

Abstract
Shiga toxin-producing E. coli (STEC) produce two types of Shiga toxin (STx): STx1 and STx2. The toxin A-subunits block protein synthesis while the B-subunits mediate retrograde trafficking. STEC infections do not have definitive treatments, and there is growing interest in generating toxin transport inhibitors for therapy. However, a comprehensive understanding of the mechanisms of toxin trafficking is essential for drug development. While STx2 is more toxic in vivo, prior studies focused on STx1 B-subunit (STx1B) trafficking. Here, we show that, compared to STx1B, trafficking of the B-subunit of STx2 (STx2B) to the Golgi occurs with slower kinetics. Despite this difference, similar to STx1B, endosome-to-Golgi transport of STx2B does not involve transit through degradative late endosomes and is dependent on dynamin II, epsinR, retromer and syntaxin5. Importantly, additional experiments show that a surface-exposed loop in STx2B (β4-β5 loop) is required for its endosome-to-Golgi trafficking. We previously demonstrated that residues in the corresponding β4-β5 loop of STx1B are required for interaction with GPP130, the STx1B-specific endosomal receptor, and for endosome-to-Golgi transport. Overall, STx1B and STx2B share a common pathway and use a similar structural motif to traffic to the Golgi, suggesting that the underlying mechanisms of endosomal sorting may be evolutionarily conserved.
Keywords: bacterial toxin, trafficking, endosome, Golgi, protein motif

Introduction
Shiga toxin (STx), produced by Shigella, and the closely-related Shiga toxin type 1 (STx1) and type 2 (STx2), produced by strains of Shiga toxin-producing E. coli (STEC), belong to the AB5 class of bacterial exotoxins (1, 2). These toxins are formed by the association of a catalytically-active A subunit with a pentameric B-subunit (1, 2). STx1 is nearly identical to STx with 100% conservation in the B-subunit and a single conservative serine to threonine substitution in the A-subunit (1, 3). In contrast, STx2 shares only 55% sequence identity with STx and STx1 (1, 3). The A-subunits of all three toxins induce toxicity by removing a specific adenine residue from the 28S ribosomal RNA in the cytosol of affected cells, which blocks protein synthesis (1, 2). The pentameric B-subunits mediate retrograde transport of the toxins from the cell exterior to the cytosol (1).
STx-producing Shigella cause massive epidemics in developing countries (4) whereas, in North America, food-borne STEC infections predominate (5). The annual incidence of STEC infections in the USA alone is >70,000 (6). Individuals infected with STx-producing Shigella or STEC initially develop gastrointestinal disease (5, 7). In a subset of patients, systemic effects of the released toxins lead to life threatening sequelae, such as hemolytic uremic syndrome (5, 7). Importantly, while antibiotic therapy is effective for the treatment of Shigella infections (7), in patients with STEC infections, usage of at least some classes of antibiotics increases STx1 and STx2 production and enhances the likelihood of developing hemolytic uremic syndrome (8-11). Consequently, antibiotic therapy is contraindicated for treatment of STEC infections, and this disease has no definitive treatment (5). As retrograde toxin trafficking is required for productive infections, there is considerable interest in generating small molecule inhibitors of toxin transport, which may be therapeutically useful (12-14).
Current understanding of the mechanisms involved in the retrograde trafficking of AB5 toxins comes largely from work performed on STx1 (1, 15, 16). Trafficking initiates with the association of the B-subunit of STx1 (STx1B) with the lipid globotriaosylceramide on the cell surface, followed sequentially by internalization to early endosomes, direct transport from early endosomes to the Golgi and delivery to the endoplasmic reticulum, from where the A subunit is translocated across the lipid bilayer to the cytosol (1). Direct early endosome-to-Golgi transport is a crucial step because it allows the toxin to bypass late endosomes/lysosomes where degradative proteolytic enzymes are active (1, 17). Until recently, the molecular mechanisms that enabled STx1B to sort into Golgi-directed membrane tubules at the level of early endosomes were not well understood. It is now clear that this direct transport step depends on a host protein, GPP130 [(1, 12, 18); also see ref.(19)]. GPP130 is a single-pass transmembrane protein that constitutively traffics between the cis-Golgi and early endosomes (20, 21). We showed that STx1B directly binds GPP130 (Kd =150 nM), which allows the toxin to “piggyback” on GPP130 and traffic to the Golgi from early endosomes (1, 12, 18). When GPP130 is depleted, STx1B still reaches early endosomes but then, instead of trafficking to the Golgi, the toxin is routed for degradation in late endosomes/lysosomes (1, 12). Thus, GPP130 functions as an endosomal receptor for STx1B. To date, GPP130 is the only endosomal receptor identified for an AB5 toxin.
While working on GPP130, we made the surprising discovery that an increase in the intracellular levels of the metal manganese (Mn) induces degradation of GPP130 (22, 23). In Mn-treated cells, as GPP130 is depleted, STx1B also gets degraded (12). Treatment with Mn confers 3800-fold protection against STx1-induced cell death in culture and complete protection against STx1-induced lethality in mice (12). These results provide as an important proof-of-concept for the effectiveness of an inhibitor of toxin transport in preventing toxin-induced disease in vivo. However, for treatment of STEC infections, it is important that the used inhibitor effectively block STx2 trafficking because, in vivo, the LD50 of STx2 is ~400 times lower than that of STx1 (24) and, in humans, severity of the disease phenotype correlates with STx2 production (25). Interestingly, the B-subunit of STx2 (STx2B) does not interact with GPP130 and treatment with Mn does not block STx2B trafficking (18). Elucidating the mechanisms involved in the retrograde trafficking of STx2B, which are poorly understood, is a prerequisite for the rational development of STx2-specific transport inhibitors.
In this study, we focus on STx2B trafficking and report that, similar to STx1B, STx2B also evades Rab7-positive late endosomes en route to the Golgi. Further, endosome-to-Golgi trafficking of STx2B requires activity of dynamin II, epsinR, Vps26 and syntaxin5, all of which are required for STx1B transport [(26-30); reviewed in (1)]. Thus, STx1B and STx2B traffic to the Golgi by a common pathway. In a separate set of experiments, we show that a surface exposed loop in STx2B (β4-β5 loop; composed of amino acid residues 72-77) is required for its transport to the Golgi and that disruption of this loop induces lysosomal degradation of the toxin. Importantly, the corresponding β4-β5 loop of STx1B contains residues required for its binding to GPP130 and early endosome-to-Golgi trafficking (18). Thus, STx1B and STx2B use a conserved structural domain to avoid trafficking to degradative late endosomes/lysosomes. Put together, our results show that there are broad similarities in the trafficking of STx1B and STx2B and suggest that the underlying mechanisms of endosomal sorting may be analogous.
Results
Variable trafficking patterns of STx1B and STx2B
In order to identify potential differences in STx1B and STx2B trafficking, we used a previously characterized trafficking assay (12, 18). Fluorescently-labeled untagged STx1B or STx2B bound the cell surface at 0 min and robustly trafficked to the Golgi within 60 min (Fig.1A&B). We then performed a time-course experiment in which the transport of STx1B and STx2B was analyzed in the same cells. In this experiment, cells were also stained to detect the Golgi. Both toxins trafficked to punctate intracellular structures, presumably endosomes, at early time-points and were clearly detected in the Golgi by 40 min (Fig.1C). However, qualitative assessment suggested that not all punctate structures were positive for both toxins, especially at the 20 min time-point (Fig.1C). To quantitatively assess overlap between STx1B, STx2B and the Golgi, we performed colocalization analyses using Pearson’s correlation co-efficient. At early time-points (0-10 min), the toxins had a high degree of colocalization with each other, suggesting that both toxins were internalized to similar membrane compartments (Fig.1C&D). However, as the assay progressed (20 min), there was a significant decrease in the colocalization between STx1B and STx2B (Fig.1C&D). At this time-point, there was also a dramatic increase in the colocalization of STx1B, but not STx2B, with the Golgi (Fig.1D). These results suggested that the decrease in STx1B-STx2B colocalization was due to faster trafficking of STx1B to the Golgi. Consistent with this, nonlinear regression analyses revealed that the time required for the Pearson’s coefficient for colocalization between STx1B-Golgi to reach its half-maximal value was 19 min (Fig.1E&F). In contrast, half-maximal overlap of STx2B with the Golgi was significantly delayed (by ~36%) and evident only at 26 min (Fig.1E&F). Note that, by the end of the time-course, maximal value for the Pearson’s co-efficient for colocalization between STx1B-Golgi (0.92 ± 0.01) was comparable to that between STx2B-Golgi (0.92 ± 0.03) (Fig.1E), implying that there was no difference in the amount of the internalized toxins that eventually trafficked to the Golgi. Thus, the kinetics of transport of STx1B and STx2B to the Golgi are different, and STx2B traffics to the Golgi at a slower rate.
