

Abstract
Neonatal seizures are commonly associated with hypoxic-ischemic encephalopathy. Phenobarbital (PB)-resistance is common and poses a serious challenge in clinical management. Using a newly characterized neonatal mouse model of ischemic seizures, this study investigated a novel strategy to rescue PB-resistance. A small-molecule TrkB antagonist, ANA12, used to selectively and transiently block post-ischemic BDNF-TrkB signaling in vivo determined whether rescuing TrkB-mediated post-ischemic degradation of KCC2 rescued PB-resistant seizures. The anti-seizure efficacy of ANA12+PB was quantitated by; 1) electrographic seizure burden using acute continuous video-electroencephalograms and 2) post-treatment expression levels of KCC2 and NKCC1 using western blot analysis in postnatal day 7 and 10 (P7, P10) CD1 pups with unilateral carotid ligation. ANA12 significantly rescued PB-resistant seizures at P7, and improved PB-efficacy at P10. Single dose of ANA12+PB prevented the post-ischemic degradation of KCC2 up to 24 hours. As anticipated, ANA12 by itself had no anti-seizure properties and was unable to prevent KCC2 degradation at 24 hours without follow-on PB. This indicates that unsubdued seizures can independently lead to KCC2 degradation by non-TrkB dependent pathways. This study, for the first time as a proof-of-concept, reports the potential therapeutic value of KCC2 modulation for the management of PB-resistant seizures in neonates. Future investigations are required to establish the mechanistic link between ANA12 and the prevention of KCC2 degradation.
Keywords: KCC2, hypoxic-ischemic encephalopathy, PB-resistance, Bumetanide, neonatal seizure
Graphical Abstract
Introduction
The neonatal period is a critical window for the increased occurrence of seizures, with a prevalence of 1 to 3.5 per 1000 newborns (Glass et al., 2009). Neonatal seizures, acquired from diverse pathologies, are associated with severe morbidity and mortality (Uria-Avellanal et al., 2013; Ronen et al., 2007; Rakhade and Jensen, 2009). Hypoxic-ischemic encephalopathy (HIE) accounts for 50–60% of the cases for neonatal seizures (Tekgul et al., 2006). HIE-associated seizures in neonates are known for their resistance to the conventional 1st-line anti-seizure drugs. GABAA agonists like phenobarbital (PB) and benzodiazepines, which are consistently efficacious in adults (Booth and Evans, 2004; Sankar and Painter, 2005; Young et al., 1990), fail as anti-seizure agents in neonates (Boylan et al., 2013). Viable alternative strategies in overcoming these hurdles are currently lacking.
The pharmaco-resistance of 1st-line GABAA agonists in immature brains has been largely attributed to the developmental physiology of [Cl−]i that is 20–40mM higher in immature neurons than in mature neurons. The age-dependent upregulation of K+ Cl− co-transporter (KCC2), operating as a Cl− extruder in an electroneutral manner (Zhu et al., 2005), has been shown to play a critical role in the shift of GABAergic signaling from depolarizing to hyperpolarizing (Blaesse et al., 2009; Morita et al., 2014; Deisz et al., 2011). The expression of KCC2, which is predominantly neuronal, increases exponentially with conceptional age, especially perinatally, starting in the second half of gestation in humans to reach the significantly higher and stable adult levels (Morita et al., 2014). Similar developmental profiles for KCC2 have been noted in mice from the age of P3 to P15 where P7 is considered as term (Dzhala et al., 2005; Stein, 2004; Morita et al., 2014; Kang et al., 2015). Recent findings in both pre-clinical and human studies highlighting the important role of KCC2 in excitotoxicity have helped shape our novel strategy of modulating KCC2 to improve the efficacy of GABAA agonists in neonatal seizures (Puskarjov et al., 2014b; Pellegrino et al., 2011; Nardou et al., 2013; Pfeffer et al., 2009; Deisz et al., 2014; Kang et al., 2015; Deisz et al., 2011).
The activation of BDNF-TrkB signaling has been associated with KCC2 downregulation in diverse pathological environments associated with excitotoxicity (Semaan et al., 2015; Dai and Ma, 2014; Choe et al., 2015). Despite this known causal-effect relationship of BDNF-TrkB activation associated with degradation of KCC2 (Rivera et al., 2002), the novel strategy of preventing post-ischemic degradation of KCC2 by using a TrkB antagonist has never been tested in vivo. This may have primarily been due to the inability of old-generation TrkB antagonists to cross blood brain barrier and remain proteolytically stable. The identification of a small-molecule TrkB antagonist, ANA12 (Cazorla et al., 2011), that can selectively block BDNF-TrkB binding directly at the extracellular domain of TrkB receptor in a non-competitive manner, allowed to test the following hypotheses: 1. ANA12 will prevent the post-ischemic degradation of KCC2; 2. The prevention of KCC2 degradation will rescue PB-resistance. This study investigated the following “proof-of-concept” question: Can the age-dependent PB-resistance for P7 seizures in a mouse model of neonatal ischemic seizures be reversed by a small-molecule TrkB antagonist? Does adding a NKCC1 antagonist, bumetanide (BTN), help improve this rescue?
Materials and Methods
Study approval
All experimental procedures were conducted in compliance with guidelines by the Committee on the Ethics of Animal Experiments, Johns Hopkins University (Permit Number: A3272-01) and all protocols were approved by the Animal Care and Use of Committee (IACUC) of Johns Hopkins. All litters of CD1 mice with dams were purchased from Charles River Laboratories Inc. (Wilmington, MA). Newly born litters of pups were delivered at postnatal 3 days old (P3) and were allowed to acclimate. Food and water were provided ad libitum. Equal numbers of male and female pups were introduced into the study. The number of pups and litters used in this study are listed in Suppl. Table 1.
Surgical procedure for ischemic insult and sub-dermal EEG electrode implantation
The surgical protocol was similar to the previously published work (Kadam et al., 2010; Kang et al., 2015). At P7 or P10, animals were subjected to permanent unilateral ligation of right common carotid artery using 6-0 surgisilk (Fine Science Tools, BC Canada) under isoflurane anesthesia. The outer skin was closed with 6-0 monofilament nylon (Covidien, MA), and lidocaine was applied as an additional local anesthetic. Under continued anesthesia, animals were then implanted with 3 sub-dermal EEG scalp electrodes: 1 reference, 1 ground, and 1 recording overlying the parietal cortex; the electrographic results reported in this study are solely from one recording site, and therefore do not reflect hippocampal seizure activity. Wire electrodes made for use in humans (IVES EEG; Model # SWE-L25 –MA, USA) were implanted sub-dermally and fixed in position with adhesive. Pups were then allowed to recover from anesthesia which took a couple of minutes. Finally, animals were tethered to a preamplifier by connecting sub-dermal electrodes within a recording chamber for 3h of video-EEG, maintained at 36°C with isothermal pads. At the end of the recording session, the animals were returned to the dam after removal of the sub-dermal electrodes. The average duration of anesthesia for both ligation and electrode implantation in this study added up to 16.18 ± 4.37 min. There is a known mortality rate of ~10–20% associated with the surgical procedure of carotid-ligation and severe seizures in the model (Kadam et al., 2009). The mortality rates for the pups 24h after surgery were n/n = 9/45 (20%) pups at P7 (6 males and 3 females) and n/n= 7/52 (13%) pups at P10 (3 males and 4 females), and were not significantly different, by age nor by sex in this study (p=0.42 and p=0.57 respectively; Fisher’s exact test, two-tailed). Mortality rates following the surgical procedure were also not significantly different by treatment (ligated control vs. treated group; p=0.26).
Experimenal paradigm
Following unilateral permanent carotid-ligation, vehicle [5% dimethyl sulfoxide (DMSO)], ANA12 (2.5mg/kg; N-[2-[[(Hexahydro-2-oxo-1H-azepin-3-yl) amino] carbonyl] phenyl] benzo[b]thiophene-2-carboxamide), or 0.9% sodium chloride (Saline) was injected intraperitoneally (IP). The general outline of the experimental paradigm is depicted in Figure 1 (top panel, I). Pups were randomly assigned to three different treatment regimens over the 3h acute and continuous EEG recording period: 1) vehicle + saline + saline, 2) ANA12 + saline + saline, and 3) ANA12 + PB + BTN. Injection of 1st drug occurred immediately after ligation, and the subsequent injections followed every hour (i.e.; Figure 1, II B&D). Each color in the bar graphs (Figure 1&3) represents the parameters (i.e.; total EEG seizure burdens) quantitated for the 60 min duration of EEG recordings during each hour: 1st h (black bars), 2nd h (gray bars), and 3rd h (white bars).
Figure 1. Effect of ANA12+PB on the total seizure burden at P7 and P10 I.

