

Abstract
Background
The proliferation of structural and functional studies of RNA has revealed an increasing range of RNA's structural repertoire. Toward the objective of systematic cataloguing of RNA's structural repertoire, we have recently described the basis of a graphical approach for organizing RNA secondary structures, including existing and hypothetical motifs.
Description
We now present an RNA motif database based on graph theory, termed RAG for RNA-As-Graphs, to catalogue and rank all theoretically possible, including existing, candidate and hypothetical, RNA secondary motifs. The candidate motifs are predicted using a clustering algorithm that classifies RNA graphs into RNA-like and non-RNA groups. All RNA motifs are filed according to their graph vertex number (RNA length) and ranked by topological complexity.
Conclusions
RAG's quantitative cataloguing allows facile retrieval of all classes of RNA secondary motifs, assists identification of structural and functional properties of user-supplied RNA sequences, and helps stimulate the search for novel RNAs based on predicted candidate motifs.
Background
Our knowledge of the functional roles of RNA molecules in the cell is increasing rapidly [1,2]. The expanding repertoire of known functional RNAs has spurred efforts to catalogue and classify RNA structures. Existing RNA databases have focused on archiving RNA primary, secondary, and tertiary structures. For example, the Nucleic Acids Database (NDB) catalogues tertiary structures [3,4], PseudoBase archives pseudoknots [5], and Gutell's database describes secondary motifs of ribosomal RNAs [6]. Rfam, an RNA family database, catalogues conserved RNA families [7]; SCOR, the Structural Classification of RNA [8], provides hierarchical classification of RNA motifs; and NCIR, a database of non-canonical interactions in RNAs [9] lists RNAs with rare, non-canonical base pairs.
We have developed an alternative approach for cataloguing and classifying all possible RNA structures based on topological properties of RNA secondary motifs (bulges, loops, junctions, stems). Classifying RNA secondary topologies is important because they are strongly correlated with their functional properties. For example, the secondary fold of the tRNA is topologically distinct from the 5S ribosomal RNA structure. Thus, cataloguing existing and hypothetical RNA topologies aids identification of novel RNAs. The graph theory concepts and techniques used for representing, analyzing, and organizing RNA secondary structures are described in the next section, as well as in our recent articles [10,11]. Our RNA-As-Graphs (RAG) web resource, or database, catalogues existing and hypothetical RNA tree structures using "tree graphs" and general RNA structures, including trees and pseudoknots, using "dual graphs". Since any RNA graph is characterized by the number of vertices (V) and the connectivity topology, we use these two basic RNA properties to quantitatively organize and archive existing and hypothetical RNA motifs. Most significantly, we now provide information about candidate novel RNA topologies, or motifs having topological properties similar to existing RNAs, allowing users to examine structures of both existing and candidate, yet unfound, RNA secondary motifs. We produce the RNA candidate motifs using clustering analysis of RNA graphs corresponding to known and hypothetical motifs.
Thus, RAG aims to: systematically catalogue all existing, candidate, and hypothetical RNAs as (graph) motif libraries; rank RNA motifs with different degrees of topological complexity; allow identification of structurally (topologically) similar RNAs; and stimulate the search for candidate RNA motifs not yet discovered in Nature or in the laboratory.
Construction and content
The key elements of RAG are: RNA graphical representations and results of graph theory; Laplacian eigenvalues for quantitative description of RNA graphs; prediction of candidate RNA topologies using a clustering algorithm; and a program for converting secondary structures to RNA graphs. Below, we discuss the integration of these elements, including statistics of existing and candidate RNAs in RAG. We also explain issues regarding compilation of existing RNA data, annotation of RNA topology entries, software development, as well as contents of RAG's tutorial pages.
