

Abstract
Clinical examination and electrodiagnostic studies remain the gold standard for diagnosis of nerve injuries. Diagnosis of chronic nerve compression (CNC) injuries may be difficult in patients with confounding factors such as diabetes. The treatment of nerve entrapment ranges from medical to surgical management depending on the nerve involved and on the severity and duration of compression. Considerable insights have been made at the molecular level differentiating between nerve crush injuries and CNC injuries. While the myelin changes after CNC injury were previously thought to be a mild form of Wallerian degeneration, recent evidence points to a distinct pathophysiology involving Schwann cell mechano-sensitivity. Future areas of research include the use of Schwann cell transplantation in the treatment regimen, the correlation between demyelination and the onset of pain, and the role of Schwann cell integrins in transducing the mechanical forces involved in nerve compression injuries to Schwann cells.
Introduction
Compressive neuropathies are common conditions that can degrade the patient’s quality of life and limit their ability to work. The symptoms may range from sensory abnormalities, paresthesias, and pain in the initial stages to motor disruption and permanent sensory loss as the injury proceeds. Chronic nerve compression (CNC) injuries are very frequent, particularly in the upper extremity. In the United States, the prevalence of carpal tunnel syndrome is estimated to be as high as 3.72 percent.1 Although certain repetitive movements and diseases have been thought to be associated with a variety of entrapment neuropathies, progress has been made into understanding the biologic processes that accompany CNC injuries. There appears to be distinct difference underlying the pathophysiology of CNC injuries compared to acute nerve injuries. While alternative diagnostic techniques such as radiologic imaging have been developed to diagnose nerve compression injuries,2,3 electrodiagnostic studies remain the most widely accepted tool for providing objective data for the diagnosis of CNC injuries.4,5 The following review will provide information regarding the current understanding of the pathogenesis, diagnosis, and treatment of compressive neuropathies.
Classification of Nerve Injuries
Compression neuropathies are commonly divided into two broad categories depending on the duration of injury. Compression of nerves may occur acutely, as with traumatic crush injuries, or may occur gradually over time as a result of increased pressures at sites of anatomic narrowing through which nerves pass. The etiology of acute nerve compression injuries is often straightforward as they are the result of a single traumatic episode. In contrast, CNC injuries are acquired neurodegenerative conditions that occur over time and have a very complex etiology involving ischemia, fibrosis, edema, and other factors.
The Seddon classification6 has been used to categorize traumatic nerve injuries as either a neurapraxia, axonotmesis, or neurotmesis depending upon which components of the nerve have been damaged. With a neurapraxia, the connective tissue of the nerve and the axons remain intact, but there is focal demyelination. With an axonotmesis, the axons are damaged, but the surrounding connective tissue remains intact. Finally, a neurotmetric injury is characterized by a nerve that has been transected, so that no components of the nerve are in continuity. The myelin remains intact in the distal segment pending the onset of Wallerian degeneration with the ensuing distal axonal degeneration. Physical examination and electrodiagnosistic studies may be misleading in differentiating between these 3 injury types, particularly in the early recovery period.
Sunderland built on the Seddon classification by classifying nerve injuries into five types based on the extent of tissue injury.7 Types 1 and 2 are equivalent to Seddon’s neuropraxia and axontmesis, respectively. Type 3, or third degree injuries result in axonal loss as well as disruption of the endoneural tubes. Further injury leads to disruption first of the perineurim (type 4 injury) and subsequently the epineurium (Type 5 injury). The Sunderland classification expands the spectrum between an axontmesis and a neurotmesis such that a Seddon type 3 is equivalent to a Sunderland type 5. Differentating between a Sunderland type 2, 3, and 4 is difficult clinically. Because of the progressive architectural disruption, however, higher order injuries may be more likely to result in a neuroma in continuity rather than spontaneous nerve regrowth and recovery as is expected of true axontmetric injuries.
Nerve injuries from CNC can separately be classified based on the extent and severity of injury. Nerve specific classification systems such as the McGowan classification have been developed to guide treatment and aid in research stratification.8,9 A Grade 1 lesion results from mild, transient compression of the ulnar nerve that induces paresthesias but does not result in weakness of the muscles innervated by the ulnar nerve. If muscle weakness is present without wasting of the interosseous muscles, the lesion is classified as a Grade 2. A Grade 3 lesion is characterized by marked weakness and wasting of the interossei, with paralysis of one or more of the ulnar innervated intrinsic muscles. Similar classification systems were later developed by others; the Dellon classification system for ulnar neuropathy also uses provocative tests and a sensory examination to help grade ulnar neuropathies, and includes two-point discrimination tests and response to vibratory stimuli.10
Pathophysiology of CNC Injury
Currently, several mechanisms have been proposed to explain the development of CNC injuries. These different processes are not exclusive, and often work together to yield the pain and paresthesias typical of CNC injuries. Increased pressure can compress blood vessels that supply the nerve and lead to epineural ischemia. At lower pressures, the reduction in venous return can lead to venous stasis, which in turn can lead to extraneural edema. If maintained over time, this process results in fibrosis and scar tissue formation around the nerve and ensuing intraneural edema.11
Although these theories seek to explain what might trigger CNC injury at a gross level, an understanding of the cellular changes involved in CNC injuries has proved less forthcoming due to the obvious inability to histologically examine chronically compressed human nerves. Until recently, most theories about the cellular mechanisms underlying CNC injury were derived by examinations of crush nerve injuries. In fact, the changes seen in CNC injury were thought to be a mild form of Wallerian degeneration. However, the recent development of various in-vivo and in-vitro models to study CNC injuries has produced several compelling insights into the cellular mechanisms underlying CNC injury.
Pathophysiology of acute nerve injuries is dependent on axonal damage
Several studies have explored the pathophysiology of acute nerve injuries such as transections and crush injuries. These injuries often occur following trauma and are characterized by Wallerian degeneration, which is a process that involves injury to both the axon and to the myelin surrounding the axon. In the initial stages of Wallerian degeneration, axonal damage is typified by loss of cell membrane integrity as well as breakdown of the axonal cytoskeleton. The axonal damage has been shown to stimulate the infiltration of macrophages from the bloodstream into the damaged nerve within one to four days following injury.12 The macrophages function to phagocytose any axonal debris. As discussed in detail in subsequent sections, given the axonal damage, compound muscle action potentials (CMAPs) are unable to be elicited during electrodiagnostic studies.
