

Abstract
Degeneration of photoreceptors (rods and cones) results in blindness. As we rely almost entirely on our daytime vision mediated by the cones, it is the loss of these photoreceptors that results in legal blindness and poor quality of life. Cone dysfunction is usually observed due to two mechanisms: non cell-autonomous due to the secondary effect of rod death if the causative gene is specifically expressed in rods, and cell autonomous, if the mutation is in a cone-specific gene. However, it is difficult to dissect cone autonomous effect of mutations in the genes that are expressed in both rods and cones. Here we report a property of murine cone photoreceptors, which is a cone-autonomous effect of the genetic perturbation of the retinitis pigmentosa 2 (Rp2) gene mutated in human X-linked RP. Constitutive loss of Rp2 results in abnormal extension of the cone outer segment (COS). This effect is phenocopied when the Rp2 gene is ablated specifically in cones but not when ablated in rods. Furthermore, the elongated COS exhibits abnormal ultrastructure with disorganized lamellae. Additionally, elongation of both the OS membrane and the microtubule cytoskeleton was observed in the absence of RP2. Taken together, our studies identify a cone morphological defect in retinal degeneration due to ablation of RP2 and will assist in understanding cone-autonomous responses during disease and develop targeted therapies.
Keywords: mouse model, photoreceptor, retina, cilia
Introduction
The eye is the most exposed and readily accessible part of the central nervous system. The retina, situated in the back of the eye, is a laminated tissue consisting of the neurons that are the first responders of light detection arranged in distinct layers [Kessel and Kardon 1979]. Of these, photoreceptors (rods and cones) form the bulk of the cell types in the retina and are the first cell types that detect light and initiate the phototransduction cascade. Photoreceptors are polarized neurons with two distinct compartments: inner segment, which contains protein synthesis transport machinery and outer segment (OS), which is the hub for proteins involved in phototransduction [Kandel 2013; Veleri et al. 2015; Wright et al. 2010].
The photosensory OS is a modified sensory cilium [Liu et al. 2007]. Cilia are microtubule-based extensions of the plasma membrane, which originate from the mother centriole (also called basal body) [Satir 2008; Satir and Christensen 2008]. Cilia maintain a unique composition of their membrane by retaining specific signaling receptor moieties, which sense the extrinsic cues to regulate intracellular signaling cascades, such as sonic hedgehog signaling, olfaction, and photoreception [Yildiz and Khanna 2012]. The function and the composition of the cilia are regulated by an elaborate and conserved protein trafficking mechanism called Intraflagellar Transport. The IFT is modulated by the activity of microtubule motor assemblies, which carry the cargo in the anterograde as well as retrograde directions [Engel et al. 2009; Pazour and Rosenbaum 2002; Pazour and Witman 2003; Rosenbaum 2002; Rosenbaum et al. 1999].
The photoreceptors develop a unique sensory cilium in the form of a light-sensing outer segment (OS) composed of membranous discs loaded with the G protein coupled receptor opsin and regulatory proteins involved in initiating the phototransduction cascade [Besharse 1986; Liu et al. 2007]. The components of the OS are transported from the inner segment to the base of the OS where they cross a narrow bridge-like structure called the connecting cilium (or transition zone; TZ) to enter the OS [Sung and Chuang 2010; Young 1968]. Rods and cones are morphologically distinct neurons. Whereas rods develop long cylindrical OS extension with membranous discs arranged in a coin-stack manner, the cones are conical-shaped structures with open OS discs [Carter-Dawson and LaVail 1979a; Carter-Dawson and LaVail 1979b; LaVail 1973]. Remarkable studies have uncovered the basic mechanisms underlying cone development and function [Livesey and Cepko 2001; Mears et al. 2001; Swaroop et al. 2010], however, we know very little about the mechanisms that regulate cone function in adulthood.
In several mammals, including mice and humans, rods outnumber the cones by a ratio of 20:1. Given that we rely on cones for our daylight and high acuity vision, it is the loss of cones that results in complete blindness and poor quality of life in retinopathies. Although cones die due to a primary defect in cone-specific genes, such as Achromatopsia due to CNGB3 mutations [Khan et al. 2007; Kohl et al. 2000; Komaromy et al. 2013; Sidjanin et al. 2002], alterations in rod-specific genes also result in secondary cone death [Leveillard et al. 2004; Punzo et al. 2009]. Further complexity is observed when the causative gene is expressed in both rods and cones. In such cases, cones are affected both due to a secondary effect of rod death as well as cone autonomous mechanisms [Wright et al. 2010]. Thus, it is difficult to dissect the cone-specific alterations that result in their dysfunction and degeneration. Lack of such knowledge has also hampered our understanding of heterogenic clinical presentation in patients with mutations in widely expressed genes, such as RPGR (retinitis pigmentosa GTPase regulator) and RP2, mutated in X-linked forms of retinal degeneration [Breuer et al. 2002; Churchill et al. 2013; Schwahn et al. 1998; Sharon et al. 2000; Wu et al. 2010]. Several patients with RP2 mutations exhibit early loss of cone function followed by rods.
We previously showed that ablation of the Rp2 gene in mice (Rp2null) (or knockdown in zebrafish) [Li et al. 2013; Patil et al. 2011], resulted in a predominant effect on cone function as compared to rods. These observations led us to further elucidate the effect of RP2 on cones with an aim to uncover new information about cone biology and maintenance. We not only utilized the Rp2null mice, but also generated and characterized cone-specific or rod-specific conditional mouse mutants of Rp2. Our studies have uncovered a novel aspect of cone photoreceptor biology and provide new knowledge, which will form the basis for further understanding these elusive sensory neurons.
