

Abstract
Background
Lipid transfer protein (LTP), an abundant protein in fruits, vegetables and nuts, is a common food allergen in Mediterranean areas causing diverse allergic reactions. Approximately 40% of food anaphylaxis induced by LTP require non-steroidal anti-inflammatory drugs (NSAIDs) as a triggering cofactor.
Objective
To better understand the determinants of NSAID-dependent (NSAID-LTP-A) and NSAID-independent LTP-anaphylaxis (LTP-A)
Methods
Selection of patients was based on a proven clinical history of NSAID-dependent or -independent anaphylaxis to LTP, positive skin prick test to LTP and serum LTP-IgE. Whole transcriptome (RNA-Seq) analysis of blood cells from 14 individuals with NSAID-LTP-A, 7 with LTP-A and 13 healthy controls was performed to identify distinct gene expression signatures.
Results
Expression of genes regulating gastrointestinal epithelium renewal was altered in both patient sets, particularly in LTP-A, who also presented gene expression profiles characteristic of an inflammatory syndrome. These included altered B cell pathways, increased neutrophil activation markers and elevated levels of reactive oxygen species. Increased expression of the IgG receptor (CD64) in LTP-A patients was mirrored by the presence of LTP-specific IgG1 and 3. Conversely, NSAID-LTP-A patients were characterized by reduced expression of IFN-γ-regulated genes and IFN-γ levels as well as up-regulated adenosine receptor 3 (ADORA3) expression and genes related to adenosine metabolism.
Conclusions
Gene ontology analysis suggests disturbances in gut epithelium homeostasis in both LTP-related anaphylaxis groups with potential integrity breaches in LTP-A that may explain their distinct inflammatory signature. Differential regulation in LTP-A and NSAID-LTP-A of the IFN-γ pathway, IgG receptors and ADORA3 may provide the pathogenic basis of their distinct responses.
Keywords: anaphylaxis, food allergy, lipid transfer protein syndrome, non-steroidal anti-inflammatory drugs, transcriptome analysis
Introduction
Anaphylaxis is a systemic allergic reaction that is rapid in onset and may cause death.1 Food allergens are the major triggers for anaphylaxis, accounting for 33-56% of all cases and up to 81% of anaphylaxis in children.2-4 Lipid transfer protein (LTP) is a relevant plant panallergen described as the most frequent cause of food-induced anaphylaxis in the Mediterranean Basin.5, 6 LTP-allergic patients are most frequently characterized by the presence of specific IgE to peach LTP (Pru p 3) but commonly show sensitization and reactions to multiple plant foods containing LTP.5, 7-9 Often, an anaphylactic episode to LTP occurs under the influence of a cofactor, such as physical exercise, alcohol and/or a medication, especially a non-steroidal anti-inflammatory drug (NSAID).6, 9-11 In an Italian cohort, 78% of patients with food-dependent exercise-induced anaphylaxis were sensitized to LTP10 and in a Spanish cohort with NSAID- and exercise-related food allergy, LTP sensitization was demonstrated in 92% of patients.11 Further, in another study almost half of LTP-induced anaphylaxis cases were related to cofactors, being NSAIDs involved in 36% of reactions.9 The enhancing effect of NSAIDs in food anaphylaxis is well documented,12, 13 and it extends beyond LTP to other common allergens such as shellfish,14 sunflower seeds,15 gliadin16, 17 and peanut.18
Some reports have associated anaphylaxis with expression of genes that regulate the innate and adaptive immune system.2, 19 However, the transcriptional profiles associated with either cofactor dependent or independent anaphylaxis to the same food allergen have not been examined. Here, we used next generation sequencing to characterize the gene expression landscape, under unchallenged conditions, of patients with LTP-related anaphylaxis that differ in their dependency on NSAID for the manifestation of anaphylaxis, to gain insights into the mechanisms influencing their distinct responses. The analysis reveals important differences in the type of immunity controlling the in vivo environment in these patient subsets and suggests that FcγRI receptors and LTP-specific IgG may play a contributory role in anaphylaxis to LTP, while adenosine receptor type 3 (ADORA3) may relate to reactions involving NSAIDs.
Methods
Further details on the methods can be found in the Online Repository.
Study population
Patients were recruited based on a clinical history of anaphylaxis (defined as in 1) elicited by LTP-containing plant foods, positive skin prick test (SPT) to LTP and/or the presence of LTP (Pru p3)-specific serum IgE (Table 1). LTP-sensitized patients were divided into two groups: 1) patients with a clinical history of anaphylaxis caused by a food source containing LTP (LTP-induced anaphylaxis, [LTP-A]); and 2) patients with a clinical history of anaphylaxis to LTP that is only observed when the culprit food was consumed in proximity to the ingestion of an NSAID (up to 2 hours before the reaction onset) (NSAID-related LTP-induced anaphylaxis [NSAID-LTP-A]). NSAID-LTP-A patients had no anaphylaxis when separately ingesting a food source containing LTP or an NSAID. Six of these patients (43%) also reported anaphylaxis to LTP after exercise (Table E1 in the Online Repository). Patients with a clinical history of NSAID-induced asthma or urticaria were excluded from the study. Oral or nasal challenge with aspirin or NSAID20 to confirm tolerance to NSAID was performed in some patients with non-conclusive clinical history. None of these patients were suffering from LTP-related reactions or consumed NSAIDs at the time samples were collected.
Table I. Population characteristics and criteria for inclusion in the study.
| NSAID-LTP-A | LTP-A | HV | |
|---|---|---|---|
| Number of subjects | 14 | 7 | 13 |
| Age (median (range)) | 32(28-36) | 32 (28-36) | 41 (38-45) |
| Females (%) | 61 | 57 | 84 |
| Positive SPT for Pru p 3 (%) | 100 | 100 | 0 |
| IgE anti-Pru p 3 (kUA/l median±SEM) | 3.3±1.9 | 7.4±1.8 | Negative |
| Total IgE (kU/l; median±SEM) | 124±55* | 81.5±128 | 25±24 |
| Reaction with LTP | No reaction/ mild urticaria | Anaphylaxis | No reaction |
| Reaction with NSAIDs | No reaction | No reaction | No reaction |
| Reaction with LTP+NSAIDs | Anaphylaxis | Anaphylaxis | No reaction |
No statistical differences between groups were observed in age, gender distribution, skin prick test and IgE anti-LTP (Pru p 3).
p<.05 compared to HV.