Figure 1.

Differences in the retrograde trafficking of STx1B and STx2B.
A and B. Retrograde trafficking of AlexaFluor-488-labeled untagged STx1B (Panel A) or AlexaFluor-555-labeled untagged STx2B (Panel B) was assayed as described in Methods. Cells were fixed at 0 or 60 min after initiation of transport, stained with an antibody against GPP130 to demarcate the Golgi and imaged to detect the labeled toxins and the Golgi. Scale bars, 10 μm.
C. Co-transport of AlexaFluor-488-labeled untagged STx1B and AlexaFluor-555-labeled untagged STx2B was assayed in the same cells as described in Methods. Beginning at time 0 min after initiation of transport, cultures were fixed every 5 min and processed to detect STx1B, STx2B and the Golgi (using an antibody against GPP130). Images for the 10, 20 and 40 min time-points are presented here and all time-points are quantified in Panels D-F. Arrows at the 10 min time-point show overlap of STx1B and STx2B. Scale bars, 10 μm for the left three columns; 5 μm for the high magnification column on the right.
D. Quantification of the Pearson’s co-efficient for colocalization between STx1B-STx2B, STx1B-Golgi, or STx2B-Golgi from Panel C (mean ± SE; n=10-18 cells per time-point; * p<0.05 for the difference between STx1B-STx2B colocalization at 20 min compared to that at all other time-points by one-way ANOVA and Tukey-Kramer post hoc test).
E. Use of non-linear regression to fit the data for the Pearson’s co-efficient for colocalization between STx1B-Golgi and between STx2-Golgi from Panel D to four parameter logistical (sigmoidal) curves. The time required for the Pearson’s co-efficient for colocalization between STx1B-Golgi to reach its half-maximal value was significantly less than that required for STx2B-Golgi (see Panel F).
F. Representation of the time required for the Pearson’s co-efficient for colocalization between STx1B-Golgi and between STx2B-Golgi to reach their respective half-maximal values from Panel E (error bars denote 95% confidence intervals; * p<0.05).
G. Transport of AlexaFluor-488-labeled untagged STx1B or AlexaFluor-555-labeled untagged STx2B was separately assayed in independent cultures, as described in Panels A and B above. Cultures were fixed every 10 min from the time transport was initiated, stained with an antibody against GPP130 to delineate the Golgi, and imaged to detect the toxins and the Golgi. Pearson’s co-efficient for colocalization between STx1B-Golgi or STx2B-Golgi for all time-points is shown here (mean ± SE; n=15 cells per time-point).
H. Time required for the Pearson’s co-efficient for colocalization between STx1B-Golgi and between STx2B-Golgi to reach half-maximal values from Panel G (error bars denote 95% confidence intervals, * p<0.05).
The above result raised the possibility that STx1B and STx2B may be competing with each other to traffic to the Golgi. To test this, we repeated the time course experiment but now assayed for the transport of STx1B or STx2B in separate cultures. We observed that, once again, transport of STx1B to the Golgi was faster than that of STx2B (Fig.1G). The time required for the Pearson’s co-efficient for colocalization between STx1B-Golgi to reach its half-maximal value was 19 min (comparable to that obtained when transport of the toxins was studied in the same cells; Fig.1F) and significantly less than that required for STx2B-Golgi (Fig.1H). Thus, the slower kinetics of transport of STx2B to the Golgi is not due to competition with STx1B.
STx2B bypasses late endosomes
We then sought to characterize the pathway used by STx2B to traffic to the Golgi. This was important because the observed kinetic difference could be because the toxins used different pathways to reach the Golgi. As the first step, we tested whether transport of STx2B to the Golgi involved transit through late endosomes by analyzing toxin trafficking in cells expressing Rab5-, Rab7- or Rab11-GFP, which demarcate early, late or recycling endosomes respectively (31, 32). This experiment was performed as a time-course and cells were fixed at 10, 15 or 20 min after initiation of toxin transport. Note that for this and all subsequent assays we used C-terminally His-tagged STx2B proteins (see Methods for details). Use of the tag was essential because it was challenging to purify untagged STx2B in amounts necessary for required assays. We confirmed that there was no detectable difference between the trafficking of His-tagged or untagged STx2B to the Golgi (compare Fig.1D to Fig.2E). In the current assay, we observed that, at early time-points (10 min), STx2B did not overlap with any Rab protein (Fig.2A&B). However, at 15 min, there was a large increase in the colocalization of STx2B with Rab5 (Fig.2A&B), suggesting that STx2B was now present in early endosomes. Consistent with transport to early endosomes, STx2B was also detected within the lumen of enlarged endosomes formed by expression of Rab5Q79L (33) (Fig.2C). The colocalization of STx2B with Rab5 decreased at 20 min (Fig.2B), and at this time, STx2B started appearing in the Golgi (Fig.1D&G). Importantly, STx2B did not noticeably overlap with Rab7 or Rab11 at any time-point prior to transport to the Golgi (Fig.2A&B). As an additional test, we assayed for the transport of STx2B in cells expressing dominant negative Rab7 (Rab7T22N). Expression of Rab7T22N does not impact STx1B transport (12), and we observed that Rab7T22N did not affect trafficking of STx2B (Fig.2D&E). This was not due to a lack of dominant negative effect because Rab7T22N robustly blocked the lysosomal degradation of GPP130 in Mn-treated cells (Fig.2F&G). Overall, prior to transport to the Golgi, STx2B overlaps with a marker of early endosomes but is not detected in compartments positive for markers of late or recycling endosomes, indicating that STx2B traffics to the Golgi directly from early endosomes.
Figure 2.

Endosome-to-Golgi transport of STx2B avoids transit through Rab7-positive late endosomes.
A. Cells were transfected with indicated GFP-tagged RabWT constructs. One day after transfection, transport of His-tagged STx2BWT was assayed as described in Methods. Cultures were fixed at 10, 15 or 20 min after initiation of transport; images for the 10 and 15 min time-points are depicted here and all time-points are quantified in Panel B. After fixation, cells were stained with an antibody against the His-tag and imaged to detect GFP and STx2B. Arrows show overlap of STx2B with Rab5. Scale bar, 10 μm.
B. Quantification of the Pearson’s co-efficient for colocalization between STx2B and Rab5, Rab7 or Rab11 from Panel A (mean ± SE; n=10 cells per time-point; * p<0.05 for the difference between STx2B-Rab5 colocalization at 15 min compared to all other conditions by one-way ANOVA and Dunnett’s post hoc test).
C. Cells were transfected with GFP-tagged Rab5Q79L. One day after transfection, transport of His-tagged STx2BWT was assayed. Cultures were fixed 15 min after initiation of transport, stained with an antibody against the His-tag and imaged to detect GFP and STx2B. Arrows show presence of STx2B in Rab5Q79L-positive endosomes. Scale bar, 10 μm.
D. Cells were transfected with GFP-tagged dominant negative Rab7 (Rab7T22N). After 24 h, transport of His-STx2BWT was assayed. Cultures were fixed 60 min after initiation of toxin transport and processed to image STx2B, using an anti-His antibody; the Golgi, using an anti-GPP130 antibody; and GFP. Asterisks denote transfected cells. Scale bar, 10 μm.