RNA graphical representation
RNA secondary structures can be represented as tree and dual graphs, although RNA pseudoknots can only be represented as dual graphs. Figure 1 illustrates the relationship between secondary structures (P5abc domain of group I intron and tRNA(leu)) and their tree and dual graphs. We summarize the rules for converting an RNA structure into either a tree or a dual graph [10]. To convert a secondary structure into a tree graph, the following four rules are utilized: (1) A bulge, hairpin loop, or internal loop is considered a vertex (•) when there is more than one unmatched nucleotide or non-complementary base pair. (2) A junction (the location where three or more stems meet) is a vertex. (3) The 3' and 5' ends of a helical stem are considered a vertex. (4) An RNA stem with more than one complementary base pair is represented as an edge (—); the complementary base pairs are AU, GC and GU. The rules for converting an RNA structure into a dual graph are as follows: (1) An RNA stem with more than one complementary base pair is represented as a vertex (•). (2) An edge (∩ or —) represents a single-strand that may occur in segments connecting the secondary elements (e.g., bulges, hairpin loops, internal loops, and junctions). (3) No representation is required for the 3' and 5' ends of an RNA molecule. As in tree graph rules, the dual graph rules require that a stem has two or more complementary base pairs and a bulge/loop/junction has more than one unmatched nucleotide or non-complementary base pairs. The tree and dual graph representations do not specify the exact sequence or the length of an RNA molecule, although the length can be approximated. Furthermore, they do not specify geometric aspects of the secondary structure, but instead give a description of the connectivity.
Figure 1.
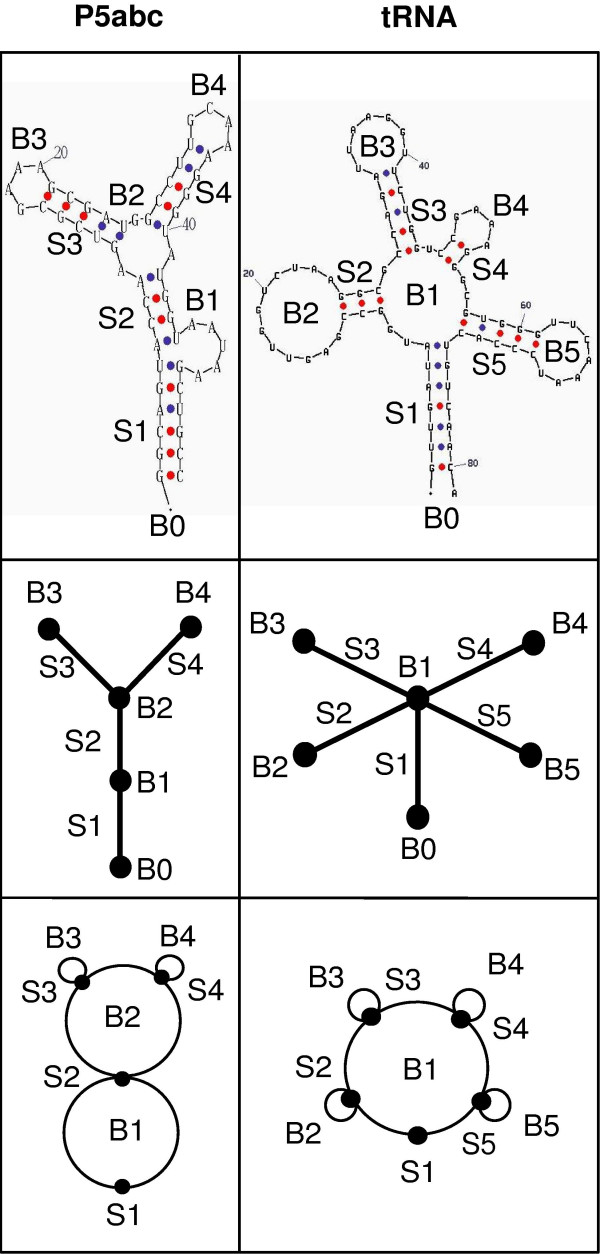
Graphical representations of RNA structures. Secondary structures of P5abc and tRNA(leu) (top row) are represented as both tree (middle) and dual (bottom) graphs. We use corresponding labels S1, S2, etc. for stems and B1, B2, etc. for bulges, loops or junctions; the chain ends (B0) are not represented in dual graphs. In tree graphs, stems are represented as edges or lines (—) and bulges/loops/junctions/chain ends as vertices (◆). In contrast, in dual graphs, bulges/loops/junctions are represented as edges or lines (—) and stems as vertices (◆).