Schematics of the experimental design. Experimental paradigm for the acute ligation surgery and synchronous video-EEG recordings with the time-course of drug administrations and the durations for each procedure. Arrowheads indicate time-points of drug administration after carotid-ligation and brain-harvesting for 24h WB data. II. A–D. Total time spent seizing on EEG was quantitated as seizure burden at P7 and P10 over a 3h period, and was represented by bar graphs (black bar=baseline, gray=2nd h, and white = 3rd h) A&C) vehicle (5% DMSO) and ANA12-alone treatment groups. The data from A&C were pooled to form a ligate-control group represented in B&D. B&D) ligate control vs. ANA12+PB+BTN. Within-group comparison was done using repeated measures ANOVA (i.e., *=P<0.05; **=P<0.01; ***=P<0.001). Brackets (@) denote significant between-group comparisons at each hour (independent sample t-tests; P<0.05).
Figure 3. The anti-seizure efficacy of ANA12+PB was not different by sex.

The efficacy of ANA12+PB on total seizure burden was evaluated by sex at P7 and P10. A&C. Males at both ages of P7 and P10 responded significantly to ANA12+PB. However, at P10, BTN administration following PB weakened the statistical significance of seizure suppression achieved by PB. B&D. ANA12+PB efficacy was significant in females at both P7 and P10. Additionally, a significant temporal increase in the total seizure burden over time-course of 3h period was detected in P7 females, but was absent in P7 males. At P7, BTN aggravated PB-subdued seizures in females such that PB-efficacy was lost. ANA12+ PB efficacy was not significantly different between males and females. (Repeated measures ANOVA; *= P<0.05; ** =P<0.01; ***=P<0.001).
The vehicle, 5% DMSO (Cat. No. 472301) was made in phosphate buffered saline (v/v; pH 7.4). ANA12 (2.5mg/kg) was dissolved in the vehicle. ANA12 was stored at −20°C in aliquots ready for use (Sigma-Aldrich; Cat. No. SML0209). PB was (25mg/kg: Sigma-Aldrich; Cat. No. P5178) dissolved in phosphate buffered saline (made on the day of experiment) injection followed 1h after the vehicle or ANA12 injection, and BTN injection [0.1–0.2mg/kg dissolved in 100% alcohol; aliquoted and stored at −20°C; protocol similar to previous study (Kang et al., 2015)] followed 1h after PB injection.
In vivo synchronous video-EEG recording and analyses
EEG recording was acquired using Sirenia Acquisition software (v 1.6.4) with synchronous video capture (Pinnacle Technology Inc. KS, USA). Data acquisition was done with sampling rates of 400Hz that had a pre-amplifier gain of 100 and the filters of 1Hz high-pass and 60Hz low-pass to remove ambient noise. The data were scored by binning the raw EEG trace in 10 sec epochs. Similar to previous study (Kang et al., 2015), seizures were defined as electrographic ictal events that consisted of rhythmic spikes of high amplitude, diffuse peak frequency of ≥ 7–8 Hz (i.e.; peak frequency detected by automated spectral power analysis) lasting ≥ 6 seconds. Short duration burst activity lasting < 6 seconds (brief runs of epileptiform discharge) was not included for seizure burden calculations in this study.
Western blot at 3h and 24h post-ligation
All animals were anesthetized with chloral hydrate (90 mg/ml; IP) before being transcardially perfused. The whole brains of P7 and P10 pups were harvested at either 3h or 24h post-ligation, and were frozen as ipsilateral and contralateral hemisphere. The brains were stored in −80°C until further use. Homogenized whole brain lysates were suspended in cell lysis buffer with 10% protease/phosphatase inhibitor cocktail. Total protein concentrations were quantified through Bradford Assay (Bio-Rad) at 570nm wavelength, and the samples were diluted for 50ug of protein at 20ul of loading volume for gel electrophoresis.
Samples were run on 4–20% gradient 1.5mm 15 wells SDS gels (Invitrogen) for 100–120 min with 130V, and were transferred onto nitrocellulose membranes for overnight wet-transfer for minimum of 18 hours at 30V. After the transfer, the nitrocellulose membranes underwent 1h blocking step in odyssey buffer, before overnight incubation in primary antibodies: rabbit α-KCC2 (1:1000, Millipore; Cat. No. 07-432); rabbit α-NKCC1 (1:500, Millipore, Cat. No. AB3560P); mouse α-actin (1:10000, LI-COR Biosciences, Cat. No. 926-42214). On the next day, nitrocellulose membranes were washed with TBS containing tween detergent, and then were incubated in chemiluminescence secondaries for 1h (1:5000 for both goat α-mouse 680LT and goat α-rabbit 800CW, LI-COR Biosciences). Chemiluminescent protein bands were analyzed using the Odyssey infrared imaging system 2.1 (LI-COR Biosciences). The optical density of each protein sample was normalized to the actin bands run on each lane for internal control. The expression levels of the proteins of interest in ipsilateral hemispheres were normalized to the same in contralateral hemispheres for each pup. The human brain was recently shown to express two splice variants of NKCC1 (a and b) and the NKCC1 probe used in this study and all previous studies referenced here can only detect the NKCC1a isoform because the targeted epitope site of this probe overlaps with exon 21 that is spliced in the dominant isoform NKCC1b (Kaila et al., 2014; Morita et al., 2014). A reliable pan-NKCC1 probe is not currently available.
A smaller sample of naïve age-matched controls (n=8, 4 each at P8 and P11, equal sexes) was run with the 24h WB data (brains harvested at P8 and P11, i.e.; 24h after P7 and P10 ligations). KCC2 and NKCC1 expression in the contralateral uninjured hemispheres from the ligate control pups (n=8, 4 each at P8 and P11, equal sexes) was not significantly different from their age-matched naïve controls (p=0.49 and p=0.23; KCC2 and NKCC1 respectively). This pilot established that contralateral hemispheres were ideal controls for normalization of KCC2 and NKCC1 expression in ipsilateral injured hemispheres for this study similar to previously reported conclusions in another ischemia model (Jaenisch et al., 2010).
Statistics
All statistical tests were performed using SPSS21 (IBM, Armonk, NY U.S). Group means of total seizure burden, number of ictal events, and ictal duration within each treatment group were compared using repeated measures ANOVA with Bonferroni’s post-hoc correlations. Differences with p value less than or equal to the alpha at 0.05 (p < 0.05) were considered statistically significant (repeated measures ANOVA; * = p < 0.05 ** = p < 0.01; *** = p < 0.001). The assumption of sphericity for data was confirmed by Mauchly’s test, similar to a previous study (Kang et al., 2015). Pairwise t-tests for group means were also reported. Independent sample t-tests were used to make comparisons between groups: i.e.; seizure burdens for P7 vs. P10. Western blot quantifications were compared among treatment groups using One-way ANOVAs. The normality of the data for t-tests was confirmed by Shapiro-Wilk test. Correlation analyses were performed using non-parametric comparisons (Spearman’s test, two-tail). For all data represented as bar graph in figures, the values of ± 1 standard error of mean (SEM) are denoted as error bars.
Results
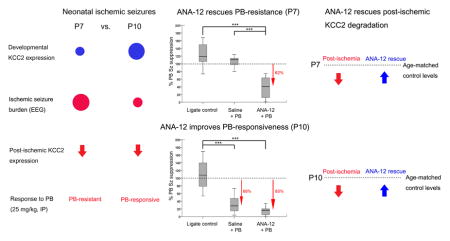
Small-molecule TrkB antagonist ANA12, rescued PB-resistance of seizures at P7 and improved PB-efficacy at P10
The effect of an acute single post-ischemic dose of ANA12 on PB-resistant seizures was evaluated at P7 and P10. PB administration at 1h following ischemic insult in the ANA12 treated group significantly reduced the seizure burden at P7 and P10 (Fig. 1B&D, P7: F1,20=91.47, P=0.001; P10: F1,20=87.75, P=0.001 repeated measures ANOVA; Suppl. Fig. 2:). ANA12+PB significantly suppressed seizures by 62% in P7 ischemic pups and by 85% in P10 ischemic pups (Fig. 2A&B, P7: F2,46=48.79, P=0.001; P10: F2,48=72.17 P=0.001 One-way ANOVA; Suppl. Fig. 2). In the absence of ANA12, PB failed to suppress seizures at P7, replicating the hallmark of the age-dependent PB-resistance in the ischemic model (Kang et al., 2015). Similar to previous findings in the model, PB by itself suppressed seizures by 68% (Fig. 2B, P=0.001) in P10 pups. ANA12+PB improved PB-efficacy to from 68% to 85%, however this improvement was not significant (P=0.08). This improvement is worth noting, as the 80% reduction in seizure burden is a standard desirable efficacy used clinically to determine a successful anti-seizure therapeutic intervention (Pressler et al., 2015). The seizure burdens quantitated here were additionally evaluated by the number of ictal events and the average ictal durations that constituted each seizure burden. The data showed that the anti-seizure efficacy of ANA12+PB at both ages was driven by a significant reduction in the number of ictal events (Suppl. Fig. 3E&F). The mean ictal durations did not change from baseline to the post-PB hour (Suppl. Fig. 3C–D and G–H). Therefore, acute TrkB inhibition significantly rescued PB-resistance at P7 and improved PB-efficacy at P10 by significantly reducing the number of ictal events post-treatment. Similar to previous study (Kang et al., 2015), PB-efficacy had no effect on ictal durations.