E. Pearson’s co-efficient for colocalization between STx2B and the Golgi from Panel D (mean ± SE; n=10 cells per group).
F and G. HeLa cells that did not over-express globotriaosylceramide were either left untransfected (Panel F) or transfected with GFP-tagged Rab7T22N (Panel G). After 24 h, cultures were treated with or without 500 μM Mn for 4 h, fixed and imaged to detect GPP130 and the Golgi, using an antibody against SPCA1, a Golgi-localized Ca/Mn transporter (Panel F) or GFP and GPP130 (Panel G). In Rab7T22N-expressing cells, after Mn-treatment, GPP130 persists in cytoplasmic punctae, as described by us previously (22). Scale bars, 10 μm.
Retrograde trafficking of STx2B requires dynamin II, epsinR, Vps26 and syntaxin5
Work over the past 15 years has revealed that the early endosome-to-Golgi transport of STx1B requires dynamin II, clathrin and its adaptor epsinR, retromer and the syntaxin5 SNARE complex [(26-30, 34, 35); reviewed in (1)]. As the next step in characterizing the pathway used by STx2B to traffic to the Golgi, we felt it was important to determine whether these factors were involved. To test for the requirement of dynamin II, we transfected cultures with a dominant negative mutant (dynamin IIK44A), which blocks the endosome-to-Golgi transport of STx1B (26). We observed that, in cells expressing this mutant, STx2B was endocytosed but its trafficking to the Golgi was strongly inhibited (Fig.3A&B). Expression of dynamin IIwild-type (WT) had no effect (Fig.3C&D). We then tested the role of the clathrin adaptor epsinR (we chose to not directly interfere with clathrin to avoid pleotropic effects on trafficking). Overexpression of epsinRWT produces a dominant negative phenotype and blocks the early endosome-to-Golgi transport of STx1B (28). In our assays, transport of STx2B to the Golgi was inhibited in cells that overexpressed epsinRWT (Fig.3E&F). To confirm the overexpression result, we depleted epsinR using a previously described siRNA that inhibits epsinR expression and blocks the early endosome-to-Golgi trafficking of STx1B (28). To assay for STx2B trafficking, we used a co-transport approach where trafficking of STx1B and STx2B was assayed in the same cells after transfection with control or anti-epsinR siRNAs. Use of the co-transport approach was necessary because we were unable to detect endogenous epsinR by immunofluorescence using available commercial antibodies; therefore, we needed an alternative means to identify cells in which knockdown of epsinR had been successful and had functionally compromised early endosome-to-Golgi trafficking. Assessment of STx1B transport provided the ideal internal positive control for this experiment. We observed that depletion of epsinR inhibited the trafficking of STx1B to the Golgi, and importantly, in these cells, STx2B also failed to traffic to the Golgi (Fig.3G-I). After this, we tested if activity of the retromer complex was required. Knockdown of the retromer component Vps26 induces Golgi fragmentation and blocks STx1B transport (27, 36). We used a previously described siRNA to target human Vps26 (27) and verified that the anti-Vps26 siRNA reduced Vps26 expression and induced Golgi fragmentation (Fig.4A). To assay for STx2B trafficking, we used the co-transport approach described for epsinR and observed that knockdown of Vps26 inhibited the trafficking of STx1B and STx2B to the Golgi (Fig.4B-D). Interestingly, in epsinR- and Vps26-depleted cells, STx1B and STx2B were detected in overlapping cytoplasmic punctae with a Pearson’s colocalization co-efficient of ~0.9 ± 0.01 (Fig.3G&4B), implying that the toxins were trapped in the same pre-Golgi compartment. Finally, we reverted to a dominant negative approach to test for the requirement of syntaxin5, which is a part of the syntaxin5/Ykt6/GS15/GS28 SNARE complex (29). Loss of syntaxin5 activity induces Golgi fragmentation and blocks STx1B trafficking (29, 30). We generated a dominant negative mutant of syntaxin5 by deleting its transmembrane domain [syntaxin5Δtransmembrane (TM)] (37). Expression of syntaxin5ΔTM induced Golgi fragmentation, confirming a dominant negative effect, and in these cells, transport of STx2B to the Golgi was strongly reduced (Fig.5A&B). Expression of syntaxin5WT had no effect on STx2B trafficking (Fig.5C&D). Thus, dynamin II, epsinR, Vps26 and syntaxin5 are all required for the optimal trafficking of STx2B from early endosomes to the Golgi. The results in Fig.2-5, put together, indicate that STx1B and STx2B use a common pathway to undergo endosome-to-Golgi transport.
Figure 3.

Transport of STx2B to the Golgi requires dynamin II and epsinR.
A. Cells were transfected with HA-tagged dominant negative dynamin II (dynamin IIK44A). One day post-transfection, transport of His-STx2BWT was assessed. Cultures were fixed 60 min after start of transport and imaged to detect HA; STx2B, using an anti-His antibody; and the Golgi, using an anti-GRASP65 antibody. Asterisks denote transfected cells. Scale bar, 10 μm.
B. Quantification of the Pearson’s co-efficient for colocalization between STx2B and the Golgi from Panel A (mean ± SE; n=10 cells per group; * p<0.05 by Student’s t test).
C. Cells were transfected with HA-tagged dynamin IIWT and subsequently processed exactly as described in Panel A. Asterisks denote transfected cells. Scale bar, 10 μm.
D. Quantification of the Pearson’s co-efficient for colocalization between STx2B and the Golgi from Panel C (mean ± SE; n=15 cells per group).
E. Cells were transfected with GFP-tagged epsinRWT. After 24 h, transport of His-STx2BWT was assessed. Cultures were fixed 60 min after initiation of transport and imaged to detect GFP, His and the Golgi (using an antibody against GRASP65). Asterisks denote transfected cells. Scale bar, 10 μm.
F. Pearson’s co-efficient for colocalization between STx2B and the Golgi from Panel E (mean ± SE; n=10 cells per group; * p<0.05 by Student’s t test).
G. Cells were transfected with control or anti-epsinR siRNAs. After three days, the co-transport of AlexaFluor 555-labeled untagged STx1B and unlabeled His-STx2BWT was assayed. Cells were fixed 60 min after start of the transport assay and imaged to detect STx1B; STx2B, using an anti-His antibody; and the Golgi, using an anti-GPP130 antibody. Scale bars, 10 μm.
H and I. Pearson’s co-efficient for co-localization between STx1B or STx2B and the Golgi from Panel G (mean ± SE; n=10 cells per group; * p<0.05 by Student’s t test).
Figure 4.

STx2B transport is Vps26-dependent.
A. Cells were transfected with control or anti-Vps26 siRNAs. After 3 days, cultures were stained to detect Vps26 or the Golgi, using an antibody against GPP130. Scale bars, 10 μm.
B. Cells were transfected with control or anti-Vps26 siRNAs as described in Methods. Three days post-transfection, cultures were used to assay for the co-transport of AlexaFluor 488-labeled untagged STx1B and unlabeled His-STx2BWT. Cells were fixed 60 min after start of the transport assay and imaged to detect STx1B; STx2B, using an anti-His antibody; and the Golgi, using an anti-GPP130 antibody. The observed fragmentation of the Golgi confirmed effective siRNA-induced Vps26 depletion. Scale bars, 10 μm.
C and D. Pearson’s co-efficient for co-localization between STx1B or STx2B and the Golgi from Panel B (mean ± SE; n=15 cells per group; * p<0.05 by Student’s t test).
Figure 5.

Syntaxin5 is required for trafficking of STx2B to the Golgi.
A. Cells were transfected with GFP-tagged dominant negative syntaxin5 (syntaxin5ΔTM). One day post-transfection, transport of His-STx2BWT was assessed. Cultures were fixed 60 min after start of transport and imaged to detect GFP; STx2B, using an anti-His antibody; and the Golgi, using an anti-GPP130 antibody. Asterisks denote transfected cells. Scale bar, 10 μm.
B. Quantification of the Pearson’s co-efficient for colocalization between STx2B and the Golgi from Panel A (mean ± SE; n=15 cells per group; * p<0.05 by Student’s t test).
C. Cells were transfected with GFP-syntaxin5WT and subsequently processed exactly as described in Panel A. Asterisks denote transfected cells. Scale bar, 10 μm.
