et al., 2008). However, these DSBs decreased at 18 h, which correlated with the decrease in oxidative stress markers. Therefore, it is likely that the DBSs were induced by oxidative stress. In addition, since neither oxidative stress nor DSBs were induced by 50 nM As+3, we conclude that As+3 induces DNA damage mainly through the inhibition of PARP at these lower concentrations leading to the increase of spontaneous strand breaks in thymus cells, while oxidative stress only occurs in cells treated with a higher dose (500 nM) of As+3.
Because of the transient nature of oxidative stress and DSB induced by 500 nM treatment with As+3 in D1 cells, we examined the expression of different As+3 efflux proteins, hypothesizing that the induction of As exporters would limit exposure and protect cells. Mrp1 and Mrp2 are well-characterized exporters of As+3 and key players in As+3 detoxification (Maciaszczyk-Dziubinska et al., 2012). Induction of Mrp1 and Mrp2 mRNA expression only occurred at the 500 nM exposure concentration of As+3 and correlated with the rapid protection of D1 cells from oxidative stress and DSBs (Figs. 4A and 4B). While the mRNA expression of Mdr1a and Mdr1b were differentially induced, with Mrp1 increased at 18 h and Mdr1a increased at 2 h, but returning to control levels at 18 h (Figs. 4C and 4D). Western blots also revealed that Mrp1 was up-regulated at 18 h by 500 nM As+3 (Figs. 5A and 5B). One major difference between these exporters is that Mdr1 exporters do not transport glutathione (GSH) conjugates (Maciaszczyk-Dziubinska et al., 2012), while Mrp1 and Mrp2 are GSH-dependent (Kala et al., 2000; Leslie et al., 2004). Therefore, it is possible that the efflux of As+3 at 18 h requires the conjugation with GSH. Besides the adaptive regulation of exporters, further studies are needed to reveal if there are changes in the expression of As+3 importers. Studies on the relationship between As+3 transporter expressions and intracellular As+3 level may further clarify the role of these proteins in regulating As+3 toxicity by cells.
Zn+2 has been demonstrated to counteract PARP inhibition following As+3 exposure (Cooper et al., 2013; Kumar et al., 2010). In our experiment, we found that Zn+2 reversed the DNA damage and PARP inhibition at 1 µM in D1 cells. However, when we increased Zn+2 concentration to 10 µM, Zn+2 itself induced DNA damage (data not shown). Therefore, Zn+2 exposure can reverse the As+3 genotoxic effects at appropriate concentrations, while excess amounts of Zn+2 are harmful to cells.
In summary, As+3 induces significant genotoxic effects in mouse thymus cells at environmentally relevant concentrations and within the nanomolar range in vitro. At low concentrations (50 nM), inhibition of DNA repair caused by PARP inhibition is mainly responsible for SSB genotoxicity in the absence of oxidative stress. At higher concentrations (500 nM), As+3 induces transient oxidative stress and DSBs that are limited by the induction of exporters that reduce concentrations of intracellular As. The dose-dependent differential genotoxicity of As+3 at environmentally relevant levels can be reversed by Zn+2 supplement.
FUNDING
National Institute of Environmental Health Sciences at the National Institutes of Health [R01 ES019968, P30 ES005022].
REFERENCES
- Ahmed S., Moore S. E., Kippler M., Gardner R., Hawlader M. D., Wagatsuma Y., Raqib R., Vahter M. (2014). Arsenic exposure and cell-mediated immunity in pre-school children in rural Bangladesh. Toxicol. Sci. 141, 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argos M., Kalra T., Rathouz P. J., Chen Y., Pierce B., Parvez F., Islam T., Ahmed A., Rakibuz-Zaman M., Hasan R., et al. (2010). Arsenic exposure from drinking water, and all-cause and chronic-disease mortalities in Bangladesh (HEALS): a prospective cohort study. Lancet 376, 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M., Goldstein M., Christmann M., Becker H., Heylmann D., Kaina B. (2011). Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl Acad. Sci. U. S. A. 108, 21105–21110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas R., Ghosh P., Banerjee N., Das J. K., Sau T., Banerjee A., Roy S., Ganguly S., Chatterjee M., Mukherjee A., et al. (2008). Analysis of T-cell proliferation and cytokine secretion in the individuals exposed to arsenic. Hum. Exp. Toxicol. 27, 381–386. [DOI] [PubMed] [Google Scholar]