Figure 2. ANA12 rescued PB-resistance at P7 and improved PB-efficacy at P10.

The effect of ANA12 on PB-efficacy was evaluated as percent PB seizure suppression over baseline: % PB seizure suppression = 100*[Post-PB seizure burden/baseline seizure burden]. A. PB by itself failed to subdue ischemic seizures at P7. ANA12+PB significantly subdued ischemic seizures by 62% (One-way ANOVA; *** = P<0.001). B. PB alone subdued seizures by 68% at P10. ANA12+PB improved this efficacy to 85%. This improved PB-efficacy however was not statistically significant when compared to the efficacy of PB alone. The sample size for saline+PB group was n=13 and n=11 for P7 and P10 respectively; n for ligate control and treated group was same as listed in Fig. 1 (see Suppl. Table 1). Sz: Seizure.
The between-group comparison of vehicle vs. ANA12-alone group did not differ significantly for any of the parameters of baseline seizure burden, the number of ictal events, and mean ictal duration at either P7 or P10 (Fig. 1A&C). Additionally, Fisher’s exact test comparing post-treatment mortality rate did not show a statistical significance (P=0.58 for P7, P=1.00 for P10). Based on these observations, the data for vehicle and ANA12-alone group were pooled to form the “ligate control” group (n= 15 and 19 for P7 and P10 respectively).
The efficacy of ANA12-alone as an anti-seizure agent was evaluated. As expected, ANA12-alone without PB administration did not have any anti-seizure effect at either P7 or P10. The baseline seizure burdens of ANA12-alone group did not differ significantly from that of vehicle group at either age (i.e. P7 F1,14=0.004 P=1.00 and P10 F1,17=0.097 P=0.63; One-way ANOVA). Additionally, ANA12 did not suppress the baseline seizure burden prior to PB administration in the treated groups at either age (Fig. 1B&D). Therefore, an acute single dose of ANA12, in the absence of subsequent PB administration, failed to suppress ischemic seizures.
To evaluate whether vehicle or ANA12-alone had any effect on seizure susceptibility, the latency to onset of ischemic seizures following unilateral carotid ligation was evaluated in each pup. Neither vehicle nor ANA12-alone significantly altered the seizure onset latency. The effect of post-ligation drugs (saline, vehicle, and ANA12) on the latency to the 1st-detected seizure were not statistically different at either age tested: P7; saline (321.7 ± 58.8 sec), vehicle (321.7 ± 171.3 sec), ANA12 (202.3 ± 38.8 sec), P10; saline (150.73 ± 40.4 sec), vehicle (164.4 ± 44.4 sec), ANA12 (245.8 ± 39.8 sec; One-way ANOVA: P7, F2,46=1.296 P=0.28; P10, F2,48=1.240 P=0.30 respectively). The latency to seizures in each treatment group did not significantly differ by age either. In summary, vehicle and ANA12 did not have any significant effects on age-dependent seizure susceptibility when evaluated by seizure onset latency in the model.
The effect of adding an adjunct NKCC1 antagonist, BTN, was also evaluated at the 3rd h of post-ischemia recording. BTN, delivered 1h after PB, failed to act as an efficacious anti-seizure adjunct at either P7 or P10, similar to a previously reported findings (Kang et al., 2015). BTN did not reduce either the total seizure burden or the number of ictal events (Fig. 1B&D, white bars; Suppl. Fig. 2) at P7. BTN administration significantly blunted PB-efficacy by increasing the seizure burden in P10 pups (Fig. 1D; white bar F1,20=9.795 P=0.01, Suppl. Fig. 2). This aggravation occurred due to an increase in the number of ictal events following BTN administration (Suppl. Fig. 3F; white bar F1,20=73.625 P=0.02). Therefore, the blockage of NKCC1 after the successful rescue of PB-resistance did not help enhance PB-efficacy; instead, it resulted in an age- and sex-dependent aggravation of PB-subdued seizures, replicating previous observations in the model (Kang et al., 2015).
ANA12 significantly reversed PB-resistance at P7 and improved PB-efficacy at P10 in both sexes
The developmental expression profile of chloride co-transporters has been reported to be sexually dimorphic (Galanopoulou, 2008; Kang et al., 2015; McCarthy et al., 2012). To evaluate the role of biological sex in the efficacy of ANA12, the data presented in Fig. 1B and D were evaluated by sex. PB-efficacy was significant for both sexes at P7 and P10 (Fig. 3A–D, Suppl. Fig. 4A–B and E–F; P<0.001 for all). In P7 pups, BTN significantly blunted ANA12+PB efficacy only in females (Fig. 3B). At P10, BTN blunted ANA12+PB anti-seizure efficacy in males but not in females. (Suppl. Fig. 4B; white bars). In summary, PB-efficacy was not different by sex and age. However, the BTN-associated aggravation of ischemic seizures was sex-dependent.
Age- and sex-dependent seizure susceptibility to ischemic seizures was unaltered by TrkB antagonist
ANA12, administered after the onset of ischemia did not alter the significant age-dependent seizure susceptibility previously documented in this model (Kang et al., 2015). Total seizure burden for ligate control group (i.e.; over 3h duration post-ischemia) was significantly higher in P7 (213%) pups compared to P10 pups (Figure 1A&C). A significantly higher seizure susceptibility was observed in P7 male pups compared to P10 male pups (F15=17.79 P=0.001; independent t-test 2-tailed), and this was not significant in females (Suppl. Fig. 5A&B). Therefore, the age- and sex-dependent seizure susceptibility to ischemic seizures established in the model was not altered by ANA12.
ANA12 followed by PB rescues the ischemic downregulation of KCC2 expression at both P7 and P10
Approximately ~20% downregulation of KCC2 expression in the ipsilateral ischemic hemisphere compared to the contralateral hemisphere was previously documented in the model when evaluated at 24h post-ischemia (Kang et al., 2015). The effect of TrkB inhibition on post-ischemic KCC2 degradation was evaluated. Quantification of KCC2 expression at 24h after an ischemic insult indicated that the post-ischemic degradation of KCC2 expression was significantly prevented in the treated group at both P7 and P10 (Fig. 4A&C). At P7, both vehicle and ANA12-alone group exhibited KCC2 degradation that was approximately 15–18% lower compared to the contralateral hemisphere; which was statistically significant between ANA12-alone and the treated group (Fig. 4A; F2,26=4.352 P<0.05). The expression levels of KCC2 in the PB-resistance rescued group was ~110%, and therefore similar to the contralateral hemisphere. At P10, both vehicle and ANA12-alone group displayed KCC2 degradation, while the treated group showed significant rescue. However, the vehicle group showed ~30% KCC2 degradation whereas ANA12-alone group showed ~12% KCC2 degradation, compared to the contralateral hemisphere. The KCC2 downregulation was only significant between the vehicle group and the treated group (Fig. 4C). The ligate control group representing the vehicle and ANA12-alone data pooled together similar to the seizure burden data in Fig. 1B&D, showed significant post-ischemic degradation of KCC2 expression (16.2% and 20.0% for P7 and P10 respectively) when compared to the ANA12+PB treated group (F1,26=4.852 P=0.04 and F1,29=12.25 P=0.002; P7 and P10 respectively; One-way ANOVA). NKCC1 expression was also quantitated in the same manner as it was done for KCC2, and no statistically significant differences were detected amongst treatment groups (Fig 4. B&D). Additionally, the overall percent NKCC1 change in the treated group was also not significantly different by age (P7 vs. P10; F1,23=1.25 P=0.28; One-way ANOVA). In summary, a single acute dose of ANA12, in the presence of follow-on PB, significantly prevented the post-ischemic degradation of KCC2 expression even at 24h post-ischemia, and did not significantly alter NKCC1 expression.
Figure 4. ANA12+PB rescued KCC2 expression but had no effect on NKCC1 expression at 24h.

A–D. Bar graphs represent mean expression of KCC2 and NKCC1 in ipsilateral hemisphere normalized to the contralateral at 24h after ischemia. β-actin was used as an internal control. The protein bands for both co-transporters were: 1) normalized to the level of actin for the same sample, 2) followed by normalization of ipsilateral co-transporter expression levels to contralateral hemisphere of the same pup A&C. The post-ischemic degradation of KCC2 expression (~20%) was prevented in the ANA12+PB+BTN treated group at P7 and P10. B&D. Post-ischemic expression levels of NKCC1 in the treated group were not significantly different from ligate controls (One-way ANOVA; *=P<0.05; ***=P<0.001).