Reagents and Instruments
Antibodies
Anti-acetylated α-tubulin and anti-KIF3A antibodies were obtained from Sigma-Aldrich (St. Louis, MO). Anti-RP1 antibody was a kind gift of Dr. Eric Pierce (Massachusetts Eye and Ear Infirmary) and anti-M-opsin antibody was procured from Dr. Cheryl M. Craft [Zhu et al. 2003]. Anti-KIF3A was procured from Abcam (Cambridge, MA) and anti-MAK was obtained from Abgent (San Diego, CA). Peanut agglutinin (PNA) and anti-Cre antibody were procured from Vector Labs (Burlingame, CA) and Millipore (Billerica, MA), respectively. Hoechst 33342 was procured from Life Technologies Corp.
Microscopy
Ultrathin sections for TEM were observed in a Zeiss EM900 electron microscope and pictures were taken with a coupled digital camera using the ImageSP software. The sections were then visualized using a scanning confocal microscope (Leica TCS SP5 II laser; Leica Microsystems).
Methods
Mice and ERG
All animal experiments were performed in accordance with the approved procedures of the Institutional Animal Care and Use Committee. All mice were maintained in the same conditions of 12-hour light to 12-hour dark, with unrestricted access to food and water. Lighting conditions were kept constant in all cages with illumination of 10 to 15 lux at the level of the cages. ERGs were recorded as described previously by using Espion e2 recording system [24].
The Rp2flox, Rp2null, iCre75, and M-Cre (HRGP-Cre; M-opsin promoter driving Cre expression in cones) mice have been previously described [Le et al. 2004; Li et al. 2013; Li et al. 2005]. The Rp2flox mice were bred to M-Cre or iCre75 mice to generate a cone- or rod-specific deletion of the Rp2 gene (Rp2MKO or Rp2iKO), respectively. Male mice of appropriate genotypes were used in these studies. All mice were also genotyped to exclude rd1 and rd8 alleles.
Immunofluorescence, Transmission Electron Microscopy (TEM) and Immunogold EM
For immunofluorescence analyses, mouse eyes (n=6 for each experiment) were enucleated and then fixed in 4% paraformaldehyde in PBS (pH 7.4) followed by cryosectioning and staining as recently described [Li et al. 2013]. Primary antibodies were prepared in blocking solution and slides were further incubated overnight at 4°C. Sections were then washed three times with PBS and incubated for 1 hour with goat anti-rabbit (or mouse) Alexa Fluor 488 nm, 546 nm or 647 nm secondary antibody (1:500) at RT.
For TEM, mouse eyes were enucleated and fixed in 2% glutaraldehyde, 2% paraformaldehyde in 0.1M sodium cacodylate buffer (pH 7.2), overnight at RT. The anterior portion was removed on the next morning, and processed as described [Li et al. 2013].
For immunoelectron microscopy, eyecups were fixed by immersion in 0.1% glutaraldehyde + 2% paraformaldehyde in 0.1M sodium cacodylate buffer (pH 7.2) and processed for embedment in LR White. LR White ultrathin sections were etched with saturated sodium periodate (Sigma, St. Louis, MO, USA), blocked with 4% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 1 hour, incubated with 1:100 M-opsin antibody in TBS + 1% BSA + 1% Tween 20 overnight at 4°C, washed and incubated with goat anti-rabbit IgG conjugated to 10 nm gold (Aurion, Electron Microscopy Sciences, Hatfield, PA, USA) in TBS + 1% BSA + 1% Tween 20 for 1 hour. EM sections were observed in a Zeiss EM900 electron microscope and pictures were taken with a coupled digital camera using the ImageSP software.
Results and Discussion
Rp2null mice possess longer COS
We previously showed that ablation of Rp2 in mice results in early loss of cone photoreceptors (as early as 1 month of age) as compared to rods [Li et al. 2013]. These studies indicated that RP2 plays a distinct role in cones. After further investigation, we observed elongation of COS in Rp2null mice. As shown in Figure 1A, M-opsin (red; marker for COS) stained OS in Rp2null retina are considerably longer than in sibling control mice, as early as 1 month of age.
Figure 1.
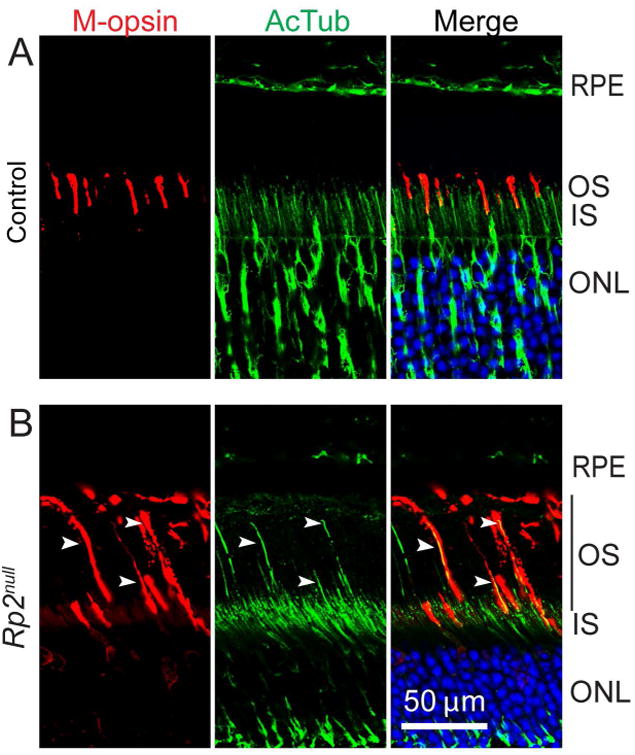
Mouse retinas from 2 months old sibling control (A) or Rp2null (B) mice were stained with anti-M-opsin (red) antibody or anti-acetylated tubulin (AcTub; green). Arrowheads indicate the elongated OS (depicted by black bar) in cones, as depicted by co-localized signal in merged images. RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; ONL, outer nuclear layer. Scale bar: 50 μm.
We then asked whether COS elongation coincides with extension of the ciliary cytoskeleton. To this end, we performed immunostaining of Rp2null retinas with anti-acetylated α-tubulin (a marker of ciliary microtubules) antibody. We found that the elongated staining of M-opsin coincided with axoneme staining (arrowheads) (Figure 1B), indicating extension of both the axoneme and ciliary membrane.
