

Summary
Protein 4.1R plays an important role in maintaining the mechanical properties of the erythrocyte membrane. We analysed the expression of Kell blood group protein in erythrocytes from a patient with hereditary elliptocytosis associated with complete 4.1R deficiency (4.1(−) HE). Flow cytometry and Western blot analyses revealed a severe reduction of Kell. In vitro pull down and co-immunoprecipitation experiments from erythrocyte membranes showed a direct interaction between Kell and 4.1R. Using different recombinant domains of 4.1R and the cytoplasmic domain of Kell, we demonstrated that the R46R motif in the juxta-membrane region of Kell binds to lobe B of the 4.1R FERM domain. We also observed that 4.1R deficiency is associated with a reduction of XK and DARC (also termed ACKR1) proteins, the absence of the glycosylated form of the urea transporter B and a slight decrease of band 3. The functional alteration of the 4.1(−) HE erythrocyte membranes was also determined by measuring various transport activities. We documented a slower rate of HCO3−/Cl− exchange, but normal water and ammonia transport across erythrocyte membrane in the absence of 4.1. These findings provide novel insights into the structural organization of blood group antigen proteins into the 4.1R complex of the human red cell membrane.
Keywords: erythrocyte membrane, macromolecular complex, 4.1R, Kell protein, blood group antigens
Introduction
Protein 4.1R is a major protein of the erythrocyte skeleton and is composed of four functional domains: the N-terminal 30 kDa domain, referred to as the FERM (F for 4.1R protein, E for ezrin, R for radixin and M for moesin) domain, the 16 kDa domain, the 10 kDa spectrin-actin binding (SAB) domain and the C-terminal 24 kDa domain (CTD) (Conboy, et al 1986, Takakuwa 2000). The analysis of the crystal structure of the FERM domain led to the identification of three globular lobes. Lobe A corresponds to the first 90 amino acids and includes the binding sites for band 3 and the Na+/H+ exchanger (NHE1). Lobe B (amino acids 91–190) contains the binding sites for GPC/D, XK and DARC (Duffy antigen receptor for chemokines, also termed ACKR1) proteins. The COOH-terminal lobe (Lobe C, amino acids 191–280) contains the binding site for p55 protein. Recently, Gauthier, et al (2011) proposed that the Kell protein is present in the 4.1R complex through its interaction with XK. Indeed, XK protein is covalently linked to the Kell glycoprotein by a single disulfide bond (XK Cys347–Kell Cys72) (Russo, et al 1998).
The Kell glycoprotein (93 kDa) is a type II single-span membrane protein that carries the Kell blood group system comprising 28 distinct antigens (Ji, et al, 2015) including the K1 (Kell) and K2 (cellano) antigens. The two distinct proteins, Kell and XK are responsible for expressing the Kell blood group antigens (Lee, et al 2001). Kell protein exhibits an ectodomain that is composed of two domains: the well-conserved membrane-proximal zinc endopeptidase domain and a more variable membrane-distal domain (Lee, et al 2003). In addition, the Kell protein shares a consensus sequence with the large family of zinc endopeptidases and has endothelin-3 converting enzyme activity of type II membrane glycoproteins. Gene disruption in mice provided evidence that cellular divalent cation regulation is functionally coupled to the Kell/XK system in erythrocytes and loss of this complex might contribute to the acanthocytosis seen in McLeod syndrome (Rivera, et al 2013). A rare phenotype termed Kellnull (Ko) is characterized by the absence of Kell protein and Kell antigens from the red cell membrane and diminished amounts of XK protein (Khamlichi, et al 1995, Redman, et al 1999). The absence of any clinical symptoms in Kellnull individuals suggest that the Kell enzyme activity is not essential for cell survival or that other metalloproteinase could compensate the lack of this protein (Lee, et al 2001).
The findings from the present study using 4.1(−) HE (4.1Rnull) human erythrocytes have enabled us to obtain novel insights into the 4.1R complex organization. Indeed, we describe a detailed novel direct interaction involving the skeletal protein 4.1R and the Kell blood group protein. Furthermore, the functional activities of AQP1, Band 3 and RhAG were measured in the 4.1(−) HE erythrocyte membrane and we show a decrease in the extent of anion exchange.
Materials and methods
Materials
Primers used in polymerase chain reaction (PCR) and mutagenesis experiments were provided from Eurogentec (Seraing, Belgium). The QuikChange Site-Directed Mutagenesis Kit was from Stratagene (La Jolla, CA, USA). The Protease Inhibitor Cocktail, the pGEX-3X-5 vector and the glutathione-sepharose 4B beads were purchased from Amersham Pharmacia Biotech, (Buckinghamshire, UK). NuPAGE® Novex Bis-Tris Gels were purchased from Invitrogen (Carlsbad, CA, USA). The Band 3 inhibitor, DIDS (4,4 2-diisothiocyanatostilbene-2,2 2-disulfonic acid disodium salt), was purchased from Sigma-Aldrich (St Louis, MO, USA).
Blood samples
Frozen red blood cell (RBC) samples from three healthy donors (normal), one 4.1R(−) one AQP1null and one Rhnull individual were obtained from the Centre National de Référence pour les Groupes Sanguins (CNRGS, Paris, France). The patient with 4.1R(−)has been previously reported (Tchernia, et al 1981).
Antibodies
Murine monoclonal antibodies (MAbs) were as follows: anti-Kell (clone 5A11) directed toward the common, intracellular, amino-terminal region of the protein (Jaber, et al 1991); anti-GPC (1F6) and anti-FY6 monoclonal antibodies (clone name: NaM185-2C3) were kindly provided by D. Blanchard (Etablissement Français du Sang, Nantes (EFS), France); anti-KEL2 (clone K2F7) was obtained from Dr P. Rubinstein (New York, NY, USA); anti-CD47, clone 3E12, was from Bioatlantic (Nantes, France); anti-RhAG LA18.18 (Dr Von dem Borne, Amsterdam, The Netherlands); anti-Band 3 (BRIC14) (sc-59476, Santa Cruz Biotechnology, Inc., Heidelberg, Germany); anti-GPA, R18 (Dr Edwards, University of Cambridge, Cambridge, UK); anti-GAPDH (cat-4300, Ambion, Austin, TX, USA). Anti-LU (clone F241) was from our Institute in collaboration with Dr D. Blanchard. Detection of urea transporter (UT-B) in erythrocyte membranes was performed with the mouse monoclonal anti-extracellular epitope of UT-B (anti-Jk3).