D. Quantification of the Pearson’s co-efficient for colocalization between STx2B and the Golgi from Panel C (mean ± SE; n=10 cells per group).
The β4-β5 loop of STx2B is required for its transport to the Golgi
The retrograde transport of STx1B from early endosomes to the Golgi is dependent on its direct interaction with GPP130 (12, 18). This binding occurs via residues in the flexible β4-β5 loop of STx1B (18). Although STx1B and STx2B share only ~55% sequence homology, a comparative view of their crystal structures [PDB:1R4Q and 1R4P; (38)] reveals an impressive similarity (Fig.6A). The structural profiles and spatial orientations of the β4-β5 loops appear nearly identical (Fig.6A). However, STx2B does not bind GPP130, and this lack of binding has been ascribed to a lack of conserved residues in the β4-β5 loop (18) (Fig.6B). In STx1B, residues H78 and N79 in the β4-β5 loop are required for GPP130 binding and endosome-to-Golgi transport (18), but the corresponding residues in STx2B are E76 and S77 (Fig.6B). Domains of STx2B required for its endosome-to-Golgi transport are unknown, and the role of its β4-β5 loop has not been investigated.
Figure 6.

The β4-β5 loop of STx2B is required for retrograde transport.
A. Crystal structure of STx1B (PDB:1R4Q) and STx2B (PDB:1R4P). Arrows denote the β4-β5 loops; boxes show an enlarged view of the loops.
B. Sequence alignment of the β4-β5 loops. Conservative substitutions are shown by a “+” sign.
C. Transport of His-STx2BWT (WT) or His-STx2B72-74;76-77A (72-74;76-77A) was assayed as described in Methods. Cells were fixed at 0, 15, 30 or 60 min after start of the transport assay. Images for the 0, 15 and 60 min time-points are depicted here and all time-points are quantified in Panel D. Toxins were detected using an anti-His antibody and the Golgi was demarcated using an anti-GPP130 antibody. Scale bars, 10 μm.
D. Quantification of the total STx2B fluorescence per cell from Panel C. For each STx2B protein, fluorescence per cell at time 0 min was adjusted to 100 and used for normalization of subsequent time-points. At time 0 min, there was no statistical difference between the fluorescence per cell for STx2BWT and STx2B72-74;76-77A (mean ± SE; n>50 cells for each STx2B protein; * p<0.05 for the difference between the fluorescence of STx2B72-74;76-77A at the 60 min time-point and all other groups using one-way ANOVA and Dunnett’s post hoc test).
Based on the structural similarities between STx1B and STx2B and the fact that both toxins share a common early endosome-to-Golgi trafficking pathway, we wondered whether the β4-β5 loop of STx2B was required for trafficking. To test this idea, we purified a STx2B mutant that had the following five mutations: S72A, S73A, T74A, E76A, and S77A (STx2B72-74;76-77A). We then compared the trafficking of this mutant to that of STx2BWT. As expected, STx2BWT robustly trafficked to the Golgi (Fig.6C). In contrast, STx2B72-74;76-77A bound cells and was internalized to punctate structures resembling endosomes at early time-points (15 min; Fig.6C). However, at 60 min, the STx2B72-74;76-77A mutant was not detected in the Golgi, and instead, there was a significant reduction in its signal intensity (Fig.6C&D), suggesting that the toxin was degraded. Blocking the endosome-to-Golgi transport of STx1B leads to re-routing of the toxin to late endosomes/lysosomes where it is degraded (12). To determine whether STx2B72-74;76-77A was similarly degraded, we pre-treated cells with the lysosomal protease inhibitors leupeptin and pepstatin (22) and repeated the transport assay. Under these conditions, STx2B72-74;76-77A still failed to traffic to the Golgi, but now, instead of being degraded, the toxin persisted in punctate cytoplasmic structures (Fig.7A&B) that partially overlapped with the lysosomal marker Lamp2 (Fig.7C). Thus, similar to STx1B, residues in the β4-β5 loop of STx2B are required for its endosome-to-Golgi transport and mutations introduced in this domain target the toxin for lysosomal degradation.
Figure 7.
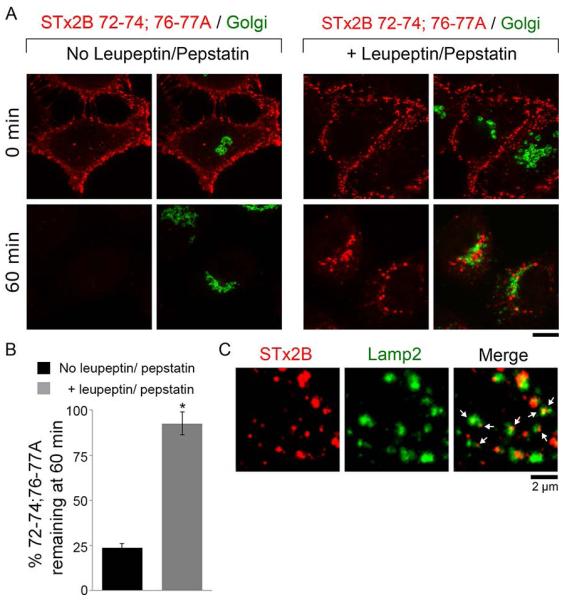
Degradation of STx2B72-74;76-77A is blocked by treatment with leupeptin and pepstatin.
A. Transport of His-STx2B72-74;76-77A was assayed in cells that had been pre-treated with or without leupeptin (100 μg/ml) and pepstatin (50 μg/ml) for 24 h as described in Methods. Cultures were fixed at indicated time-points after initiation of transport and stained to detect the STx2B protein, using an anti-His antibody, and the Golgi, using an antibody against GPP130. Scale bar, 10 μm.
B. Quantification of total STx2B72-74;76-77A fluorescence per cell remaining at 60 min from Panel A. For each treatment condition, fluorescence per cell at 0 min was normalized to 100% (mean ± SE; n=10-15 cells per group; data for the “no leupeptin/pepstatin” group is re-plotted from Fig.6D; * p<0.05 using Student’s t test).
C. Transport of His-STx2B72-74;76-77A was assayed after pre-treatment with leupeptin and pepstatin as described in Panel A. Cultures were fixed 60 min after start of transport and stained with antibodies against His, to detect the STx2B protein, and Lamp2. Arrows show presence of STx2B in Lamp2-positive structures. Scale bar, 2 μm.
Identification of additional STx2B residues that are involved in its endosome-to-Golgi transport
While working on the leupeptin/pepstatin assay, in a separate set of experiments, we transfected cells with Rab7T22N to block lysosomal degradation of proteins (12, 39), and then analyzed the transport of STx2BWT or STx2B72-74;76-77A. Surprisingly, in cells transfected with Rab7T22N, STx2B72-74;76-77A robustly trafficked to the Golgi (Fig.8). Indeed, in Rab7T22N expressing cells, the Pearson’s co-efficient for colocalization between STx2B72-74;76-77A and the Golgi (0.91 ± 0.01; data shown as part of Fig.9E) was comparable to that observed for STx2BWT-Golgi (0.90 ± 0.01; data from Fig.2E). Importantly, this effect was not seen in cells that did not express Rab7T22N in the same culture and, in these untransfected cells, STx2B72-74;76-77A was degraded (Fig.8).
Figure 8.
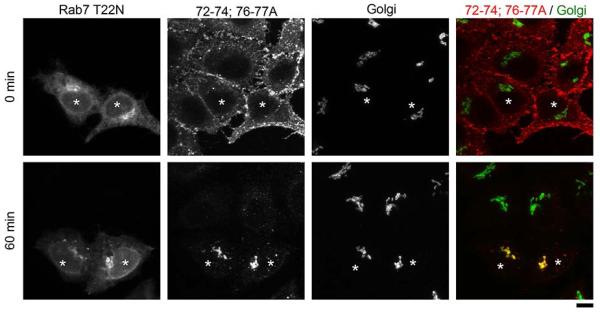
STx2B72-74;76-77A traffics to the Golgi in Rab7T22N expressing cells but gets degraded in untransfected cells.