Enumeration of RNA tree and pseudoknot structure libraries
The enumeration and generation of tree and dual graphs form the basis of RAG. RAG's RNA motif libraries are derived from the exact enumeration formula [12,13] for (unlabeled) tree topologies and from computational enumeration techniques for dual graph topologies, which can represent both RNA tree and pseudoknot motifs [11]. For a given vertex number V, a library of possible RNA motifs is generated, with size depending on V and the motif type (tree or pseudoknot). For example, the tree-motif libraries for V = 2, 3, 4, 5, 6, 7 and 8 contain 1, 1, 2, 3, 6, 11 and 23 distinct motifs, respectively. In contrast, for dual-graph motif libraries, there are 3, 8 and 30 distinct motifs for V = 2, 3 and 4, respectively. Tree libraries are smaller than dual-graph libraries because the latter cover tree, pseudoknot, and other motif types. Furthermore, the number of pseudoknots in any given library V is larger than that of trees, and this discrepancy increases with V [10]. We automate the process of cataloguing RNA motifs through quantitative characterization of RNA graphs to provide an easy access to, and search of, existing and hypothetical RNA motifs.
Quantitative description of RNA topologies
Every RNA secondary structure is mapped onto a 2D graph or topology and catalogued according to its V value and eigenvalue spectrum (λ1,λ2,…,λV). The eigenvalues of an RNA motif are obtained from the Laplacian matrix representation of its RNA graph [14]. In particular, the second eigenvalue λ2 measures a motif's topological complexity; a linear-like RNA motif (e.g., 70S(F)) has a smaller λ2 value than that of a highly-branched RNA motif (e.g., tRNA); we reference all RNA motifs by (V, λ2). For easy reference, we further index each RNA motif as (V, n), where n represents an integer corresponding to the λ2 ranking. Our cataloguing scheme allows RNA motifs of varying degrees of topological complexity to be distinguished, except for a small percentage of motifs that are co-spectral.
Prediction of novel RNA topologies using clustering analysis
Significantly, RNA's topological properties as described by Laplacian eigenvalues can be exploited to predict RNA topologies that are likely to exist in Nature. We use RNA topological descriptors and the method of Partitioning Around Medoids (PAM) to partition the enumerated RNA (tree and dual) graphs into RNA-like and non-RNA clusters or groups [15]. PAM partitions a set of data into k groups by using the Euclidean distance to assign objects closest to the k centers called medoids [16,17]. We choose k = 2 to partition theoretical RNA graphs into RNA-like and non-RNA topologies. The RNA-like cluster must contain predominantly existing RNA topologies and the non-RNA cluster contains few or no natural RNA motifs in it. We consider unidentified RNA motifs in the RNA-like cluster as RNA candidates. This analysis can be performed for various tree and dual graph libraries, yielding predictions of RNA's structural repertoire for different RNA sizes. The accuracy of our analysis depends on the number of existing RNA motifs. We used a total of 26 motifs representing 2 to 6 vertex dual graphs; there are fewer known motifs with higher vertex numbers (V > 6).
RNA tree and dual graph libraries
Our current RAG version archives tree graphs having up to 10 vertices and dual graphs up to 4 vertices to cover RNA topologies up to about 200 nt; already, a library of 10-vertex tree graphs has 106 motifs and a 4-vertex dual graph library has 30 motifs. Figure 2 shows examples of RAG's tree libraries (A) and general RNA motif libraries (or dual graphs), including tree and pseudoknot motifs (B), organized by V and λ2. Each library lists existing, candidate, and hypothetical RNA motifs, accompanied by available sequence, structural (2D and 3D) and functional data for natural motifs (about 26 found) through links to other databases (NDB, PseudoBase, 5S, etc.). As λ2 increases, motifs with higher-order junctions are formed. For example, the V = 5 tree library with 3 distinct motifs is represented by the 70S (chain F) RNA with no junction (λ2 = 0.3820), by the P5abc domain of group I intron containing a 3-stem junction (λ2 = 0.5188), and by the tRNA with a maximum of 4-stem junction (λ2 = 1.000). Though most of the motifs in small libraries (V < 6) exist in Nature, the many possible but yet unobserved candidate motifs for larger graphs listed in RAG are likely to stimulate the search for novel RNA motifs. To facilitate finding novel RNA motifs, we indicate in our tree (from V = 3 to V = 8) and dual graph (for V = 3 and V = 4) libraries which motifs are most likely to exist in Nature. The predicted candidate motif numbers for 2-, 3-,..., 8-vertex tree libraries are 1, 1, 1, 0, 3, 4, 8, respectively; for 2-, 3- and 4-vertex dual graph libraries, the candidate motif numbers are 0, 2 and 8.
Figure 2.