Polyclonal antibodies (PAbs): The rabbit antibodies raised against the 43 kDa N-terminal cytoplasmic domain of human Band 3 and against protein 4.1R (el Ouggouti, et al 1992) were generous gifts of Dr D. Dhermy (INSERM, U665, Paris, France); anti-4.2 was described previously (Hayette, et al 1995, Jons and Drenckhahn 1992); MPC8 raised against the C-terminal region of the Rh polypeptides was described previously (Hermand, et al 1993); rabbit anti-Kx2 obtained by immunization against a synthetic peptide containing XK residues 111 to 127 located in the large extracellular loop of the protein XK (Carbonnet, et al 1997); anti- aquaporin (AQP1) antibody (AB3065, Chemicon, Billerica, MA, USA); anti-Co3 (Sar serum) was from the CNRGS (Institut National de la Transfusion Sanguine, Paris, France). For UT-B, the antibody was raised against a 20-amino acid peptide corresponding to the C-terminal sequence of rat UT-B (Trinh-Trang-Tan, et al 2002). Anti-GLUT-1 was from Interchim (1:100 dilution; Montluçon, France; cat. U47950).
Flow Cytometry Analysis
All blood samples were analysed using a FACSCanto II (BD Biosciences, San Jose, CA) flow cytometer as previously described (Mouro-Chanteloup, et al 2003). When using anti-mouse antibodies, the cell surface antigen expression was quantified using calibration mouse IgG-coated beads (Qifikit; DAKO, Glostrup, Denmark). When detected with human MAbs, antigen surface expression was estimated as specific Mean Fluorescence Intensity (MFI), which was calculated by subtracting the background fluorescence observed with the isotypic control from that observed with the specific antibody. For intracellular epitope analysis (GLUT-1), RBCs were fixed in formaldehyde-glutaraldehyde and permeabilized with octyl-glucoside, before analysing by flow cytometry as previously described (Gane, et al 2001).
Western blot analysis
Membrane proteins from whole ghost lysates or Triton X-100 soluble and insoluble fractions separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) were transferred to nitrocellulose membrane and incubated with relevant primary antibodies. Following washing, the nitrocellulose membrane was incubated with the appropriate peroxidase-conjugated secondary antibody (anti-rabbit or anti-mouse, IgG) (Biosys, Compiegne, France). Immunoblots were visualized using the enhanced chemiluminescent (ECL) system (Amersham Pharmacia Biotech, Buckinghamshire, UK).
Preparation of erythrocyte membranes
Erythrocyte ghosts were prepared by hypotonic lysis of washed cells in Na2HPO4 5 mM, EDTA 0.35 mM pH 8.0, with protease inhibitor cocktail followed by six to eight washes in the same buffer until the ghosts became white. All membranes were prepared from erythrocytes that had been deep-frozen using preservative medium, a procedure generally used to store erythrocytes with rare antigen expression for extended periods of time for transfusion use. Protein extraction from erythrocyte membranes was performed by adding 1 volume of packed ghosts to 4 volumes of Na2HPO4 5 mM, NaCl 150 mM pH 8.0, Triton X-100 1% with protease inhibitor cocktail and gentle shaking at 4°C for 1 h. Supernatant and pellet were separated by centrifugation (15,000 g 15 min at 4°C) and the pellet was resuspended in 4 volumes of Laemmli buffer. Equal volumes of supernatant and pellet fractions were loaded on a 4–12% NuPAGE® Novex Bis-Tris Gels under reducing conditions. Proteins were probed with specific antibodies.
Co-immunoprecipitation assay
Erythrocyte ghosts were lysed for 1 h at 4°C with gentle shaking in Na2HPO4 5 mM, NaCl 150 mM pH 8.0, Triton X-100 1% with protease inhibitor cocktail. Supernatant and pellet were separated by centrifugation (15,000 g 15 min at 4°C). Supernatant was incubated overnight at 4°C with the anti-Kell (clone 5A11), or anti-4.1, or an irrelevant antibody (anti-VCAM-1) bound to protein-A-Sepharose (Amersham Pharmacia Biotech, Buckinghamshire, UK) or incubated only with protein-A-Sepharose as negative control. After extensive washing in lysis buffer, the co-immunoprecipitated proteins were eluted from the protein A sepharose in Laemmli buffer at 100°C for 5 min, and analysed by Western blot.
Preparation of recombinant proteins
PCR amplified cDNAs fragments encoding the N-terminal end of the Kell protein (wt-Kell) (AA 1 to 47, starting from the ATG codon) were fused in frame with the glutathione S-transferase protein in the pGEX-5X-3 plasmid using the sense primer, 5′-TGCGGAATTCCATGGAAG GTGGGGACCAAAGTG -3′; antisense primer, 5′-TGCGCTCGAGCTACACCCGTCTGG CCACTGCCC -3′. A mutant bearing the RR46–47AA (mt-Kell) was obtained by in vitro mutagenesis as described above. The glutathione s transferase (GST)-fusion proteins expressed in E. Coli BL21 were purified by elution from glutathione-sepharose beads (NaCl 150 mM, Tris-HCl 50 mM pH 8.0, glutathione 20 mM), and quantified by absorption at 280 nm.
His-tagged peptides 30 and 16 kDa domains and different MBP-tagged sub-domains of the 30 kD domain have been previously described (An, et al 2001, Gauthier, et al 2011). The His-tagged 30 kDa membrane binding domain of 4.1R was cloned into the pET31b(+) vector (Novagene, Madison, MI) using NsiI and XhoI sites upstream and downstream respectively. His-tagged 16 kDa fragments of 4.1R was sub-cloned into pET28b(+) vector (Novagene, Madison, MI) using NcoI and XhoI. MBP-tagged lobe A, lobe B, and lobe C of the 30 kDa domain were subcloned into pMAL-p2x vector using EcoRI and SalI upstream and downstream, respectively. The GST-tagged recombinant proteins were purified using glutathione-Sepharose 4B beads. The MBP-tagged recombinant protein was purified on an amylose resin affinity column. Proteins were dialysed against binding buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% Tween-20).
GST – pull down assays
GST fusion proteins bound to glutathione-sepharose beads were incubated in Low Ionic Strength (LIS) buffer (0.3 mM Na2HPO4, 0.1 mM phenylmethylsulfonyl fluoride, 0.1 mM EDTA, pH 8) with erythrocyte extract supernatant (Kroviarski, et al 2004) or purified protein 4.1R (kind gift from Dr Y. Kroviarsky, Hopital Bichat, Paris) for 2 h at 4°C with gentle shaking. The supernatant was discarded after centrifugation and beads were then washed extensively with phosphate-buffered saline and once with Tris-HCl 50 mM pH 8.0 at 4°C. Elution of proteins bound to glutathione-sepharose 4B beads were performed with NaCl 150 mM, Tris-HCl 50 mM pH 8.0, glutathione 20 mM at 4°C. Eluants were quantified by absorption at 280 nm and equal amounts of proteins were analysed by SDS-PAGE.
To measure the interaction of 4.1R functional domain 30 kDa domains and sub-domains of the 30 kDa to the cytoplasmic tail of Kell, domains and sub-domains were incubated with GST-fusion proteins. Glutathione-Sepharose 4B beads were first incubated with the GST-fusion proteins at room temperature for 30 min, pelleted and washed. Domains and subdomains were then added to the coupled beads. The mixture was incubated for 1 h at room temperature, pelleted, washed and eluted with 10% SDS. The pellet was analysed by SDS-PAGE. The binding of His-tagged domains to GST-fusion proteins was analysed by Western blot using anti-Penta-His antibody (Qiagen, Courtaboeuf, France). The binding of the MBP-tagged sub-domains was analysed using the MAb anti-MBP (New England Biolabs, Evry, France).