Rp2 conditional mutant mice
To assess whether the elongated COS phenotype is due to a cone-autonomous effect or a secondary effect of dying rods, we generated conditional mutants of Rp2, in which the Rp2 gene is specifically ablated in cone or rod photoreceptors. We bred the Rp2flox mice in which exon 2 of Rp2 is flanked by loxp sites [Li et al. 2013] to previously described transgenic mice expressing the Cre gene under the control of human red-green opsin promoter (Medium wave-length (M) cone specific expression) [Le et al. 2004] (for cone specific expression; Rp2MKO:M-opsin promoter-driven Cre knock out) or rhodopsin promoter (iCre75; for rod specific expression of Cre; Rp2iko: iCre75-driven Cre knock out) [Li et al. 2005]. Expression of the CRE protein was confirmed by immunofluorescence analysis using the anti-CRE antibody. As shown in Supplementary Figure S1A and B, CRE expression in Rp2MKO mice is confined to the top layers of the outer nuclear layer (ONL), which consist of mostly cone photoreceptor nuclei whereas CRE-staining in the Rp2iko mice is detected in majority of photoreceptor nuclei, which belong to rod photoreceptors. We first assessed the effect of conditional inactivation of Rp2 on photoreceptor function by electroretinography (ERG). ERG analysis of 1 month and 3 months old mice revealed an early progressive cone dysfunction, as determined by decline in the photopic a-wave amplitude (p<0.05 and p<0.01) (Figure 2 A, B). On the other hand, inactivation of Rp2 in rods (Rp2iko) resulted in a delayed and relatively less severe dysfunction of scotopic a- and b-wave responses (rod-mediated) with no detectable effect on photopic (cone-derived) signals (Figure 2 C, D). Taken together, we found that the conditional ablation of Rp2 in cones phenocopies the early cone dysfunction in the Rp2null mice.
Figure 2.
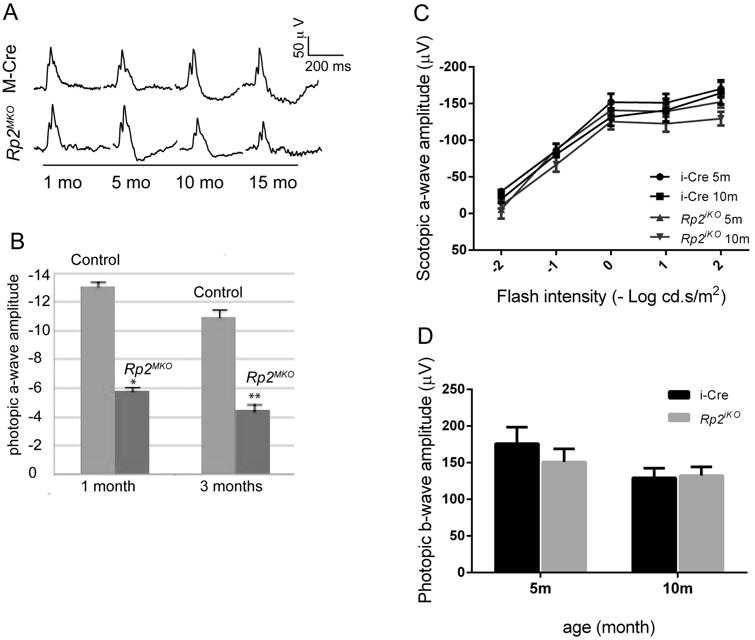
A and B. Photopic ERG analysis represented as wave forms (A) and quantitative analysis (B) of the a-wave amplitude of the Rp2MKO mice at indicated ages as compared to control M-Cre mice show a progressive decline in cone function. *: p<0.05; **: p<0.01. C, D show the scotopic (C) ERG and photopic (D) ERG recordings of the Rp2iko and control mice. All data represent analysis of 6 mice of each genotype.
COS in the Rp2MKO and Rp2iko mice
We then investigated the localization of M-opsin in Rp2MKO and Rp2iko mice. Immunofluorescence analysis revealed that the M-opsin stained OS is considerably longer than the sibling control mice in Rp2MKO mice (Figure 3A). Our analysis of Rp2iko mice did not reveal COS extension even at 6 months of age (Figure 3B), indicating that Rp2 ablation in rods does not affect COS extension. We did not detect a change in the length of the rod OS in the absence of RP2, as determined by staining with anti-rds (retinal degeneration slow; rod OS marker) antibody (Supplementary Figure S2).
Figure 3.
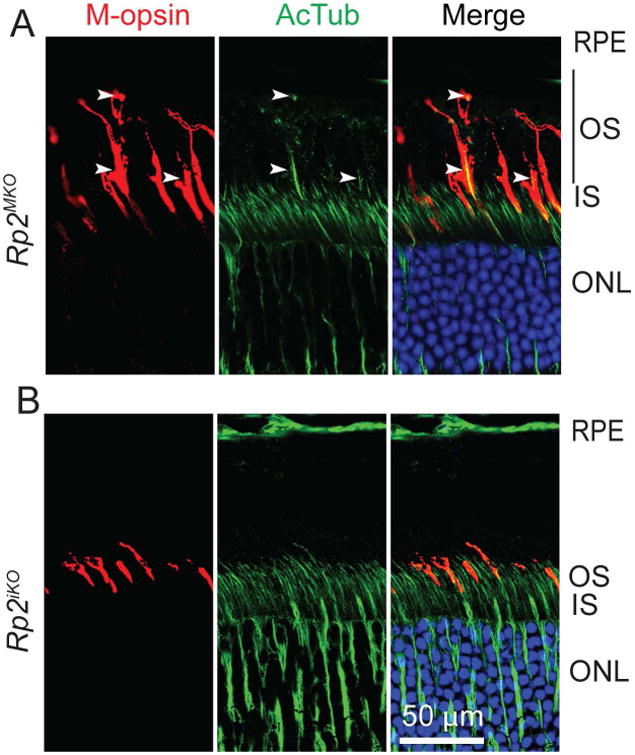
Mouse retinas from 2 months old Rp2MKO (A) or Rp2iko (B) were stained with anti-M-opsin (red) antibody or anti-acetylated tubulin (AcTub; green). Arrowheads indicate the elongated OS (depicted by black bar) in cones, as depicted by co-localized signal in merged images. RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; ONL, outer nuclear layer. Scale bar: 50 μm.