KCC2 expression levels were also evaluated acutely at 3h post-ischemia. An additional group of saline treatment was included in this experiment, to establish the KCC2 expression in untreated ischemic pups. The results were similar to the findings for the 24h data. At P7, all groups except the ANA12+PB treated group underwent approximately 15–20% downregulation of KCC2 expression (Suppl. Fig. 6A). A similar 20% loss of KCC2 surface expression has been shown to decrease KCC2 function by 50% (Gilad et al., 2015). The KCC2 expression levels for saline group and ANA12-alone group significantly differed from the treated group, but the vehicle group did not (F3,25=4.583 P<0.05; One-way ANOVA). At P10, the vehicle and ANA12-alone groups also underwent degradation of KCC2, however no statistical significance was detected when compared to the ANA12+PB treated group (Suppl. Fig. 6C). Similar to the 24h results, NKCC1 expression among groups was not statistically different (Suppl. Fig. 6B&D).
Since severe seizures can independently degrade KCC2 in a non-TrkB dependent pathway (Puskarjov et al., 2012), a pilot experiment to evaluate the efficacy of ANA12 to rescue PB-resistance after the occurrence of status-like seizures was additionally conducted. In contrast to experimental paradigm described in Fig. 1, ANA12 was administered 1h after PB in P7 seizing pups (i.e.; 2h after repetitive ischemic seizures, PB+ANA12). The post-PB administration of ANA12 protocol, failed to subdue the PB-resistant seizures at P7 (P=1.00; n=3). Additionally, this treatment protocol also failed to rescue post-ischemic KCC2 degradation when evaluated at 24h (p=1.00; n=5) similar to the vehicle-treated pups. These data indicate that the TrkB-pathway - mediated KCC2 degradation (~20%) starts early after ischemia and is irreversible by administration of ANA-12 after emergence of PB-resistance.
Correlation of PB-efficacy and KCC2 expression
Correlations between seizure suppression and KCC2 degradation were evaluated to determine whether the PB-efficacy was driven by the prevention of KCC2 degradation. P10 pups showed a significant correlation between efficacious seizure suppression and rescue of KCC2 expression at P10 (Fig. 5D; P=0.04, r=0.6). This correlation was not significant at P7 (Fig 5A–D; P=0.35, r=0.2). This differential correlation may be in part due to age-dependent upregulation of KCC2 in naïve brains during this developmental window (Kang et al., 2015). Naïve P10 pups have been reported to have a higher age-dependent expression of KCC2 compared to P7 (Kang et al., 2015; Dzhala et al., 2005). However, the percentage of KCC2 degradation following ischemia at both P7 and P10 were similar (i.e.; ~20%) and not significantly different (F1,50=0.04 P=0.84; One-way ANOVA). No significant correlations between seizure suppression and NKCC1 expression were detected at P7 or P10 (Fig. 5E–H), which was expected from the inconsistent (Fig. 5E&G) post-ischemic expression levels of NKCC1 detected at both ages for all treatment groups (Fig. 4B&D and Suppl. Fig. 6B&D).
Figure 5. Significant correlation between rescue of KCC2 degradation at 24h and seizure suppression was detected.

The percent change of KCC2 expression of an ipsilateral hemisphere [% change = 100*(ipsi - contra/contra KCC2 expression)] was correlated to the percent seizure suppression [% baseline seizure burden = 100*(baseline seizure burden - post-PB seizure burden)/baseline seizure burden)]. The color gradient (blue or red) applied to the background represents the data distribution in the positive (blue) or negative (red) direction of KCC2 expression after ligation/treatment. The dotted gray lines within each scatter plot represent the mean values for percent seizure suppression in each group (x axis). A&C. The ligated control group at both ages of P7 and P10 showed the post-ischemic degradation of KCC2 expression evident by predominant distribution of dots on the red gradient. B&D. The post-ischemic KCC2 expression of the treated group was rescued at both ages of P7 and P10, as seen in the positive-shift of the dots towards the blue gradient (red arrows). The correlation between the percent KCC2 expression and percent seizure suppression (black arrows) was significant at P10 (Spearman’s test; P=0.04) but not at P7. When P7 data were binned by baseline seizure burden (<1200sec vs. ≥1200sec), the correlation became significant for the pups that seized <1200sec (p=0.02; data not shown).
The efficacy of PB seizure suppression was significantly dependent on the baseline seizure burden at P7 but not at P10 (Spearman’s test: i.e.; P7; P=0.001, P10; P=0.30). P7 pups with severe baseline seizure burden (i.e.; >1200/3600 sec) were less responsive to ANA12+PB with a 54.9±6.6% seizure suppression (n=10, P<0.001) in contrast to their age-matched ligated-pups with lower baseline seizure burden (<1200/3600 sec) that responded with a 68.5±8.3% seizure suppression to ANA12+PB (n=11, P<0.001). Using this binning for moderate and severe baseline seizure severity at P7 (i.e.; seizing for < or > 1/3 of the total baseline hour), correlations between seizure suppression and rescue of KCC2 degradation became evident. Ligated pups at P7 that seized for less than 1/3rd of the hour before PB administration, showed a significant positive correlation with rescue of KCC2 degradation (P=0.02, r=0.8) similar to that detected in P10 rescued pups. In contrast, in pups which seized for >1/3rd of the hour, this correlation was not only lost significance but became negative (P=0.31, r=−0.4). In summary, if the baseline seizure burden was status-like at P7 (i.e.; >1200/3600 sec), the ANA12+PB anti-seizure efficacy did not correlate significantly with percent KCC2 expression (Fig. 5B). In contrast, the percent change in the expression levels of NKCC1 did not correlate significantly with the percent post-PB seizure suppression (Fig. 5F&H), and further binning by baseline seizure severity did not reveal any significant correlations either. Ligated pups at P10 responded significantly to PB regardless of the severity of their baseline seizures burden. The age-dependent seizure susceptibility and PB-resistance has been previously reported and this study replicated that hallmark characteristic for the model (Kang et al., 2015).
Discussion
Using a small-molecule TrkB antagonist and a single systemic (IP) dosing protocol, this study reports for the first time, the proof-of-concept results for the pharmaco-modulation of KCC2 expression as a novel strategy for preventing the emergence of PB-resistant seizures in neonatal ischemia. The salient findings are: 1) a single post-insult dose of TrkB antagonist, ANA12, significantly rescued the PB-resistance of ischemic seizures at P7; 2) the combination of a single dose ANA12+PB at 1h, not only prevented the acute post-ischemic degradation of KCC2 expression at 3h but also maintained age-dependent KCC2 expression 24h later; 3) the TrkB antagonist by itself could neither prevent the occurrence of ischemic seizures nor rescue post-ischemic KCC2 degradation, suggesting that PB-resistant seizures can independently degrade KCC2 by non-TrkB dependent pathways; 4) At P10, ANA12 helped improve PB-efficacy to >80%; and 5) NKCC1 antagonist BTN failed to further improve PB-efficacy as an adjunct therapy at both ages. These findings highlight the pivotal role of KCC2 in seizure susceptibility and the emergence of PB-resistant seizures in neonatal ischemia. Therefore, transient pharmaco-modulation of KCC2 with the goal to restore the developmental profile of KCC2 upregulation may be a promising strategy for preventing the emergence of PB-resistant seizures in HIE.
A single dose of ANA12 reversed PB-resistance at P7
Age-dependent PB-resistance of ischemic seizures was the hallmark of the neonatal ischemic seizure model used here (Kang et al., 2015). ANA12 successfully reversed PB-resistance at P7 at a dose of 2.5mg/kg. Therefore, the inability of old-generation TrkB antagonists crossing the blood brain barrier was successfully circumvented by the small molecule TrkB antagonist, ANA12. The brain bioavailability of ANA12 after systemic injection, examined in adult mouse brains indicates that ANA12 administered IP at 0.5mg/kg dose was active as early as 0.5h [400nM] up to 6h [10nM] (Cazorla et al., 2011). Additionally, a single dose of 0.5mg/kg ANA12, in the same study, suppressed the ratio of phospho-/total TrkB activity in the whole brain by 8% at 2h and 25% at 4h. The post-insult transient opening of blood brain barrier in the immature brain has been shown to result in the increased bioavailability of small and large molecules (Ek et al., 2015), and may suggest a similar enhancement in the pharmacokinetics of the single dose ANA12 given acutely in this model of neonatal ischemia. In summary, acute TrkB inhibition by a single dose of ANA12, achieved significant rescue of PB-resistance even when PB was given 1h after the occurrence of severe recurrent seizures.
ANA12 required 5% DMSO to stay in solution. DMSO can independently act as an anti-epileptic drug by modifying brain bioelectric activity, specifically at above 50% concentration but not at below 10% (Carletti et al., 2013). To evaluate the anti-seizure effect of 5% DMSO used as vehicle for ANA12 in this study, a separate vehicle-treatment group of 5% DMSO was investigated. 5% DMSO administered following ischemia did not have any anti-seizure effect, and the baseline seizure burdens remained unaltered and similar to previously published reports for the model (Kang et al., 2015).