Transport activity experiments
Water, HCO3−/Cl− and ammonia permeabilities of ghosts from human erythrocyte variants were determined by using stopped-flow spectrophotometer (SFM74, BioLogic, Grenoble, France) as previously described (Azouzi, et al 2013, Frumence, et al 2013, Genetet, et al 2012).
RESULTS
Flow cytometric analysis of 4.1(−) HE erythrocytes
Expression levels of 13 transmembrane proteins on the surface of the human 4.1(−) erythrocytes were determined by flow cytometry experiments. Normal and 4.2(−) hereditary spherocytosis (HS) (Mouro-Chanteloup, et al 2003) erythrocytes were used as controls. As shown in Table I, this analysis revealed that Kell and UT–B were severely reduced in 4.1(−) erythrocytes (by 70 and 79%, respectively). In 4.2(−) erythrocytes, the amount of Kell protein was unchanged, suggesting that the decreased expression observed in 4.1(−) erythrocytes is specific and is not due to skeletal network disorganization (data not shown). As expected, GPC was also decreased by 90%. In addition, a 25% reduction of DARC [Fy phenotype of the patient and controls: Fy(a+b+)] and band 3 was also observed. The slight decrease of band 3 expression was also confirmed by labelling with eosin-5′-maleimide (EMA), a reagent commonly used for the diagnosis of erythrocyte membrane disorders (Table I). Lu/BCAM was normally expressed in the 4.1(−) erythrocyte, which is in agreement with its direct interaction with spectrin (Kroviarski, et al 2004). The expression levels of CD47, CD44, Rh, RhAG, AQP1 and GLUT-1 were unchanged in 4.1(−) erythrocytes.
Table I.
Human erythrocyte antigen and protein expression. Specific antibody-binding capacity, as determined by indirect immunofluorescence using QIFIKIT calibrated beads (see material and methods) and given as the mean fluorescence intensity ± SD (arbitrary units). All experiments were repeated a minimum of three times.
| Protein | Antibodies and ligand | Normal (SD) | 4.1(−) HE (SD) |
|---|---|---|---|
| GPC | MR4-130 | 54075 (± 1075) | 4000 (± 500) |
| Band 3 | BRIC14 | 415000(±7000) | 317000(±6000) |
| EMA | 5216 (± 97) | 3812 (± 39) | |
| Kell | F7 | 4150 (±66) | 1220 (±35) |
| CD47 | 3E12 | 21300 (±1556) | 21000 (± 2350) |
| Rh | BRIC69 | 135200 (±283) | 121000 (±113) |
| RhAG | LA18.18 | 90450 (±2192) | 71950 (±13150) |
| GPA | R18 | 357500 (±7072) | 310500(±12500) |
| DARC | anti-Fy6 2C3 | 2500 (±283) | 1850 (±250) |
| CD44 | 9D6 | 4150 (±71) | 4100 (±200) |
| UT-B | anti-JK3 | 13125 (± 1968) | 2702 (±142) |
| Lu/BCAM | F241 | 1300 (±141) | 1624 (±153) |
| AQP1 | anti-Co3 | 256 (±15) | 202 (± 22) |
| GLUT-1 | GT11-A | 10961 (± 239) | 11542 (± 806) |
SD, standard deviation; HE, hereditary elliptocytosis
Western blot analysis of 4.1(−) HE erythrocytes
As expected, no protein 4.1R was detected in 4.1(−) erythrocytes (Fig 1). Expression level of various membrane proteins was monitored by quantitative densitometry scanning of the signals corresponding to these proteins using GADPH as reference (Fig 1). In agreement with the flow cytometric analysis, a clear reduction of Kell, XK and GPC/D, a slight reduction of band 3 and normal amounts of Rh, RhAG, and AQP1 were noted in membrane preparation from 4.1(−) erythrocytes. In contrast, the reduction of DARC, as assessed by Western blotting, was more pronounced than that indicated by flow cytometry. The total amount of UT-B was markedly reduced, with a total absence of the major N-glycosylated form and slight decrease of the minor unglycosylated form. The noted reduction in UT-B is consistent with flow cytometry data.
Fig. 1.
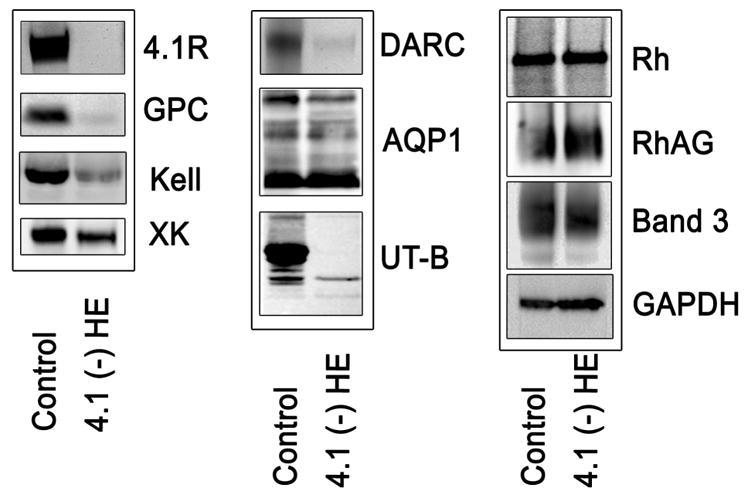
Western blotting analysis of membranes proteins from normal and 4.1 (−) HE erythrocytes. Membrane proteins from whole ghost lysates separated by SDS-PAGE were transferred to a nitrocellulose membrane and incubated with relevant primary antibodies against the indicated proteins. The following antibodies were used: anti 4.1R (PAb); anti-GPC (MAb 1F6); anti-Kell (MAb 5A11); anti XK (rabbit PAb); anti DARC (MAb NaM185-2C3); anti AQP1 (PAb AB3065); anti Rh (PAb MPC8); anti UT-B (rabbit PAb); anti RhAG (MAb LA18.18); anti band 3 (rabbit PAb); anti GAPDH (MAb). GADPH was used as loading control to show that similar amounts of membrane proteins were present on each lane. All experiments were repeated at least three times. HE, hereditary elliptocytosis; MAb, monoclonal antibody; PAb, polyclonal antibody.
