

Abstract
Background
A novel member of the Wnt signalling pathway, Chibby, was recently identified. This protein inhibits Wnt/β-catenin mediated transcriptional activation by competing with Lef-1 (the transcription factor and target of β-catenin) to bind to β-catenin. This suggests that Chibby could be a tumour suppressor protein. The C22orf2 gene coding Chibby is located on chromosome 22, a region recurrently lost in colorectal cancer. Activation of the Wnt pathway is a major feature of colorectal cancer and occurs through inactivation of APC or activation of β-catenin. All of this led us to analyse the possible implication of Chibby in colorectal carcinogenesis.
Methods
First, 36 tumour and matched normal colonic mucosa DNA were genotyped with five microsatellite markers located on chromosome 22 to search for loss of heterozygosity. Then, mutation screening of the C22orf2 coding sequence and splice sites was performed in the 36 tumour DNA. Finally, expression of Chibby was analysed by quantitative RT-PCR on 10 patients, 4 with loss of heterozygosity (LOH) on chromosome 22.
Results
Loss of heterozygosity involving the C22orf2 region was detected in 11 out of 36 patients (30%). Sequencing analysis revealed a known variant, rs3747174, in exon 5: T321C leading to a silent amino acid polymorphism A107A. Allelic frequencies were 0.69 and 0.31 for T and C variants respectively. No other mutation was detected. Among the 10 patients studied, expression analysis revealed that Chibby is overexpressed in 2 tumours and underexpressed in 1. No correlations were found with 22q LOH status.
Conclusion
As no somatic mutation was detected in C22orf2 in 36 colorectal tumour DNA, our results do not support the implication of Chibby as a tumour suppressor in colorectal carcinogenesis. This was supported by the absence of underexpression of Chibby among the tumour samples with 22q LOH. The implication of other Wnt pathway members remains to be identified to explain the part of colorectal tumours without mutation in APC and β-catenin.
Background
Identifying components of the Wnt signalling pathway has been at the forefront of cancer biology since a link was made between Wnt, the mammalian homologue of the fruitfly Wingless (Wg), and the development of cancer. Acting through a core set of proteins that are highly conserved in evolution, this pathway regulates the ability of the oncoprotein β-catenin to activate transcription of specific target genes. This regulation, in turn, results in changes in expression of genes that modulate cell fate, proliferation and apoptosis [1]. Recently, Takemaru et al. identified a novel human protein, named Chibby, that interacts with the carboxy-terminal transcription activation domain of β-catenin [2]. Chibby is a nuclear protein of 126 amino acids with coiled-coil domains and is conserved from Drosophila to Human. It has been shown that Chibby antagonizes the Wnt signalling pathway by inhibition of the transcription protein complex comprising β-catenin. This result suggests that Chibby could act as a tumour suppressor protein.
In colorectal cancer, the activation of the Wnt signalling pathway occurs in more than 60% of tumours through the inactivation of the APC tumour suppressor gene by mutations and allelic losses, or through the presence of β-catenin activating mutations [3]. The C22orf2 gene encoding Chibby is located on 22q13.1. This chromosome region is frequently lost in colorectal cancer suggesting the existence of a tumour suppressor gene that remains to be identified.
The putative function and the location of the C22orf2 gene led us to analyse the possible implication of C22orf2 as a tumour suppressor gene in colorectal carcinogenesis. First, the allelic status of chromosome 22 was established on 36 colorectal tumour and matched normal colonic mucosa DNA, second, mutation analysis of the C22orf2 gene was performed on tumour DNA, and third, expression analysis of Chibby was studied in few patients.