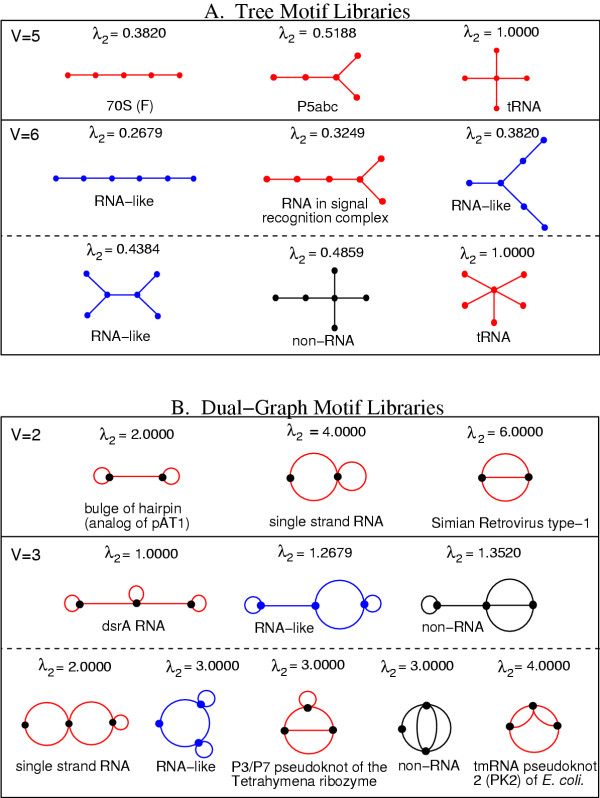
Entries in tree and dual graph libraries. The tree graph (A) and dual graph (B) motifs are ordered according to the vertex number V and the second smallest Laplacian eigenvalue λ2. Existing (red color), candidate (blue) and hypothetical (black) RNA motifs.
RNA Matrix Program
In addition, RAG contains an RNA Matrix Program to assist structural and functional identification of RNA motifs. It converts a user-supplied secondary structure file (in 'ct' format) into its graphical representation (at present limited to tree graphs) and calculates the RNA graph's topological characteristics (vertex number V, eigenvalues, order of junctions or degree of vertices, etc.). Such information directs the user to the corresponding existing (or hypothetical) RNA motif in the database, with links to other RNA sequence, structure (2D and 3D) and function databases.
Compilation of existing RNA data
We collected data for existing RNAs from the literature and other databases. We conducted a thorough search of distinct RNA topologies in NDB and PseudoBase; RNA structure data in these databases are derived from experiments. We also used structural information from the Rfam database and sequences from the 5S rRNA database. The sequences in the 5S database were converted to secondary structures using a folding program (e.g., Mfold [18], Vienna RNAfold [19]) and then to RNA graphs using our RNA Matrix program. Since Mfold predictions are not exact, we indicate the source of the secondary structure in each topology entry where applicable. Thus, RAG represents a compilation of topologies from RNA primary, secondary and tertiary structures, as well as from enumerated structures. The compiled natural RNAs include RNA domains, whole RNAs and RNAs complexed with proteins.
Annotation of RNA topology entries
Each RNA topology is annotated by the vertex number (V), graph ID (combination of vertex number and second eigenvalue), entire Laplacian eigenvalue spectrum, and status of topology (existing, candidate, hypothetical). For an existing RNA topology, its biological function is indicated together with an image of its secondary structure and hyperlinks to other databases providing further information about sequence, structure, and function. We also provide an internal link to other members of the same functional class with distinct secondary topologies.
Software development
RAG is freely available as a web-based platform written in HTML to archive RNA motif libraries. The RNA Matrix Program module is written in the C language and its executable is embedded in RAG using PHP, an open source, server-side scripting language for creating dynamic Web pages. The Matrix Program converts a user-supplied secondary structure file in the .ct format. The .ct file is generated automatically by the Mfold program when an RNA sequence is folded. Since Mfold is not integrated into RAG, the .ct file must be saved by the user and then uploaded to our RNA Matrix Program server. In future versions of RAG, we plan to integrate the RNA folding program so that the only input from the user is the RNA sequence. We also plan to employ database technology to allow efficient storage and retrieval of many large candidate RNA motifs.
RNA tutorial pages
The tutorial pages are an integral part of RAG. These pages concisely explain all key concepts and methods used to construct RAG: RNA structure; graph theory and RNA structures; rules for representing RNA structures (trees and pseudoknots) as graphs, illustrated with examples; Laplacian matrix, spectral graph analysis; graph isomorphism; clustering of RNA motifs; and a glossary of technical terms. The tutorials are intended to aid users from both biological and mathematical backgrounds to navigate through the RAG database.