NSAIDs: non-steroidal anti-inflammatory drugs; Pru p 3: peach lipid transfer protein (LTP); NSAID-LTP-A: NSAID-related LTP-induced anaphylaxis, LTP-A: LTP-induced anaphylaxis (without NSAIDs involvement); HV: healthy volunteers; SPT: skin prick test.
Healthy volunteers (HV) had no clinical history of food allergy or drug reactions and had negative SPTs to LTP-containing plant foods and other common food allergens. Informed consent was obtained from all participating subjects. The study was approved by the Local Ethics Committee of the Hospital Clinic (Barcelona, Spain).
Transcriptome sequencing and analysis
Whole blood samples were used as a non-invasive source for RNA as it has been demonstrated that whole blood cell RNA is an adequate genomic tool to investigate organ biomarkers21 and disease pathogenesis22. Blood was collected in PAXgene Blood RNA or DNA tubes and processed as described in Methods in the Online Repository. Poly-A RNA was reverse-transcribed into cDNA and approximately 200 bp fragments ligated to sequencing adapters using the TruSeq™ RNA (Illumina Inc., San Diego, CA, USA) system followed by 15 cycles of PCR amplification as per manufacturer's specifications. The cDNA libraries were sequenced using the Genome Analyzer HiSeq 2000 (Illumina Inc. San Diego, CA, USA) using 50 bp read length. The RNA-Seq data were aligned to the hg18 reference genome with Tophat software, using options (-no-coverage-search and –G) to specify RefSeq gene model. FastQC was used to evaluate sequence quality. Transcript expression levels were estimated using Partek GS to determine the reads per kilobase of exon per million mapped reads (RPKM). A gene was considered expressed if the RPKM value was ≥ 0.1. Ingenuity Pathway Analysis (IPA) (Mountain View, CA, USA) was used for pathway analysis. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health, Bethesda, Md. (http://biowulf.nih.gov).
Statistical analysis
Sequencing results were analyzed using Partek GS 6.6 software and expressed in RPKM. RPKMs were log2 transformed with an offset by 0.001 and then ANOVA was performed to determine differential gene expression comparing patient groups vs. HV (Partek GS 6.6). Statistical analyses in all other measurements were performed with GraphPad Prism (La Jolla, CA, USA). All measurements other than RNA-Seq were done in duplicates or triplicates. Comparison between groups was performed using a non-parametric test (Mann-Whitney test) unless otherwise indicated. P values < .05 were considered statistically significant.
Results
Study population
A total of 21 patients and 13 healthy individuals with ages ranging from 28 to 45 were recruited (Table I). All patients showed positive SPT to peach LTP (Pru p3) and presented LTP (Pru p3)-specific IgE in serum as measured by ImmunoCAP™ (Table I and Table E1 in the Online Repository).
Whole-transcriptome analysis
Alignment of sample reads to the human genome database using Tophat showed that 75-92% of reads from each sample were properly mapped to the human genome. Sequencing resulted in an average 40,821,991 reads mapped per subject that corresponded to a total of 20,650 expressed genes. Transcritptome comparison between the different groups indicated broader expression changes in LTP-A than in NSAID-LTP-A. Further details on these comparisons can be found in the Online Repository. Of note, transcript abundance in blood may reflect differences in transcriptional regulation or in blood cell composition, both however valuable predictors of underlying pathology.22, 23 The numbers of blood cell subtypes in both patient cohorts were in the normal range and no differences were found between LTP-A and NSAID-LTP-A (Table E2 in the Online Repository).
Differentially expressed genes in both patient subsets are associated with gastrointestinal diseases and epithelial alterations
Cancer and Gastrointestinal Diseases were identified as the main Diseases/Disorders associated with differentially expressed genes in both NSAID-LTP-A- (Table II) and LTP-A- specific transcripts (Table III), although the degree of confidence (p<10-12) and the number of molecules involved (>654) were higher in the LTP-A group. In both, this disease category included Disease/Function Annotations mostly related to adenocarcinomas and epithelial neoplasias (Table II and III). However, this is unlikely to reflect gastrointestinal neoplasias since the analysis predicted that Disease/Function Annotations such as tumor cell invasion, migration, proliferation and colony formation, were inhibited (not shown). Instead, the association with Cancer/Gastrointestinal Diseases may suggest changes in the regulation of cell growth/death in gastrointestinal epithelium as the site of the alterations in these patients.
Table II.
Association of differentially expressed genes in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A) patients with Diseases and Disorders, Canonical pathways and Networks.
| NSAID-LTP-A- Diseases and Disorders | P-value | Num. Molec. | Top Disease/Function Annotation | |||
|---|---|---|---|---|---|---|
| Cancer | 0.03-3.4×10-9 | 445 | Adenocarcinoma; carcinoma; epithelial neoplasia; abdominal neoplasm; abdominal cancer; breast or colorectal cancer; gastrointestinal adenocarcinoma; colorectal adenocarcinoma | |||
| Gastrointestinal Disease | 0.03-5.7×10-7 | 232 | ||||
| NSAID-LTP-A -Top Canonical Pathways | P-value | Ratio | Molecules up- or downregulated in pathway | |||
| Death Receptor Signaling | 0.0067 | 0.088 | ↑DIABLO, ↓PARP15, ↑ACTA2, ↑APAF1, ↓ TNFSF10, ↓BIRC3, ↓PARP9, ↓XIAP | |||
| The Visual Cycle (and Retinoate biosynthesis I) | ↓ | 0.0098 (0.04) | 0.2 (0.1) | DHRS9, RDH5, RBP5 | ||
| Sonic Hedgehog Signaling | ↓ | 0.0112 | 0.138 | PRKACB, PTCH1, GLI1, DYRK1A | ||
| ERK5 Signaling | ↓ | 0.0123 | 0.095 | IL6ST, MEF2A, GNAQ, FOSL1, WNK1, ELK4 | ||
| p53 Signaling | ↓ | 0.0186 | 0.072 | PIK3R3, HDAC9, MDM4, PLAGL1, APAF1, BRCA1, SERPINE2 | ||
| NSAID-LTP-A - Top Networks | Score | Molecules in pathway | ||||
| Cell Morphology, Embryonic Development, Connective Tissue Disorders | 40 | 30 | ||||
| Cell Death and Survival, Embryonic Development, Renal Necrosis/Cell Death | 33 | 27 | ||||
| Cell Movement, Cardiov. System Development anci Function, Organ Morphology | 29 | 25 | ||||
| Cell-To-Cell Signaling and Interaction, Organismal Development, Cell Morphology | 26 | 23 | ||||
Shown are the most significant identified pathways/networks. Arrows indicate the predicted state of activation for the pathway considering the changes in the majority genes involved.