Cells were transfected with GFP-Rab7T22N, and one day after transfection, the transport of His-STx2B72-74;76-77A was assayed. Cultures were fixed at indicated times after initiation of transport and imaged to detect GFP; STx2B, using an anti-His antibody; and the Golgi, using an anti-GPP130 antibody. Asterisks denote transfected cells. Quantification for data shown here is presented in Fig.9E. Scale bar, 10 μm.
Figure 9.

Combinatorial mutations in the N87 residue and the β4-β5 loop are required to block the transport of STx2B to the Golgi in Rab7T22N expressing cells.
A. Cartoon depiction of the crystal structure of STx2B (PDB:1R4P) showing location of the N87 residue relative to the β4-β5 loop.
B. Trafficking of His-STx2B72-74;76-77A;N87R was assayed in untransfected cells. Cells were fixed at 0 or 60 min after initiation of transport and imaged to detect the STx2B protein, using an anti-His-antibody and the Golgi, using an anti-GPP130 antibody. Scale bar, 10 μm.
C and D. Cells were transfected with GFP-Rab7T22N. One day post-transfection, transport of His-STx2B72-74;76-77A;N87R (Panel C) or His-STx2BN87R (Panel D) was assayed. Cells were fixed at the 60 min time-point and stained with an antibody against the His-tag, to detect STx2 proteins, and GPP130, to demarcate the Golgi. Asterisks denote transfected cells. Scale bars, 10 μm.
E. Pearson’s co-efficient for colocalization with the Golgi for indicated His-tagged STx2B proteins at the 60 min time-point (mean ± SE; n=10-15 cells per group; * p<0.05 for the difference between STx2B72-74;76-77A;N87R and all other groups using one-way ANOVA and Dunnett’s post hoc test).
The above result raised the possibility that one or more additional domains/sites of STx2B played a role in its endosome-to-Golgi trafficking and that the requirement of this site became evident when normal endosomal dynamics was altered by Rab7T22N expression. If this idea was correct, eliminating the additional site would be expected to inhibit the transport of STx2B72-74;76-77A to the Golgi in Rab7T22N transfected cells. We investigated the surface-exposed residue N87, which is proximal to the β4-β5 loop (Fig.9A). The corresponding residue in STx1B is an arginine. We mutated the N87 residue to arginine in STx2B72-74;76-77A to generate STx2B72-74;76-77A;N87R and verified that, similar to STx2B72-74;76-77A, STx2B72-74;76-77A;N87R was also degraded in a transport assay in untransfected cells (Fig.9B). Importantly, after transfection with Rab7T22N, unlike STx2B72-74;76-77A, STx2B72-74;76-77A;N87R failed to traffic to the Golgi and, instead, remained trapped in perinuclear punctate structures (Fig.9C&E). To assess the effect of the N87R mutation alone, we generated STx2BN87R and repeated the transport assay. The N87R mutation by itself did not affect the trafficking of STx2B to the Golgi in cells that expressed Rab7T22N or in untransfected cells in the same culture (Fig.9D&E). Thus, under normal conditions, the β4-β5 loop is required for the endosome-to-Golgi transport of STx2B while the N87 residue is not. However, when the dynamics of intracellular trafficking are compromised by Rab7T22N expression, the β4-β5 loop and the N87 residue together are required.
β4-β5 loop of STx1B is not sufficient to confer GPP130-dependence to STx2B transport
The importance of the respective β4-β5 loops of STx1B and STx2B in mediating retrograde toxin transport led us to determine whether this loop was sufficient to alter the transport specificity of the toxins. That is, we decided to test if substitution of the β4-β5 loop of STx1B with that of STx2B would allow STx1B to traffic to the Golgi in a GPP130-independent manner and vice versa. We could not purify a mutated form of STx1B in which the β4-β5 loop was substituted with that of STx2B. However, we managed to successfully purify a STx2B mutant containing a substitution of its β4-β5 loop with that of STx1B (designated STx2Bhybrid). We then performed a transport assay to test whether retrograde trafficking of STx2Bhybrid was GPP130-dependent. For this assay, we induced GPP130 degradation by transfecting cells with SPCA1Q747A (23). SPCA1 is a Golgi-localized Ca/Mn pump that transports Mn from the cytosol to the Golgi (23). We previously demonstrated that SPCA1 activity is required for the Mn-induced loss of GPP130 (23). We also showed that the Q747A mutation increases the Mn transport activity of SPCA1, and due to this, GPP130 is degraded in cells expressing SPCA1Q747A even when the growth medium is not supplemented with additional Mn (23). For the current study, as control, we first assayed the transport of STx1BWT in cultures transfected with SPCA1Q747A. Consistent with our previous work (12, 23), GPP130 was degraded in cells expressing SPCA1Q747A, and in these cells, STx1BWT failed to traffic to the Golgi and was also degraded (Fig.10A). Importantly, untransfected cells in the same culture had strong GPP130 expression and exhibited robust trafficking of STx1BWT to the Golgi (Fig.10A). Moreover, also similar to our previous results (18), we verified that loss of GPP130 did not affect transport of STx2BWT to the Golgi (Fig.10B&D). Finally, we tested STx2Bhybrid and were surprised to find that, although this mutant contained the β4-β5 loop of STx1B, it robustly trafficked to the Golgi in cells that did or did not express GPP130 (Fig.10C&D). Thus, substitution of the β4-β5 loop of STx2B with that of STx1B did not make the retrograde trafficking of STx2B GPP130-dependent. These results indicate that residues in the β4-β5 loop of STx1B, while required, are not sufficient to mediate GPP130-dependent trafficking.
Figure 10.

The β4-β5 loop of STx1B is not sufficient to confer GPP130-dependence to STx2B transport.
A-C. Cells were transfected with SPCA1Q747A. One day post-transfection, transport of AlexaFluor-488-labeled untagged STx1B (Panel A), His-tagged STx2BWT (Panel B) or His-tagged STx2Bhybrid (Panel C) was assayed. Cells were fixed at the 60 min time-point and stained using antibodies against GPP130 and GRASP65 (Panel A) or GPP130, GRASP65 and the His-tag (Panels B and C). Asterisks denote cells lacking detectable GPP130 due to expression of the transfected construct. Scale bars, 10 μm.
D. Pearson’s coefficient for colocalization between STx2BWT-Golgi or STx2Bhybrid-Golgi in cells that did or did not express GPP130 from Panels B and C. GRASP65 staining was used to demarcate the Golgi (mean ± SE; n=14 cells per group).
In sum, we show that the kinetics of the transport of STx1B and STx2B to the Golgi are different. In spite of this, both toxins traffic to the Golgi via a common pathway. Moreover, the toxins use a structurally conserved domain to sort out of the endosomal compartment. Thus, there are extensive conceptual and thematic similarities in STx1B and STx2B trafficking, raising the possibility that the molecular mechanisms of endosomal sorting of the two toxins may be comparable.
Discussion
Based on our current and prior work, the primary difference in the endosome-to-Golgi transport of STx1B and STx2B is that STx1B trafficking is GPP130-dependent, but STx2B is not (1, 12, 18). This molecular divergence may contribute to the observed difference in the kinetics of transport of the two toxins. The mechanisms that lead to the increased toxicity of STx2 in vivo are not yet clear. It will be important to determine whether differences in retrograde trafficking enhance the efficiency of STx2 transport in a manner that increases its toxicity in vivo.
Our results are consistent with an earlier study in Vero cells showing that, during retrograde trafficking and prior to detection in the Golgi, STx1B and STx2B only partially colocalize in punctate endosome-like structures (40). Furthermore, it is important to note that the observed block in the endosome-to-Golgi transport of STx2B (Fig.3A&B) and STx1B (26) in cells expressing dominant negative dynamin does not imply that dynamin directly acts at the level of endosomes to mediate toxin transport to the Golgi. Available evidence suggests that dynamin primarily acts at the plasma membrane (41). In dominant negative dynamin-expressing cells, a block in transport at other subcellular locations, including endosome-to-Golgi, may be due to indirect effects, such as sequestration of clathrin or other trafficking factors at the plasma membrane (41).