Detergent extractability of Kell and GPC/D from normal and, 4.1(−) HE erythrocytes
As Kell protein is greatly reduced in red cells in the absence of 4.1R protein, Triton X-100 extractability of Kell from ghost membranes of 4.1R-deficient HE erythrocytes was compared to that from normal red cells. As expected, 4.1R was predominantly found in the insoluble fraction (P) from normal ghost membranes (92%) and was totally absent in both soluble and insoluble fractions in 4.1R-deficient erythrocytes (Fig 2). Kell was distributed equally between soluble and insoluble fractions in control erythrocyte samples whereas it was found predominantly in the soluble fraction and barely detectable in the insoluble fraction in the 4.1(−) red cells. Of note, the change of the extractability pattern of GPC, well known to interact with 4.1, was identical to that observed for Kell.
Fig. 2.
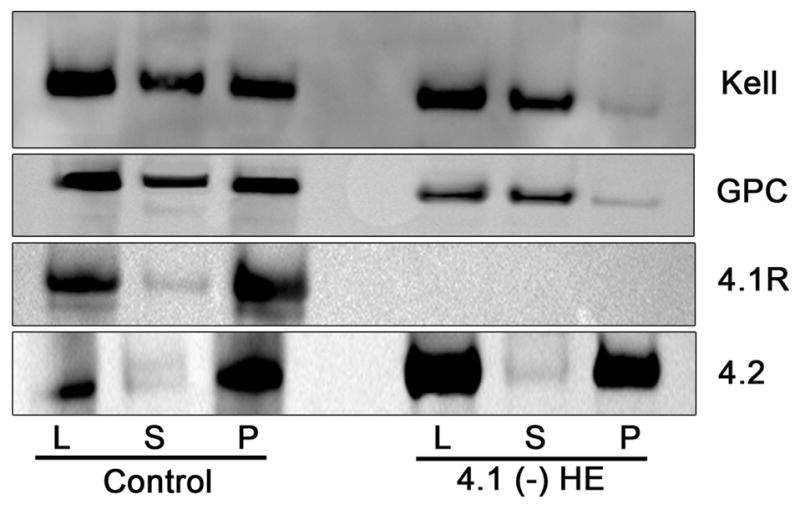
Comparison of Triton X-100 extractability of the Kell protein from normal and 4.1 (−) HE erythrocytes. Erythrocyte ghosts (L) were extracted with 1% Triton-X-100, and the soluble (S) and insoluble (Pellet, P) fractions were analysed by immunoblotting. Immunostaining of Kell, GPC and 4.1R proteins was performed as described in the legend to Figure 1. Anti- 4.2 protein, which is not decreased in 4.1 (−) HE, was used to control proteins extraction for each normal and variant sample.
Co-immunoprecipitation of Kell with protein 4.1R
We sought direct evidence for the association of Kell with the 4.1R complex in mature erythrocyte membranes by performing co-immunoprecipitation studies. As shown in Fig 3A, immunoprecipitation of Kell by its specific antibody in normal red cells followed by Western blotting using anti 4.1R revealed the presence of protein 4.1R. No signal was detected when this experiment was performed with 4.1(−) erythrocytes or when an irrelevant (IR) anti-VCAM1 (VCAM1 is not present in red cells) was used in the immunoprecipitation protocol. Conversely, immunoprecipitation of 4.1 by its specific antibody followed by Western blotting using anti Kell revealed the presence of Kell in normal erythrocytes (Fig 3B). These results imply an association between Kell and 4.1R in normal red cells.
Fig. 3.

Co-immunoprecipitation of 4.1R and Kell. Erythrocytes were lysed (L) and extracts were subjected to immunoprecipitation with specific antibody (IP) or mouse irrelevant antibody (IR, anti-VCAM1 antibody). (A) Immunoprecipitation with anti-Kell monoclonal antibodies. (B) Immunoprecipitation with anti-4.1 polyclonal antibodies. Presence of the Kell protein or 4.1R protein was studied by Western blot (WB) in lysates (L) and IP eluates (IR and IP).
In vitro interaction of Kell with 4.1R
Our findings with normal and 4.1R-deficient erythrocytes strongly imply the association between Kell and 4.1R proteins. In vitro GST pull-down assays were used to explore the possibility that Kell interacts directly with 4.1R. As shown in Fig 4B, 4.1R purified from erythrocyte membranes binds to the cytoplasmic domain of Kell (wt-Kell). As ionic interactions have been described to be responsible for the binding of cytoplasmic domains of transmembrane proteins with 4.1R (Jons and Drenckhahn 1992, Nunomura, et al 2012), we chose to introduce the RR/AA mutation at the juxta-membrane positively charged residues 46–47 of Kell. The R-R/46–47/A-A mutant cytoplasmic tail of Kell failed to bind to 4.1R present in LIS red cell membrane extracts in the pull down assay (Fig 4B), although equal amounts of GST recombinant proteins were obtained (Fig 4A). This indicates that RR46–47 motif is part of the Kell binding site for 4.1R. To define the domain of 4.1R that interacts with Kell, we performed pull down assays using different recombinant peptides of 4.1R. The 16 kDa and 30 kDa domains were produced as His-tagged peptides. The pull down assay demonstrated that the cytoplasmic domain of Kell specifically binds to the 30 kDa membrane-binding domain of 4.1R (Fig 5B). The 30 kDa domain is composed of 3 lobes that bind to different transmembrane proteins (Fig 5A). As shown in Fig 5C, lobe B but not lobes A and C bind the cytoplasmic tail of Kell. It is noteworthy that lobe B has been previously described to bind XK and DARC proteins in murine red cells (Salomao, et al 2008).
Fig. 4.

Association of 4.1R with the cytoplasmic domain of Kell. (A) Coomassie blue staining of GST, GST-wt-Kell and GST-mt-Kell aliquots before the pull down assays (12.5% acrylamide gel). Vertical line indicates grouping of images from two different gels. (B) GST- pull down was performed using purified protein 4.1R or low ionic strength red cell membrane extracts with GST, GST-wt-Kell and GST-mt-Kell. Eluted proteins were revealed with anti-4.1R polyclonal antibodies. SM: starting material; RBC, red blood cell.
Fig. 5.

Direct binding of Kell protein to FERM binding domain of 4.1R. The binding of 4.1R functional domains to cytoplasmic tails of Kell, was assessed by pull down assay. (A) Crystal structure of the FERM domain (30 kDa domain) of 4.1R (PDB accession ID: 1gg3). The FERM domain consists of three lobes (A, B and C), which bind to different transmembrane proteins. The binding of His-tagged domains (16 and 30 kDa domains) of 4.1R (B) and MBP-tagged subdomains of FERM domain (C) to the cytoplasmic domains of Kell, was detected by using anti- His and anti-MBP antibodies, respectively. L.A, lobe A; L.B, lobe B; L.C lobe C.