Quantitative analysis of elongated COS
We then investigated the extent of elongation of the COS and the corresponding axoneme by using two methods: (i) quantifying the distance between the base and the tip of the COS in the Rp2null and Rp2MKO mice and (ii) measuring the distance between the outer limiting membrane (base of the inner segment) and the base of the RPE (retinal pigmented epithelium) after staining with anti-M-opsin antibody and anti-acetylated α-tubulin antibodies. Analysis of the COS length revealed significant (p<0.01 and p<0.0001) increase in the length of the COS in Rp2null and Rp2MKO retina respectively compared to age-matched control) (Supplementary Figure S1C). As another validation for COS extension, we calculated the relative intensity in 10 equally distributed regions in the photoreceptor layer (1-close to outer limiting membrane (OLM) and 10: close to RPE for wild type, Rp2MKO and Rp2null retinas (Supplementary Figure S1 D-F). Increased intensity of acetylated α-tubulin and M-opsin was consistently detected in regions 6-9 of mutant retinas whereas majority of the signal in wild type retina was concentrated in regions 3-5.
Composition of Rp2 mutant COS
Previous studies have indicated a role of RP2 in regulating protein trafficking and cilia maintenance [Chapple et al. 2000; Evans et al. 2006; Holopainen et al. 2010; Hurd et al. 2010; Li et al. 2013; Patil et al. 2011; Schwarz et al. 2012; Wright et al. 2011]. We therefore, asked whether trafficking of key OS proteins is perturbed in the absence of RP2 to gain insights into the mode of COS elongation in Rp2MKO and Rp2null mice. Photoreceptor sensory cilium contains doublet microtubules in the TZ and singlet microtubules in the distal axoneme. The elongated COS showed normal localization of distal axonemal protein RP1 in both mutants (Figure 4 A-C). Previous reports showed that MAK (male germ cell associated kinase) negatively regulates cilia length in photoreceptors [Omori et al. 2010]. We tested whether absence of MAK in Rp2null retina mediates the long COS phenotype. Contrary to our hypothesis, our analysis revealed localization of MAK in the axoneme of the elongated COS (Figure 4 D-F). However, KIF3A, a microtubule motor previously shown to be associated with doublet microtubules of the TZ of cilia, did not localize in the long cone axoneme (Figure 4 G, H). Although subtle differences in the amount of MAK or other axonemal proteins may account for long COS, such changes in relative amounts of COS proteins might be below the detection levels of analyses due to paucity of cones in mice.
Figure 4.
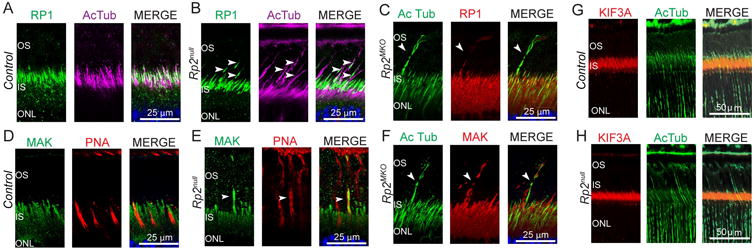
Retinas from control, Rp2null and Rp2MKO mice were stained with anti- RP1 (A, B, green and C, red), MAK (D, E, green; F, red) or KIF3A (G, H, red) antibodies. Anti-acetylated α-tubulin (AcTub) antibody was used as axoneme marker. Arrowheads indicate the presence of RP1-specific and MAK-specific signal in the elongated cone outer segment of mutant retinas. G, H: Immunostaining of control and Rp2null mouse retina sections did not show KIF3A positive signal in elongated COS. Nuclei are stained with Hoechst (blue).
We then examined the ultrastructure of the COS in Rp2-mutant mice. Transmission electron microscopy (TEM) analysis of Rp2null mice revealed that the abnormally long COS have disorganized OS morphology (Figure 5 A, B). Cones were identified by their unique morphology, nucleus and electron density of the inner segment, and the COS was stained with anti-M-opsin antibody (Figure 5 C, C′).
Figure 5.
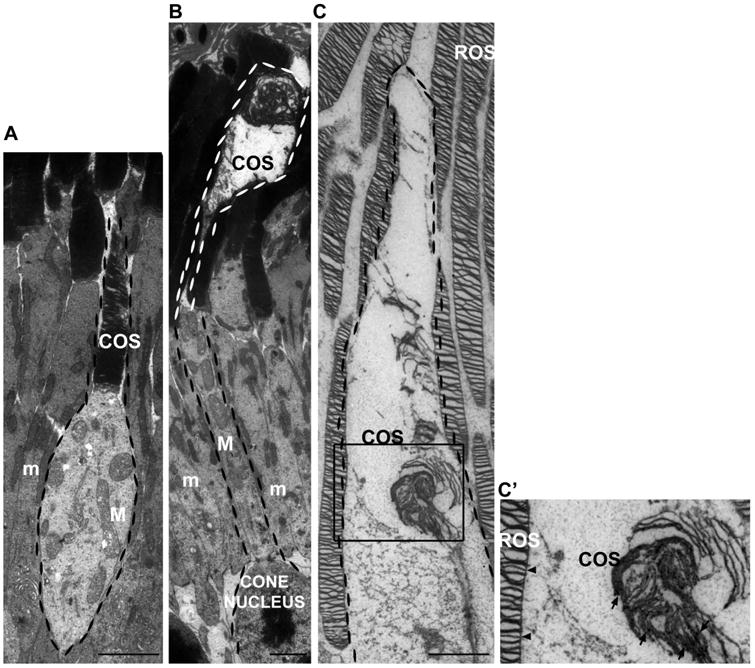
TEM analysis of the control and the Rp2null mouse retina was performed. The cone photoreceptors are highlighted with a dotted border. Inner segments of cones are distinguishable since at the EM level, cone mitochondria (M) are less electron-dense, larger and more distended than rod mitochondria (m). As compared to the sibling control mice (A), the Rp2null mouse retinas showed extension of COS and disorganized discs (B). The OS of Rp2null cone extended much longer than control with disorganized lamellae. C: Immunogold-labeling of cone opsin in an Rp2null cone photoreceptor (square enlarged in C′;), showing extension of the outer segment and disorganization of disks. C′: Gold particles are present in COS (arrows) but not in ROS (rod outer segment) (arrowheads). Scale bars: 2500 nm.