As both STx1B and STx2B use a conserved domain and a common pathway to traffic to the Golgi, there is a possibility that STx2B trafficking depends on interaction of the toxin with an as yet unidentified endosomal receptor. If such a receptor exists, it may have similarities to the GPP130-STx1B paradigm. That is, the putative receptor may be an endogenous protein that constitutively traffics between early endosomes and the Golgi, has at least one transmembrane domain, and uses its intra-lumenal domain to directly interact with STx2B. The β4-β5 loop of STx2B may form the primary high affinity site for interaction with this hypothetical receptor. Furthermore, the receptor may also interact with additional sites on STx2B, such as the N87 residue, with lower affinity. The requirement of these secondary sites may become apparent when dynamics of intracellular trafficking change, as after Rab7T22N expression. The existence of a multimodal strategy to traffic to the Golgi may be an evolutionary feature that increases the chances of cellular intoxication after STx2 internalization under different physiologic and pathologic conditions. Large-scale proteomic and genetic studies are now required to determine whether such a STx2 receptor exists; these studies are an important aspect of current research in our laboratory, and we have already identified several promising candidates through an on-going unbiased proteomic screen. As other AB5 toxins, such as cholera toxin, share structural similarities with STx1 and STx2 (1), the use of endosomal receptors may be a universal strategy utilized by this class of toxins to avoid lysosomal degradation.
STx2B transport may also occur by a receptor-independent mechanism. This mechanism may still be protein-based in that, instead of interacting with one receptor protein, STx2B may use its β4-β5 loop and perhaps additional residues, such as N87, to interact with an endosomal protein complex that sorts to the Golgi. Membrane lipids may also contribute to STx2B trafficking. Prior studies have shown that lipid-based sorting is important for the retrograde transport of AB5 toxins (1, 16). The β4-β5 loop of STx2B, either independently or in concert with other sites on the toxin, may recruit STx2B to an endosomal microdomain that also recruits factors required for endosome-to-Golgi transport (e.g. epsinR and retromer) on its cytoplasmic face. Recruitment of STx2 to this microdomain may occur by a direct toxin-lipid interaction or by the interaction of the toxin with proteins that also sort into the microdomain. Clearly, further studies are essential to better understand the molecular details of STx2 transport.
Residues H78 and N79 on the β4-β5 loop of STx1B are required for binding GPP130 (18). However, whether additional residues/domains of STx1B are also involved is not clear. Our current observations show that the β4-β5 loop of STx1B is not sufficient to allow STx2B, a related homolog, to traffic in a GPP130-dependent manner raising the possibility that the β4-β5 loop of STx1B is not sufficient to mediate direct interaction with GPP130. It will be important to carefully analyze the STx1B-GPP130 interaction and identify additional toxin residues/domains that may be required. Such analyses will likely improve our understanding of receptor-based endosomal sorting because GPP130 residues that are required and sufficient for this interaction have already been discovered (18).
Our finding that STx2B uses its β4-β5 loop to traffic to the Golgi is important from a therapeutic perspective. Drug discovery approaches may succeed in generating small molecule inhibitors that simultaneously target the β4-β5 loops of STx1 and STx2 and block the transport of both toxins. Alternatively, it may be more feasible to design inhibitors that target the β4-β5 loop of STx2 only. STx2-specific inhibitors, either by themselves or in combination with Mn to block STx1, may provide a viable strategy for the management of STEC infections. Transport inhibitors may also enable antibiotic administration to rapidly clear STEC bacteria in patients, as these inhibitors may protect against antibiotic-induced increase in toxin release.
Methods
Constructs, antibodies and chemicals
Plasmids encoding untagged STx1B and STx2B have been previously described by us (12, 18). Histidine (His)-tagged STx2BWT was made by introducing the amino acid sequence GSGHHHHHHH to the C-terminus of untagged STx2B by the loop-in modification of the QuikChange protocol (Agilent Technologies, Santa Clara, CA). Mutations into His-tagged STx2B were introduced using QuikChange. Plasmids encoding GFP-tagged Rab5WT, Rab5Q79L, Rab7WT and Rab7T22N; and HA-tagged SPCA1Q747A, dynamin IIWT and dynamin IIK44A have been previously described by us (22, 23). Plasmid encoding GFP-tagged Rab11WT was a gift of Dr. Marino Zerial (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany) and has been previously described (42). Plasmid for GFP-tagged epsinRWT was a gift of Dr. Margaret Robinson (University of Cambridge, Cambridge, England) and has also been described (43). Construct for GFP-tagged rat syntaxin5WT was a gift of Dr. Vladimir Lupashin (University of Arkansas for Medical Sciences, Little Rock, AR) and has been reported in ref. (44). The dominant negative mutant (syntaxin5ΔTM) was created by introducing a stop codon at amino acid 334, which deleted the C-terminal transmembrane domain. His-tagged STx2B proteins were detected using a rabbit polyclonal antibody against the His-tag (Bethyl Laboratories, Montgomery, TX). GRASP65 was detected using a goat polyclonal antibody (Santa Cruz Biotechnology Inc., Dallas, TX). Previously described mouse monoclonal antibodies were used to detect GPP130, Lamp2 and the HA-tag (22, 23). A rabbit polyclonal antibody was used to detect Vps26 (Abcam, Cambridge, MA). Unless otherwise specified, chemicals were from Sigma Aldrich (St. Louis, MO) or Thermo Fisher Scientific (Waltham, MA).
Cell culture, DNA transfections and microscopy
Unless otherwise specified, assays were performed using a HeLa cell line retrovirally transfected with globotriaosylceramide synthase to induce over-expression of globotriaosylceramide (45), which is the cell surface glycosphingolipid receptor of STx1B and STx2B (2). This cell line was a gift of Dr. Toshiyuki Yamaji (National Institute of Infectious Diseases, Tokyo, Japan) (45). Cells were cultured in Dulbecco’s Modified Eagle Medium (Mediatech, Inc., Manassas, VA) supplemented with 10% fetal bovine serum (Atlanta Biologicals, Norcross, GA), 100 IU/ml penicillin-G, 100 μg/ml streptomycin (both Mediatech) and 200 μg/ml G418 (Thermo Fisher). Treatment with Mn, when required, was performed by the addition of freshly-prepared MnCl2 to a final concentration of 500 μM, as described by us previously (22, 23, 46). DNA transfections were performed using JetPEI (VWR International, Randor, PA), as described by us previously (46). Cells were routinely transfected when 60%-70% confluent and used for experiments 24 h after transfection. For immunofluorescence, cultures were fixed with 3.7% formaldehyde for 10 min at room temperature. Further processing was exactly as described by us previously (46). Images were captured using a Nikon swept field confocal equipped with a four-line high-power laser launch and a 100×, 1.45 numerical aperture oil-immersion objective (Nikon, Tokyo, Japan). An iXon3 X3 DU897 EM-CCD camera (Andor Technology, Belfast, Ireland) was used for image capture. All images were captured as Z-stacks with 0.2-0.3 μm spacing between individual frames. Images depicted in the figures are maximum intensity projections of the stacks.
Small interfering RNA (siRNA) transfections
These were performed essentially as described by us previously (23, 46). The control siRNA used does not target any human gene and has been extensively characterized by us (23, 46). The siRNA against Vps26 targeted the sequence AAC TCT ATT AAG ATG GAA GTG in the human Vps26 gene and has been previously used to knockdown Vps26 (27). The siRNA against epsinR targeted the sequence AAG TGC CAG AGA ACA CAT TTA in the human epsinR gene and has also been previously described (28). Cells were transfected 24 h after plating, siRNAs were added at a final concentration of 120 nM using Oligofectamine reagent (Life Technologies, Carlsbad, CA), and cultures were used for assays 72-96 h post-transfection.
Protein purification and labeling
Purification of untagged STx1B was exactly as described by us previously (12). Untagged STx2B was obtained from BEI Resources (Manassas, VA). His-tagged STx2BWT and mutants were purified using Ni-NTA Agarose beads (Molecular Cloning Laboratories, South San Francisco, CA) using previously described protocols (18). Untagged STx1B and untagged STx2B were labeled with AlexaFluor-488 or AlexaFluor-555 (Life Technologies) using manufacturer’s instructions.