Structural changes in acute crush injuries are not limited to the axon. The neuromuscular junction (NMJ) exhibits alterations that range from presynaptic terminal degeneration to increases in the number of postsynaptic receptors, as well as the insertion of receptors in locations outside the NMJ.13,14 The muscles also begin to exhibit the classic signs of denervation atrophy, which include small and angularly-shaped muscle fibers, variations in muscle fiber diameter, and muscle fiber atrophy.15
Consequent to Wallerian degeneration of the axon, the Schwann cells that previously myelinated the receding axon exhibit altered expression of various proteins involved in myelination and proliferation. Chief among these proteins are krox-20 and c-jun. Krox-20 has previously been shown to promote myelination,16 while c-jun has been shown to promote Schwann cell de-differentiation.17 In injured nerves, krox-20 is downregulated while c-jun is upregulated, leading to Schwann cell de-differentiation and consequent demyelination. This de-differentiation ironically serves a regenerative purpose; the de-differentiated Schwann cells form tubes that guide the path of subsequent axonal regeneration and remyelination.
Pathophysiology of chronic nerve compression injuries is independent of axonal damage
Until recently, CNC injuries were also characterized as a mild form of Wallerian degeneration. However, in stark contrast to acute crush injuries, CNC injuries do not involve damage to the axon itself until far later in the disease process, when motor weakness begins to appear clinically. Several pieces of data support this concept. Light and electron microscopy images of nerves subjected to CNC failed to show any changes to the form or shape of the axons in CNC-injured nerves, which contrasts with the axonal degradation seen in crush injuries.18,19 Furthermore, the NMJ exhibits none of the morphological changes seen in acute crush injuries.20 Moreover, studies of the soleus muscle following sciatic nerve compression reveal intact muscle fiber integrity in CNC injuries, which argues against any sort of denervation atrophy. Electrodiagnostic findings further support a lack of axonal injury in CNC injuries. Although nerve conduction velocity is decreased in CNC injury, CMAPs of normal amplitude can still be elicited, in direct contrast to acute nerve compression injuries where CMAP’s are unable to be elicited. This data infers preservation of axonal integrity in the early phases of CNC injuries.
The decreased nerve conduction velocity seen with CNC injuries suggests demyelination followed by remyelination. Histological analysis of compressed nerves lends credence to such a scenario. The pioneering efforts of Drs. Susan Mackinnon and A. Lee Dellon provided some of the earliest evidence that compressed human nerve segments show markedly thinner myelin following injury.21 In animal models, the appearance of such new, thinner myelin has since been definitively linked to remyelination by Schwann cells that follows demyelination. The appearance of thin myelin is further corroborated by recent studies that show an increase in g-ratios (an objective measure of myelination defined as the ratio of axon diameter to total fiber diameter) increase from 0.6 to 0.8 in CNC injured nerve fibers. Closer examination of these compressed animal nerve fibers reveals demyelination immediately adjacent to the Node of Ranvier. This demyelination proceeds towards the internode and, as it does so, oval structures typical of myelin degeneration are seen in areas that are about to undergo demyelination. Some areas will also show proliferating Schwann cells. In these areas, thinner myelin typical of remyelinating axons and decreased internodal length are also seen, along with an increase in Schmidt-Lantermann incisures (SLIs). SLIs are theorized to be responsible for helping maintain metabolic activity of the myelin sheath; thus an increase in Schmidt-Lantermann incisures would likely be instrumental in meeting the increased metabolic demands required by the demyelination and remyelination that occurs in CNC injuries. Taken together, these findings strongly suggest a wave of demyelination followed by remyelination following CNC injury.22 This demyelination, however, cannot be triggered by axonal damage, since axonal damage is not seen until far later time points of CNC injuries.
Schwann cell proliferation in CNC injuries is macrophage-independent, and induced by mechanical forces
Given the lack of axonal damage in CNC injury, another mechanism must account for the observed Schwann cell changes and the ensuing decreased nerve conduction velocities. Initially, it was postulated that perhaps macrophages were responsible for triggering the Schwann cell proliferation that leads to thinner myelin and decreased nerve conduction velocities. In acute crush injuries, macrophages invade compressed nerves from 1-4 days post-injury in an effort to clean up axonal debris. In CNC injuries, however, the macrophage infiltration is far more gradual, taking place over several weeks. This slower rate of macrophage infiltration makes sense given the lack of axonal damage seen in CNC injuries. Given that the Schwann cell proliferation involved in remyelination also takes place after several weeks, it was postulated that macrophages might produce Schwann cell mitogens that induce Schwann cell proliferation and remyelination in CNC injured nerves.
Schwann cells have been shown to proliferate at 2 weeks following CNC injury, with peak proliferation taking place at 4 weeks. The Schwann cell proliferation is accompanied, paradoxically, by an increase in Schwann cell apoptosis. Apoptosis seen later in CNC injuries can theoretically be explained by overgrowth of connective tissue that can cause ischemia and induce axonal death; this late stage of CNC injury is often accompanied by motor loss. Early stages of proliferation and apoptosis of Schwann cells, however, cannot be explained by this mechanism since hypertrophy has not yet occurred. For this reason, and because macrophages are known to secrete transcription factors that induce Schwann cell mitosis (interleukins 1, 6, 10, and 12; transforming growth factor beta; and tumor necrosis factor alpha), attention was turned to macrophages as a possible stimulator of Schwann cell proliferation in CNC injuries. Recent studies have shown that Schwann cells are still induced to proliferate following CNC injury even when macrophages are depleted using clondronate liposomes, thus ruling out the possibility that Schwann cell proliferation is induced by macrophage-derived mitogenic factors.23
Most recently, mechanical forces such as shear stress has been proposed as the causative factor inducing Schwann cell proliferation following CNC injury. In vitro cultures of Schwann cells subjected to laminar fluid flows exhibit significantly increased proliferation following just two hours of mechanical stimulation.24 Proliferation can be verified via down regulation of mRNA and increased uptake of radiolabeled bromodeoxyuridine.24 These types of studies provide the clearest evidence yet of what causes Schwann cells to proliferate following CNC injury for two reasons. First, the laminar fluid flows applied to the pure Schwann cell cultures approximate defined mechanical loading of nerves during CNC injuries. Furthermore, the lack of neurons in these cultures makes it more likely that Schwann cells are able to respond to mechanical stress directly, independent of any of the axonal input that appears to drive Schwann cell dedifferentiation in acute crush injuries.24
Recently, an even more sophisticated in vitro co-culture model has been developed that allow Schwann cell-DRG co-cultures to be subjected to mechanical stresses; this new in vitro chamber helps to eliminate cytotoxicity as a confounding variable because it allows the researcher to control for oxygen tension, pH and pressure.25 As such, co-cultures grown in the chamber can be subjected to purely hydrostatic (e.g. mechanical) stresses. As with previous models, co-cultures grown in this chamber exhibited robust proliferation of Schwann cells following just 24 hours of compression, even when subjected to pressures far lower than those typically assumed to exist in entrapment sites during pathological states.25 After just three days of compression at these lower physiologic pressures, however, the cell membrane exhibited marked disintegration.25 In addition, when the co-cultures were subjected to pressures typical of those found in entrapment neuropathies, the cells exhibited a markedly increase in the release of stress markers such as lactate dehydrogenase (LDH).25 Still, the pressures required to obtain LDH release from the co-cultures were significantly less than the pressures needed to elicit LDH release from other, non-neural types of cells.25 Taken together, these data provide a strong indication that myelinated neurons may be exquisitely sensitive to mechanical stress as compared to other types of cells.25 However, since lower physiologic pressures did not result in an increase in cell death, it is also possible that lower ambient physiologic pressures on nerve serve a neuroprotective function.25