A single dose of ANA12 with PB prevented both early and late downregulation of KCC2 expression after ischemia
The BDNF-TrkB pathway has been shown to be responsible for excitoxicity related degradation of KCC2 in in vitro experiments (Rivera et al., 2002). This degradation has been shown to occur within minutes of an ischemic insult in vitro (Puskarjov et al., 2012; Rivera et al., 2004). BDNF-TrkB pathway has also been implicated in epileptogenesis, neuropathic pain, and psychiatric disorders (Andero et al., 2011; Heinrich et al., 2011; Ferrini and De, 2013; Doyon et al., 2013; Zhou et al., 2012; McNamara and Scharfman, 2012; Mao et al., 2010). Ischemic insults resulted in the downregulation of KCC2 expression levels in several studies (Rivera et al., 2002; Rivera et al., 2004; Jaenisch et al., 2010; Kang et al., 2015). Neonatal ischemia resulted in a transient downregulation of KCC2 expression that recovered over the period of a few days and caught up with the age-dependent KCC2 upregulation occurring during this developmental window (Kang et al., 2015). Similar KCC2 degradation has also been reported to occur hours after the onset of status epilepticus (Puskarjov et al., 2012; Puskarjov et al., 2014a). However, those studies used chemoconvulsants to induce seizures that also resulted in an early upregulation of KCC2 similar to a hypoxia-only model of neonatal seizures (Cleary et al., 2013). The mechanism underlying this early upregulation of KCC2 was not explored by either group (Puskarjov et al., 2014a). However, recent research has reported that the kainate-dependent upregulation of KCC2 was a homeostatic response to increased neuronal firing during early seizures and occurs via the extracellular signal-regulated kinase (ERK)1/2 signaling cascade (Besser et al., 2009; Gilad et al., 2015; Chorin et al., 2011) that may help improve inhibition.
In contrast, when status epilepticus continued to occur over a period of >240min in the Puskarjov et al., study (Puskarjov et al., 2014a), KCC2 degradation via the calpain-mediated pathway became evident. These findings indicate that early KCC2 homeostatic upregulation may fail in etiologies associated with energy deprivation like ischemia. Additionally, after severe and repeated seizures have occurred, seizures alone may degrade KCC2 function through non-TrkB mediated pathways. However, our results also indicate that if the PB-resistant seizures are subdued early by blocking TrkB, the late onset KCC2 degradation can also be prevented. The effect of ANA12 on seizure suppression may also occur through other downstream effects triggered by TrkB inhibition, as TrkB activation is associated with multiple pathways such as ERK and JAK/STAT.
Repeated seizures can independently lead to KCC2 degradation
Prevention of KCC2 degradation only occurred in the presence of both TrkB inhibition and efficacious seizure suppression. In contrast, the vehicle and ANA12-alone groups both underwent significant KCC2 downregulation in the ipsilateral hemisphere (Fig. 4) similar to untreated pups (Kang et al., 2015). ANA12-alone failed to prevent the post-ischemic KCC2 degradation in pups where ischemic seizures continued. This indicates that prolonged recurrent seizures are capable of degrading KCC2 expression likely through a non-TrkB mediated pathway (Puskarjov et al., 2012). Calpain-dependent downregulation of KCC2 had been shown to occur following 3h of status epilepticus induced seizures. Calpain is a calcium-sensitive protease which is activated in conditions associated with long duration status epilepticus (Puskarjov et al., 2012). Similarly, the failure of ANA12 to improve PB-efficacy when given 1h after PB showed that when many repeated ischemic seizures have already occurred (i.e.; 2 hours of repetitive post-ischemic seizures), ANA12 failed to reverse KCC2 downregulation even in the presence of PB, indicating that TrkB pathway related KCC2 degradation starts early after ischemia. Both of these observations support the previous conclusions that severe seizures are independently detrimental in ischemia and can lead to KCC2 downregulation initiated by both TrkB and non-TrkB dependent pathways (Puskarjov et al., 2012; Lee et al., 2011). Since the calpain pathway has been shown to be triggered later during a severe status-like seizure cascade in a slice culture model (Puskarjov et al., 2012) and in acute slices derived from mice after kainate induced status(Puskarjov et al., 2014a), it may be temporally different from the BDNF/TrkB pathway in the sequence of initiation post-insult. Inability of calpain inhibitors to cross the BBB is currently prohibitive to test the hypothesis in an in vivo model. Additionally, other pathways may be related to the finding that ANA12-alone failed to prevent KCC2 downregulation. One possible example that can explain the effect of ANA12 on seizures can be JAK/STAT pathway activated by BDNF that results in altered subunits of GABAA receptors (Lund et al., 2008), and thus altered neuronal response to anti-seizure agents.
At P10, with the age-dependent lower seizure burden as compared to P7, and developmentally higher expression profile of KCC2 in naïve brains, the ANA12-alone group showed a trend towards the recovery of KCC2 expression compared to the vehicle groups at both 3h and 24h, however it was not statistically significant. This indicates that TrkB inhibition, in the absence of PB, may be able to marginally rescue KCC2 degradation by itself at P10 but not at P7. This difference may be due to the age-dependent susceptibility to ischemic seizure burden and the developmental profile of KCC2 upregulation (i.e.; P10>P7).
Correlation between rescue of KCC2 downregulation and efficacious seizure suppression
The results of this study indicated that when seizures were efficaciously suppressed, there was an associated significant rescue of KCC2 degradation at both P7 and P10 (Fig. 5). This was not true for NKCC1, highlighting the critical role of Cl− extrusion played by KCC2 in determining the efficacy of GABAA agonists like PB. However, it is important to note that the currently available NKCC1 probes can only detect the non-dominant isoform NKCC1a (Morita et al., 2014). The dominant isoform NKCC1b expressed in the brain is a spice variant with missing exon 21 that remains undetected with currently available probes. Unlike NKCC1a, NKCC1b has been shown to have stable expression throughout development and adulthood in human brains (Kaila et al., 2014): Human Brain Transcriptome website. These findings from human brain research, indicate that the previous interpretations related to the developmental downregulation of the immature form Cl− transporter, NKCC1 with age, in animal research models may not be accurate (Dzhala et al., 2005; Jensen, 2009). A reliable pan-NKCC1 probe is not currently available, but is an area of active research. Therefore, in the absence of a probe capable of detecting simultaneous NKCC1a and NKCC1b expression levels, interpretations related to the expression profile of NKCC1 and the BTN-inefficacy replicated in this study, remain elusive. In contrast, the KCC2 probe used in this study, detects both brain isoforms with rigorous specificity (Williams et al., 1999). In addition, KCC2 protein can stay internalized in cytosolic vesicles or remain inactive in the plasmalemma (Kovacs et al., 2014; Gulyas et al., 2001), therefore, additional strategies to increase the membrane insertion kinetics of KCC2 proteins may also be an appropriate alternative to test the proof-of-concept approach investigated here.
The significant positive correlation of seizure suppression to rescue of KCC2 degradation at P10 was absent at P7. However, when data were binned by baseline seizure severity (< or ≥1200 sec), there was a strong positive correlation of the rescue of KCC2 degradation to the severity of the baseline seizure burden before PB was administered at P7. This indicated that when a P7 ligated pup seized for >1/3rd of the first hour (i.e.; status-like severity) before PB administration, non-TrkB related pathways like calpain-mediated KCC2 degradation, may get initiated earlier, weakening the significant correlation detected in pups with relatively lower seizure burdens. This observation highlights the important role of seizure severity in the emergence of refractoriness. Severe repeated seizures can degrade KCC2 by multiple pathways (Rivera et al., 2002; Puskarjov et al., 2012). Therefore, both the etiology and severity of seizures dictate the pharmacological consequences in neonates (Kang SK and Kadam SD, 2014; Cleary et al., 2013; Puskarjov et al., 2014b).
Role of KCC2 in brain development and long-term sequelae of transient KCC2 down-regulation
KCC2 is known to play a role in early development, spine formation, interneuron migration, and synaptogenesis (Stein et al., 2004; Bortone and Polleux, 2009; Sun et al., 2013; Li et al., 2007). KCC2 is robustly expressed in the dendritic spines of cortical neurons (Kovacs et al., 2014; Gauvain et al., 2011; Baldi et al., 2010; Gulyas et al., 2001). KCC2 also plays a crucial role in development of interneurons and their response to injury (Inamura et al., 2012; Bortone and Polleux, 2009). The stronger tolerance of CA1 interneurons to ischemic injury compared to pyramidal cells reported in the hippocampus, may suggest that the higher density of KCC2 in CA1 interneurons may drive the exceptional resistance of parvalbumin-positive interneurons to excitotoxic injury associated with ischemia (Papp et al., 2008). Ischemic insults can result in long-term damage to interneurons that become evident after months (Arabadzisz and Freund, 1999; Fukuda et al., 1993). The transient ischemia-related KCC2 degradation during early development detected in this model may have neurological consequences that are not evident in the acute stages but may manifest as late-onset co-morbidities (Robinson et al., 2010; Tao et al., 2012; Hyde et al., 2011).