Effect of 4.1R deficiency on the activity of membrane transporters
To assess the role of 4.1R on the transport activities of AQP1, Band 3 and RhAG proteins, water permeabilities, Cl−/HCO3− exchange and ammonia transports were measured in ghosts from WT and 4.1(−) erythrocytes. The diameters of ghosts were assessed by fluorescence microscopy. No difference between the ghost diameters from control (6.1± 0.3 μm), 4.1(−) HE (5.9 μm), Rhnull (5.8 ± 0.1 μm), and AQP1null (5.8 ± 0.3 μm) samples was observed. Fig 6 depicts the averaged time courses of fluorescence changes obtained from three different experiments. The osmotic water permeability was determined from a stopped-flow analysis of control, 4.1(−) and AQP1null erythrocytes that exhibit no expression of AQP1. A clear difference was observed between AQP1null ghosts and the two other ghosts (Fig 6A). In contrast, no difference was observed between control and 4.1(−) HE ghosts expressing similar levels of AQP1 (Table I and Fig 1). This indicates that the absence of 4.1R does not affect water transport across erythrocyte membrane. HCO3−/Cl− exchange was measured indirectly by the variation of intracellular pH generated by HCO3− influx, which actually reflects band 3 activity. The fluorescence variation for control and 4.1(−) erythrocytes exhibited a rapid increase corresponding to intracellular alkalinization (Fig 6B). To ascertain that these results were directly correlated to band 3 activities, control ghosts were incubated with 30 μM DIDS, a band 3 inhibitor. Indeed, the rapid phase of alkalinization was totally abolished, confirming the inhibition of HCO3−/Cl− exchange. Interestingly, band 3 activity was reduced in 4.1(−) erythrocytes compared to control.
Fig. 6.

Comparison of water, bicarbonate and ammonia transport in ghosts from erythrocyte variants. (A) Time courses of fluorescence quenching corresponding to osmotic water permeability of ghost from control (black), 4.1Rnull (blue) and AQP1null erythrocytes (red). Ghosts were resealed in presence of 8 mM 6-carboxyfluorescein and submitted to an osmotic gradient (150 mosm mannitol/kg H2O) at 15°C in the stopped-flow spectrofluorometer. (B) Time courses of intracellular pH increase in ghosts corresponding to HCO3−/Cl− exchange. Ghosts derived from control (black) and 4.1Rnull (blue) erythrocytes resealed in the presence of pyranine, a fluorescent pH-sensitive probe, and bovine carbonic anhydrase at 2 mg/ml, were rapidly mixed with an equal volume of buffer containing KHCO3, generating inwardly-directed 50 MEq HCO3−/CO2 and outwardly-directed 50 MEq Cl− gradients. Ghost from control was incubated with the anion exchanger inhibitor DIDS (30 μM) for 20 min (red curve). (C) Time course of intracellular pH increase corresponding to ammonia transport in ghosts from control (black), 4.1Rnull (blue) and RhAGnull erythrocytes (red). Ghost resealed in the presence of pyranine were submitted to 10 mM inwardly-directed gradient of ammonia at 15°C followed by stopped-flow analysis. All experiments were repeated twice. Each kinetic corresponds to three averaged time courses of fluorescence changes.
To test the impact of the absence of 4.1R on the ammonia permeability of red cell membranes, ghosts prepared from control and 4.1(−) erythrocytes were resealed in the presence of pyranine, a fluorescent pH-sensitive dye, and submitted to an ammonium gradient. As a control, ammonia permeability in Rhnull was decreased compared to control (Fig 6C), in agreement with previous studies indicating that the Rh-associated glycoprotein (RhAG) mediates facilitated transport of ammonia into human erythrocytes (Ripoche, et al 2004). However, we found that 4.1(−) erythrocytes showed no reduction of ammonia permeability compared to the control. Thus the RhAG-mediated ammonia transport is not altered in the absence of 4.1R protein.
Discussion
Protein 4.1R is a cytoskeletal adaptor protein that is responsible for the control of the mechanical stability of erythrocyte membranes, and for the proper anchoring of diverse transmembrane proteins to the membrane skeletal network (Nunomura and Takakuwa 2006, Takakuwa, et al 1986). Analysis of 4.1R-deficient human and murine erythrocytes revealed the complex array of membrane proteins that bind 4.1R and link these proteins to the spectrin-based skeletal network. Salomao et al (2008) showed that nearly all the FERM domain-binding proteins are lost in 4.1R-deficient mouse red cells, including DARC, XK, Rh and GPC (Salomao, et al 2008). It has been also demonstrated that 4.1R regulates the activity of the Na+/H+ exchanger (NHE1) through a direct protein-protein interaction. In fact, 4.1Rnull murine erythrocytes are characterized by cell dehydration and high intracellular concentration of Na+ as a result of NHE1 hyperactivity (Nunomura, et al 2012, Rivera, et al 2006). However, the 4.1R-based multiprotein complex has not been previously defined in detail in human erythrocytes (Jeremy, et al 2009). In the present study, we provide the first evidence for a direct interaction between human Kell and 4.1R. We also demonstrated that protein 4.1R binds specifically to Kell by electrostatic interaction involving two positively charged residues in the juxta-membrane region of Kell cytoplasmic domain. This type of interaction involving positively charged binding sequence has already been shown for the binding of band 3 (I386RRRY and L343RRRY) (Jons and Drenckhahn 1992), GPC/D (R82HK) (Anderson and Lovrien 1984), and NHE1 (K519R and R556FNKKYVKK) (Nunomura, et al 2012). It has also been shown that the E38ED motif located in lobe A of the 4.1R FERM binding domain, mediates 4.1R binding to NHE1 and band 3 (Jons and Drenckhahn 1992, Nunomura, et al 2012). By examining subdomains of the 4.1R FERM domain, we found that the binding site for Kell is localized in lobe B. Salomao et al (2008) documented that lobe B also interacts in vitro to other erythrocyte transmembrane proteins, such as XK and DARC. Indeed, we observed a marked reduction of Kell, XK and DARC in the erythrocytes from the patient with 4.1(−) HE. These results are largely in agreement with those obtained in 4.1R−/− mice erythrocytes. However, we observed that, in contrast with murine erythrocytes, Rh and RhAG were not reduced in human 4.1-deficient erythrocytes. Accordingly, functional studies showed a normal ammonia transport activity in 4.1(−) HE human erythrocytes. Therefore, it is likely that, in contrast to mice, the 4.1R junctional complex in human red cells does not include Rh/RhAG proteins. In mice, Rh but not RhAG is linked to the junctional complex, suggesting that parts of Rh and RhAG traffic independently of each other to the cell surface (Salomao, et al 2008). In contrast, our previous findings in Rhnull RBCs and with in vitro culture systems revealed that in humans the expression of Rh is strictly dependent on RhAG (Cherif-Zahar, et al 1996, Cherif-Zahar, et al 1993) (Mouro-Chanteloup, et al 2002, van den Akker, et al 2010a). Recently, RhAG knockdown experiments showed that the Rh dependency on RhAG is established early during erythropoiesis, at the basophilic stage (Satchwell, et al 2011). In summary, several differences in multiprotein complex composition are observed between humans and mice RBCs (van den Akker, et al 2010b). Our present findings lend further support to the thesis that it is not always possible to extrapolate the results from mouse red cell membranes to human red cell membranes, as reflected by the abundant expression of GLUT-1 transporter in human red cells but its complete absence in mouse red cells (Montel-Hagen, et al 2008).