Methods
Patients, sample collection and nucleic acid extraction
Tumour samples and matched normal colonic mucosa were collected from 36 patients (23 women, 13 men) hospitalised in the surgical department of Laennec Hospital in Paris between 1997 and 1999. The mean age of patients was 69.9 years old, range [56.4–83.4]. An histological HES staining was performed before DNA extraction and only tumour fragments with more than 70% of tumour cells were retained. Tumours were all classified as adenocarcinoma. Twenty-nine tumours were classified as well differentiated, and 7 as poor differentiated. No tumour samples showed a microsatellite instability phenotype. Tumours were located in proximal colon in 9 cases, in distal colon in 21 cases and 6 were located in rectum. According to TNM classification, tumours were classified in stage I in 1 case, stage II in 17, stage III in 8 and stage IV in 10 cases. Samples were immediately frozen in liquid nitrogen and stored at -80°C. Informed consent was signed according to French laws. DNA extraction was performed using the QIAamp® DNA Mini Kit (Qiagen, Courtaboeuf, France) for all patients in 1999, and stored at -20°C. Tumour samples and matched normal colonic mucosa were available for RNA extraction for 10 patients. RNA isolation was performed recently with RNeasy® Mini Kit (Qiagen) and stored at -80°C. Quality of RNA was determined by electrophoresis through agarose gel stained with ethidium bromide. Intensity of 18S and 28S RNA bands was estimated under UV light. Measuring UV absorbance at 260 nm was performed to quantify RNA.
Genotyping
Thirty-six tumour and matched normal colonic mucosa DNA were genotyped with five microsatellite markers located on chromosome 22. Amplification of the five markers was performed by multiplex PCR using 50 ng DNA in a final reaction volume of 12.5 μL using 6.25 μL of 2X Qiagen Multiplex PCR Master Mix (Qiagen) and variable concentration of primers as given in Table 1 (Goasguen et al, unpublished data). Primers are from Linkage Mapping Set (Applied Biosystems, Courtaboeuf, France) and labelled on forward primer by VIC, NED or 6-FAM. Amplified fragments were analysed after dilution on an ABI 310 Genetic Analyzer (Applied Biosystems). Allelic imbalance (AI) was analysed by Genotyper 2.1® software (Applied Biosystems) with an automatic data-processing trace program described previously [4]. AI was defined for each tumor as α = (NL/NS)/(TL/TS) where L is the intensity of the larger allele and S the intensity of the smaller allele in normal DNA (N) or tumour DNA (T). When α(loss of the small allele) or 1/α (loss of the large allele) was ≤ 0.5, LOH was ascertained.
Table 1.
Microsatellite markers analysed on chromosome 22
| Markers | 5'location on chromosome 22 (bp) | label (on forward primer) | concentration for Multiplex PCR (μM) | PCR product (bp) |
| D22S420 | 16133834 | VIC | 0,125 | 153–169 |
| D22S315 | 24240393 | 6-FAM | 0,5 | 180–210 |
| D22S283 | 34922517 | NED | 0,125 | 127–155 |
| C22orf2 | 37295719 | |||
| D22S423 | 38525224 | VIC | 0,125 | 287–309 |
| D22S274 | 43445681 | NED | 0,25 | 276–298 |
Sequencing analysis
PCR amplification of the four C22orf2 coding exons and intron-exon junctions was performed as described in Table 2. Due to the small size of intron 3, exons 3 and 4 were amplified together. 100 ng of tumour DNA were used in a final reaction volume of 30 μL. For exons 2 and 3–4, 0.75 U of AmpliTaq® polymerase (Applied Biosystems) were used with 3 μL of 10X corresponding buffer, 1.5 mM MgCl2, 800 μM of dNTP mix and 0.3 μM of each forward and reverse primers. Program was as follows : initial step at 94°C 5 min, 35 cycles of 94°C 30 s, 58°C 30 s and 72°C 30 s, and final step 72°C 7 min. For exon 5, 0.75 U of HotStarTaq® polymerase (Qiagen) were used with the same protocol except the initial step at 95°C 10 min and the annealing temperature at 56°C. PCR products were purified by centrifugation on a Sephadex G-100® (Amersham Biosciences, Orsay, France) filter loaded in a MultiScreen® plate (Millipore, Molsheim, France). Then direct sequencing was performed in both forward and reverse orientation from 2 μL of purified products using 2 μL of 2.5X BigDye™ Terminator v3.1 Ready Reaction Cycle Kit (Applied Biosystems) with 3 μL of 5X corresponding buffer and 0.5 μM of primer in a final reaction volume of 20 μL. Products were purified on Sephadex G-50® (Amersham Biosciences) and analysed on an ABI 310 Genetic Analyzer (Applied Biosystems). Sequence data were aligned using AutoAssembler™ 2.0 software (Applied Biosystems).