If mechanical loading of Schwann cells induces proliferation, it still remains unanswered precisely how this stimuli is transmitted to the Schwann cells to induce the ensuing signaling pathways. Recent studies have pointed to integrin alpha6beta4 (α6β4) as the possible protein involved in translating mechanical stress to Schwann cells.26 Integrins are cell membrane proteins that have long been known to facilitate cellular adhesion to extracellular matrix components; they have previously been demonstrated to act as cell-signaling molecules and transducers of mechanical forces in other contexts.27 In animal models, beta4 (β4) integrin relocalizes to Schmidt-Lanterman incisures (SLIs) as early as 1 week following CNC injury.26 (Fig. 1) Because SLIs contain several cell-signaling molecules, relocation of β4 integrin to the incisures following CNC injury may facilitate cellular interaction with the extracellular matrix, in an effort to satisfy the increased nutritive demands of cells during the processes of demyelination and remyelination.26 In addition, β4 integrin exhibits decreased expression at 2 and 4 weeks following chronic nerve compression, at the same time that changes in the expression levels of that krox-20, a positive regulator of myelination, and c-jun, a negative regulator of myelination, are observed.26 As β4 integrin expression is decreased, c-jun expression increases and krox-20 expression decreases.26 This temporal correlation lends credence to the idea that integrins may be involved in transmitting the mechanical stress of chronic nerve compression to Schwann cells, and thus may be crucial players in the process of demyelination following CNC injury. Such a hypothesis is supported by the fact that integrin proteins are known to connect the extracellular matrix with intracellular signaling pathways, and integrin-linked kinases have recently been shown to be crucial to remyelination in crush injuries.27,28
FIGURE 1.
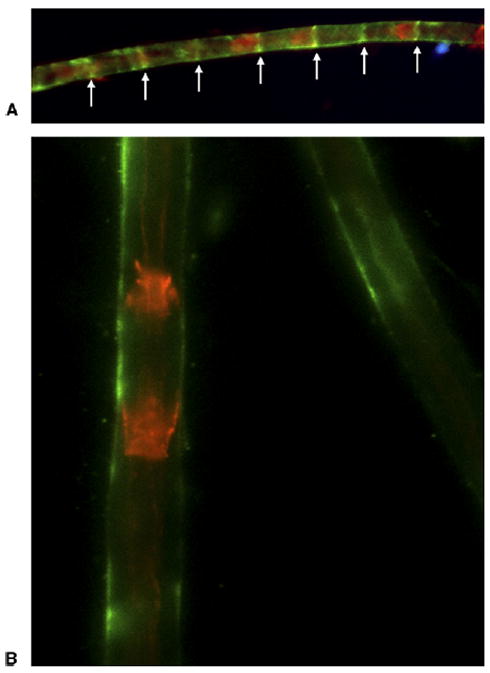
Integrin β4 relocalization following CNC injury. CNC-injured nerves show localization of integrin β4 (arrows) to SLIs as early as one week following injury. A Immunostaining of CNC-injured rat nerve at one week postcompression. B Normal nerves at the same timepoint. Red staining (phalloidin-TRITC) reveals F-actin localization at SLI’s, and green staining indicates integrin β4. Reprinted with permission from Pham K, Nassiri N, Gupta R. c-Jun, krox-20, and integrin β4 expression following chronic nerve compression injury. Neurosci Lett 2009;465:194–198. Copyright 2009, with permission from Elsevier.
Electrodiagnosis of CNC injuries
Patients with compressive neuropathies often present with varying symptoms, depending in part on the location, extent, and duration of compression. Given the wide variety of symptoms, there have been many attempts at creating a standard diagnostic approach. For example, consensus groups have tried to establish a definitive constellation of symptoms associated with carpal tunnel syndrome (e.g. pain, burning, numbness, and tingling; particularly at night).29 Others have long suggested that so-called “provocative” tests such as Phalen’s test, Tinel’s test, 2-point discrimination tests, and carpal compression tests may be helpful in diagnosing carpal tunnel syndrome and other compression neuropathies.30 Some authors have gone as far as to suggest that compressive neuropathies cannot definitively be diagnosed based on clinical criteria alone, and that the positive predictive value of provocative tests is minimal at best.29,31,32,33 The ASSH has recently withheld recommending widespread use of electrodiagnostic studies for the routine diagnosis of CTS. However, when the clinical picture is atypical or clouded by comorbid conditions, clinical criteria and provocative tests can be combined with electrodiagnostic studies to assist with diagnosis.34,35
There are two types of electrodiagnostic studies commonly utilized in the diagnosis of compression neuropathies: nerve conduction studies and electromyography. Of the two, nerve conduction studies, which are also referred to as neurodiagnostic studies, are particularly useful in diagnosing compressive neuropathies. Neurodiagnosis involves placing two electrodes on the skin, along the length of a nerve. The first electrode is used to stimulate a peripheral nerve above the threshold values required for it to fire, while the second electrode is used to record the characteristics of the generated action potential at some distance away from the point of stimulation. The recording electrodes used in neurodiagnostic studies typically detect only larger, faster-conducting fibers as opposed to smaller, slower conducting fibers; as such, if even just a few thick, fast fibers remain, nerve conduction study measurements will remain normal. However, since thick fibers are more vulnerable to the compression and ischemia seen in entrapment neuropathies, neurodiagnostic studies are often quite helpful in diagnosing compression neuropathies. Noninvasive nerve conduction testing has recently been introduced into the marketplace with portable electrodiagnostic devices such as NC-Stat (NeuroMetrix, Inc. Waltham, MA); these devices employ biosensors that detect temperature fluctations on the skin surface that occur when nerves are stimulated. These devices are increasingly being used in orthopedic and neurologic ambulatory settings, with some limited evidence of their clinical utility in screening for compression neuropathies.36
Neurodiagnostic studies yield a number of clinically useful measurements. For purely sensory nerves, recording electrodes are commonly placed proximally along the nerve towards the spinal cord yielding a sensory nerve action potential (SNAP). For mixed motor and sensory nerves, the recording electrodes are commonly placed distally along the nerve, towards the target muscle, yielding a compound nerve action potential (CNAP). However, since stimulation of a nerve at a particular site produces impulses in both expected the physiologic (or orthodromic) and non-physiologic (or antidromic) directions, SNAPs may also be obtained by placing recording electrodes distally, while CNAPs can also be obtained by placing recording electrodes proximally. Alternatively, motor nerve recordings may be obtained at the target muscle itself, yielding a compound muscle action potential (CMAP), which reflects the summed activity of all the muscle fibers innervated by a single nerve (e.g. a motor unit). In this manner, nerve conduction studies elicit information about the amplitude (the size of a response, which is roughly proportional to the number of depolarizing axons), latency (delay in response following stimulation), and conduction velocity (latency divided by distance).37,38,39
Another parameter that may be elicited by neurodiagnostic studies is the F-wave. F-waves tend to follow CMAPs in time; they are waves produced by antidromic conduction of the artificially stimulated action potential back to the anterior horn cells, which then discharge and send a volley of impulses back down the nerve toward the muscle. Because only a small percentage of anterior horn cells will discharge in this manner, F-wave amplitudes are typically very small. Despite their seeming usefulness in diagnosing entrapment neuropathies, F-waves are typically not used for this purpose. The focal demyelination seen in most entrapment neuropathies occurs at a far distance from the anterior horn cell; as such it is unlikely to see F-waves exhibiting significantly decreased latencies. Moreover, since only a few anterior horn cells are typically activated by the antidromic conduction of the action potential, if there are even a few axons remaining that have not undergone focal demyelination as a result of the entrapment, these few axons will give rise to normal F-wave latencies.40 As such, F-wave studies are typically used less in the diagnosis of entrapment neuropathies and more in the diagnosis of proximal neuropathies such as plexopathies and radiculopathies,41 as well as multifocal neuropathies.