Table III.
Association of differentially expressed genes in LTP-induced anaphylaxis (LTP-A) patients with Diseases and Disorders, Canonical pathways and Networks.
| LTP-A- Diseases and Disorders | P-value | Num. Molec. | Top Disease/Function Annotation | |||
|---|---|---|---|---|---|---|
| Cancer | 0.03-1×10-13 | 1305 | Adenocarcinoma; gastrointestinal adenocarcinoma; colorectal adenocarcinoma; transcription of RNA (↓); colorectal carcinoma; epithelial neoplasia; transcription (↓); colon adenocarcinoma | |||
| Gastrointestinal Disease | 0.03-1×10-12 | 654 | ||||
| LTP-A- Top Canonical Pathways | P value | Ratio | Molecules up- or downregulated in pathway | |||
| B Cell Receptor Signaling | ↓ | 0.00001 | 0.187 | BLNK, PTK2B, PIK3R1, ABL1, PDPK1, PTPRC, PTK2, PAX5, IKBKB, CAMK2D, CFL2, CD22, AKT3, MAP3K2, ATM, ETS1, MAP3K9, CD19, PIK3C2A, MAPK8, IKBKE, ATF2, SYNJ2, PIK3R3, EBF1, RRAS2, INPP5F, VAV3, NFATC2, MEF2C, INPP5K, ELK1 | ||
| Molecular Mechanisms of Cancer | ↓ | 0.00049 | 0.136 | TGFBR1, ARHGEF7, SMAD3, ADCY4, PIK3R1, ABL1, BRAF, PTK2, E2F6, ITGA3, CAMK2D, RHOU, E2F5, AKT3, PLCB1, CASP8, BRCA1, SMAD1, BIRC3, RASA1, TAB1, ATM, ITGA4, CASP10, LRP5, CCNE2, PIK3C2A, PTCH1, GNAQ, PRKAR2A, SMAD7, MAPK8, APAF1, XIAP, APC, PIK3R3, RASGRF2, RRAS2, MAX, RASGRP1, IRS1, GNAT2, RBPJ, LEF1, ELK1, GLI1, ARHGEF9, BCL2L11, CTNND1 | ||
| Sphingosine-1-phosphate Signaling | ↓ | 0.00022 | 0.194 | ASAH2B, PIK3C2A, PTK2B, PIK3R1, ACER2, ADCY4, GNAQ, PLCL2, PDGFC, PDGFB, PIK3R3, PTK2, SMPD4, CASP2, CASP1, RHOU, PLCB1, AKT3, CASP8, ATM, CASP10 | ||
| PI3K Signaling in B Lymphocytes | ↓ | 0.00021 | 0.187 | BLNK, IL4R, CD19, PIK3R1, ABL1, PDPK1, IKBKE, ITPR1, PLCL2, ATF2, PTPRC, IKBKB, TLR4, RRAS2, CAMK2D, VAV3, IRS1, PLCB1, AKT3, NFATC2, PLEKHA1, ELK1, CR2 | ||
| Top Networks | Score | Molecules in pathway | ||||
| Cancer, Dermatological Diseases and Conditions, Tumor Morphology | 35 | 35 | ||||
| Gene Expression, Cancer, Gastrontestinal Disease | 33 | 34 | ||||
| Cell Signaling, Hereditary Disorcer, Inflammatory Disease | 31 | 33 | ||||
| Carbohydrate Metabolism, Lipid Metabolism, Small Molecue Biochemistry | 29 | 32 | ||||
Shown are the most significant identified pathways/networks. Arrows indicate the predicted state of activation for the pathway considering the changes in the majority genes involved.
In keeping with this interpretation, the top canonical pathways in NSAID-LTP-A included those related to survival and proliferation processes (Table II, first, fourth and fifth canonical pathways) and the regulation of epithelial cell differentiation/proliferation and epithelial-mesenchymal transitions (Table II, second and third pathways), all important processes for Organ/Cell Development and Morphology (see Top Networks of gene connections in Table II). Furthermore, among the most changed transcripts in NSAID-LTP-A there were modulators of growth in normal or malignant epithelial cells (AREG, mir-50), stem cell proliferation and renewal (MICU1, NR2C1) and the epithelial-mesenchymal transition (SIVA1) (Table E3 in the Online Repository).
In LTP-A, the association of differentially expressed genes with cell growth/survival and gastrointestinal tract was not only reflected in the top Diseases and the main Canonical Pathways (Molecular Mechanisms of Cancer) and Networks (Cancer, Tumor Morphology, Gastrointestinal Disease) (Table III), but also in the most affected individual gene transcripts related to cell cycle and cell death (Table E3 in the Online Repository) as described for NSAID-LTP-A. Overall the analysis suggests disturbances in the dynamic renewal and turnover of the intestinal epithelium in both sets of patients.