These data indicate that (i) the elongated COS contains long distal axoneme and (ii) elongation of COS is not due to the absence of MAK, a negative regulator of photoreceptor cilia length. It is possible that other MAK-like cilia length regulators are present in cone photoreceptors, whose activity and/or localization is mediated by RP2. RP2 may also be involved in modulating the turnover of tubulin required for the extension of OS axonemal microtubules. RP2 shows homology to tubulin-binding cofactor C, which, along with other cofactors, is an activator of GTPase activity of tubulin [Bartolini et al. 2002; Kuhnel et al. 2006]. Consistent with this, RP2 was found to stimulate the activity of tubulin as well as another small GTPase ARL3 (Arf-GTPase like-3) [Bartolini et al. 2002; Grayson et al. 2002; Kuhnel et al. 2006]. However, if such activity were to be involved in COS extension it should be regulated by cone-specific modulators of microtubule extension.
Photoreceptor cilia display a unique characteristic of periodic shedding of their distal tips, which are engulfed by the overlaying retinal pigment epithelium (RPE). It is estimated that about 10% of the distal OS is shed each day and the renewal of OS involved trafficking of immense load of opsins and other OS proteins [Besharse and Hollyfield 1976; Besharse and Hollyfield 1979; Besharse et al. 1977; Young 1968]. Thus, even subtle defects in the regulation of OS formation and function are associated with severe retinal degenerative diseases, such as retinitis pigmentosa (RP), which results from dysfunction and degeneration of photoreceptors [Deretic et al. 1995; Deretic et al. 2005; Heckenlively et al. 1988; Mazelova et al. 2009; Pazour et al. 2002]. An alternative explanation for long COS is that RP2 regulates the shedding of the COS tips. Elegant previous studies revealed that COS shedding occurs similarly to that of the rod OS and likely by a conserved mechanism [Anderson et al. 1978]. It has been shown that asymmetric distribution of anionic phospholipids such as phosphoserine (PS), redistributed to the external surface of the OS, is the main ‘eat me’ signal recognized by the RPE [Ruggiero et al. 2012]. Hyperelongation of the COS may indicate a defect in this recognition process. As RP2 is shown to associate with membranes and phospholipids in vitro [Demers et al. 2015], we suggest that RP2 is involved in regulating the trafficking of moieties involved in facilitating OS shedding.
Taken together, our data provide a platform for further studies to unravel not only the mechanism of cone sensory cilium elongation but also to understand the mechanism by which RP2 specifically regulates COS extension. However, a roadblock to testing such hypotheses is the paucity of cones in conventional animal models. Recently, a gene-trap mouse model of Rp2 ablation was reported to have a relatively delayed defect in both cones and rods and associated with defective trafficking of prenylated proteins [Zhang et al. 2015]. Moreover, a zebrafish model of rp2 ablation was recently reported [Liu et al. 2015]. This model develops an early rod and cone dysfunction but a preservation of the morphology of the retina up to 7 months of age. Both the zebrafish and the gene-trap mouse models show defects in the trafficking of prenylated proteins. However, no effect of the loss of RP2 on the extension of COS was reported in these studies. The progression of photoreceptor dysfunction in the Rp2-gene trap mice as well as decrease in cone PDE6 levels were similar to that of the Rp2null mice used in this study [Mookherjee 2015]. It would however, be interesting to delineate the effect of loss of RP2 on the COS of the gene trap Rp2-ko mice and the rp2-ko zebrafish. Taken together, these studies further attest to the complexities and clinical heterogeneity of RP2-associated disease and warrant the development of amenable animal models to examine the underlying mechanisms of cell-autonomous effects of widely expressed genes. Such information also holds key to delineating the pathogenesis of pleiotropic ciliary disorders.
Supplementary Material
Supplementary Figure S1: A, B: Immunostaining of cryosections of mouse retinas of indicated genotypes was performed using anti-CRE antibody (green). Scale bar: 50 μm. C. To quantify COS length, 246μm × 246μm images were taken 300-500 μm away from the optic nerve head of the control, Rp2null and Rp2MKO mice. Maximum projection from a 4 μm Z-stack series was used to quantify the distance between the base and the tip of the COS. Statistical analysis was performed from 5 mice in each group. *: p<0.01; **: p<0.0001. D-F: Quantitative analysis of the M-opsin and acetylated α-tubulin positive signal in the control, Rp2null or Rp2MKO retina was performed. 2-3 months old mice retina cryosections were stained with antibodies against M-opsin (red) and acetylated α-tubulin (green). A representative image of a wild type mouse retina is shown (D). The region from the outer limiting membrane (OLM) to the RPE is equally divided into 10 parts. Fluorescence intensity of each fraction using anti-acetylated α-tubulin (E) or M-opsin (F) is quantified as percentage of total intensity of the area between OLM and RPE. The results are analyzed from 6 images of 3 mice for each group. RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; OLM, outer limiting membrane; ONL, outer nuclear layer.