Electromyography studies (EMG), in contrast to nerve conduction studies, help to analyze the integrity of muscle function by analyzing the electrical activity in the muscles during contraction or when the muscle membranes are manipulated. Because muscle integrity is heavily dependent on innervation, EMG studies are particularly important to determine whether axonal damage has occurred. EMG studies typically involve insertion of an electrode in a muscle, at which observation of slight activity is expected as the muscle membranes are induced to depolarize by the electrode. Then, the needle is moved from location to location within a muscle to check for fibrillation potentials, which signify twitching of individual muscle fibers that are abnormal and often occur in the absence of any innervation. Next, the needle is kept in a single place to check for fasciculations, which are random action potentials generated by a motor unit. Fasciculations can occur when there is normal neuronal input to a muscle, but as with fibrillation potentials they more commonly occur in cases of traumatic denervation. Finally, the integrity of the neuromuscular junction and the nerve is checked by recording motor unit potentials (MUPs), whose amplitude is proportional to the number of muscle fibers in a given motor unit that are activated as the muscle is voluntarily contracted.
Electrodiagnostic findings in traumatic injuries
As discussed earlier, traumatic crush and transection injuries often incite Wallerian degeneration, characterized by distal axonal disintegration in conjunction with myelin fragmentation. Because of the axonal loss distal to the injury, traumatic crush or transection injuries will typically exhibit a markedly diminished amplitude or altogether absence of CMAP or CNAP as soon as Wallerian degeneration occurs at the nerve segment to be tested. SNAPs or CNAPs are also typically diminished in amplitude distal to the injury site in humans 7-10 days following injury due to Wallerian degeneration.42
Moreover, with considerable amounts of damage to the axon following traumatic crush injuries, the axon is no longer able to provide input to its target muscle, which can result in denervation atrophy of the muscle at later stages.43 This is best seen with electromyography, which will show fibrillations and positive sharp waves by days 10-14 post-injury in humans.42 Within one year of the onset of denervation of a muscle, fibrillation potentials are typically over 100 millivolts; subsequently, fibrillation potential magnitudes become smaller; as such, the magnitude of the fibrillation potentials can be used to some extent to determine how long ago the onset of denervation began.44 Fibrillations should not be confused with fasciculations, which are large enough to be seen on the skin and typically do not occur with compression injuries. Following injury, the proximal portion of the damaged axon will attempt to reestablish its connection with the target muscle or sensory organ by growing at about 1 mm per day. Even if the Schwann cell corridor remains intact, not all regenerating axons will reach their target organ. Conduction amplitude will therefore be permanently decreased. Moreover, when reinnervation does occur, nerve conduction velocities typically never return to their preinjury levels.
Electrodiagnostic findings in CNC injury
Conversely, CNC injuries usually do not result in axonal damage in their early stages,45 and thus can be categorized as neuropraxias. Focal demyelination occurs at 7-10 days, with remyelination beginning around 14-28 days. The myelin produced, however, is thin with decreased intermodal lengths.21,46,47,48 When myelin is damaged, the electrical current leaks in demyelinated areas, which not only causes a loss of saltatory conduction but also causes an increase in the amount of time it takes for action potentials to develop.38 As such, the major hallmarks of entrapment neuropathies early on are an increase in latency and a decrease in nerve conduction velocity (Figs. 2A, 2B) that is caused by focal demyelination.5,37,38 Some studies cite a decrease in conduction velocity of at least 70% and an increase in latency of at least 150% as necessary for the diagnosis of entrapment neuropathy.5 In contrast, CMAPs and SNAPs elicited distal to the injury site generally exhibit little to no decrease in the initial stages of entrapment neuropathy (Fig. 5C) given the lack of axonal damage.
FIGURE 2.
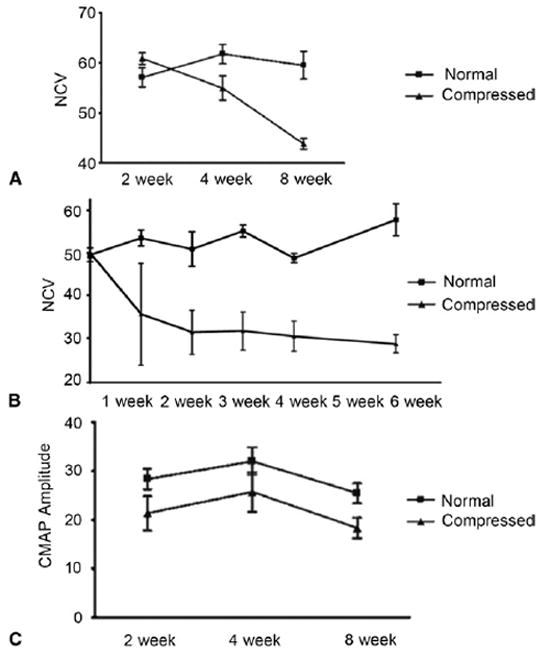
Decreased nerve conduction velocity (NCV) in compressed nerves. Electrodiagnostic studies in both A rat models, and B murine models reveal decreased NCV (in m/s) following chronic nerve compression injury as compared to controls. C CMAP amplitudes are decreased slightly in CNC-injured nerves at 2 weeks, but the difference in CMAP amplitudes is not statistically significant, and the CMAP amplitudes recorded do not exhibit further declines. A and C are reproduced with permission from Mozaffar T, Strandberg E, Abe K, Hilgenberg LG, Smith MA, Gupta R. Neuromuscular junction integrity after chronic nerve compression injury. J Orthop Res 2009;27:114–119. Copyright 2009, with permission from Elsevier.