Utility and discussion
Our cataloguing of existing, candidate and hypothetical RNA motifs is intended as a tool for searching existing RNAs and for discovering novel RNA molecules. RAG's organization of RNAs according to topological motifs rather than detailed sequence and structural features, as in current databases, will enable users to easily navigate through the space of RNA structural classes or libraries, which are catalogued by vertex number and second eigenvalue. The user starts with a higher-level topological description (i.e., size and complexity as quantified by vertex number and second eigenvalue) but is directed to specific sequence and structure information via links to other RNA databases; see Fig. 3. Our approach emphasizes global aspects of the RNA universe, an approach which is likely to be increasingly important in the genomic era, for example, in the design of sequences that fold into desired motifs [20].
Figure 3.
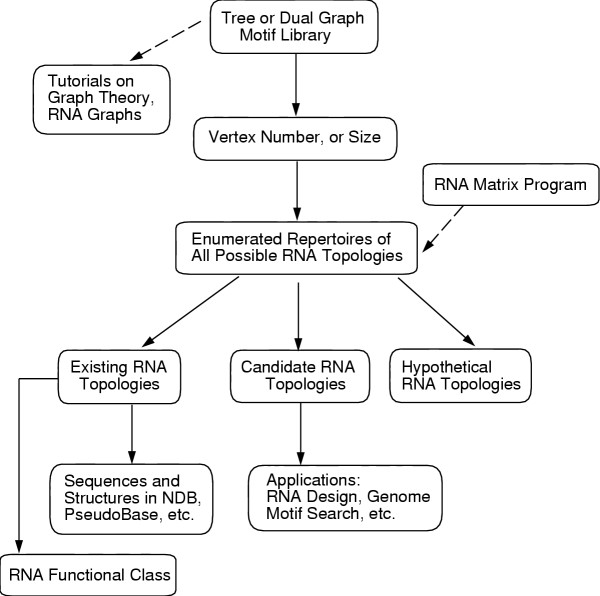
Organization of the RAG database. Information about RNA's topological libraries in RAG is organized hierarchically according to graph motif type (tree or dual), vertex number, library of topologies, and status of topology (existing, candidate, or hypothetical), with hyperlinks to other databases providing sequence, structure, and function data of existing RNAs. The existing RNA topologies are also catalogued according to functional class. Other components of RAG include an RNA Matrix Program for converting a secondary structure into a tree graph and RNA tutorials on concepts and techniques employed.
RAG effectively lists RNA motifs by topological similarity, which may imply structural and functional similarities between neighboring motifs. Although our motif-ordering scheme suggests such a relation, the utility of our database does not depend on its existence. Many non-existing tree and pseudoknot topologies are similar to existing motifs, suggesting that they may be potential functional motifs. For example, most candidate motifs in V = 4 dual graph library resemble existing motifs.
Searching for structural neighbors of an existing RNA
RAG is useful when the user is searching for structural neighbors of a known RNA. Knowing structural neighbors helps to identify RNAs having potentially related functions. Figure 4 illustrates the steps in this search with a star-shaped, five-stem tRNA(leu) structure. First, the RNA sequence is folded using either Mfold or Vienna RNAfold program to produce a .ct file for the tRNA fold. If an experimental structure is available, the user can also construct a .ct file without using RNA folding programs to avoid structural inaccuracies. Second, the RNA Matrix Program converts the tRNA's .ct file into a 6-vertex tree graph and reports its topological properties (e.g., Laplacian eigenvalue spectrum). Third, information about the tree graph directs the user to the specific structure library (vertex number) and location or neighbor (eigenvalue spectrum) in which the tRNA graph is found. We find that tRNA(leu) has two 6-vertex closest structural neighbors, one of which is a candidate RNA and the other is a hypothetical RNA (see Fig. 4). A similar search using a 5S rRNA leads to a cluster of 5S structures.
Figure 4.