- Burchiel S. W., Lauer F. T., Beswick E. J., Gandolfi A. J., Parvez F., Liu K. J., Hudson L. G. (2014). Differential susceptibility of human peripheral blood T cells to suppression by environmental levels of sodium arsenite and monomethylarsonous acid. PLoS One 9, e109192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza M. J., Stefanick D. F., Horton J. K., Kedar P. S., Wilson S. H. (2009). PARP inhibition during alkylation-induced genotoxic stress signals a cell cycle checkpoint response mediated by ATM. DNA Repair 8, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins A. R. (2004). The comet assay for DNA damage and repair: principles, applications, and limitations. Mol. Biotechnol. 26, 249–261. [DOI] [PubMed] [Google Scholar]
- Cooper K. L., Liu K. J., Hudson L. G. (2009). Enhanced ROS production and redox signaling with combined arsenite and UVA exposures: contributions of NADPH oxidase. Free Radic. Biol. Med. 47, 381–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper K. L., King B. S., Sandoval M. M., Liu K. J., Hudson L. G. (2013). Reduction of arsenite-enhanced ultraviolet radiation-induced DNA damage by supplemental zinc. Toxicol. Appl. Pharmacol. 269, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangleben N. L., Skibola C. F., Smith M. T. (2013). Arsenic immunotoxicity: a review. Environ. Health 12, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeh P. C., Lauer F. T., MacKenzie D., McClain S., Liu K. J., Hudson L. G., Gandolfi A. J., Burchiel S. W. (2014). Arsenite selectively inhibits mouse bone marrow lymphoid progenitor cell development in vivo and in vitro and suppresses humoral immunity in vivo. PLoS One 9, e93920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faita F., Cori L., Bianchi F., Andreassi M. G. (2013). Arsenic-induced genotoxicity and genetic susceptibility to arsenic-related pathologies. Int. J. Environ. Res. Public Health 10, 1527–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonsebatt M. E., Vega L., Montero R., Garcia-Vargas G., Del Razo L. M., Albores A., Cebrian M. E., Ostrosky-Wegman P. (1994). Lymphocyte replicating ability in individuals exposed to arsenic via drinking water. Mutat. Res. 313, 293–299. [DOI] [PubMed] [Google Scholar]
- Jiang Y. J., Lu B., Kim P., Elias P. M., Feingold K. R. (2006). Regulation of ABCA1 expression in human keratinocytes and murine epidermis. J. Lipid Res. 47, 2248–2258. [DOI] [PubMed] [Google Scholar]
- Kaina B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem. Pharmacol. 66, 1547–1554. [DOI] [PubMed] [Google Scholar]
- Kala S. V., Neely M. W., Kala G., Prater C. I., Atwood D. W., Rice J. S., Lieberman M. W. (2000). The MRP2/cMOAT transporter and arsenic-glutathione complex formation are required for biliary excretion of arsenic. J. Biol. Chem. 275, 33404–33408. [DOI] [PubMed] [Google Scholar]
- Kim K., Khaled A. R., Reynolds D., Young H. A., Lee C. K., Durum S. K. (2003). Characterization of an interleukin-7-dependent thymic cell line derived from a p53−/− mouse. J. Immunol. Methods 274, 177–184. [DOI] [PubMed] [Google Scholar]
- Kumar A., Malhotra A., Nair P., Garg M., Dhawan D. K. (2010). Protective role of zinc in ameliorating arsenic-induced oxidative stress and histological changes in rat liver. J. Environ. Pathol. Toxicol. Oncol. 29, 91–100. [DOI] [PubMed] [Google Scholar]
- Leslie E. M., Haimeur A., Waalkes M. P. (2004). Arsenic transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J. Biol. Chem. 279, 32700–32708. [DOI] [PubMed] [Google Scholar]
- Li D., Morimoto K., Takeshita T., Lu Y. (2001). Arsenic induces DNA damage via reactive oxygen species in human cells. Environ. Health Prev. Med. 6, 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q., Lauer F. T., Liu K. J., Hudson L. G., Burchiel S. W. (2010). Low-dose synergistic immunosuppression of T-dependent antibody responses by polycyclic aromatic hydrocarbons and arsenic in C57BL/6J murine spleen cells. Toxicol. Appl. Pharmacol. 245, 344–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M. R. (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 79, 181–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Chen H., Miller D. S., Saavedra J. E., Keefer L. K., Johnson D. R., Klaassen C. D., Waalkes M. P. (2001). Overexpression of glutathione S-transferase II and multidrug resistance transport proteins is associated with acquired tolerance to inorganic arsenic. Mol. Pharmacol. 60, 302–309. [DOI] [PubMed] [Google Scholar]
- Liu J., Liu Y., Powell D. A., Waalkes M. P., Klaassen C. D. (2002). Multidrug-resistance mdr1a/1b double knockout mice are more sensitive than wild type mice to acute arsenic toxicity, with higher arsenic accumulation in tissues. Toxicology 170, 55–62. [DOI] [PubMed] [Google Scholar]