Over time, however, as the CNC injury becomes more severe, axonal damage ensues causing decreases in CMAPs even distal to the lesion. Because sensory axons are more susceptible to compression, SNAPs typically exhibit decreased amplitudes prior to CMAPs. In these later stages where end-stage axonal dropout ensues, EMG studies may also reveal increased insertional activity, fibrillation potentials, positive sharp waves, and/or fasciculations. Axonal sprouting may also occur as a response to axonal loss, where the surviving axons develop sprouts that attempt to reconnect to denervated end organs.19 As such, the surviving axons take over some of the innervation territory of the lost axons. This collateral reinnervation may show on electromyography as high amplitude, short duration MUP’s (otherwise known as giant MUP’s). Recent studies of animal models of CNC injury reveal that this axonal regeneration occurs prior to any axonal degeneration;20 thus with CNC injury giant MUP’s may be seen on electromyography prior to any decline in CMAP amplitude. Moreover, longer periods of compression tend to produce even more marked decreases in conduction velocities due to repeated episodes of demyelination and remyelination. If remyelination is complete, nerve conduction velocity should typically return to normal.
To fully understand these concepts, one may consider the electrodiagnostic findings seen in carpal tunnel syndrome. Carpal tunnel syndrome will typically manifest with slowing of NCV and an increase in latency across the wrist demonstrative of focal demyelination. In some patients, particularly those in the later stages who have progressed to axonal loss, sensory deficits may be evidenced by decreased amplitudes of SNAPs while motor deficits are evidenced by decreased CMAP amplitude when recording from the thenar muscles. Oftentimes, latencies of the median nerve are compared to latencies of another nerve, such as the ulnar nerve or radial nerve. The accuracy of this comparison is unclear.
CNC Injury in diabetic patients
Given the rapidly increasing incidence of diabetes worldwide, a brief treatment of diabetic-related compression neuropathy is warranted. Some studies have postulated that nearly one out of three diabetic patients suffers from a compression neuropathy.49 Although it is often difficult to distinguish between compression neuropathies and diabetic neuropathies in diabetic patients,50 some studies have suggested that diabetic patients tend to suffer from compression neuropathies at a much higher incidence than the general population.51 Additionally, the rate of progression of nerve injury due to compression, the severity of pain resulting from compression, and the susceptibility to nerve compression are often increased in diabetic patients; these differences may be due to the altered metabolic milieu that exists in the cells of diabetic patients.52,53 The loss of sensation that accompanies compression neuropathies can put many diabetic patients at risk of severe complications, including ulcerations secondary to undiscovered injuries of the extremities. Early studies on rats showed that the sciatic nerves of rats with hyperglycemia demonstrated significantly decreased nerve conduction velocities and amplitudes through their sciatic nerve as compared to nondiabetic rats.52
Several theories exist regarding the unique pathophysiology of CNC injury in diabetic patients. Recent histological studies indicate that diabetes may produce significant metabolic derangements in the endoneurium and perineurium of nerves. Segments of entrapped nerve in diabetic rats exhibit increases in both tenascin-C and alpha-smooth muscle actin (SMA) positive myofibroblasts in their endoneurium as compared to normal rats.54 Since both tenascin-C and SMA-positive fibroblasts have been implicated in perineurial fibrosis and collagen contraction, these molecules may be of particular interest in outlining a unique pathogenesis of and developing targeted treatments for diabetic compression neuropathy.
Because diabetic patients are at increased risk not just for focal entrapment neuropathies but also for several other types of diffuse neuropathies, electrodiagnostic studies are of particular importance in diabetic patients. In particular, electromyography can help to distinguish focal neuropathies such as carpal tunnel syndrome from other diffuse neuropathies commonly suffered by diabetics, such as distal symmetric polyneuropathy and double-crush syndrome, which is a radiculopathy involving the C7-C8 nerve roots. Neurodiagnostic studies can similarly help to distinguish combined median and ulnar nerve entrapment from distal symmetric polyneuropathy.
Current theories of treatment of diabetic compression neuropathies do not differ radically from methods of treating compression neuropathies in nondiabetics.55 Surgical decompression is recommended in severe cases, just as in non-diabetic patients.56 However, recovery of sensation and motor function surgical decompression is often slower in diabetic patients, perhaps due to accumulation of sorbitol and glycosylated end products, as well as due to myoinositol deficiencies in diabetic patients.57 Recent studies have suggested insulin injections at points of entrapment may help relieve symptoms particularly in non insulin-dependent patients.58,59 Predictably, general improvement of glycemic control has also been shown to reduce symptoms of compression neuropathy in diabetic patients.60
Future Directions
To develop improved non-operative therapeutic regimens for CNC injuries, elucidation of the mechanisms involved with the transduction of the biophysical stimuli into the pathology of CNC injuries. Mechanical loading of neural tissue induces CNC injuries are directly stimulating Schwann cells to proliferate; precisely which molecules are responsible for transducing the mechanical stimulus to Schwann cells still remain unanswered. Increasing evidence points to Schwann cell integrins as the probable transducer of mechanical stress to Schwann cells. If changes in α6β4 integrin can be shown to correlate spatially and temporally to the myelination changes seen in CNC injuries, and if α6β4 integrin can be definitively linked to the transcription factors responsible for regulating myelination such as c-jun and krox-20, this will provide powerful evidence that integrins play a crucial role in the pathophysiology of CNC injuries.
As many of the clinical consequences that result from CNC injuries can be attributed to demyelination rather than axonal damage, it is of particular interest to further explore the relationship between demyelination and the onset of pain in CNC injuries. CNC injuries exhibit early alterations in ion channel distribution that correlate temporally to increases in c-fos expression, which is a marker for pain and neural sensitivity. 61 These changes do not correlate with inflammatory changes, as indicated by the fact that the increase in c-fos expression following CNC injury is not accompanied by increases in pro-inflammatory cytokines such as IL-1 and IL-6.61 In addition, certain sodium channels are upregulated in endoneurial Schwann cells subjected to CNC injury; taken together, these data suggest that CNC injuries cause pain via novel and as yet unexplored molecular mechanisms.61 Moreover, these changes are preceded by myelin degeneration at the Node of Ranvier, which results in breakdown of separation of sodium and potassium channels. As such, it is quite plausible that the development of mechanisms to stabilize myelin stabilization could greatly aid prevention of pain in CNC injuries via stabilization of ion channels.