Supplementary Figure S2: Mouse retina cryosections of indicated genotypes were stained with anti-rds (retinal degeneration slow; a rod marker; green) and anti-M-opsin (red) antibodies. Although the rod OS length does not vary, increase in COS staining (white arrowheads) was detected in the Rp2null and Rp2MKO mice, but not in the control mice. Scale bar: 50 μm.
Acknowledgments
This work is supported by grants from Foundation Fighting Blindness, UMCCTS, and National Eye Institute (EY022372); UMASS Cell Biology Confocal Core and Electron Microscopy Core (Award # S10RR027897). The authors thank Dr. Gregory Pazour and Dr. George Witman (UMASS Medical School) for helpful discussions.
Footnotes
Author Contributions: L.L., K.N.R., and C.L.D.: performed experiments
TWH and YL: provided reagents
H.K.: conceived the study and wrote the paper
Competing Financial Interests: The authors declare no conflict of interest.
References
- Anderson DH, Fisher SK, Steinberg RH. Mammalian cones: disc shedding, phagocytosis, and renewal. Invest Ophthalmol Vis Sci. 1978;17:117–33. [PubMed] [Google Scholar]
- Bartolini F, Bhamidipati A, Thomas S, Schwahn U, Lewis SA, Cowan NJ. Functional overlap between retinitis pigmentosa 2 protein and the tubulin-specific chaperone cofactor C. J Biol Chem. 2002;277:14629–34. doi: 10.1074/jbc.M200128200. [DOI] [PubMed] [Google Scholar]
- Besharse JC. In: The Retina: A Model for Cell Biological Studies Part I. Adler R, Farber D, editors. Academic; New York: 1986. pp. 297–352. [Google Scholar]
- Besharse JC, Hollyfield JG. Renewal of normal and degenerating photoreceptor outer segments in the Ozark cave salamander. J Exp Zool. 1976;198:287–302. doi: 10.1002/jez.1401980302. [DOI] [PubMed] [Google Scholar]
- Besharse JC, Hollyfield JG. Turnover of mouse photoreceptor outer segments in constant light and darkness. Invest Ophthalmol Vis Sci. 1979;18:1019–24. [PubMed] [Google Scholar]
- Besharse JC, Hollyfield JG, Rayborn ME. Photoreceptor outer segments: accelerated membrane renewal in rods after exposure to light. Science. 1977;196:536–8. doi: 10.1126/science.300504. [DOI] [PubMed] [Google Scholar]
- Breuer DK, Yashar BM, Filippova E, Hiriyanna S, Lyons RH, Mears AJ, Asaye B, Acar C, Vervoort R, Wright AF, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–54. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979a;188:245–62. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. II. Autoradiographic analysis of cell generation using tritiated thymidine. J Comp Neurol. 1979b;188:263–72. doi: 10.1002/cne.901880205. [DOI] [PubMed] [Google Scholar]
- Chapple JP, Hardcastle AJ, Grayson C, Spackman LA, Willison KR, Cheetham ME. Mutations in the N-terminus of the X-linked retinitis pigmentosa protein RP2 interfere with the normal targeting of the protein to the plasma membrane. Hum Mol Genet. 2000;9:1919–26. doi: 10.1093/hmg/9.13.1919. [DOI] [PubMed] [Google Scholar]
- Churchill JD, Bowne SJ, Sullivan LS, Lewis RA, Wheaton DK, Birch DG, Branham KE, Heckenlively JR, Daiger SP. Mutations in the X-linked retinitis pigmentosa genes RPGR and RP2 found in 8.5% of families with a provisional diagnosis of autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54:1411–6. doi: 10.1167/iovs.12-11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demers E, Boisselier E, Horchani H, Blaudez D, Calvez P, Cantin L, Belley N, Champagne S, Desbat B, Salesse C. Lipid Selectivity, Orientation, and Extent of Membrane Binding of Nonacylated RP2. Biochemistry. 2015;54:2560–70. doi: 10.1021/bi501517r. [DOI] [PubMed] [Google Scholar]
- Deretic D, Huber LA, Ransom N, Mancini M, Simons K, Papermaster DS. rab8 in retinal photoreceptors may participate in rhodopsin transport and in rod outer segment disk morphogenesis. J Cell Sci. 1995;108(Pt 1):215–24. doi: 10.1242/jcs.108.1.215. [DOI] [PubMed] [Google Scholar]
- Deretic D, Williams AH, Ransom N, Morel V, Hargrave PA, Arendt A. Rhodopsin C terminus, the site of mutations causing retinal disease, regulates trafficking by binding to ADP-ribosylation factor 4 (ARF4) Proc Natl Acad Sci U S A. 2005;102:3301–6. doi: 10.1073/pnas.0500095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel BD, Ludington WB, Marshall WF. Intraflagellar transport particle size scales inversely with flagellar length: revisiting the balance-point length control model. J Cell Biol. 2009;187:81–9. doi: 10.1083/jcb.200812084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RJ, Hardcastle AJ, Cheetham ME. Focus on molecules: X-linked Retinitis Pigmentosa 2 protein, RP2. Exp Eye Res. 2006;82:543–4. doi: 10.1016/j.exer.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Grayson C, Bartolini F, Chapple JP, Willison KR, Bhamidipati A, Lewis SA, Luthert PJ, Hardcastle AJ, Cowan NJ, Cheetham ME. Localization in the human retina of the X-linked retinitis pigmentosa protein RP2, its homologue cofactor C and the RP2 interacting protein Arl3. Hum Mol Genet. 2002;11:3065–74. doi: 10.1093/hmg/11.24.3065. [DOI] [PubMed] [Google Scholar]
- Heckenlively JR, Yoser SL, Friedman LH, Oversier JJ. Clinical findings and common symptoms in retinitis pigmentosa. Am J Ophthalmol. 1988;105:504–11. doi: 10.1016/0002-9394(88)90242-5. [DOI] [PubMed] [Google Scholar]