Another interesting finding is that UT-B was severely decreased in 4.1 (−) human erythrocytes. In particular, the glycosylated form of UT-B was not detected (Olives, et al 1995), suggesting either that (i) one or several proteins of 4.1R complex play a critical role in the translocation of UT-B to the plasma membrane or (ii) UT-B is directly or indirectly associated with the 4.1R protein. On the basis of sequence homology with the motif in the cytoplasmic domain of band 3 and NHE1, we identified a positively charged candidate binding sequence (A380KKR) in the cytoplasmic domain of UT-B that could mediate direct interaction between UT-B and 4.1R. Recently, we have shown that UT-B and AQP1 are the major water channels in the mature human erythrocytes (Azouzi, et al 2013). In the present study, we showed that AQP1 expression and function were not altered in the absence of 4.1R, suggesting that it is not present in the 4.1R complex. Similarly the normal level of Lu/BCAM in 4.1(−) erythrocytes confirms the hypothesis that Lu/BCAM interacts directly with spectrin independent of protein 4.1R (Kroviarski, et al 2004). On the other hand, we observed a slight reduction of band 3 in 4.1(−) erythrocytes by both flow cytometric and Western blot analyses using antibodies that recognize external and intracellular domains of band 3, respectively. This result is in agreement with a previous study in 4.1R knockout mice (Salomao, et al 2008) but not with the recent study of two 4.1(−) patients which reported a normal expression of band 3 (Jeremy, et al 2009, Salomao, et al 2008). Supporting our present findings, stopped-flow analysis of HCO3−/Cl− exchange showed a clear decrease of band 3 activity in 4.1(−) erythrocytes as compared to controls. This result is in favour of a decreased band 3 expression in the absence of 4.1R. However, it cannot be ruled out that the decrease of band 3 activity could result from the change of its organization on the erythrocyte membrane (unpublished data). It is assumed that the erythrocyte membrane skeleton is tethered to the lipid bilayers via immobile band 3 at the ankyrin and 4.1R complexes. The remaining band 3 molecules move relatively freely in the membrane (Burton and Bruce 2011, Kodippili, et al 2009). Thus, we suggest that more mobile band 3 may be present at the red cell membrane in the absence of 4.1R protein. Furthermore, it has been shown that the increase of the mobile fraction of band 3 can lead to the formation of band 3 clusters (Arashiki, et al 2013). Using the band 3 antibody that specifically recognizes clustered band 3 (Arashiki, et al 2013), we explored the extent of band 3 clustering in 4.1(−) erythrocytes. No clustered band 3 could be detected in either the control or in the patient with 4.1 (−) HE (data not shown). These results suggest that the alteration in the rate of HCO3−/Cl− exchange across the 4.1(−) erythrocyte membranes is caused by the decreased expression of band 3.
The current model of the human erythrocyte membrane suggests that the spectrin-actin junctions are linked to the plasma membrane via protein 4.1 and GPC. In this study, we provide evidence for a direct interaction between Kell and 4.1R and propose an updated model for the 4.1R- multiprotein complex in human erythrocyte (Fig 7). Lobe A in the 4.1R FERM domain binds to protein transporters, such as band 3, NHE1 and UT-B. Functional and structural experiments are required to confirm the presence of UT-B in this complex. The transmembrane proteins GPC, XK, DARC and Kell bind to the lobe B and the binding site of p55 is located in lobe C. Understanding the structure and organization of the erythrocyte membrane is mainly derived from analyses of (i) mouse knockout model systems, (ii) human variant blood samples and (iii) erythrocyte membrane disorders in which particular membrane proteins are deficient (HS and HE) (Mouro-Chanteloup, et al 2003, van den Akker, et al 2010b). The deficiency of blood group antigens carrying proteins in HS and HE erythrocytes can be explained by various molecular mechanisms including: perturbed trafficking to the erythroblast membrane, aberrant protein sorting during erythroblast enucleation and selective loss during reticulocyte membrane remodelling (Bell, et al 2013, Liu, et al 2010, Salomao, et al 2010, Satchwell, et al 2011, van den Akker, et al 2010a). Establishing when and where these proteins associate during erythroid differentiation should provide mechanistic insights into membrane multi-protein complex formation in both normal and abnormal erythropoiesis.
Fig. 7.

Proposed model of the 4.1R- multiprotein complex in human erythrocyte. The novel 4.1R-based complex contains transmembrane proteins GPC/D, XK, Kell, DARC, band 3, NHE1 and UT-B. Kell, DARC, XK and GPC/D bind to lobe B. Transporters such as NHE1, band 3 and UT-B bind to lobe A and p55 to lobe C. Our schematic representation summarizes all possible protein interactions. Note that all these interactions do not necessarily occur at the same time. SABD, specific antibody-binding capacity.
Acknowledgments
We are grateful to Dr Yolande Kroviarski for helpful advice on the GST pull down experiments and to Pierre Gane for flow cytometric analyses. The authors thank Dr. Thierry Peyrard and Eliane Vera at the CNRGS (Paris, France) for providing rare human blood samples. We thank Pr. Y. Takakuwa and Dr. N. Arashiki from Tokyo Women’s Medical University (Japan) for providing the band 3-cluster antibody.
Funding
This study was supported by grants from Laboratory of Excellence GR-Ex, reference ANR-11-LABX-0051 and by NIH DK26263. The LabEx GR-Ex is funded by the program “Investissements d’avenir” of the French National Research Agency, reference ANR-11-IDEX-0005-02.
Footnotes
Authorship
SA, EC, NM, YC and CLVK designed the protocol. SA, EC and XA performed the experiments. SA, EC, YC and CLVK analysed and interpreted the data. SA, EC, NM, YC and CLVK wrote the article.