Table 2.
PCR conditions for sequencing analysis of the C22orf2 gene
| Exon | Primers (5'-3') | Annealing temperature (°C) | PCR product (bp) |
| 2 | ex2F: GGCATAAGGTCAGTGATCCAG | 58 | 236 |
| ex2R: TTCAGAGACACGCCTCAGCAC | |||
| 3–4 | ex3F: GGTCATATTGTTGGCGGAAG | 58 | 490 |
| ex4R: CCAAACTGGCATTGAGGAGA | |||
| 5 | ex5F: CCAGCAGCATCAGAGAAGTG | 56 | 245 |
| ex5R: AAAAGCACTCAGCCACATCC |
Quantitative RT-PCR
Three μg of RNA were reverse transcribed in a final volume of 50 μL using the High Capacity cDNA Archive Kit with random primers (Applied Biosystems). Reverse transcribed samples were diluted 20 fold in water and stored at -20°C. For each sample, 5 μL of cDNA, corresponding to 15 ng of reverse transcribed RNA, were analysed by SYBR Green PCR, in triplicate, using the ABI PRISM® 7900HT Sequence Detection System (Applied Biosystems). Q-PCR was performed in a final volume of 12 μL comprising 1X Master Mix SYBR Green (ABgene, Courtaboeuf, France) and 0.3 μM of forward and reverse primers for Chibby (Fwd: 5'-GGAGAAAACCAAGATTCCAG-3' ; Rev: 5'-CCAACACAACCCAACAGAG-3'). Levels of RNA expression were determined using the SDS software version 2.1 (Applied Biosystems) according to the 2-ΔΔCt method. Briefly, expression results of a gene were normalised to internal control ribosomal 18S and relatively to a calibrator, consisting in the mean expression level of the corresponding gene in normal colonic mucosa samples as follows : 2-ΔΔCt = 2-((Ct Chibby - Ct 18S)tumour sample) - (Ct Chibby - Ct 18S) calibrator)). Results are given in Table 3, they express the n-fold ratio of the gene expression in a sample compared to the mean of normal tissues. A ratio ≤ 0.66 has been considered as underexpression of Chibby and ≥ 1.5 as overexpression.
Table 3.
Expression analysis of Chibby by Quantitative RT-PCR
| Patient | 2-ΔΔCt | Expression of Chibby in tumour tissues compared to normal tissues | LOH on chromosome 22 |
| 20 | 0.46 | underexpressed | no |
| 27 | 0.78 | similar | no |
| 33 | 1.23 | similar | no |
| 26 | 1.43 | similar | no |
| 2 | 1.48 | similar | no |
| 19 | 1.57 | overexpressed | no |
| 22 | 0.93 | similar | yes |
| 24 | 1.24 | similar | yes |
| 14 | 1.34 | similar | yes |
| 3 | 2.26 | overexpressed | yes |
Results
Loss of heterozygosity analysis on chromosome 22
Five dinucleotide microsatellite markers spread over the entire chromosome 22 were genotyped in order to determine the existence of a loss of heterozygosity (LOH). The human gene coding Chibby, C22orf2, is localised between the markers D22S283 and D22S423 (Table 1). Thirty-six tumour and matched normal colonic mucosa DNA were amplified by multiplex PCR. A deletion of the entire chromosome 22 arm was observed in 9 cases, a distal deletion including C22orf2 was observed in 2 cases (Figure 1) and an interstitial deletion encompassing the D22S315 was observed in one case. Thus, among the 36 tumours, 11 (30%) demonstrated LOH involving the C22orf2 region.