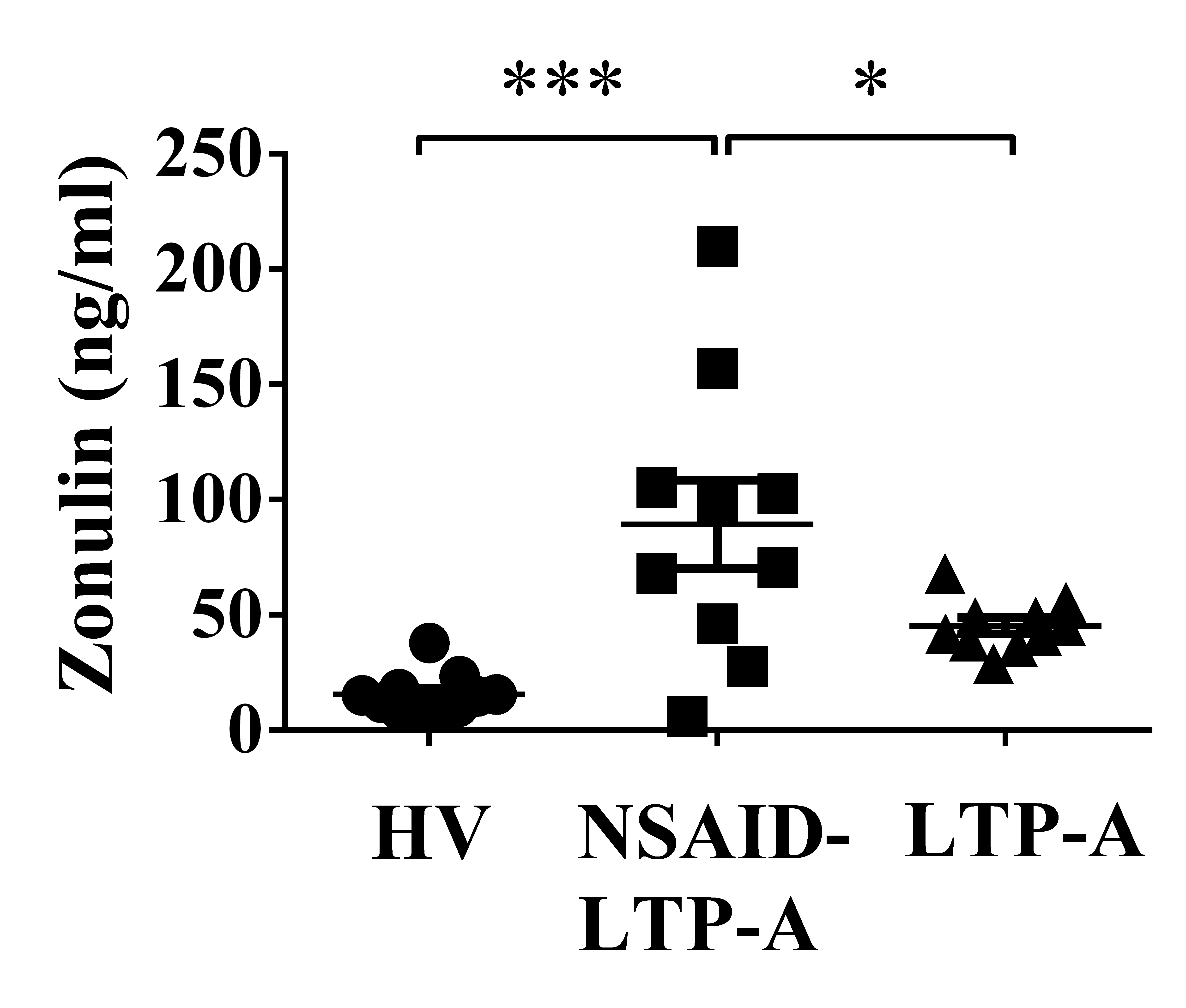
Changes in intestinal epithelium turnover can associate with altered permeability. Consistent with this concept, a protein involved in the regulation of tight junctions that has been described as a marker of intestinal permeability in patients with celiac disease and diabetes mellitus type 1,24 was significantly increased in both groups of patients compared to HV (Figure E1 in the Online Repository)
Associations with inflammatory conditions exclusively in LTP-A
A distinct aspect of the transcriptome in LTP-A patients was its association with Inflammatory Disease Networks and Canonical Pathways related to immune regulation (B Cell Receptor Signaling and PI3K Signaling in B Lymphocytes) (Table III and Table E4 in the Online Repository). Another Canonical Pathway exclusively affected in LTP-A was Sphingosine-1-phosphate signaling, important for immunity,25 sensitization to food allergens,26 recovery from anaphylactic shock27 and cell cycle regulation28 (Table III).
Approximately 24% of all significantly affected Canonical Pathways in LTP-A were linked to immune cell function (Table E4 in the Online Repository), while the remaining 76% related mostly to cell cycle regulation, survival, stress response and cancer (not shown). The main Diseases/Disorders associated with all the up-regulated genes were Infectious Diseases (p=2×10-9-10-2; 38 molecules) and Immunological and Inflammatory Diseases (p=1.4×10-8-10-2; 48 molecules). Of interest, the annotations linked with those categories included Bacterial Infections, Autoimmunity, Reactive Oxygen Species Production as well as Immune Cell Migration (Table E5 in the Online Repository). When considering all the upregulated genes, IPA predicted an activated state of IFNG (activation z-score: 3.39; p=6.5×10-5) and IFNA2 (activation z-score: 2.41; p=2.4×10-4), which are important for the activation of multiple immune cells and immunity to infectious agents. Furthermore, 18% of the increased transcripts in LTP-A compared to HV were markers of neutrophil subsets or genes related to neutrophil function (Table IV). The increase in the abundance of neutrophil markers is unlikely to represent an increase in total neutrophil numbers since these transcripts were not significantly changed in NSAID-LTP-A, which had similar neutrophil counts than LTP-A (Table E2 in the Online Repository). Instead it suggests that LTP-A may be enriched in a distinct neutrophil subset with possible functional consequences. Indeed, the levels of reactive oxygen species (ROS), a measurement of neutrophil oxidative outburst, were significantly elevated in the serum of LTP-A compared to those in HV and NSAID-LTP-A (Fig 1, A). Thus, LTP-A but not NSAID-LTP-A patients show a steady-state inflammatory condition with neutrophilic involvement and extensive control of the adaptive immune response.
Table IV.