Transport assays
Transport assays using untagged STx1B, untagged STx2B, His-tagged STx2BWT or His-tagged STx2B mutants were performed essentially as described by us previously (12, 18). Briefly, cells were washed 3-times with ice-cold phosphate buffered saline and then incubated with 2 μg/ml of the toxin in transport media (Dulbecco’s Modified Eagle Medium supplemented with 10% fetal bovine serum, 100 IU/ml penicillin-G and 100 μg/ml streptomycin) for 30 min on ice at 4°C. After this, cells were washed 3-times with ice-cold phosphate buffered saline and transferred to toxin-free transport media at 37°C to initiate toxin transport. At times indicated in each figure, cultures were fixed and processed for microscopy.
When the transport of STx1B and STx2B was analyzed in the same cells, cultures were incubated with 2 μg/ml of each toxin and then processed as described above.
When the transport of STx2B was assayed after treatment with protease inhibitors, cultures were treated with 100 μg/ml leupeptin and 50 μg/ml pepstatin for 24 h (22). After this, the transport assay was performed as described above. Leupeptin and pepstatin were not present in the transport media at any point in the assay.
Image analyses
All analyses were performed using Image J (National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/index.html). For colocalization analyses, average value projections were created from individual Z-stacks. The cell was then outlined using the freehand tool, and the Pearson’s co-efficient was determined using the Coloc2 plugin. For measurement of total cellular fluorescence, average projections were created as described above. The background for each image was calculated for an area outside the cells using Analyze-Measure and subtracted using Process-Math-Subtract. Total fluorescence per cell was then measured by outlining the cell using the freehand tool and using Analyze-Measure.
Statistical analyses
Statistical analyses were performed using Prism 6 (GraphPad, La Jolla, CA). All experiments were repeated at least 3 times independently. Comparisons between two groups were performed using Student’s t-test assuming equal variances. Comparisons between multiple groups at the same time were performed using one-way ANOVA followed by Tukey-Kramer or Dunnett’s post hoc tests. In general, p<0.05 was considered to be statistically significant. Asterisks in graphs, wherever present, denote statistically significant differences.
Synopsis.
E. coli bacteria that produce Shiga toxin 2 (STx2) cause untreatable and life-threatening human disease. We show that a small, surface-exposed part of STx2 (named “β4-β5 loop”) enables the toxin to successfully intoxicate human cells. Our results imply that drugs targeting the β4-β5 loop of STx2 may be therapeutically useful.
Acknowledgements
We thank Dr. Toshiyuki Yamaji (National Institute of Infectious Diseases, Tokyo, Japan) for providing us with the HeLa cell line used in this study; and Dr. Marino Zerial (Max Planck Institute of Molecular Cell Biology and Genetics, Dresden, Germany), Dr. Margaret Robinson (University of Cambridge, Cambridge, England) and Dr. Vladimir Lupashin (University of Arkansas for Medical Sciences, Little Rock, AR) for sharing plasmids. We also thank Dr. Manojkumar Puthenveedu (Carnegie Mellon University, Pittsburgh, PA), Dr. Jennifer Maynard, Steven Hutchens, Charles Zogzas, and Jonathan Mercado (all from The University of Texas at Austin) for helpful discussions and critical reading of the manuscript. This work was supported in part by NIH grant R00-ES020844 and start-up funds from UT Austin (both to S.M.).
Abbreviations used
- Mn
manganese
- STx
Shiga toxin
- STx1
Shiga toxin type 1
- STx2
Shiga toxin type 2
- STx1B
Shiga toxin type 1 B-subunit
- STx2B
Shiga toxin type 2 B-subunit
- STEC
Shiga toxin-producing E. coli
- His
histidine
- WT
wild-type
Footnotes
The authors declare that they have no conflicts of interest with this study.
References
- 1.Mukhopadhyay S, Linstedt AD. Retrograde trafficking of AB(5) toxins: mechanisms to therapeutics. Journal of molecular medicine. 2013;91(10):1131–1141. doi: 10.1007/s00109-013-1048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beddoe T, Paton AW, Le Nours J, Rossjohn J, Paton JC. Structure, biological functions and applications of the AB5 toxins. Trends in biochemical sciences. 2010;35(7):411–418. doi: 10.1016/j.tibs.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strockbine NA, Jackson MP, Sung LM, Holmes RK, O'Brien AD. Cloning and sequencing of the genes for Shiga toxin from Shigella dysenteriae type 1. Journal of bacteriology. 1988;170(3):1116–1122. doi: 10.1128/jb.170.3.1116-1122.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotloff KL, Winickoff JP, Ivanoff B, Clemens JD, Swerdlow DL, Sansonetti PJ, Adak GK, Levine MM. Global burden of Shigella infections: implications for vaccine development and implementation of control strategies. Bulletin of the World Health Organization. 1999;77(8):651–666. [PMC free article] [PubMed] [Google Scholar]
- 5.Nataro JP, Pickering LK. Diarrheagenic Escherichia Coli. In: McMillan JA, Feigin RD, DeAngelis C, Jones MD Jr, editors. Oski’s Paediatrics: Principles and Practice. Lippincott Williams & Wilkins; Philadelphia, PA: 2006. pp. 1063–1068. [Google Scholar]
- 6.Mohawk KL, O'Brien AD. Mouse models of Escherichia coli O157:H7 infection and shiga toxin injection. Journal of biomedicine & biotechnology. 2011;2011:258185. doi: 10.1155/2011/258185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ochoa TJ, Cleary TG. Shigellosis. In: McMillan JA, Feigin RD, DeAngelis C, Jones MD Jr, editors. Oski’s Paediatrics: Principle and Practice. Lippincott, Williams & Wilkins; Philadelphia, PA: 2006. pp. 1116–1120. [Google Scholar]
- 8.Matsushiro A, Sato K, Miyamoto H, Yamamura T, Honda T. Induction of prophages of enterohemorrhagic Escherichia coli O157:H7 with norfloxacin. Journal of bacteriology. 1999;181(7):2257–2260. doi: 10.1128/jb.181.7.2257-2260.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGannon CM, Fuller CA, Weiss AA. Different classes of antibiotics differentially influence shiga toxin production. Antimicrobial agents and chemotherapy. 2010;54(9):3790–3798. doi: 10.1128/AAC.01783-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walterspiel JN, Ashkenazi S, Morrow AL, Cleary TG. Effect of subinhibitory concentrations of antibiotics on extracellular Shiga-like toxin I. Infection. 1992;20(1):25–29. doi: 10.1007/BF01704889. [DOI] [PubMed] [Google Scholar]
- 11.Zhang X, McDaniel AD, Wolf LE, Keusch GT, Waldor MK, Acheson DW. Quinolone antibiotics induce Shiga toxin-encoding bacteriophages, toxin production, and death in mice. The Journal of infectious diseases. 2000;181(2):664–670. doi: 10.1086/315239. [DOI] [PubMed] [Google Scholar]
- 12.Mukhopadhyay S, Linstedt AD. Manganese blocks intracellular trafficking of Shiga toxin and protects against Shiga toxicosis. Science. 2012;335(6066):332–335. doi: 10.1126/science.1215930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saenz JB, Doggett TA, Haslam DB. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infection and immunity. 2007;75(9):4552–4561. doi: 10.1128/IAI.00442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stechmann B, Bai SK, Gobbo E, Lopez R, Merer G, Pinchard S, Panigai L, Tenza D, Raposo G, Beaumelle B, Sauvaire D, Gillet D, Johannes L, Barbier J. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell. 2010;141(2):231–242. doi: 10.1016/j.cell.2010.01.043. [DOI] [PubMed] [Google Scholar]
- 15.Johannes L, Popoff V. Tracing the retrograde route in protein trafficking. Cell. 2008;135(7):1175–1187. doi: 10.1016/j.cell.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 16.Sandvig K, van Deurs B. Membrane traffic exploited by protein toxins. Annual review of cell and developmental biology. 2002;18:1–24. doi: 10.1146/annurev.cellbio.18.011502.142107. [DOI] [PubMed] [Google Scholar]