Furthermore, given that many of the clinical consequences of CNC injury stem from Schwann cell dedifferentiation, it is plausible that transplantation of Schwann cells into lesioned nerve segments could have significant therapeutic benefit in the treatment of CNC injury. To date, little study has been done on the applicability of Schwann cell transplantation to compressive neuropathies, but animal studies on Schwann cell transplantation to treat spinal cord injuries are encouraging. Animal studies where preconditioned Schwann cells were transplanted in peripheral nerve grafts after spinal cord contusions and spinal cord hemisections resulted in significant improvement of recovery.62,63 Scientists in Miami have begun further exploring the use of Schwann cell transplantation in the treatment of spinal cord injury; these scientists are finding new methods of ensuring Schwann cell survival in transplanation sites, including inhibition of a substance called calpain that induces the death of Schwann cells and other tissues,64 as well as supplementation with growth factors65 to create a favorable environment for Schwann cell survival and axonal and myelin regeneration.
Conclusion
In recent, years, considerable insights have also been made into elucidating how nerve crush injuries differ from chronic nerve compression injuries at the molecular level. The myelin changes seen in chronic nerve compression injuries exhibit distinct pathophysiology which does not involve damage to the axon itself and is not precipitated by macrophage-mediated induction of Schwann cell proliferation. Rather, recent studies done in in vitro compression chambers that subject Schwann cell-DRG neuron co-cultures to isolated hydrostatic stress indicate that mechanical stress may be the major causative factor driving Schwann cell proliferation following compression injuries. The question still remains, however, how mechanical forces are transmitted to Schwann cells to stimulate this proliferation. Recent studies in animal models note spatial and quantitative changes in expression in β4 integrins that correlate temporally with demyelination following chronic nerve compression injury, and thus point to β4 integrins as potential mediators of myelin changes following compression injuries.
Despite these recent major breakthroughs in the diagnosis, treatment, and pathophysiology of chronic nerve compression injuries, much work remains to be done. Current topical areas of exploration in the field include the use Schwann cell transplantation in treatment, the correlation between demyelination and the onset of pain, and the role of integrins in transducing the mechanical forces involved in nerve compression injuries to Schwann cells.
Acknowledgments
We are grateful to the members of the Peripheral Nerve Research Lab for all of their help.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Papanicolaou GD, McCabe SJ, Firrell J. The prevalence and characteristics of nerve compression symptoms in the general population. J Hand Surg Am. 2001;26:460–466. doi: 10.1053/jhsu.2001.24972. [DOI] [PubMed] [Google Scholar]
- 2.Andreisek G, Crook DW, Burg D, Marincek B, Weishaupt D. Peripheral neuropathies of the median, radial, and ulnar nerves: MR imaging features. Radiographics. 2006;26:1267–1287. doi: 10.1148/rg.265055712. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Choi JY, Huh YM, Song HT, Lee SA, Kim SM, et al. Role of magnetic resonance imaging in entrapment and compressive neuropathy--what, where, and how to see the peripheral nerves on the musculoskeletal magnetic resonance image: part 2, upper extremity. Eur Radiol. 2007;17:509–522. doi: 10.1007/s00330-006-0180-y. [DOI] [PubMed] [Google Scholar]
- 4.Graham B. The value added by electrodiagnostic testing in the diagnosis of carpal tunnel syndrome. J Bone Joint Surg Am. 2008;90:2587–2593. doi: 10.2106/JBJS.G.01362. [DOI] [PubMed] [Google Scholar]
- 5.Strandberg EJ, Mozaffar T, Gupta R. The role of neurodiagnostic studies in nerve injuries and other orthopedic disorders. J Hand Surg Am. 2007;32:1280–1290. doi: 10.1016/j.jhsa.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 6.Seddon H. Three types of nerve injury. Brain. 1943;66:247–288. [Google Scholar]
- 7.Sunderland S. A classification of peripheral nerve injury produced by loss of function. 1951;74:491–495. doi: 10.1093/brain/74.4.491. [DOI] [PubMed] [Google Scholar]
- 8.McGowan A. The results of transposition of the ulnar nerve for traumatic ulnar neuritis. J Bone Joint Surg Br. 1950;32B:293–301. doi: 10.1302/0301-620X.32B3.293. [DOI] [PubMed] [Google Scholar]
- 9.Shin R, Ring D. The ulnar nerve in elbow trauma. J Bone Joint Surg Am. 2007;89:1108–1116. doi: 10.2106/JBJS.F.00594. [DOI] [PubMed] [Google Scholar]
- 10.Dellon AL. Clinical grading of peripheral nerve problems. Neurosurg Clin N Am. 2001;12:229–240. [PubMed] [Google Scholar]
- 11.Rempel DM, Diao E. Entrapment neuropathies: pathophysiology and pathogenesis. J Electromyogr Kinesiol. 2004;14:71–75. doi: 10.1016/j.jelekin.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 12.Taskinen HS, Roytta M. The dynamics of macrophage recruitment after nerve transection. Acta Neuropathol. 1997;93:252–259. doi: 10.1007/s004010050611. [DOI] [PubMed] [Google Scholar]
- 13.Burden SJ, Sargent PB, McMahan UJ. Acetylcholine receptors in regenerating muscle accumulate at original synaptic sites in the absence of the nerve. J Cell Biol. 1979;82:412–425. doi: 10.1083/jcb.82.2.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pestronk A, Drachman DB. Motor nerve terminal outgrowth and acetylcholine receptors: inhibition of terminal outgrowth by alpha-bungarotoxin and anti-acetylcholine receptor antibody. J Neurosci. 1985;5:751–758. doi: 10.1523/JNEUROSCI.05-03-00751.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter AJ, Kristmundsdottir F, Gilmour J, Glasby MA. Changes in muscle cytoarchitecture after peripheral nerve injury and repair A quantitative and qualitative study. J Hand Surg Br. 1998;23:365–369. doi: 10.1016/s0266-7681(98)80059-4. [DOI] [PubMed] [Google Scholar]