References
- An XL, Takakuwa Y, Manno S, Han BG, Gascard P, Mohandas N. Structural and functional characterization of protein 4.1R-phosphatidylserine interaction: potential role in 4.1R sorting within cells. J Biol Chem. 2001;276:35778–35785. doi: 10.1074/jbc.M101364200. [DOI] [PubMed] [Google Scholar]
- Anderson RA, Lovrien RE. Glycophorin is linked by band 4.1 protein to the human erythrocyte membrane skeleton. Nature. 1984;307:655–658. doi: 10.1038/307655a0. [DOI] [PubMed] [Google Scholar]
- Arashiki N, Kimata N, Manno S, Mohandas N, Takakuwa Y. Membrane peroxidation and methemoglobin formation are both necessary for band 3 clustering: mechanistic insights into human erythrocyte senescence. Biochemistry. 2013;52:5760–5769. doi: 10.1021/bi400405p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouzi S, Gueroult M, Ripoche P, Genetet S, Colin Aronovicz Y, Le Van Kim C, Etchebest C, Mouro-Chanteloup I. Energetic and molecular water permeation mechanisms of the human red blood cell urea transporter B. PLoS One. 2013;8:e82338. doi: 10.1371/journal.pone.0082338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AJ, Satchwell TJ, Heesom KJ, Hawley BR, Kupzig S, Hazell M, Mushens R, Herman A, Toye AM. Protein distribution during human erythroblast enucleation in vitro. PLoS One. 2013;8:e60300. doi: 10.1371/journal.pone.0060300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton NM, Bruce LJ. Modelling the structure of the red cell membrane. Biochem Cell Biol. 2011;89:200–215. doi: 10.1139/o10-154. [DOI] [PubMed] [Google Scholar]
- Carbonnet F, Hattab C, Collec E, Le van Kim C, Cartron JP, Bertrand O. Immunochemical analysis of the Kx protein from human red cells of different Kell phenotypes using antibodies raised against synthetic peptides. Br J Haematol. 1997;96:857–863. doi: 10.1046/j.1365-2141.1997.d01-2110.x. [DOI] [PubMed] [Google Scholar]
- Cherif-Zahar B, Raynal V, Gane P, Mattei MG, Bailly P, Gibbs B, Colin Y, Cartron JP. Candidate gene acting as a suppressor of the RH locus in most cases of Rh-deficiency. Nat Genet. 1996;12:168–173. doi: 10.1038/ng0296-168. [DOI] [PubMed] [Google Scholar]
- Cherif-Zahar B, Raynal V, Le Van Kim C, D’Ambrosio AM, Bailly P, Cartron JP, Colin Y. Structure and expression of the RH locus in the Rh-deficiency syndrome. Blood. 1993;82:656–662. [PubMed] [Google Scholar]
- Conboy J, Kan YW, Shohet SB, Mohandas N. Molecular cloning of protein 4.1, a major structural element of the human erythrocyte membrane skeleton. Proc Natl Acad Sci U S A. 1986;83:9512–9516. doi: 10.1073/pnas.83.24.9512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Ouggouti S, Bournier O, Boivin P, Bertrand O, Dhermy D. Purification of erythrocyte protein 4.1 by selective interaction with inositol hexaphosphate. Protein Expr Purif. 1992;3:488–496. doi: 10.1016/1046-5928(92)90066-6. [DOI] [PubMed] [Google Scholar]
- Frumence E, Genetet S, Ripoche P, Iolascon A, Andolfo I, Le Van Kim C, Colin Y, Mouro-Chanteloup I, Lopez C. Rapid Cl−/HCOFormula exchange kinetics of AE1 in HEK293 cells and hereditary stomatocytosis red blood cells. Am J Physiol Cell Physiol. 2013;305:C654–662. doi: 10.1152/ajpcell.00142.2013. [DOI] [PubMed] [Google Scholar]
- Gane P, Le Van Kim C, Bony V, El Nemer W, Mouro I, Nicolas V, Colin Y, Cartron JP. Flow cytometric analysis of the association between blood group-related proteins and the detergent-insoluble material of K562 cells and erythroid precursors. Br J Haematol. 2001;113:680–688. doi: 10.1046/j.1365-2141.2001.02757.x. [DOI] [PubMed] [Google Scholar]
- Gauthier E, Guo X, Mohandas N, An X. Phosphorylation-dependent perturbations of the 4.1R-associated multiprotein complex of the erythrocyte membrane. Biochemistry. 2011;50:4561–4567. doi: 10.1021/bi200154g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genetet S, Ripoche P, Picot J, Bigot S, Delaunay J, Armari-Alla C, Colin Y, Mouro-Chanteloup I. Human RhAG ammonia channel is impaired by the Phe65Ser mutation in overhydrated stomatocytic red cells. Am J Physiol Cell Physiol. 2012;302:C419–428. doi: 10.1152/ajpcell.00092.2011. [DOI] [PubMed] [Google Scholar]
- Hayette S, Dhermy D, dos Santos ME, Bozon M, Drenckhahn D, Alloisio N, Texier P, Delaunay J, Morle L. A deletional frameshift mutation in protein 4.2 gene (allele 4.2 Lisboa) associated with hereditary hemolytic anemia. Blood. 1995;85:250–256. [PubMed] [Google Scholar]
- Hermand P, Mouro I, Huet M, Bloy C, Suyama K, Goldstein J, Cartron JP, Bailly P. Immunochemical characterization of rhesus proteins with antibodies raised against synthetic peptides. Blood. 1993;82:669–676. [PubMed] [Google Scholar]
- Jaber A, Loirat MJ, Willem C, Bloy C, Cartron JP, Blanchard D. Characterization of murine monoclonal antibodies directed against the Kell blood group glycoprotein. Br J Haematol. 1991;79:311–315. doi: 10.1111/j.1365-2141.1991.tb04539.x. [DOI] [PubMed] [Google Scholar]
- Jeremy KP, Plummer ZE, Head DJ, Madgett TE, Sanders KL, Wallington A, Storry JR, Gilsanz F, Delaunay J, Avent ND. 4.1R-deficient human red blood cells have altered phosphatidylserine exposure pathways and are deficient in CD44 and CD47 glycoproteins. Haematologica. 2009;94:1354–1361. doi: 10.3324/haematol.2009.006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Veldhuisen B, Ligthart P, Haer-Wigman L, Jongerius J, Boujnan M, Ait Soussan A, Luo G, Fu Y, van der Schoot CE, de Haas M. Novel alleles at the Kell blood group locus that lead to Kell variant phenotype in the Dutch population. Transfusion. 2015;55:413–421. doi: 10.1111/trf.12838. [DOI] [PubMed] [Google Scholar]
- Jons T, Drenckhahn D. Identification of the binding interface involved in linkage of cytoskeletal protein 4.1 to the erythrocyte anion exchanger. EMBO J. 1992;11:2863–2867. doi: 10.1002/j.1460-2075.1992.tb05354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamlichi S, Bailly P, Blanchard D, Goossens D, Cartron JP, Bertrand O. Purification and partial characterization of the erythrocyte Kx protein deficient in McLeod patients. Eur J Biochem. 1995;228:931–934. [PubMed] [Google Scholar]
- Kodippili GC, Spector J, Sullivan C, Kuypers FA, Labotka R, Gallagher PG, Ritchie K, Low PS. Imaging of the diffusion of single band 3 molecules on normal and mutant erythrocytes. Blood. 2009;113:6237–6245. doi: 10.1182/blood-2009-02-205450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroviarski Y, El Nemer W, Gane P, Rahuel C, Gauthier E, Lecomte MC, Cartron JP, Colin Y, Le Van Kim C. Direct interaction between the Lu/B-CAM adhesion glycoproteins and erythroid spectrin. Br J Haematol. 2004;126:255–264. doi: 10.1111/j.1365-2141.2004.05010.x. [DOI] [PubMed] [Google Scholar]