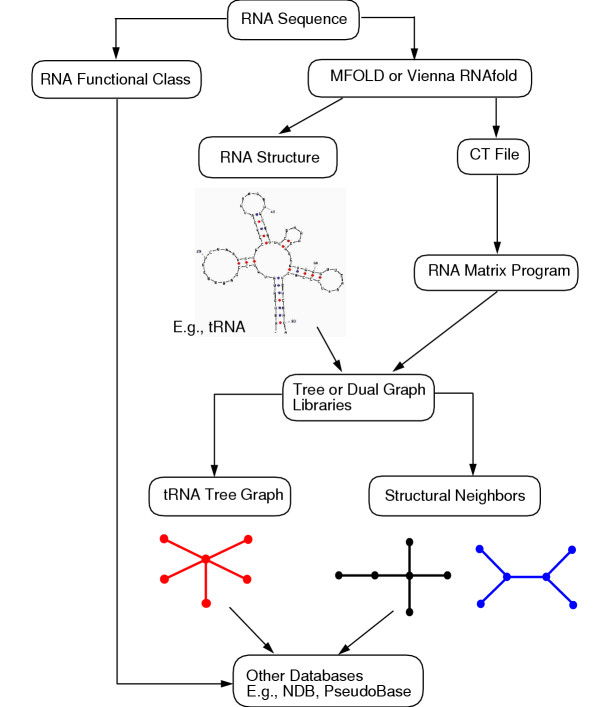
Searching for structural neighbors of a natural RNA in RAG. The search steps are illustrated using a star-shaped, five-stem tRNA(leu) structure, starting from its sequence to structural neighbors. The search utilizes an RNA folding program, RNA Matrix Program, and archived libraries of topologies.
Applications in RNA design and motif searches
Significantly, access to such candidate RNA motifs in RAG could stimulate both theoretical and experimental search for novel functional RNA molecules for various applications in biotechnology, chemistry and medicine. For example, novel motifs in RAG can be designed theoretically and their functional properties verified experimentally. Our group has already initiated research in this direction by coupling computational design with experimental in vitro selection method for identifying novel functional RNAs [21] (Gan and Schlick, in preparation). Another emerging application of candidate RNA topologies is in the computational search for novel RNA genes. We have begun using novel topologies in RAG as templates in our search for RNA-like genes in bacterial genomes via RNA motif scanning and folding algorithms. Other uses of the RAG database are anticipated in the near future.
We plan to enhance RAG in several significant ways. First, we will use graphs with labeled vertices and directed edges to allow differentiation of specific loops/bulges/junctions and determination of strand directions in RNA secondary motifs. Second, we will exploit database technologies for storage and retrieval to greatly expand the number of RNA graphs available for analysis and application. Third, we plan to classify existing RNA topologies into functional categories to complement our mathematical cataloguing scheme.
Conclusions
Both natural and synthetic RNAs have diverse functional roles. The range of their structural motifs is rapidly increasing, especially from genomics projects for identifying novel non-coding RNAs [1,2]. The RAG database uniquely organizes all known and hypothetical RNA motifs by schematic graphical representations. Although the size of natural RNAs' structural repertoire is not known, some of our predicted candidate motifs may be found in novel RNA genes or synthesized in the laboratory. Perhaps, the quest for RAG's missing motifs may echo the search for missing elements in the early days of the chemical periodic table. We invite users to explore RAG and send us their comments to RAG@biomath.nyu.edu.
Availability
The database is accessible on the web at
Contact
RAG@biomath.nyu.edu.
Authors' Contributions
DF was responsible for organizing the tree and dual graph libraries as well as writing and editing the manuscript. NK performed the PAM clustering analyses of RNA graphs. NS wrote a program for enumerating dual graphs using a probabilistic method. JZ wrote and incorporated the RNA Matrix Program in RAG. UL used an exact method for the enumeration of small dual graphs. HHG and TS conceived and co-supervised the whole project, including writing and editing the database and manuscript.
Acknowledgments
Acknowledgments
This work was supported by a Joint NSF/NIGMS Initiative to Support Research Grants in the Area of Mathematical Biology (DMS-0201160) as well as Human Frontier Science Program (HFSP). D. Fera acknowledges support from the Dean's Undergraduate Research Fund and a summer fellowship from the Department of Chemistry.
Contributor Information
Daniela Fera, Email: df448@nyu.edu.
Namhee Kim, Email: nk401@cims.nyu.edu.
Nahum Shiffeldrim, Email: nshiffeld@biomath.nyu.edu.
Julie Zorn, Email: jz290@nyu.edu.
Uri Laserson, Email: ul212@nyu.edu.
Hin Hark Gan, Email: hgan@biomath.nyu.edu.
Tamar Schlick, Email: schlick@nyu.edu.