Figure 1.
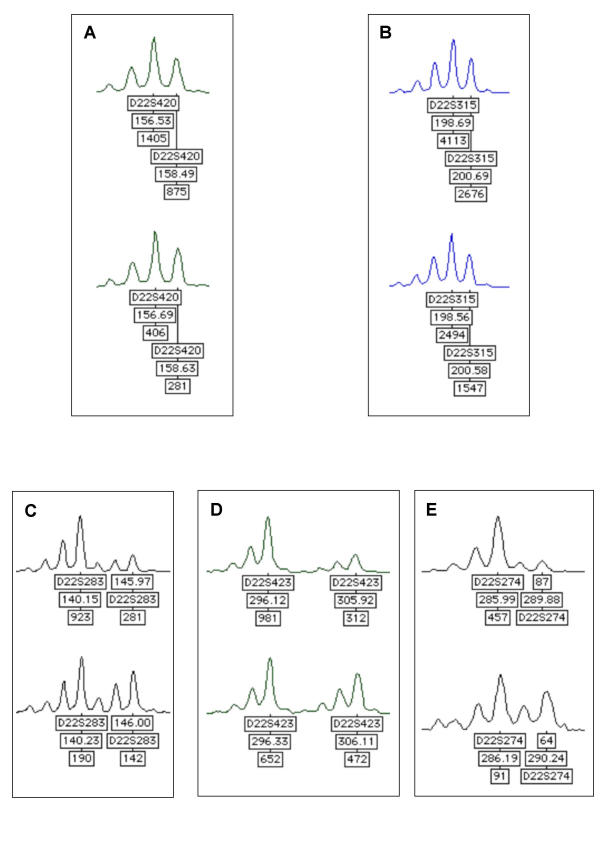
Microsatellite markers used for genotyping of chromosome 22. Genotyper® graphs obtained after multiplex PCR of the five microsatellite markers on chromosome 22 are shown on A, B, C, D, E. In each case the upper graph corresponds to the tumour DNA and the lower graph to the matched normal colonic mucosa DNA. Peaks are labelled with: name of markers, size of PCR products (bp) and peak intensity. A and B show no allelic loss for D22S420 and D22S315, whereas C, D and E show allelic loss for DS22S283, D22S423, D22S274. These results suggest a distal deletion of chromosome 22 including the C22orf2 gene.
Sequencing analysis of C22orf2
The C22orf2 gene spreads over 17.2 kb and comprises five exons. ORF starts at exon 2 leading to a 381 bp cDNA. Coding sequence and intron exon junctions were analysed by direct sequencing on the 36 tumour DNA to search for sequence variations. A known variant, rs3747174, was observed in exon 5 leading to a T321C transition, corresponding to a silent polymorphism A107A. Allelic frequencies were 0.69 and 0.31 for T and C variants respectively. The distribution of the different genotypes is in agreement with Hardy and Weinberg equilibrium. No other somatic mutation was detected either in coding exons or in splice sites.
Expression analysis of Chibby
The difference of the level of expression of Chibby between tumour samples and normal colonic mucosa samples was analysed by quantitative RT-PCR, using ribosomal 18S as internal control. Both samples were available for 10 patients, 4 of which presented LOH on chromosome 22. Q-PCR expression analysis revealed that Chibby is overexpressed in 2 tumours and underexpressed in 1 (Table 3). No correlations were found with 22q LOH status.