- 16.Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, et al. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- 17.Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta R, Steward O. Chronic nerve compression induces concurrent apoptosis and proliferation of Schwann cells. J Comp Neurol. 2003;461:174–186. doi: 10.1002/cne.10692. [DOI] [PubMed] [Google Scholar]
- 19.Gupta R, Rummler LS, Palispis W, Truong L, Chao T, Rowshan K, et al. Local down-regulation of myelin-associated glycoprotein permits axonal sprouting with chronic nerve compression injury. Exp Neurol. 2006;200:418–429. doi: 10.1016/j.expneurol.2006.02.134. [DOI] [PubMed] [Google Scholar]
- 20.Mozaffar T, Strandberg E, Abe K, Hilgenberg LG, Smith MA, Gupta R. Neuromuscular junction integrity after chronic nerve compression injury. J Orthop Res. 2009;27:114–119. doi: 10.1002/jor.20704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mackinnon SE, Dellon AL, Hudson AR, Hunter DA. Chronic human nerve compression--a histological assessment. Neuropathol Appl Neurobiol. 1986;12:547–565. doi: 10.1111/j.1365-2990.1986.tb00159.x. [DOI] [PubMed] [Google Scholar]
- 22.Ludwin SK, Maitland M. Long-term remyelination fails to reconstitute normal thickness of central myelin sheaths. J Neurol Sci. 1984;64:193–198. doi: 10.1016/0022-510x(84)90037-6. [DOI] [PubMed] [Google Scholar]
- 23.Gray M, Palispis W, Popovich PG, van Rooijen N, Gupta R. Macrophage depletion alters the blood-nerve barrier without affecting Schwann cell function after neural injury. J Neurosci Res. 2007;85:766–777. doi: 10.1002/jnr.21166. [DOI] [PubMed] [Google Scholar]
- 24.Gupta R, Truong L, Bear D, Chafik D, Modafferi E, Hung CT. Shear stress alters the expression of myelin-associated glycoprotein (MAG) and myelin basic protein (MBP) in Schwann cells. J Orthop Res. 2005;23:1232–1239. doi: 10.1016/j.orthres.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 25.Frieboes LR, Gupta R. An in-vitro traumatic model to evaluate the response of myelinated cultures to sustained hydrostatic compression injury. J Neurotrauma. 2009;26:2245–2256. doi: 10.1089/neu.2009.0973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pham K, Nassiri N, Gupta R. c-Jun, krox-20, and integrin beta4 expression following chronic nerve compression injury. Neurosci Lett. 2009;465:194–198. doi: 10.1016/j.neulet.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vogel V. Mechanotransduction involving multimodular proteins: converting force into biochemical signals. Annu Rev Biophys Biomol Struct. 2006;35:459–488. doi: 10.1146/annurev.biophys.35.040405.102013. [DOI] [PubMed] [Google Scholar]
- 28.Pereira JA, Benninger Y, Baumann R, Goncalves AF, Ozcelik M, Thurnherr T, et al. Integrin-linked kinase is required for radial sorting of axons and Schwann cell remyelination in the peripheral nervous system. J Cell Biol. 2009;185:147–161. doi: 10.1083/jcb.200809008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rempel D, Evanoff B, Amadio PC, de Krom M, Franklin G, Franzblau A, et al. Consensus criteria for the classification of carpal tunnel syndrome in epidemiologic studies. Am J Public Health. 1998;88:1447–1451. doi: 10.2105/ajph.88.10.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuschner SH, Ebramzadeh E, Johnson D, Brien WW, Sherman R. Tinel’s sign and Phalen’s test in carpal tunnel syndrome. Orthopedics. 1992;15:1297–1302. doi: 10.3928/0147-7447-19921101-08. [DOI] [PubMed] [Google Scholar]
- 31.Beekman R, Schreuder AH, Rozeman CA, Koehler PJ, Uitdehaag BM. The diagnostic value of provocative clinical tests in ulnar neuropathy at the elbow is marginal. J Neurol Neurosurg Psychiatry. 2009;80:1369–1374. doi: 10.1136/jnnp.2009.180844. [DOI] [PubMed] [Google Scholar]
- 32.D’Arcy CA, McGee S. The rational clinical examination. Does this patient have carpal tunnel syndrome? J Am Med Assn. 2000;283:3110–3117. doi: 10.1001/jama.283.23.3110. [DOI] [PubMed] [Google Scholar]
- 33.El Miedany Y, Ashour S, Youssef S, Mehanna A, Meky FA. Clinical diagnosis of carpal tunnel syndrome: old tests, new concepts. Joint Bone Spine. 2008;75:451–457. doi: 10.1016/j.jbspin.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 34.Keith MW, Masear V, Chung K, Maupin K, Andary M, Amadio PC, et al. Diagnosis of carpal tunnel syndrome. J Am Acad Orthop Surg. 2009;17:389–396. doi: 10.5435/00124635-200906000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacDermid JC, Doherty T. Clinical and electrodiagnostic testing of carpal tunnel syndrome: a narrative review. J Orthop Sports Phys Ther. 2004;34:565–588. doi: 10.2519/jospt.2004.34.10.565. [DOI] [PubMed] [Google Scholar]
- 36.Elkowitz SJ, Dubin NH, Richards BE, Wilgis EF. Clinical utility of portable versus traditional electrodiagnostic testing for diagnosing, evaluating, and treating carpal tunnel syndrome. Am J Orthop. 2005;34:362–364. [PubMed] [Google Scholar]
- 37.Lee DH, Claussen GC, Oh S. Clinical nerve conduction and needle electromyography studies. J Am Acad Orthop Surg. 2004;12:276–287. doi: 10.5435/00124635-200407000-00008. [DOI] [PubMed] [Google Scholar]
- 38.Robinson LR. Role of neurophysiologic evaluation in diagnosis. J Am Acad Orthop Surg. 2000;8:190–199. doi: 10.5435/00124635-200005000-00006. [DOI] [PubMed] [Google Scholar]
- 39.Slutsky D. Electrodiagnostic Testing of the Upper Extremity. In: Slutsky D, editor. Upper Extremity Nerve Repair: Tips and Techniques, A Master Skills Publication. 1. Rosemont: American Society for Surgery of the Hand; 2008. pp. 521–535. [Google Scholar]