Increased expression of neutrophil-related genes in LTP-anaphylaxis (LTP-A) patients. Genes are sorted by fold change
| Gene | Protein name | p-value | Fold Change |
|---|---|---|---|
| CD177 | CD177 molecule | 0.0289 | 4.69 |
| LTF | Lactotransferrin | 0.0037 | 4.309 |
| PRTN3 | Proteinase 3 | 0.0303 | 4.191 |
| SCO2 | SCO2 cytochrome c oxidase assembly protein | 0.0047 | 4.144 |
| OLFM4 | Olfactomedin 4 | 0.0308 | 4.089 |
| LCN2 | Lipocalin 2 | 0.0073 | 3.928 |
| DEFA4 | Defensin, alpha 4, corticostatin | 0.0125 | 3.920 |
| DEFA1 | Defensin, alpha 1 | 0.0196 | 3.627 |
| IFI6 | Interferon, alpha-inducible protein 6 | 0.0039 | 3.531 |
| MMP8 | Matrix metallopeptidase 8 (neutrophil collagenase) | 0.0247 | 3.447 |
| ELANE | Elastase, neutrophil expressed | 0.0149 | 3.033 |
| CAMP | Cathelicidin antimicrobial peptide | 0.0014 | 2.847 |
| AZU1 | Azurocidin 1 | 0.0180 | 2.768 |
| RETN | Resistin | 0.0054 | 2.570 |
| FCGR1 | Fc fragment of IgG, high affinity 1B or C), receptor (CD64) | 0.0164 | 2.442 |
| MX1 | myxovirus (influenza virus) resistance 1, interferon-inducible protein p78 (mouse) | 0.0037 | 2.345 |
| CEACAM8 | Carcinoembryonic antigen-related cell adhesion molecule 8 | 0.0311 | 2.270 |
| CEACAM6 | Carcinoembryonic antigen-related cell adhesion molecule 6 (non-specific cross reacting antigen) | 0.0442 | 2.192 |
| TYROBP (DP12) | TYRO protein tyrosine kinase binding protein | 0.0005 | 2.182 |
| GSR | Glutathione reductase | 0.0048 | 2.124 |
| VNN2 | Vanin 2 | 0.0004 | 2.083 |
| ARG1 | Arginase | 0.0229 | 2.064 |
Note: Ingenuity Pathway Analysis using the genes shown above indicated that the top Network association representing their functional connection was Infectious Disease, Cellular Movement and Immune Cell Trafficking.
Fig 1. Increased reactive oxygen species and Top scored gene network in the comparison NSAID-LTP-A vs. LTP-A.

A) Serum levels of free radicals (reactive oxygen species, ROS, and reactive nitrogen species, RNS) in the indicated groups. Data are expressed as mean±SEM. **p<.01, ***p<.001 compared to HV. B) Functional relationships of differentially expressed genes in NSAID-LTP-A vs. LTP-A in the top scored network “Cell death and Survival, Inflammatory response, Cancer”. The intensity of the node color indicates the degree of up-regulation (red) or down-regulation (green). Genes in uncolored nodes were of relevance for this network and integrated into the computationally generated networks on the basis of the evidence stored in the IPA knowledge memory, but they were not identified as differentially expressed. Lines indicate molecular interactions.
Alterations in adenosine metabolism exclusively in NSAID-LTP-A
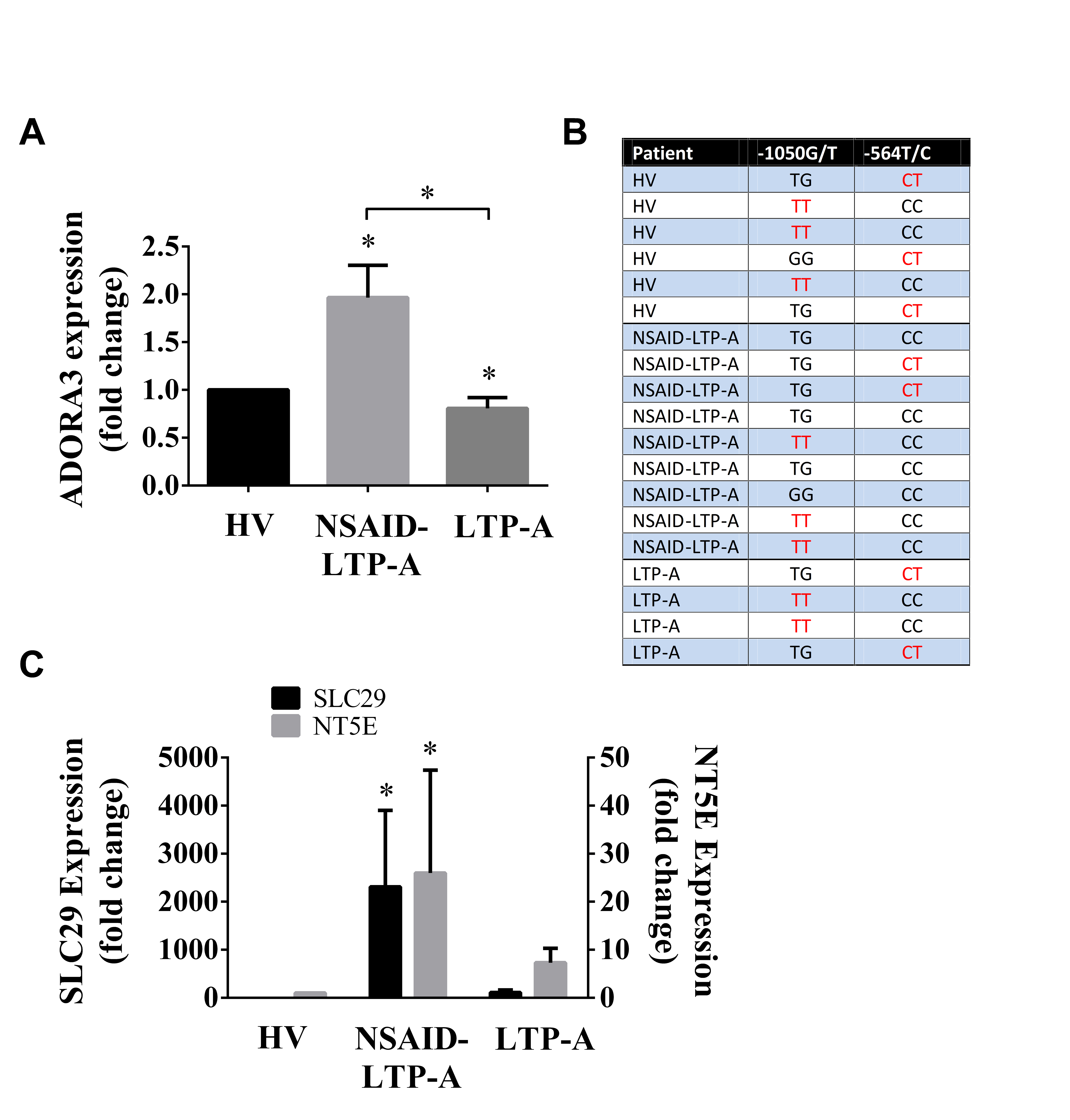
We compared the transcriptomes of NSAID-LTP-A vs. LTP-A seeking unique transcriptional differences that may explain their differential susceptibility to LTP or the need for NSAID in NSAID-LTP-A patients. Fig 1,B represents the main Network Functions associated with this and shows several genes regulated by IFN-γ (IFI6, EIF2AK2, MX1, CD274, TNFSF10, KLRC4-KLRK1/KLRK1, FAS and FCGR1) and genes related to mast cell function/differentiation and allergic disease, airway hypersensitivity and Th2-type responses (including GATA2, KIT, ADORA3, MAPK8, FCGRI, IL1RL1 and NCR1). Among these, ADORA3 was of interest because a polymorphism in the promoter of ADORA3 at -1050G/T and a higher frequency in the high-transcript haplotype ht1 (T-1050C-564), have been previously associated with aspirin-induced urticaria.29 Although we confirmed a higher expression of ADORA3 in the NSAID-LTP-A group by qRT-PCR (Figure E2,A in the Online Repository), we could not demonstrate an association with the polymorphism at -1050G/T or the haplotype ht1 (T-1050C-564) in these patients (Figure E2,B in the Online Repository). Genes involved in the metabolism and function of adenosine, including ENTPD2 and ADK (encoding, respectively, for an ectonucleotidase that degrades ATP into AMP and a kinase that phosphorylates adenosine to AMP) (Fig 3 and those shown in Figure E2,C in the Online Repository) were also expressed at higher levels in NSAID-LTP-A relative to LTP-A. The data is suggestive of an altered nucleotide/nucleoside turnover with a favored expression of ADORA3 in NSAID-LTP-A patients, and observation that may be of interest since NSAID causes an increase in ATP metabolism.30, 31
Fig 3.