- Lee S, Russo DC, Reiner AP, Lee JH, Sy MY, Telen MJ, Judd WJ, Simon P, Rodrigues MJ, Chabert T, Poole J, Jovanovic-Srzentic S, Levene C, Yahalom V, Redman CM. Molecular defects underlying the Kell null phenotype. J Biol Chem. 2001;276:27281–27289. doi: 10.1074/jbc.M103433200. [DOI] [PubMed] [Google Scholar]
- Lee S, Debnath AK, Redman CM. Active amino acids of the Kell blood group protein and model of the ectodomain based on the structure of neutral endopeptidase 24.11. Blood. 2003;102:3028–3034. doi: 10.1182/blood-2003-05-1564. [DOI] [PubMed] [Google Scholar]
- Liu J, Guo X, Mohandas N, Chasis JA, An X. Membrane remodeling during reticulocyte maturation. Blood. 2010;115:2021–2027. doi: 10.1182/blood-2009-08-241182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montel-Hagen A, Blanc L, Boyer-Clavel M, Jacquet C, Vidal M, Sitbon M, Taylor N. The Glut1 and Glut4 glucose transporters are differentially expressed during perinatal and postnatal erythropoiesis. Blood. 2008;112:4729–4738. doi: 10.1182/blood-2008-05-159269. [DOI] [PubMed] [Google Scholar]
- Mouro-Chanteloup I, D’Ambrosio AM, Gane P, Le Van Kim C, Raynal V, Dhermy D, Cartron JP, Colin Y. Cell-surface expression of RhD blood group polypeptide is posttranscriptionally regulated by the RhAG glycoprotein. Blood. 2002;100:1038–1047. [PubMed] [Google Scholar]
- Mouro-Chanteloup I, Delaunay J, Gane P, Nicolas V, Johansen M, Brown EJ, Peters LL, Van Kim CL, Cartron JP, Colin Y. Evidence that the red cell skeleton protein 4.2 interacts with the Rh membrane complex member CD47. Blood. 2003;101:338–344. doi: 10.1182/blood-2002-04-1285. [DOI] [PubMed] [Google Scholar]
- Nunomura W, Takakuwa Y. Regulation of protein 4.1R interactions with membrane proteins by Ca2+ and calmodulin. Front Biosci. 2006;11:1522–1539. doi: 10.2741/1901. [DOI] [PubMed] [Google Scholar]
- Nunomura W, Denker SP, Barber DL, Takakuwa Y, Gascard P. Characterization of cytoskeletal protein 4.1R interaction with NHE1 (Na(+)/H(+) exchanger isoform 1) Biochem J. 2012;446:427–435. doi: 10.1042/BJ20120535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olives B, Mattei MG, Huet M, Neau P, Martial S, Cartron JP, Bailly P. Kidd blood group and urea transport function of human erythrocytes are carried by the same protein. J Biol Chem. 1995;270:15607–15610. doi: 10.1074/jbc.270.26.15607. [DOI] [PubMed] [Google Scholar]
- Redman CM, Russo D, Lee S. Kell, Kx and the McLeod syndrome. Baillieres Best Pract Res Clin Haematol. 1999;12:621–635. doi: 10.1053/beha.1999.0045. [DOI] [PubMed] [Google Scholar]
- Ripoche P, Bertrand O, Gane P, Birkenmeier C, Colin Y, Cartron JP. Human Rhesus-associated glycoprotein mediates facilitated transport of NH(3) into red blood cells. Proc Natl Acad Sci U S A. 2004;101:17222–17227. doi: 10.1073/pnas.0403704101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera A, De Franceschi L, Peters LL, Gascard P, Mohandas N, Brugnara C. Effect of complete protein 4.1R deficiency on ion transport properties of murine erythrocytes. Am J Physiol Cell Physiol. 2006;291:C880–886. doi: 10.1152/ajpcell.00436.2005. [DOI] [PubMed] [Google Scholar]
- Rivera A, Kam SY, Ho M, Romero JR, Lee S. Ablation of the Kell/Xk complex alters erythrocyte divalent cation homeostasis. Blood Cells Mol Dis. 2013;50:80–85. doi: 10.1016/j.bcmd.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo D, Redman C, Lee S. Association of XK and Kell blood group proteins. J Biol Chem. 1998;273:13950–13956. doi: 10.1074/jbc.273.22.13950. [DOI] [PubMed] [Google Scholar]
- Salomao M, Zhang X, Yang Y, Lee S, Hartwig JH, Chasis JA, Mohandas N, An X. Protein 4.1R-dependent multiprotein complex: new insights into the structural organization of the red blood cell membrane. Proc Natl Acad Sci U S A. 2008;105:8026–8031. doi: 10.1073/pnas.0803225105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomao M, Chen K, Villalobos J, Mohandas N, An X, Chasis JA. Hereditary spherocytosis and hereditary elliptocytosis: aberrant protein sorting during erythroblast enucleation. Blood. 2010;116:267–269. doi: 10.1182/blood-2010-02-264127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satchwell TJ, Bell AJ, Pellegrin S, Kupzig S, Ridgwell K, Daniels G, Anstee DJ, van den Akker E, Toye AM. Critical band 3 multiprotein complex interactions establish early during human erythropoiesis. Blood. 2011;118:182–191. doi: 10.1182/blood-2010-10-314187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakuwa Y. Protein 4.1, a multifunctional protein of the erythrocyte membrane skeleton: structure and functions in erythrocytes and nonerythroid cells. Int J Hematol. 2000;72:298–309. [PubMed] [Google Scholar]
- Takakuwa Y, Tchernia G, Rossi M, Benabadji M, Mohandas N. Restoration of normal membrane stability to unstable protein 4.1-deficient erythrocyte membranes by incorporation of purified protein 4.1. J Clin Invest. 1986;78:80–85. doi: 10.1172/JCI112577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tchernia G, Mohandas N, Shohet SB. Deficiency of skeletal membrane protein band 4.1 in homozygous hereditary elliptocytosis. Implications for erythrocyte membrane stability. J Clin Invest. 1981;68:454–460. doi: 10.1172/JCI110275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh-Trang-Tan MM, Lasbennes F, Gane P, Roudier N, Ripoche P, Cartron JP, Bailly P. UT-B1 proteins in rat: tissue distribution and regulation by antidiuretic hormone in kidney. Am J Physiol Renal Physiol. 2002;283:F912–922. doi: 10.1152/ajprenal.00359.2001. [DOI] [PubMed] [Google Scholar]
- van den Akker E, Satchwell TJ, Pellegrin S, Flatt JF, Maigre M, Daniels G, Delaunay J, Bruce LJ, Toye AM. Investigating the key membrane protein changes during in vitro erythropoiesis of protein 4.2 (−) cells (mutations Chartres 1 and 2) Haematologica. 2010a;95:1278–1286. doi: 10.3324/haematol.2009.021063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Akker E, Satchwell TJ, Williamson RC, Toye AM. Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol Dis. 2010b;45:1–8. doi: 10.1016/j.bcmd.2010.02.019. [DOI] [PubMed] [Google Scholar]