- 17.Mallard F, Antony C, Tenza D, Salamero J, Goud B, Johannes L. Direct pathway from early/recycling endosomes to the Golgi apparatus revealed through the study of shiga toxin B-fragment transport. The Journal of cell biology. 1998;143(4):973–990. doi: 10.1083/jcb.143.4.973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukhopadhyay S, Redler B, Linstedt AD. Shiga toxin-binding site for host cell receptor GPP130 reveals unexpected divergence in toxin-trafficking mechanisms. Molecular biology of the cell. 2013;24(15):2311–2318. doi: 10.1091/mbc.E13-01-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Natarajan R, Linstedt AD. A cycling cis-Golgi protein mediates endosome-to-Golgi traffic. Molecular biology of the cell. 2004;15(11):4798–4806. doi: 10.1091/mbc.E04-05-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachert C, Lee TH, Linstedt AD. Lumenal endosomal and Golgi-retrieval determinants involved in pH-sensitive targeting of an early Golgi protein. Molecular biology of the cell. 2001;12(10):3152–3160. doi: 10.1091/mbc.12.10.3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Linstedt AD, Mehta A, Suhan J, Reggio H, Hauri HP. Sequence and overexpression of GPP130/GIMPc: evidence for saturable pH-sensitive targeting of a type II early Golgi membrane protein. Molecular biology of the cell. 1997;8(6):1073–1087. doi: 10.1091/mbc.8.6.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukhopadhyay S, Bachert C, Smith DR, Linstedt AD. Manganese-induced trafficking and turnover of the cis-Golgi glycoprotein GPP130. Molecular biology of the cell. 2010;21(7):1282–1292. doi: 10.1091/mbc.E09-11-0985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mukhopadhyay S, Linstedt AD. Identification of a gain-of-function mutation in a Golgi P-type ATPase that enhances Mn2+ efflux and protects against toxicity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(2):858–863. doi: 10.1073/pnas.1013642108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tesh VL, Burris JA, Owens JW, Gordon VM, Wadolkowski EA, O'Brien AD, Samuel JE. Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infection and immunity. 1993;61(8):3392–3402. doi: 10.1128/iai.61.8.3392-3402.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boerlin P, McEwen SA, Boerlin-Petzold F, Wilson JB, Johnson RP, Gyles CL. Associations between virulence factors of Shiga toxin-producing Escherichia coli and disease in humans. Journal of clinical microbiology. 1999;37(3):497–503. doi: 10.1128/jcm.37.3.497-503.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lauvrak SU, Torgersen ML, Sandvig K. Efficient endosome-to-Golgi transport of Shiga toxin is dependent on dynamin and clathrin. Journal of cell science. 2004;117(Pt 11):2321–2331. doi: 10.1242/jcs.01081. [DOI] [PubMed] [Google Scholar]
- 27.Popoff V, Mardones GA, Tenza D, Rojas R, Lamaze C, Bonifacino JS, Raposo G, Johannes L. The retromer complex and clathrin define an early endosomal retrograde exit site. Journal of cell science. 2007;120(Pt 12):2022–2031. doi: 10.1242/jcs.003020. [DOI] [PubMed] [Google Scholar]
- 28.Saint-Pol A, Yelamos B, Amessou M, Mills IG, Dugast M, Tenza D, Schu P, Antony C, McMahon HT, Lamaze C, Johannes L. Clathrin adaptor epsinR is required for retrograde sorting on early endosomal membranes. Developmental cell. 2004;6(4):525–538. doi: 10.1016/s1534-5807(04)00100-5. [DOI] [PubMed] [Google Scholar]
- 29.Tai G, Lu L, Wang TL, Tang BL, Goud B, Johannes L, Hong W. Participation of the syntaxin 5/Ykt6/GS28/GS15 SNARE complex in transport from the early/recycling endosome to the trans-Golgi network. Molecular biology of the cell. 2004;15(9):4011–4022. doi: 10.1091/mbc.E03-12-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amessou M, Fradagrada A, Falguieres T, Lord JM, Smith DC, Roberts LM, Lamaze C, Johannes L. Syntaxin 16 and syntaxin 5 are required for efficient retrograde transport of several exogenous and endogenous cargo proteins. Journal of cell science. 2007;120(Pt 8):1457–1468. doi: 10.1242/jcs.03436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell. 1990;62(2):317–329. doi: 10.1016/0092-8674(90)90369-p. [DOI] [PubMed] [Google Scholar]
- 32.Ullrich O, Reinsch S, Urbe S, Zerial M, Parton RG. Rab11 regulates recycling through the pericentriolar recycling endosome. The Journal of cell biology. 1996;135(4):913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stenmark H, Parton RG, Steele-Mortimer O, Lutcke A, Gruenberg J, Zerial M. Inhibition of rab5 GTPase activity stimulates membrane fusion in endocytosis. The EMBO journal. 1994;13(6):1287–1296. doi: 10.1002/j.1460-2075.1994.tb06381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bujny MV, Popoff V, Johannes L, Cullen PJ. The retromer component sorting nexin-1 is required for efficient retrograde transport of Shiga toxin from early endosome to the trans Golgi network. Journal of cell science. 2007;120(Pt 12):2010–2021. doi: 10.1242/jcs.003111. [DOI] [PubMed] [Google Scholar]
- 35.Popoff V, Mardones GA, Bai SK, Chambon V, Tenza D, Burgos PV, Shi A, Benaroch P, Urbe S, Lamaze C, Grant BD, Raposo G, Johannes L. Analysis of articulation between clathrin and retromer in retrograde sorting on early endosomes. Traffic. 2009;10(12):1868–1880. doi: 10.1111/j.1600-0854.2009.00993.x. [DOI] [PubMed] [Google Scholar]
- 36.Seaman MN. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. The Journal of cell biology. 2004;165(1):111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y, Martin S, James DE, Hong W. GS15 forms a SNARE complex with syntaxin 5, GS28, and Ykt6 and is implicated in traffic in the early cisternae of the Golgi apparatus. Molecular biology of the cell. 2002;13(10):3493–3507. doi: 10.1091/mbc.E02-01-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fraser ME, Fujinaga M, Cherney MM, Melton-Celsa AR, Twiddy EM, O'Brien AD, James MN. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. The Journal of biological chemistry. 2004;279(26):27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 39.Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Molecular biology of the cell. 2000;11(2):467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tam P, Mahfoud R, Nutikka A, Khine AA, Binnington B, Paroutis P, Lingwood C. Differential intracellular transport and binding of verotoxin 1 and verotoxin 2 to globotriaosylceramide-containing lipid assemblies. Journal of cellular physiology. 2008;216(3):750–763. doi: 10.1002/jcp.21456. [DOI] [PubMed] [Google Scholar]
- 41.Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nature reviews Molecular cell biology. 2012;13(2):75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Feng Y, Chen D, Wandinger-Ness A. Rab11 is required for trans-golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Molecular biology of the cell. 1998;9(11):3241–3257. doi: 10.1091/mbc.9.11.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirst J, Motley A, Harasaki K, Peak Chew SY, Robinson MS. EpsinR: an ENTH domain-containing protein that interacts with AP-1. Molecular biology of the cell. 2003;14(2):625–641. doi: 10.1091/mbc.E02-09-0552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willett R, Kudlyk T, Pokrovskaya I, Schonherr R, Ungar D, Duden R, Lupashin V. COG complexes form spatial landmarks for distinct SNARE complexes. Nature communications. 2013;4:1553. doi: 10.1038/ncomms2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamaji T, Nishikawa K, Hanada K. Transmembrane BAX inhibitor motif containing (TMBIM) family proteins perturbs a trans-Golgi network enzyme, Gb3 synthase, and reduces Gb3 biosynthesis. The Journal of biological chemistry. 2010;285(46):35505–35518. doi: 10.1074/jbc.M110.154229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leyva-Illades D, Chen P, Zogzas CE, Hutchens S, Mercado JM, Swaim CD, Morrisett RA, Bowman AB, Aschner M, Mukhopadhyay S. SLC30A10 is a cell surface-localized manganese efflux transporter, and parkinsonism-causing mutations block its intracellular trafficking and efflux activity. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2014;34(42):14079–14095. doi: 10.1523/JNEUROSCI.2329-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]