Differentially expressed genes in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A) patients compared to LTP-anaphylaxis (LTP-A) patients. Genes in grey boxes indicate their association with Th2-type of immunity while genes in blue boxes indicate association with Th1-type of immunity. Differences between groups were statistically significant (p<0.05) unless otherwise indicated. NS: not significant. Columns 3-5 were color-coded in accordance with the fold change
IFN-γ is repressed in NSAID-LTP-A compared to LTP-A
The network shown in Fig 1,B hinted at differences between NSAID-LTP-A and LTP-A in Th1- and Th2-regulated genes. Indeed, IPA predicted an inhibited state of IFNG (activation z-score: -1.77; p= 2.4×10-3) and STAT1 (p=7.94×10-4) as top Upstream Regulators in NSAID-LTP-A (as compared to LTP-A) (Fig 2,A). Examination of all differentially expressed genes in NSAID-LTP-A vs. LTP-A confirmed the notion that gene transcripts directly or indirectly linked to Th2- responses or Th2-effector cells (including cytokine receptors, chemokines and genes that regulate eosinophil, mast cell and basophil function/differentiation), were more represented in NSAID-LTP-A than in LTP-A (shown in grey in Fig 3). In contrast, gene transcripts controlled by IFN-γ, including FAS, ARG1, IFI6, MX1, FcGR1, CASP7 and TNFSF10 were under-represented in NSAID-LTP-A compared to LTP-A (shown in blue in Fig 3 and Fig 2,A).
Fig 2. LTP-specific IgG1/IgG3 in LTP-A serums and repressed IFN-γ and IFN-γ regulated genes in NSAID-LTP-A.

A) Representation of IFNG as an upstream regulator of differentially regulated genes in NSAID-LTP-A compared to LTP-A, as identified by IPA. The arrows connect genes whose expression is altered and may be regulated by IFNG. The style of the lines defines the direction of change. The color of the node indicates the direction of the gene regulation. Grey: down-regulated; blank: up-regulated. B) Serum levels of IFN-γ and C) IgG1 and IgG3 anti-Pru p 3 in the indicated groups. Data are expressed as mean±SEM. *p<.05, **p<.01, ***p<.001 compared to HV unless otherwise indicated.
In agreement, the serum levels of IFN-γ in NSAID-LTP-A were significantly reduced compared to LTP-A and HV (Fig 2,B). The circulating levels of IFN-γ in LTP-A were not significantly higher than those in HV, although a higher percentage of LTP-A samples had detectable IFN-γ levels (71%) than in HV (55%). Other than an apparent repression of Th-1 immunity in NSAID-LTP-A (Fig 2,A and B), no major systemic cytokine profile alterations were found in these patients (see Results in the Online repository). The overall analysis suggests prominent differences between NSAID-LTP-A and LTP-A in the transcripts that regulate their type of immunity.
LTP-A patients show increased FCGR1 transcripts and LTP-specific IgG1/IgG3 levels
FCGR1 expression, which is regulated by IFN-γ, 31, 32 was increased but FCER1 decreased in LTP-A compared to NSAID-LTP-A (Fig 3). In addition, the ratio of expression of FCGR1 to FCER1 was almost 4 fold higher in LTP-A compared to NSAID-LTP-A, a result that was confirmed by qRT-PCR (not shown).
If FcγRI had any role in LTP-induced anaphylaxis as shown in animal models,33 then LTP (Pru p 3)-specific IgG1/3, which bind FcγRI and can be crosslinked by LTP, should be present in the serum of these individuals. LTP-specific IgG1 and especially IgG3 were found in LTP-A and to a much lesser extent in NSAID-LTP-A (Fig 2,C). LTP-specific IgG2 was very low and indistinguishable among the groups (data not shown). Thus, another fundamental difference in LTP-A is the higher ratio of expression of FcγRI to FcεRI messages concomitant with elevated levels of LTP-specific IgG1 and IgG3 isotypes that can engage these receptors.
Discussion
Although individuals sensitized to LTP suffer from varied allergic manifestations after ingestion of LTP-containing food, anaphylactic episodes are restricted to a small percentage of patients unless a cofactor -such as NSAID intake- precipitates this event. This poses the conundrum of what are the mechanisms leading to the provocation of anaphylaxis to LTP. Our characterization of the transcriptome by RNA-Seq in NSAID-dependent or -independent LTP-related anaphylaxis indicates that distinct transcriptional profiles define these patients even before anaphylaxis is manifested. The data presented herein contributes to a better understanding of the similarities and differences between two groups of patients with dramatically distinct responses to LTP but indistinguishable sensitization profiles and provides a first step in the elucidation of mechanisms of cofactors in food allergy.