KCC2 expression was significantly lost in the brain samples of preterm infants with white matter damage (Robinson et al., 2010). Therefore, KCC2 degradation may be a common pathology associated with perinatal brain insults that are excitotoxic (Jantzie et al., 2014; Kang et al., 2015). Pharmaco-modulation of KCC2 with the goal of restoring normal expression levels, therefore, could have both acute and long-term beneficial effects. The known action of ANA12 as an anti-depressant and anxiolytic in rodent studies (Cazorla et al., 2011) supports the further evaluation of the long-term effects of ANA12 following neonatal use in this model. The prevention of KCC2 degradation documented in this study should be clearly differentiated from KCC2 overexpression, because upregulation of KCC2, above its normal developmental expression levels, that is not physiological, may lead to a precocious maturation of neuronal circuits in immature brains. Ischemic seizures are transient during the neonatal period (Boylan et al., 2013), and therefore a single acute intervention to prevent KCC2 degradation similar to that reported here during the critical stage may be sufficient.
Blocking NKCC1 fails to improve PB-efficacy
The efficacy of BTN as an adjuvant anti-seizure agent has been controversial (Vanhatalo et al., 2009; Tollner et al., 2015; Puskarjov et al., 2014b; Kang SK and Kadam SD, 2014; Kang et al., 2015; Chabwine and Vanden Eijnden, 2011). The recent termination of NEMO clinical trial [NCT01434225] reported the inefficacy of BTN for HIE seizures (Pressler et al., 2015). Our previous studies have also reported a similar BTN inefficacy for acute ischemic seizures and detected an age- and sex-specific aggravation of PB-subdued seizures in P10 CD1 pups (Kang et al., 2015). These findings were replicated in the current study. In spite of the improved PB-efficacy elicited by acute TrkB inhibition, BTN still failed to act as an adjunct therapy. The expression level of NKCC1a was not significantly altered regardless of the treatment paradigm or anti-seizure efficacy, further supporting the importance of KCC2 rather than NKCC1 in regulating Cl− gradient and modulating seizures (Brandt et al., 2010; Deisz et al., 2014; Deisz et al., 2011). BTN has also been reported to have poor brain bioavailability and short half-life (Cleary et al., 2013). However, recent studies evaluating the efficacy of a BTN pro-drug designed to have improved brain bioavailability have reported no clear anti-seizure effect (Tollner et al., 2015). BTN also has been shown to have non-safety issues associated with ototoxicity detected in the NEMO trial (Pressler et al., 2015), adding to an unfavorable risk-benefit ratio as an adjuvant in neonates. NKCC1 serves a critical function in the circuit formation during early development (Wang and Kriegstein, 2011), and is also expressed systemically (Kaila et al., 2014). The long-term side-effects to blocking NKCC1 function during development, although not fully investigated, indicate potential pitfalls. Although early literature on the subject touted the important role of NKCC1 for Cl− regulation (Dzhala et al., 2005; Kahle and Staley, 2008), a lot of follow-on research has shown that to not be true. NKCC1 KO mice have neurons that regulate Cl− very well (Nardou et al., 2011), indicating that NKCC1 by itself has a non-significant role in neuronal Cl− gradients. In sharp contrast, KCC2 KO mice have severe seizures at birth and high mortality (Woo et al., 2002). The results of our study show that in ischemia where energy deprivation occurs early, KCC2, rather than NKCC1 plays the critical role in chloride regulation. This finding matches the new emergent data in the field (Deisz et al., 2011).
Conclusion
This study reports the successful rescue of PB-resistance associated with the rescue of KCC2 degradation in a model of neonatal ischemic seizures. Pre-clinical studies of neonatal seizures utilizing various translational models have reported: a) differential alteration of KCC2 expression, and b) differential severity of the seizure burdens in response to various seizure induction methods (Kang SK and Kadam SD, 2014). The main findings of this “proof-of-concept” study suggest a critical association between the anti-seizure efficacy of GABA-agonists and post-ischemic KCC2 degradation; however, further studies are required to confirm the causal link of TrkB antagonism to the rescue of the KCC2 degradation following excitotoxic injuries. An ongoing debate in clinical management of neonatal seizures is whether they can independently harm the developing brain and how aggressively do they need to be managed?, When compared to the results from other pre-clinical models (Loscher et al., 2013; Cleary et al., 2013; Kang SK and Kadam SD, 2014; Dzhala et al., 2008; Puskarjov et al., 2012), studies in this model suggest that the etiology and severity of neonatal seizures dictate their deleterious effects. Therefore, severe seizure burdens commonly reported in HIE (Boylan et al., 2013) may independently cause KCC2 degradation that leads to the emergence of refractory seizures. This study highlights the potential of KCC2 pharmaco-modulation as a novel therapeutic strategy in treating PB-resistant seizures in neonates.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health under Award Number R21HD073105 (SDK).
Abbreviations
- BDNF
Brain-derived neurotrophic factor
- BTN
Bumetanide
- HIE
hypoxic-ischemic encephalopathy
- KCC2
K+ Cl− co-transporter
- NKCC1
Na+K+ 2Cl− co-transporter
- PB
phenobarbital
- TrkB
Tropomyosin receptor kinase B
Footnotes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Reference List
- (2009a) BTN clinical trial in Europe
- (2009b) BTN clinical trial in USA
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am J Psychiatry. 2011;168:163–172. doi: 10.1176/appi.ajp.2010.10030326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arabadzisz D, Freund TF. Changes in excitatory and inhibitory circuits of the rat hippocampus 12–14 months after complete forebrain ischemia. Neuroscience. 1999;92:27–45. doi: 10.1016/s0306-4522(98)00736-2. [DOI] [PubMed] [Google Scholar]
- Baldi R, Varga C, Tamas G. Differential distribution of KCC2 along the axo-somato-dendritic axis of hippocampal principal cells. Eur J Neurosci. 2010;32:1319–1325. doi: 10.1111/j.1460-9568.2010.07361.x. [DOI] [PubMed] [Google Scholar]
- Besser L, Chorin E, Sekler I, Silverman WF, Atkin S, Russell JT, Hershfinkel M. Synaptically released zinc triggers metabotropic signaling via a zinc-sensing receptor in the hippocampus. J Neurosci. 2009;29:2890–2901. doi: 10.1523/JNEUROSCI.5093-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaesse P, Airaksinen MS, Rivera C, Kaila K. Cation-chloride cotransporters and neuronal function. Neuron. 2009;61:820–838. doi: 10.1016/j.neuron.2009.03.003. [DOI] [PubMed] [Google Scholar]
- Booth D, Evans DJ. Anticonvulsants for neonates with seizures. Cochrane Database Syst Rev. 2004:CD004218. doi: 10.1002/14651858.CD004218.pub2. [DOI] [PubMed] [Google Scholar]
- Bortone D, Polleux F. KCC2 expression promotes the termination of cortical interneuron migration in a voltage-sensitive calcium-dependent manner. Neuron. 2009;62:53–71. doi: 10.1016/j.neuron.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan GB, Stevenson NJ, Vanhatalo S. Monitoring neonatal seizures. Semin Fetal Neonatal Med. 2013;18:202–208. doi: 10.1016/j.siny.2013.04.004. [DOI] [PubMed] [Google Scholar]
- Brandt C, Nozadze M, Heuchert N, Rattka M, Loscher W. Disease-Modifying Effects of Phenobarbital and the NKCC1 Inhibitor Bumetanide in the Pilocarpine Model of Temporal Lobe Epilepsy. J Neurosci. 2010;30:8602–8612. doi: 10.1523/JNEUROSCI.0633-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carletti F, Ferraro G, Rizzo V, Cannizzaro C, Sardo P. Antiepileptic effect of dimethyl sulfoxide in a rat model of temporal lobe epilepsy. Neurosci Lett. 2013;546:31–35. doi: 10.1016/j.neulet.2013.04.031. [DOI] [PubMed] [Google Scholar]
- Cazorla M, Premont J, Mann A, Girard N, Kellendonk C, Rognan D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121:1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabwine JN, Vanden Eijnden S. A claim for caution in the use of promising bumetanide to treat neonatal seizures. J Child Neurol. 2011;26:657–658. doi: 10.1177/0883073811401395. [DOI] [PubMed] [Google Scholar]