- Los M., Mozoluk M., Ferrari D., Stepczynska A., Stroh C., Renz A., Herceg Z., Wang Z. Q., Schulze-Osthoff K. (2002). Activation and caspase-mediated inhibition of PARP: a molecular switch between fibroblast necrosis and apoptosis in death receptor signaling. Mol. Biol. Cell. 13, 978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciaszczyk-Dziubinska E., Wawrzycka D., Wysocki R. (2012). Arsenic and antimony transporters in eukaryotes. Int. J. Mol. Sci. 13, 3527–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah L. J., El-Osta A., Karagiannis T. C. (2010). GammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686. [DOI] [PubMed] [Google Scholar]
- Nadeau K. C., Li Z., Farzan S., Koestler D., Robbins D., Fei D. L., Malipatlolla M., Maecker H., Enelow R., Korrick S., et al. (2014). In utero arsenic exposure and fetal immune repertoire in a US pregnancy cohort. Clin. Immunol. 155, 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X. J., Hudson L. G., Liu W., Ding W., Cooper K. L., Liu K. J. (2008). Dual actions involved in arsenite-induced oxidative DNA damage. Chem. Res. Toxicol. 21, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X. J., Liu W., Li Y. N., Sun X., Hai C. X., Hudson L. G., Liu K. J. (2012). Poly(ADP-ribose) polymerase-1 inhibition by arsenite promotes the survival of cells with unrepaired DNA lesions induced by UV exposure. Toxicol. Sci. 127, 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher-Wolz U., Dieter H. H., Klein D., Schneider K. (2009). Oral exposure to inorganic arsenic: evaluation of its carcinogenic and non-carcinogenic effects. Crit. Rev. Toxicol. 39, 271–298. [DOI] [PubMed] [Google Scholar]
- Shi H., Hudson L. G., Liu K. J. (2004). Oxidative stress and apoptosis in metal ion-induced carcinogenesis. Free Radic. Biol. Med. 37, 582–593. [DOI] [PubMed] [Google Scholar]
- Soto-Peña G. A., Luna A. L., Acosta-Saavedra L., Conde P., López-Carrillo L., Cebrián M. E., Bastida M., Calderón-Aranda E. S., Vega L. (2006). Assessment of lymphocyte subpopulations and cytokine secretion in children exposed to arsenic. FASEB J. 20, 779–781. [DOI] [PubMed] [Google Scholar]
- Sun X., Zhou X., Du L., Liu W., Liu Y., Hudson L. G., Liu K. J. (2012). Arsenite binding-induced zinc loss from PARP-1 is equivalent to zinc deficiency in reducing PARP-1 activity, leading to inhibition of DNA repair. Toxicol. Appl. Pharmacol. 274, 313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenberg J. A., Lu K., Moeller B. C., Gao L., Upton P. B., Nakamura J., Starr T. B. (2011). Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol. Sci. 120(Suppl. 1), S130–S145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahter M. (2008). Health effects of early life exposure to arsenic. Basic Clin. Pharmacol. Toxicol. 102, 204–211. [DOI] [PubMed] [Google Scholar]
- Vile G. F., Basu-Modak S., Waltner C., Tyrrell R. M. (1994). Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl Acad. Sci. U. S. A. 91, 2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiseman H., Halliwell B. (1996). Damage to DNA by reactive oxygen and nitrogen species: role in inflammatory disease and progression to cancer. Biochem. J. 313, 17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamos J., Farres J., Llacuna L., Ampurdanes C., Martin-Caballero J. (2011). PARP-1 and PARP-2: New players in tumour development. Am. J. Cancer Res. 1, 328–346. [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Nakatsukasa H., Tu E., Kasagi S., Cui K., Ishikawa M., Konkel J. E., Maruyama T., Wei G., Abbatiello B., et al. (2013). PARP-1 regulates expression of TGF-β receptors in T cells. Blood. 122, 2224–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Wang X., Cheng S., Sun L., Son Y. O., Yao H., Li W., Budhraja A., Li L., Shelton B. J., et al. (2011). Reactive oxygen species mediate arsenic induced cell transformation and tumorigenesis through Wnt/β-catenin pathway in human colorectal adenocarcinoma DLD1 cells. Toxicol. Appl. Pharmacol. 256, 114–121. [DOI] [PubMed] [Google Scholar]
- Zharkov D. O. (2008). Base excision DNA repair. Cell. Mol. Life Sci. 65, 1544–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Sun X., Cooper K. L., Wang F., Liu K. J., Hudson L. G. (2011). Arsenite interacts selectively with zinc finger proteins containing C3H1 or C4 motifs. J. Biol. Chem. 286, 22855–22863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Sun X., Mobarak C., Gandolfi A. J., Burchiel S. W., Hudson L. G., Liu K. J. (2014). Differential binding of monomethylarsonous acid compared to arsenite and arsenic trioxide with zinc finger peptides and proteins. Chem. Res. Toxicol. 27, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]