Transcriptome analysis of both NSAID-LTP-A and particularly LTP-A identified an association with gastrointestinal diseases and multiple genes and pathways involved in cell growth/differentiation/death of the gut epithelium, suggesting defects in the gut epithelia turnover. Changes in the normal turnover of the intestinal epithelium may breach the epithelial continuity and barrier function, which can be a critical factor predisposing patients to food allergies and intestinal inflammation.32, 33 Although increased intestinal permeability is apparent during food allergic reactions, it is unclear whether these changes in permeability are constitutive.33 Our findings of increased serum zonulin, a marker for intestinal permeability,24 support the notion of constitutive permeability dysfunction in these patients. However, it is unclear whether this defect precedes the sensitization to the food allergen or it represents a defense mechanism to the inevitable constant exposure to food antigens.24, 34
LTP-A patients showed significant increases in the expression of markers of neutrophil activation and trafficking that are characteristic of inflammatory and infectious diseases. No blood neutrophilia was found in these patients but enhanced neutrophilic activity was suggested by their elevated ROS/RNS, a measure of oxidative outburst. A possible interpretation is that a prominent breach in epithelial barrier continuity and exposure to gut microbiota in LTP-A causes the activation and trafficking of neutrophils into the gastrointestinal tract, which may partly explain their enhanced sensitivity to LTP. Confirmation of a chronic gastrointestinal inflammatory condition in LTP-A may be of importance for disease management options.
The data herein also raise a provocative link between anaphylaxis to LTP in LTP-A and a non-classical pathway for anaphylaxis whereby an increased presence of IgG1/3 and the increased expression of the three genes coding for FcγRI (CD64) may contribute substantially to the response. The high affinity receptor for IgG, FcγRI, has been involved in systemic anaphylaxis in rodents. Even though these receptors are normally occupied by serum monomeric IgG35, 36, this does not prevent their activation by IgG immunocomplexes since these have higher binding affinity and can rapidly displace monomeric IgG, 35, 36 triggering hypersensitivity responses. 37, 38 The displacement of IgG by immunocomplexes is particularly favored in the presence of cytokines, such as IFN-γ, which seem to regulate not only the levels of FcγRI but its ligand binding characteristics.36 Recently, it was demonstrated that human FcγRI expressed in neutrophils induces systemic anaphylaxis in immunized mice.37 There is little information about the involvement of allergen specific IgG on human anaphylaxis, although some observations may link IgG to anaphylaxis. For instance, IgG1 anti-α-gal, a carbohydrate epitope responsible for anaphylaxis to red meat, was found elevated together with α-gal-IgE in sensitized individuals in higher proportion than α-gal-IgG2 (normally found in all humans);39 mediators of IgG-induced anaphylaxis40 are better markers of human anaphylaxis than classical IgE mediators41; furthermore, analysis of gene networks during anaphylaxis in humans showed evidence of neutrophil activation, consistent with an IgG-mediated response.19 With those considerations, our findings suggest a contributory role for FcγRI pathway, in addition to the classical IgE/FcεRI, to LTP-induced anaphylaxis, a hypothesis that deserves further investigation.
Unique features of the transcriptome in NSAID-LTP-A included a repression in IFN-γ production and IFN-γ-regulated genes as well as increased expression of ADORA3. IFN-γ is a cytokine usually regarded as a marker of Th1 immunity that antagonizes Th-2 type of responses as those to food allergens. It is thus unexpected that patients with allergic responses to LTP requiring a cofactor show repressed IFN-γ transcriptional activity compared to LTP-A. A role for IFN-γ in allergic inflammation, however, is not unprecedented since IFN-γ and its receptors have been reported to contribute to some aspects of tissue inflammation, including recruitment of neutrophils and eosinophils, and tissue pathology in models of chronic airway inflammation.42, 43 IFN-γ has also been shown to promote secretion of ROS upon FcεRI activation, expression of FcγRI, and neutrophil and mast cell activation.44, 45 Thus, the repression of IFN-γ in NSAID-LTP-A may represent a mechanism to prevent recruitment and responsiveness of immune cells to an altered gut epithelium. The mechanism regulating IFN-γ levels in these patients is unclear, but it might be linked to ADORA3 overexpression. ADORA3 agonists have shown anti-inflammatory effects in several mouse models of inflammation46, 47 due to inhibition of pro-inflammatory cytokines such as IFN-γ and immune cell activation.48
Despite the anti-inflammatory effects of ADORA3, activation of this receptor can also potentiate FcεRI-induced degranulation in human mast cells and thus contribute to allergic inflammation.49-51 A link between NSAIDs and ADORA3 to allergic inflammation was suggested by the findings that NSAIDs inhibit oxidative phosphorylation and promotes ATP hydrolysis with release of adenosine30, 31, 52 and that NSAID-dependent urticaria associates with a high transcript variant of ADORA3. We found a higher transcription of ADORA3 and enzymes involved in adenosine metabolism in NSAID-LTP-A, but not the ADORA3 variant described in NSAID-induced urticaria, which is consistent with the sensitivity to NSAID alone. Although further studies are needed, one possible explanation is that increased expression of ADORA3 and enzymes involved in nucleotide metabolism in NSAID-LTP-A may normally support an anti-inflammatory environment by repressing IFN-γ production; following NSAID and LTP intake, adenosine production may be favored due to the increased expression of ATP exonucleotidases, and robust activation of ADORA3 may synergize with LTP-engaged IgE receptors, promoting anaphylaxis.
In summary, despite the limitation of the sample size, this study underlines for the first time the similarities as well as fundamental differences in the transcriptional profile of NSAID-LTP-A and LTP-A and it provides important clues for the understanding of the pathogenic mechanisms underlying LTP-related anaphylaxis, which might be applicable to other allergen-induced food anaphylaxis.
Supplementary Material
Supplemental Figure E1- Zonulin levels in serum are increased in NSAID-LTP-A and LTP-A patients. Zonulin levels were measured by specific ELISA. Statistical analysis was performed using non-parametric tests. *p<.05, ***p<.001 compared to HV unless otherwise indicated.