- Choe KY, Han SY, Gaub P, Shell B, Voisin DL, Knapp BA, Barker PA, Brown CH, Cunningham JT, Bourque CW. High salt intake increases blood pressure via BDNF-mediated downregulation of KCC2 and impaired baroreflex inhibition of vasopressin neurons. Neuron. 2015;85:549–560. doi: 10.1016/j.neuron.2014.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorin E, Vinograd O, Fleidervish I, Gilad D, Herrmann S, Sekler I, Aizenman E, Hershfinkel M. Upregulation of KCC2 activity by zinc-mediated neurotransmission via the mZnR/GPR39 receptor. J Neurosci. 2011;31:12916–12926. doi: 10.1523/JNEUROSCI.2205-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary RT, Sun H, Huynh T, Manning SM, Li Y, Rotenberg A, Talos DM, Kahle KT, Jackson M, Rakhade SN, Berry G, Jensen FE. Bumetanide enhances phenobarbital efficacy in a rat model of hypoxic neonatal seizures. PLoS One. 2013;8:e57148. doi: 10.1371/journal.pone.0057148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai S, Ma Z. BDNF-trkB-KCC2-GABA pathway may be related to chronic stress-induced hyperalgesia at both the spinal and supraspinal level. Med Hypotheses. 2014;83:772–774. doi: 10.1016/j.mehy.2014.10.008. [DOI] [PubMed] [Google Scholar]
- Deisz RA, Lehmann TN, Horn P, Dehnicke C, Nitsch R. Components of neuronal chloride transport in rat and human neocortex. J Physiol. 2011;589:1317–1347. doi: 10.1113/jphysiol.2010.201830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deisz RA, Wierschke S, Schneider UC, Dehnicke C. Effects of VU0240551, a novel KCC2 antagonist, and DIDS on chloride homeostasis of neocortical neurons from rats and humans. Neuroscience. 2014;277:831–841. doi: 10.1016/j.neuroscience.2014.07.037. [DOI] [PubMed] [Google Scholar]
- Doyon N, Ferrini F, Gagnon M, De KY. Treating pathological pain: is KCC2 the key to the gate? Expert Rev Neurother. 2013;13:469–471. doi: 10.1586/ern.13.40. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Brumback AC, Staley KJ. Bumetanide enhances phenobarbital efficacy in a neonatal seizure model. Ann Neurol. 2008;63:222–235. doi: 10.1002/ana.21229. [DOI] [PubMed] [Google Scholar]
- Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, Delpire E, Jensen FE, Staley KJ. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11:1205–1213. doi: 10.1038/nm1301. [DOI] [PubMed] [Google Scholar]
- Ek CJ, D’Angelo B, Baburamani AA, Lehner C, Leverin AL, Smith PL, Nilsson H, Svedin P, Hagberg H, Mallard C. Brain barrier properties and cerebral blood flow in neonatal mice exposed to cerebral hypoxia-ischemia. J Cereb Blood Flow Metab. 2015;35:818–827. doi: 10.1038/jcbfm.2014.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrini F, De KY. Microglia control neuronal network excitability via BDNF signalling. Neural Plast. 2013;2013:429815. doi: 10.1155/2013/429815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, Nakano S, Yoshiya I, Hashimoto PH. Persistent degenerative state of non-pyramidal neurons in the CA1 region of the gerbil hippocampus following transient forebrain ischemia. Neuroscience. 1993;53:23–38. doi: 10.1016/0306-4522(93)90281-j. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res. 2008;80:99–113. doi: 10.1016/j.eplepsyres.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvain G, Chamma I, Chevy Q, Cabezas C, Irinopoulou T, Bodrug N, Carnaud M, Levi S, Poncer JC. The neuronal K-Cl cotransporter KCC2 influences postsynaptic AMPA receptor content and lateral diffusion in dendritic spines. Proc Natl Acad Sci U S A. 2011;108:15474–15479. doi: 10.1073/pnas.1107893108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad D, Shorer S, Ketzef M, Friedman A, Sekler I, Aizenman E, Hershfinkel M. Homeostatic regulation of KCC2 activity by the zinc receptor mZnR/GPR39 during seizures. Neurobiol Dis. 2015 doi: 10.1016/j.nbd.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass HC, Pham TN, Danielsen B, Towner D, Glidden D, Wu YW. Antenatal and intrapartum risk factors for seizures in term newborns: a population-based study, California 1998–2002. J Pediatr. 2009;154:24–28. doi: 10.1016/j.jpeds.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyas AI, Sik A, Payne JA, Kaila K, Freund TF. The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci. 2001;13:2205–2217. doi: 10.1046/j.0953-816x.2001.01600.x. [DOI] [PubMed] [Google Scholar]
- Heinrich C, Lahteinen S, Suzuki F, Anne-Marie L, Huber S, Haussler U, Haas C, Larmet Y, Castren E, Depaulis A. Increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol Dis. 2011;42:35–47. doi: 10.1016/j.nbd.2011.01.001. [DOI] [PubMed] [Google Scholar]
- Hyde TM, Lipska BK, Ali T, Mathew SV, Law AJ, Metitiri OE, Straub RE, Ye T, Colantuoni C, Herman MM, Bigelow LB, Weinberger DR, Kleinman JE. Expression of GABA signaling molecules KCC2, NKCC1, and GAD1 in cortical development and schizophrenia. J Neurosci. 2011;31:11088–11095. doi: 10.1523/JNEUROSCI.1234-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura N, Kimura T, Tada S, Kurahashi T, Yanagida M, Yanagawa Y, Ikenaka K, Murakami F. Intrinsic and extrinsic mechanisms control the termination of cortical interneuron migration. J Neurosci. 2012;32:6032–6042. doi: 10.1523/JNEUROSCI.3446-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch N, Witte OW, Frahm C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke. 2010;41:e151–e159. doi: 10.1161/STROKEAHA.109.570424. [DOI] [PubMed] [Google Scholar]
- Jantzie LL, Getsy PM, Firl DJ, Wilson CG, Miller RH, Robinson S. Erythropoietin attenuates loss of potassium chloride co-transporters following prenatal brain injury. Mol Cell Neurosci. 2014;61:152–162. doi: 10.1016/j.mcn.2014.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen FE. Neonatal seizures: an update on mechanisms and management. Clin Perinatol. 2009;36:881–900. vii. doi: 10.1016/j.clp.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam SD, Mulholland JD, Smith DR, Johnston MV, Comi AM. Chronic brain injury and behavioral impairments in a mouse model of term neonatal strokes. Behav Brain Res. 2009;197:77–83. doi: 10.1016/j.bbr.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadam SD, Smith-Hicks CL, Smith DR, Worley PF, Comi AM. Functional integration of new neurons into hippocampal networks and poststroke comorbidities following neonatal stroke in mice. Epilepsy Behav. 2010;18:344–357. doi: 10.1016/j.yebeh.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Price TJ, Payne JA, Puskarjov M, Voipio J. Cation-chloride cotransporters in neuronal development, plasticity and disease. Nat Rev Neurosci. 2014;15:637–654. doi: 10.1038/nrn3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SK, Kadam SD. Pre-clinical models of acquired neonatal seizures: Differential effects of of injury on function of chloride co-transporters. Austin Journal of Cerebrovas Dis & Stroke. 2014;1:1–6. [PMC free article] [PubMed] [Google Scholar]
- Kang SK, Markowitz GJ, Kim ST, Johnston MVD, Kadam SD. Age-dependent susceptibility to phenobarbital-resistant neonatal seizures: role of chloride co-transporters. Frontiers in Cellular Neuroscience. 2015;9 doi: 10.3389/fncel.2015.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs K, Basu K, Rouiller I, Sik A. Regional differences in the expression of K(+)-Cl(−) 2 cotransporter in the developing rat cortex. Brain Struct Funct. 2014;219:527–538. doi: 10.1007/s00429-013-0515-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Deeb TZ, Walker JA, Davies PA, Moss SJ. NMDA receptor activity downregulates KCC2 resulting in depolarizing GABAA receptor-mediated currents. Nat Neurosci. 2011;14:736–743. doi: 10.1038/nn.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Khirug S, Cai C, Ludwig A, Blaesse P, Kolikova J, Afzalov R, Coleman SK, Lauri S, Airaksinen MS, Keinanen K, Khiroug L, Saarma M, Kaila K, Rivera C. KCC2 interacts with the dendritic cytoskeleton to promote spine development. Neuron. 2007;56:1019–1033. doi: 10.1016/j.neuron.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Lin G, Bella AJ, Lue TF, Lin CS. Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: part 2. J Sex Med. 2006;3:821–827. doi: 10.1111/j.1743-6109.2006.00292.x. [DOI] [PubMed] [Google Scholar]
- Loscher W, Puskarjov M, Kaila K. Cation-chloride cotransporters NKCC1 and KCC2 as potential targets for novel antiepileptic and antiepileptogenic treatments. Neuropharmacology. 2013;69:62–74. doi: 10.1016/j.neuropharm.2012.05.045. [DOI] [PubMed] [Google Scholar]