- 40.Perry JD. Electrodiagnosis in musculo-skeletal disease. Best Pract Res Clin Rheumatol. 2005;19:453–466. doi: 10.1016/j.berh.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 41.Eisen A, Schomer D, Melmed C. The application of F-wave measurements in the differentiation of proximal and distal upper limb entrapments. Neurol. 1977;27:662–668. doi: 10.1212/wnl.27.7.662. [DOI] [PubMed] [Google Scholar]
- 42.Chaudhry V, Cornblath DR. Wallerian degeneration in human nerves: serial electrophysiological studies. Muscle Nerve. 1992;15:687–693. doi: 10.1002/mus.880150610. [DOI] [PubMed] [Google Scholar]
- 43.Adams JE, Steinmann SP. Nerve injuries about the elbow. J Hand Surg Am. 2006;31:303–313. doi: 10.1016/j.jhsa.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 44.Kraft GH. Fibrillation potential amplitude and muscle atrophy following peripheral nerve injury. Muscle Nerve. 1990;13:814–821. doi: 10.1002/mus.880130907. [DOI] [PubMed] [Google Scholar]
- 45.Pham K, Gupta R. Understanding the mechanisms of entrapment neuropathies. Neurosurg Focus. 2009;26:E7,1–8. doi: 10.3171/FOC.2009.26.2.E7. [DOI] [PubMed] [Google Scholar]
- 46.Gupta R, Rowshan K, Chao T, Mozaffar T, Steward O. Chronic nerve compression induces local demyelination and remyelination in a rat model of carpal tunnel syndrome. Exp Neurol. 2004;187:500–508. doi: 10.1016/j.expneurol.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 47.Gupta R, Rummler L, Steward O. Understanding the biology of compressive neuropathies. Clin Orthop Relat Res. 2005:251–260. doi: 10.1097/01.blo.0000164354.61677.f5. [DOI] [PubMed] [Google Scholar]
- 48.O’Brien JP, Mackinnon SE, MacLean AR, Hudson AR, Dellon AL, Hunter DA. A model of chronic nerve compression in the rat. Ann Plast Surg. 1987;19:430–435. doi: 10.1097/00000637-198711000-00008. [DOI] [PubMed] [Google Scholar]
- 49.Vinik A, Mehrabyan A, Colen L, Boulton A. Focal entrapment neuropathies in diabetes. Diabetes Care. 2004;27:1783–1738. doi: 10.2337/diacare.27.7.1783. [DOI] [PubMed] [Google Scholar]
- 50.Bales JG, Meals R. Peripheral neuropathy of the upper extremity: medical comorbidity that confounds common orthopedic pathology. Orthopedics. 2009;32 doi: 10.3928/01477447-20090818-19. [DOI] [PubMed] [Google Scholar]
- 51.Stamboulis E, Vassilopoulos D, Kalfakis N. Symptomatic focal mononeuropathies in diabetic patients: increased or not? J Neurol. 2005;252:448–452. doi: 10.1007/s00415-005-0672-8. [DOI] [PubMed] [Google Scholar]
- 52.Dellon AL, Mackinnon SE, Seiler WAt. Susceptibility of the diabetic nerve to chronic compression. Ann Plast Surg. 1988;20:117–119. doi: 10.1097/00000637-198802000-00004. [DOI] [PubMed] [Google Scholar]
- 53.Siemionow M, Zielinski M, Sari A. Comparison of clinical evaluation and neurosensory testing in the early diagnosis of superimposed entrapment neuropathy in diabetic patients. Ann Plast Surg. 2006;57:41–49. doi: 10.1097/01.sap.0000210634.98344.47. [DOI] [PubMed] [Google Scholar]
- 54.Nishimura T, Hirata H, Tsujii M, Iida R, Hoki Y, Iino T, et al. Pathomechanism of entrapment neuropathy in diabetic and nondiabetic rats reared in wire cages. Histol Histopathol. 2008;23:157–166. doi: 10.14670/HH-23.157. [DOI] [PubMed] [Google Scholar]
- 55.Mondelli M, Aretini A, Rossi S. Ulnar neuropathy at the elbow in diabetes. Am J Phys Med Rehabil. 2009;88:278–285. doi: 10.1097/PHM.0b013e318190b89d. [DOI] [PubMed] [Google Scholar]
- 56.Melenhorst WB, Overgoor ML, Gonera EG, Tellier MA, Houpt P. Nerve decompression surgery as treatment for peripheral diabetic neuropathy: literature overview and awareness among medical professionals. Ann Plast Surg. 2009;63:217–221. doi: 10.1097/SAP.0b013e31818ba768. [DOI] [PubMed] [Google Scholar]
- 57.Ozkul Y, Sabuncu T, Kocabey Y, Nazligul Y. Outcomes of carpal tunnel release in diabetic and non-diabetic patients. Acta Neurol Scand. 2002;106:168–172. doi: 10.1034/j.1600-0404.2002.01320.x. [DOI] [PubMed] [Google Scholar]
- 58.Ashraf A, Moghtaderi AR, Yazdani AH, Mirshams S. Evaluation of effectiveness of local insulin injection in none insulin dependent diabetic patient with carpal tunnel syndrome. Electromyogr Clin Neurophysiol. 2009;49:161–166. [PubMed] [Google Scholar]
- 59.Ozkul Y, Sabuncu T, Yazgan P, Nazligul Y. Local insulin injection improves median nerve regeneration in NIDDM patients with carpal tunnel syndrome. Eur J Neurol. 2001;8:329–334. doi: 10.1046/j.1468-1331.2001.00240.x. [DOI] [PubMed] [Google Scholar]
- 60.Wein TH, Albers JW. Diabetic neuropathies. Phys Med Rehabil Clin N Am. 2001;12:307–320. [PubMed] [Google Scholar]
- 61.Frieboes LR, Palispis WA, Gupta R. Nerve compression activates selective nociceptive pathways and upregulates peripheral sodium channel expression in Schwann cells. J Orthop Res. 2009 doi: 10.1002/jor.21047. in print. [DOI] [PubMed] [Google Scholar]
- 62.Rasouli A, Bhatia N, Suryadevara S, Cahill K, Gupta R. Transplantation of preconditioned schwann cells in peripheral nerve grafts after contusion in the adult spinal cord. Improvement of recovery in a rat model. J Bone Joint Surg Am. 2006;88:2400–2410. doi: 10.2106/JBJS.E.01424. [DOI] [PubMed] [Google Scholar]
- 63.Dinh P, Bhatia N, Rasouli A, Suryadevara S, Cahill K, Gupta R. Transplantation of preconditioned Schwann cells following hemisection spinal cord injury. Spine. 2007;32:943–949. doi: 10.1097/01.brs.0000261408.61303.77. [DOI] [PubMed] [Google Scholar]
- 64.Hill CE, Hurtado A, Blits B, Bahr BA, Wood PM, Bartlett Bunge M, et al. Early necrosis and apoptosis of Schwann cells transplanted into the injured rat spinal cord. Eur J Neurosci. 2007;26:1433–1445. doi: 10.1111/j.1460-9568.2007.05771.x. [DOI] [PubMed] [Google Scholar]
- 65.Golden KL, Pearse DD, Blits B, Garg MS, Oudega M, Wood PM, et al. Transduced Schwann cells promote axon growth and myelination after spinal cord injury. Exp Neurol. 2007;207:203–217. doi: 10.1016/j.expneurol.2007.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]