Discussion
The aim of this study was to analyse the possible implication of Chibby in colorectal carcinogenesis. Genotyping analysis was performed on chromosome 22 to search for loss of heterozygosity. Among the 36 patients, 12 (33%) showed LOH including the C22orf2 region. Mutation analysis was performed on C22orf2 coding sequence and splice sites: no somatic mutation was detected. However, a known variant, rs3747174, was observed in exon 5: T321C, corresponding to A107A. In the dbSNP database http://www.ncbi.nlm.nih.gov/SNP/, this variant was detected with a set of 52 chromosomes and allelic frequencies were estimated from 1496 chromosomes at 0.63 and 0.37 respectively which is similar to that observed in this present series, i.e. 0.67 and 0.31 respectively [5]. Furthermore, quantitative RT-PCR expression analysis showed that Chibby is overexpressed in 2 tumours, 1 of which showing LOH at C22orf2 locus, and Chibby is underexpressed in 1 tumour showing no 22q LOH. Taking together, these results do not support a putative epigenetic modification, i.e. methylation of the C22orf2 promoter, that could repress gene expression as another mechanism of gene inactivation than mutation. Thus, Chibby does not seem implicated as a tumour suppressor in colorectal carcinogenesis.
Conclusions
The APC gene was found mutated in several series of colorectal tumours with a frequency of 60%. Furthermore, in a series of tumours lacking APC mutations, 48% presented a mutation in the β-catenin regulatory domain [3]. Thus, more than one gene could be implicated in colorectal carcinogenesis through the activation of the Wnt signalling pathway. The recently identified function of Chibby in this pathway supports the idea that it could act as a tumour suppressor [2]. However, we did not detect mutation in a series of 36 colorectal tumours, suggesting that the role of Chibby in colorectal carcinogenesis is probably weak. Other genes remain to be studied to explain the part of colorectal tumours without mutation in APC and β-catenin.
Competing interests
None declared.
Authors' contributions
SG carried out genotyping and expression analysis, trained DT and drafted the manuscript. DT carried out the sequencing analysis. AL and NG have developed multiplex PCR with microsatellite markers. AB provided patient samples. PB and PLP conceived of the study, participated in its design and coordination, and drafted the manuscript. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
This study was supported by La Région Ile de France and La Ligue Nationale de Lutte Contre le Cancer.
Contributor Information
Sophie Gad, Email: Sophie.Gad@biomedicale.univ-paris5.fr.
David Teboul, Email: dteboul@noos.fr.
Astrid Lièvre, Email: Astrid.Lievre@biomedicale.univ-paris5.fr.
Nicolas Goasguen, Email: ngoa@9online.fr.
Anne Berger, Email: Anne.Berger@hop.egp.ap-paris.fr.
Philippe Beaune, Email: Philippe.Beaune@biomedicale.univ-paris5.fr.
Pierre Laurent-Puig, Email: Pierre.Laurent-Puig@biomedicale.univ-paris5.fr.
References
- Moon RT, Bowerman B, Boutros M, Perrimon N. The promise and perils of Wnt signaling through beta-catenin. Science. 2002;296:1644–1646. doi: 10.1126/science.1071549. [DOI] [PubMed] [Google Scholar]
- Takemaru K, Yamaguchi S, Lee YS, Zhang Y, Carthew RW, Moon RT. Chibby, a nuclear beta-catenin-associated antagonist of the Wnt/Wingless pathway. Nature. 2003;422:905–909. doi: 10.1038/nature01570. [DOI] [PubMed] [Google Scholar]
- Laurent-Puig P, Blons H, Cugnenc PH. Sequence of molecular genetic events in colorectal tumorigenesis. Eur J Cancer Prev. 1999;8 Suppl 1:S39–47. [PubMed] [Google Scholar]
- Bluteau O, Legoix P, Bayer J, Bioulac-Sage P, Flejou JF, Capron F, Monges G, Brechot C, Thomas G, Laurent-Puig P, Zucman-Rossi J. [Semi-automated quantitative method for detecting the loss of heterozygosity at the long arm of chromosome 4 in hepatocellular carcinoma] Gastroenterol Clin Biol. 1999;23:1225–1232. [PubMed] [Google Scholar]
- Haga H, Yamada R, Ohnishi Y, Nakamura Y, Tanaka T. Gene-based SNP discovery as part of the Japanese Millennium Genome Project: identification of 190,562 genetic variations in the human genome. Single-nucleotide polymorphism. J Hum Genet. 2002;47:605–610. doi: 10.1007/s100380200092. [DOI] [PubMed] [Google Scholar]