References
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Storz G. An expanding universe of noncoding RNAs. Science. 2002;296:1260–1263. doi: 10.1126/science.1072249. [DOI] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Iype L, Schneider B, Zardecki C. The Nucleic Acid Database. Acta Crystallogr D Biol Crystallogr. 2002;58:889–898. doi: 10.1107/S0907444902003487. [DOI] [PubMed] [Google Scholar]
- Berman HM, Olson WK, Beveridge DL, Westbrook J, Gelbin A, Demeny T, Hsieh SH, Srinivasan AR, Schneider B. The Nucleic-Acid Database - A Comprehensive Relational Database of 3-Dimensional Structures of Nucleic-Acids. Biophysical Journal. 1992;63:751–759. doi: 10.1016/S0006-3495(92)81649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Batenburg FHD, Gultyaev AP, Pleij CWA, Ng J, Oliehoek J. PseudoBase: a database with RNA pseudoknots. Nucleic Acids Research. 2000;28:201–204. doi: 10.1093/nar/28.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannone JJ, Subramanian S, Schnare MN, Collett JR, D'Souza LM, Du YS, Feng B, Lin N, Madabusi LV, Muller KM, Pande N, Shang ZD, Yu N, Gutell RR. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics. 2002;3 doi: 10.1186/1471-2105-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura M, Hendrix DK, Klosterman PS, Schimmelman NR, Brenner SE, Holbrook SR. SCOR: Structural Classification of RNA, version 2.0. Nucleic Acids Res. 2004;32 Database issue:D182–D184. doi: 10.1093/nar/gkh080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaswamy U, Larios-Sanz M, Hury J, Collins S, Zhang ZD, Zhao Q, Fox GE. NCIR: a database of non-canonical interactions in known RNA structures. Nucleic Acids Research. 2002;30:395–397. doi: 10.1093/nar/30.1.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan HH, Pasquali S, Schlick T. Exploring the repertoire of RNA secondary motifs using graph theory; implications for RNA design. Nucleic Acids Res. 2003;31:2926–2943. doi: 10.1093/nar/gkg365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan HH, Fera D, Zorn J, Shiffeldrim N, Tang M, Laserson U, Kim N, Schlick T. RAG: RNA-As-Graphs Database - Concepts, Analysis, and Features. Bioinformatics. 2004;20:1285–1291. doi: 10.1093/bioinformatics/bth084. [DOI] [PubMed] [Google Scholar]
- F Harary, Prins G. The number of homeomorphically irreducible trees and other species. In: PrinsG, editor. Acta Math. Vol. 101. 1959. pp. 141–162. [Google Scholar]
- Harary F. Graph theory. Reading, Mass., Addison-Wesley; 1969. [Google Scholar]
- Mohar B. The Laplacian spectrum of graphs. In: AlaviY, CharthrandG, OellermannOR and SchwenkJA, editor. Graph theory, combinatorics, and applications. Wiley; 1991. pp. 871–899. [Google Scholar]
- Kim N, Shiffeldrim N, Gan HH, Schlick T. Novel candidates for RNA topologies. J Mol Biol (In Press) 2004. [DOI] [PubMed]
- Kaufman Leonard, Rousseeuw Peter J. Finding groups in data: an introduction to cluster analysis. New York, Wiley; 1990. (Wiley series in probability and mathematical statistics Applied probability and statistics). [Google Scholar]
- The R Project for Statistical Computing, http://www.r-project.org/ 2004.
- Zuker M, Mathews DH, Turner DH. Algorithms and thermodynamics for RNA secondary structure prediction: A practical guide. In: BarciszewskiJ and ClarkBFC, editor. RNA Biochemistry and Biotechnology. Dordrecht, Kluwer Academic Publishers; 1999. pp. 11–43. (NATO ASI Series). [Google Scholar]
- Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Research. 2003;31:3429–3431. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andronescu M, Fejes AP, Hutter F, Hoos HH, Condon A. A new algorithm for RNA secondary structure design. Journal of Molecular Biology. 2004;336:607–624. doi: 10.1016/j.jmb.2003.12.041. [DOI] [PubMed] [Google Scholar]
- Wilson DS, Szostak JW. In vitro selection of functional nucleic acids. Annu Rev Biochem. 1999;68:611–647. doi: 10.1146/annurev.biochem.68.1.611. [DOI] [PubMed] [Google Scholar]