- Mao LM, Fibuch EE, Wang JQ. Decoding BDNF-LTP coupling in cocaine addiction. Neuron. 2010;67:679–681. doi: 10.1016/j.neuron.2010.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy MM, Arnold AP, Ball GF, Blaustein JD, de Vries GJ. Sex differences in the brain: the not so inconvenient truth. J Neurosci. 2012;32:2241–2247. doi: 10.1523/JNEUROSCI.5372-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Scharfman HE. Temporal Lobe Epilepsy and the BDNF Receptor, TrkB. 2012 [PubMed] [Google Scholar]
- Morita Y, Callicott JH, Testa LR, Mighdoll MI, Dickinson D, Chen Q, Tao R, Lipska BK, Kolachana B, Law AJ, Ye T, Straub RE, Weinberger DR, Kleinman JE, Hyde TM. Characteristics of the cation cotransporter NKCC1 in human brain: alternate transcripts, expression in development, and potential relationships to brain function and schizophrenia. J Neurosci. 2014;34:4929–4940. doi: 10.1523/JNEUROSCI.1423-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardou R, Ferrari DC, Ben-Ari Y. Mechanisms and effects of seizures in the immature brain. Semin Fetal Neonatal Med. 2013;18:175–184. doi: 10.1016/j.siny.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Papp E, Rivera C, Kaila K, Freund TF. Relationship between neuronal vulnerability and potassium-chloride cotransporter 2 immunoreactivity in hippocampus following transient forebrain ischemia. Neuroscience. 2008;154:677–689. doi: 10.1016/j.neuroscience.2008.03.072. [DOI] [PubMed] [Google Scholar]
- Pellegrino C, Gubkina O, Schaefer M, Becq H, Ludwig A, Mukhtarov M, Chudotvorova I, Corby S, Salyha Y, Salozhin S, Bregestovski P, Medina I. Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J Physiol. 2011;589:2475–2496. doi: 10.1113/jphysiol.2010.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezet S, Malcangio M, Lever IJ, Perkinton MS, Thompson SW, Williams RJ, McMahon SB. Noxious stimulation induces Trk receptor and downstream ERK phosphorylation in spinal dorsal horn. Mol Cell Neurosci. 2002;21:684–695. doi: 10.1006/mcne.2002.1205. [DOI] [PubMed] [Google Scholar]
- Pfeffer CK, Stein V, Keating DJ, Maier H, Rinke I, Rudhard Y, Hentschke M, Rune GM, Jentsch TJ, Hubner CA. NKCC1-dependent GABAergic excitation drives synaptic network maturation during early hippocampal development. J Neurosci. 2009;29:3419–3430. doi: 10.1523/JNEUROSCI.1377-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressler RM, Boylan GB, Marlow N, Blennow M, Chiron C, Cross JH, De Vries LS, Hallberg B, Hellstrom-Westas L, Jullien V, Livingstone V, Mangum B, Murphy B, Murray D, Pons G, Rennie J, Swarte R, Toet MC, Vanhatalo S, Zohar S. Bumetanide for the treatment of seizures in newborn babies with hypoxic ischaemic encephalopathy (NEMO): an open-label, dose finding, and feasibility phase 1/2 trial. Lancet Neurol. 2015 doi: 10.1016/S1474-4422(14)70303-5. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Ahmad F, Kaila K, Blaesse P. Activity-dependent cleavage of the K-Cl cotransporter KCC2 mediated by calcium-activated protease calpain. J Neurosci. 2012;32:11356–11364. doi: 10.1523/JNEUROSCI.6265-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puskarjov M, Ahmad F, Khirug S, Sivakumaran S, Kaila K, Blaesse P. BDNF is required for seizure-induced but not developmental up-regulation of KCC2 in the neonatal hippocampus. Neuropharmacology. 2014a doi: 10.1016/j.neuropharm.2014.09.005. [DOI] [PubMed] [Google Scholar]
- Puskarjov M, Kahle KT, Ruusuvuori E, Kaila K. Pharmacotherapeutic targeting of cation-chloride cotransporters in neonatal seizures. Epilepsia. 2014b;55:806–818. doi: 10.1111/epi.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakhade SN, Jensen FE. Epileptogenesis in the immature brain: emerging mechanisms. Nat Rev Neurol. 2009;5:380–391. doi: 10.1038/nrneurol.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M. BDNF-induced TrkB activation down-regulates the K+-Cl− cotransporter KCC2 and impairs neuronal Cl− extrusion. J Cell Biol. 2002;159:747–752. doi: 10.1083/jcb.200209011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Thomas-Crusells J, Li H, Emri Z, Sipila S, Payne JA, Minichiello L, Saarma M, Kaila K. Mechanism of activity-dependent downregulation of the neuron-specific K-Cl cotransporter KCC2. J Neurosci. 2004;24:4683–4691. doi: 10.1523/JNEUROSCI.5265-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S, Mikolaenko I, Thompson I, Cohen ML, Goyal M. Loss of cation-chloride cotransporter expression in preterm infants with white matter lesions: implications for the pathogenesis of epilepsy. J Neuropathol Exp Neurol. 2010;69:565–572. doi: 10.1097/NEN.0b013e3181dd25bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronen GM, Buckley D, Penney S, Streiner DL. Long-term prognosis in children with neonatal seizures: a population-based study. Neurology. 2007;69:1816–1822. doi: 10.1212/01.wnl.0000279335.85797.2c. [DOI] [PubMed] [Google Scholar]
- Sankar R, Painter MJ. Neonatal seizures: after all these years we still love what doesn’t work. Neurology. 2005;64:776–777. doi: 10.1212/01.WNL.0000157320.78071.6D. [DOI] [PubMed] [Google Scholar]
- Semaan S, Wu J, Gan Y, Jin Y, Li GH, Kerrigan JF, Chang YC, Huang Y. Hyperactivation of BDNF-TrkB signaling cascades in human hypothalamic hamartoma (HH): a potential mechanism contributing to epileptogenesis. CNS Neurosci Ther. 2015;21:164–172. doi: 10.1111/cns.12331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack SE, Pezet S, McMahon SB, Thompson SW, Malcangio M. Brain-derived neurotrophic factor induces NMDA receptor subunit one phosphorylation via ERK and PKC in the rat spinal cord. Eur J Neurosci. 2004;20:1769–1778. doi: 10.1111/j.1460-9568.2004.03656.x. [DOI] [PubMed] [Google Scholar]
- Stein V. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Stein V, Hermans-Borgmeyer I, Jentsch TJ, Hubner CA. Expression of the KCl cotransporter KCC2 parallels neuronal maturation and the emergence of low intracellular chloride. J Comp Neurol. 2004;468:57–64. doi: 10.1002/cne.10983. [DOI] [PubMed] [Google Scholar]
- Sun C, Zhang L, Chen G. An unexpected role of neuroligin-2 in regulating KCC2 and GABA functional switch. Mol Brain. 2013;6:23. doi: 10.1186/1756-6606-6-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao R, Li C, Newburn EN, Ye T, Lipska BK, Herman MM, Weinberger DR, Kleinman JE, Hyde TM. Transcript-specific associations of SLC12A5 (KCC2) in human prefrontal cortex with development, schizophrenia, and affective disorders. J Neurosci. 2012;32:5216–5222. doi: 10.1523/JNEUROSCI.4626-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekgul H, Gauvreau K, Soul J, Murphy L, Robertson R, Stewart J, Volpe J, Bourgeois B, du Plessis AJ. The current etiologic profile and neurodevelopmental outcome of seizures in term newborn infants. Pediatrics. 2006;117:1270–1280. doi: 10.1542/peds.2005-1178. [DOI] [PubMed] [Google Scholar]
- Tollner K, Brandt C, Erker T, Loscher W. Bumetanide is not capable of terminating status epilepticus but enhances phenobarbital efficacy in different rat models. Eur J Pharmacol. 2015;746:78–88. doi: 10.1016/j.ejphar.2014.10.056. [DOI] [PubMed] [Google Scholar]
- Uria-Avellanal C, Marlow N, Rennie JM. Outcome following neonatal seizures. Semin Fetal Neonatal Med. 2013;18:224–232. doi: 10.1016/j.siny.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Vanhatalo S, Hellstrom-Westas L, de Vries LS. Bumetanide for neonatal seizures: Based on evidence or enthusiasm? Epilepsia. 2009;50:1292–1293. doi: 10.1111/j.1528-1167.2008.01894.x. [DOI] [PubMed] [Google Scholar]
- Wang DD, Kriegstein AR. Blocking early GABA depolarization with bumetanide results in permanent alterations in cortical circuits and sensorimotor gating deficits. Cereb Cortex. 2011;21:574–587. doi: 10.1093/cercor/bhq124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA. The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem. 1999;274:12656–12664. doi: 10.1074/jbc.274.18.12656. [DOI] [PubMed] [Google Scholar]
- Young GB, Gilbert JJ, Zochodne DW. The significance of myoclonic status epilepticus in postanoxic coma. Neurology. 1990;40:1843–1848. doi: 10.1212/wnl.40.12.1843. [DOI] [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Byun HS, Chen H, Li L, Han HD, Lopez-Berestein G, Sood AK, Pan HL. N-methyl-D-aspartate receptor- and calpain-mediated proteolytic cleavage of K+-Cl− cotransporter-2 impairs spinal chloride homeostasis in neuropathic pain. J Biol Chem. 2012;287:33853–33864. doi: 10.1074/jbc.M112.395830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Lovinger D, Delpire E. Cortical neurons lacking KCC2 expression show impaired regulation of intracellular chloride. J Neurophysiol. 2005;93:1557–1568. doi: 10.1152/jn.00616.2004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.