- Holopainen JM, Cheng CL, Molday LL, Johal G, Coleman J, Dyka F, Hii T, Ahn J, Molday RS. Interaction and localization of the retinitis pigmentosa protein RP2 and NSF in retinal photoreceptor cells. Biochemistry. 2010;49:7439–47. doi: 10.1021/bi1005249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd T, Zhou W, Jenkins P, Liu CJ, Swaroop A, Khanna H, Martens J, Hildebrandt F, Margolis B. The retinitis pigmentosa protein RP2 interacts with polycystin 2 and regulates cilia-mediated vertebrate development. Hum Mol Genet. 2010;19:4330–44. doi: 10.1093/hmg/ddq355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. Principles of neural science. Vol. 1. New York: McGraw-Hill; 2013. p. 1709. [Google Scholar]
- Kessel RG, Kardon RH. Tissues and organs : a text-atlas of scanning electron microscopy. ix. San Francisco: W. H. Freeman; 1979. p. 317. [Google Scholar]
- Khan NW, Wissinger B, Kohl S, Sieving PA. CNGB3 achromatopsia with progressive loss of residual cone function and impaired rod-mediated function. Invest Ophthalmol Vis Sci. 2007;48:3864–71. doi: 10.1167/iovs.06-1521. [DOI] [PubMed] [Google Scholar]
- Kohl S, Baumann B, Broghammer M, Jagle H, Sieving P, Kellner U, Spegal R, Anastasi M, Zrenner E, Sharpe LT, et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet. 2000;9:2107–16. doi: 10.1093/hmg/9.14.2107. [DOI] [PubMed] [Google Scholar]
- Komaromy AM, Rowlan JS, Corr AT, Reinstein SL, Boye SL, Cooper AE, Gonzalez A, Levy B, Wen R, Hauswirth WW, et al. Transient photoreceptor deconstruction by CNTF enhances rAAV-mediated cone functional rescue in late stage CNGB3-achromatopsia. Mol Ther. 2013;21:1131–41. doi: 10.1038/mt.2013.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnel K, Veltel S, Schlichting I, Wittinghofer A. Crystal structure of the human retinitis pigmentosa 2 protein and its interaction with Arl3. Structure. 2006;14:367–78. doi: 10.1016/j.str.2005.11.008. [DOI] [PubMed] [Google Scholar]
- LaVail MM. Kinetics of rod outer segment renewal in the developing mouse retina. J Cell Biol. 1973;58:650–61. doi: 10.1083/jcb.58.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le YZ, Ash JD, Al-Ubaidi MR, Chen Y, Ma JX, Anderson RE. Targeted expression of Cre recombinase to cone photoreceptors in transgenic mice. Mol Vis. 2004;10:1011–8. [PubMed] [Google Scholar]
- Leveillard T, Mohand-Said S, Lorentz O, Hicks D, Fintz AC, Clerin E, Simonutti M, Forster V, Cavusoglu N, Chalmel F, et al. Identification and characterization of rod-derived cone viability factor. Nat Genet. 2004;36:755–9. doi: 10.1038/ng1386. [DOI] [PubMed] [Google Scholar]
- Li L, Khan N, Hurd T, Ghosh AK, Cheng C, Molday R, Heckenlively JR, Swaroop A, Khanna H. Ablation of the X-linked retinitis pigmentosa 2 (Rp2) gene in mice results in opsin mislocalization and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2013;54:4503–11. doi: 10.1167/iovs.13-12140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chen D, Sauve Y, McCandless J, Chen YJ, Chen CK. Rhodopsin-iCre transgenic mouse line for Cre-mediated rod-specific gene targeting. Genesis. 2005;41:73–80. doi: 10.1002/gene.20097. [DOI] [PubMed] [Google Scholar]
- Liu F, Chen J, Yu S, Raghupathy RK, Liu X, Qin Y, Li C, Huang M, Liao S, Wang J, et al. Knockout of RP2 decreases GRK1 and rod transducin subunits and leads to photoreceptor degeneration in zebrafish. Hum Mol Genet. 2015;24:4648–59. doi: 10.1093/hmg/ddv197. [DOI] [PubMed] [Google Scholar]
- Liu Q, Tan G, Levenkova N, Li T, Pugh EN, Jr, Rux JJ, Speicher DW, Pierce EA. The proteome of the mouse photoreceptor sensory cilium complex. Mol Cell Proteomics. 2007;6:1299–317. doi: 10.1074/mcp.M700054-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livesey FJ, Cepko CL. Vertebrate neural cell-fate determination: lessons from the retina. Nat Rev Neurosci. 2001;2:109–18. doi: 10.1038/35053522. [DOI] [PubMed] [Google Scholar]
- Mazelova J, Astuto-Gribble L, Inoue H, Tam BM, Schonteich E, Prekeris R, Moritz OL, Randazzo PA, Deretic D. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28:183–92. doi: 10.1038/emboj.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, Swaroop A. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–52. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- Mookherjee S, Hiriyanna S, Kaneshiro K, Li L, Li Y, Li W, Qian H, Li T, Khanna H, Colosi P, Swaroop A, Wu Z. Long-term rescue of cone photoreceptor degeneration in Retinitis Pigmentosa 2 (RP2) Knockout mice by gene replacement therapy. Hum Mol Genet. 2015;187:81–9. doi: 10.1093/hmg/ddv354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omori Y, Chaya T, Katoh K, Kajimura N, Sato S, Muraoka K, Ueno S, Koyasu T, Kondo M, Furukawa T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci U S A. 2010;107:22671–6. doi: 10.1073/pnas.1009437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil SB, Hurd TW, Ghosh AK, Murga-Zamalloa CA, Khanna H. Functional analysis of retinitis pigmentosa 2 (RP2) protein reveals variable pathogenic potential of disease-associated missense variants. PLoS One. 2011;6:e21379. doi: 10.1371/journal.pone.0021379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–13. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Rosenbaum JL. Intraflagellar transport and cilia-dependent diseases. Trends Cell Biol. 2002;12:551–5. doi: 10.1016/s0962-8924(02)02410-8. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Witman GB. The vertebrate primary cilium is a sensory organelle. Curr Opin Cell Biol. 2003;15:105–10. doi: 10.1016/s0955-0674(02)00012-1. [DOI] [PubMed] [Google Scholar]
- Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12:44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum J. Intraflagellar transport. Curr Biol. 2002;12:R125. doi: 10.1016/s0960-9822(02)00703-0. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Cole DG, Diener DR. Intraflagellar transport: the eyes have it. J Cell Biol. 1999;144:385–8. doi: 10.1083/jcb.144.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero L, Connor MP, Chen J, Langen R, Finnemann SC. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5-/- or Mfge8-/- mouse retina. Proc Natl Acad Sci U S A. 2012;109:8145–8. doi: 10.1073/pnas.1121101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satir P. Primary cilia: integral to development and disease. Dev Dyn. 2008;237:1953–4. doi: 10.1002/dvdy.21470. [DOI] [PubMed] [Google Scholar]
- Satir P, Christensen ST. Structure and function of mammalian cilia. Histochem Cell Biol. 2008;129:687–93. doi: 10.1007/s00418-008-0416-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–32. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- Schwarz N, Novoselova TV, Wait R, Hardcastle AJ, Cheetham ME. The X-linked retinitis pigmentosa protein RP2 facilitates G protein traffic. Hum Mol Genet. 2012;21:863–73. doi: 10.1093/hmg/ddr520. [DOI] [PubMed] [Google Scholar]
- Sharon D, Bruns GA, McGee TL, Sandberg MA, Berson EL, Dryja TP. X-linked retinitis pigmentosa: mutation spectrum of the RPGR and RP2 genes and correlation with visual function. Invest Ophthalmol Vis Sci. 2000;41:2712–21. [PubMed] [Google Scholar]
- Sidjanin DJ, Lowe JK, McElwee JL, Milne BS, Phippen TM, Sargan DR, Aguirre GD, Acland GM, Ostrander EA. Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum Mol Genet. 2002;11:1823–33. doi: 10.1093/hmg/11.16.1823. [DOI] [PubMed] [Google Scholar]
- Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol. 2010;190:953–63. doi: 10.1083/jcb.201006020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaroop A, Kim D, Forrest D. Transcriptional regulation of photoreceptor development and homeostasis in the mammalian retina. Nat Rev Neurosci. 2010;11:563–76. doi: 10.1038/nrn2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veleri S, Lazar CH, Chang B, Sieving PA, Banin E, Swaroop A. Biology and therapy of inherited retinal degenerative disease: insights from mouse models. Dis Model Mech. 2015;8:109–29. doi: 10.1242/dmm.017913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11:273–284. doi: 10.1038/nrg2717. [DOI] [PubMed] [Google Scholar]
- Wright KJ, Baye LM, Olivier-Mason A, Mukhopadhyay S, Sang L, Kwong M, Wang W, Pretorius PR, Sheffield VC, Sengupta P, et al. An ARL3-UNC119-RP2 GTPase cycle targets myristoylated NPHP3 to the primary cilium. Genes Dev. 2011;25:2347–60. doi: 10.1101/gad.173443.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DM, Khanna H, Atmaca-Sonmez P, Sieving PA, Branham K, Othman M, Swaroop A, Daiger SP, Heckenlively JR. Long-term follow-up of a family with dominant X-linked retinitis pigmentosa. Eye (Lond) 2010;24:764–74. doi: 10.1038/eye.2009.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz O, Khanna H. Ciliary signaling cascades in photoreceptors. Vision Res. 2012;75:112–6. doi: 10.1016/j.visres.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW. Passage of newly formed protein through the connecting cilium of retina rods in the frog. J Ultrastruct Res. 1968;23:462–73. doi: 10.1016/s0022-5320(68)80111-x. [DOI] [PubMed] [Google Scholar]
- Zhang H, Hanke-Gogokhia C, Jiang L, Li X, Wang P, Gerstner CD, Frederick JM, Yang Z, Baehr W. Mistrafficking of prenylated proteins causes retinitis pigmentosa 2. FASEB J. 2015;29:932–42. doi: 10.1096/fj.14-257915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Brown B, Li A, Mears AJ, Swaroop A, Craft CM. GRK1-dependent phosphorylation of S and M opsins and their binding to cone arrestin during cone phototransduction in the mouse retina. J Neurosci. 2003;23:6152–60. doi: 10.1523/JNEUROSCI.23-14-06152.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1: A, B: Immunostaining of cryosections of mouse retinas of indicated genotypes was performed using anti-CRE antibody (green). Scale bar: 50 μm. C. To quantify COS length, 246μm × 246μm images were taken 300-500 μm away from the optic nerve head of the control, Rp2null and Rp2MKO mice. Maximum projection from a 4 μm Z-stack series was used to quantify the distance between the base and the tip of the COS. Statistical analysis was performed from 5 mice in each group. *: p<0.01; **: p<0.0001. D-F: Quantitative analysis of the M-opsin and acetylated α-tubulin positive signal in the control, Rp2null or Rp2MKO retina was performed. 2-3 months old mice retina cryosections were stained with antibodies against M-opsin (red) and acetylated α-tubulin (green). A representative image of a wild type mouse retina is shown (D). The region from the outer limiting membrane (OLM) to the RPE is equally divided into 10 parts. Fluorescence intensity of each fraction using anti-acetylated α-tubulin (E) or M-opsin (F) is quantified as percentage of total intensity of the area between OLM and RPE. The results are analyzed from 6 images of 3 mice for each group. RPE, retinal pigment epithelium; OS, outer segment; IS, inner segment; OLM, outer limiting membrane; ONL, outer nuclear layer.
Supplementary Figure S2: Mouse retina cryosections of indicated genotypes were stained with anti-rds (retinal degeneration slow; a rod marker; green) and anti-M-opsin (red) antibodies. Although the rod OS length does not vary, increase in COS staining (white arrowheads) was detected in the Rp2null and Rp2MKO mice, but not in the control mice. Scale bar: 50 μm.