{kind=link}
Supplemental figure E2- Increased expression of enzymes in adenosine metabolism and adenosine type 3 receptor in NSAID-LTP-A patients Quantitative PCR measurements of ADORA3 (A) SCL29 (nucleoside transporter or Solute carrier family 29, a transporter for adenosine) and NTSE (5-Nucleotidase, ecto (CD73), that converts AMP into Adenosine) (B) were obtained from whole blood RNA. Values were corrected for GAPDH expression for each sample and expressed as a fold change (compared to HV) using ΔΔCt method. Statistical analysis was performed using non-parametric tests. *p<0.05 compared to HV unless otherwise indicated. C) Two polymorphisms at the promoter of ADORA3 (-1050G/T and -564T/C) previously associated to NSAID-induced urticaria were evaluated. The variants for each site are indicated in red. No statistical differences were found among the groups.
{kind=link}
Supplemental Table E1- Pollen and food sensitization profile in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A) and LTP-anaphylaxis (LTP-A) patients.
Supplemental Table E2- Counts of blood cells in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A), LTP-anaphylaxis (LTP-A) patients and healthy volunteers (HV).
Supplemental Table E3- Top up- or down regulated gene transcripts in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A) and LTP-anaphylaxis (LTP-A) patients compared to healthy volunteers (HV) as identified by Ingenuity Pathway Analysis (IPA).
Supplemental Table E4- List of canonical pathways related to immune cell functions significantly affected in LTP-A patients compared to HV. Up-regulated and down-regulated gene transcripts (p<0.05; ≤2 or ≥2) were used for analysis using Ingenuity Pathway Analysis (IPA). Canonical pathways were sorted according to their p values.
Supplemental Table E5. Diseases or Function Annotations under the category of “Infectious, Immunological and Inflammatory Diseases” associated with upregulated gene transcripts in LTP-anaphylaxis patients (LTP-A).
Supplemental Table E6. ADORA3 PCR and Sequencing Primers
Key Messages.
Gene expression patterns define patients with LTP-induced anaphylaxis associated or not to NSAIDs prior to allergen provocation, suggesting predisposing influences into the triggering of anaphylaxis.
Transcriptome profiling suggests disturbances in gut epithelium homeostasis in both groups, likely with intestinal barrier compromise in patients with LTP-induced anaphylaxis, which might be a factor in allergic sensitization and/or the anaphylaxis prone phenotype of these patients.
While NSAID-LTP-A patients were defined by a baseline repression of IFN-γ-regulated genes and IFN-γ levels, LTP-A patients showed an inflammatory-like syndrome with apparent neutrophilic involvement and increased expression of FcγRI. LTP-specific IgG1 and IgG3, ligands for FcγRI, were also elevated in LTP-A, which might be considered as a potential diagnostic tool.
Acknowledgments
We thank Dr. Kenneth A. Jacobson for his scientific advice, Mireya Fuentes, Gemma Lopez and Elizabeth Baskin for technical support and Dr. Barbara Dema and Dr. Jorg Scheffel for scientific and technical support.
Funding: This work was supported by the Division of Intramural Research Programs within NIAID and NIAMS, NIH; and by the Ministerio de Economia y Competitividad – Instituto de Salud Carlos III (FIS- PI11/01326), Spain. The first author was recipient of the “Rio Hortega” Fellowship– Ministerio de Economia y Competitividad – Instituto de Salud Carlos III, Spain.
Abbreviations
- ADORA3
adenosine receptor 3
- FcεR1A
IgE high affinity receptor
- FcγR1
IgG high affinity receptor
- HV
healthy volunteer
- IFN-γ
interferon gamma
- IPA
Ingenuity Program Analysis
- LTP
lipid transfer protein
- LTP-A
LTP-induced anaphylaxis (NSAID independent)
- NSAIDs
non-steroidal anti-inflammatory drugs
- NSAID-LTP-A
NSAID-related LTP-induced anaphylaxis,
- Pru p 3
peach lipid transfer protein
- STAT
Signal transducer and activator of transcription
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sampson HA, Muñoz-Furlong A, Campbell RL, Adkinson NF, Bock SA, Branum A, et al. Second symposium on the definition and management of anaphylaxis: Summary report—Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposium. Journal of Allergy and Clinical Immunology. 2006;117:391–7. doi: 10.1016/j.jaci.2005.12.1303. [DOI] [PubMed] [Google Scholar]
- 2.Ben-Shoshan M, Clarke AE. Anaphylaxis: past, present and future. Allergy. 2011;66:1–14. doi: 10.1111/j.1398-9995.2010.02422.x. [DOI] [PubMed] [Google Scholar]
- 3.Cianferoni A, Novembre E, Mugnaini L, Lombardi E, Bernardini R, Pucci N, et al. Clinical features of acute anaphylaxis in patients admitted to a university hospital: an 11-year retrospective review (1985-1996) Ann Allergy Asthma Immunol. 2001;87:27–32. doi: 10.1016/S1081-1206(10)62318-6. [DOI] [PubMed] [Google Scholar]
- 4.Wang J, Sampson HA. Food anaphylaxis. Clin Exp Allergy. 2007;37:651–60. doi: 10.1111/j.1365-2222.2007.02682.x. [DOI] [PubMed] [Google Scholar]
- 5.Pastorello EA, Robino AM. Clinical role of lipid transfer proteins in food allergy. Mol Nutr Food Res. 2004;48:356–62. doi: 10.1002/mnfr.200400047. [DOI] [PubMed] [Google Scholar]
- 6.Asero R, Antonicelli L, Arena A, Bommarito L, Caruso B, Colombo G, et al. Causes of Food-Induced Anaphylaxis in Italian Adults: A Multi-Centre Study. International Archives of Allergy and Immunology. 2009;150:271–7. doi: 10.1159/000222679. [DOI] [PubMed] [Google Scholar]
- 7.Palacin A, Bartra J, Munoz R, Diaz-Perales A, Valero A, Salcedo G. Anaphylaxis to wheat flour-derived foodstuffs and the lipid transfer protein syndrome: a potential role of wheat lipid transfer protein Tri a 14. Int Arch Allergy Immunol. 2010;152:178–83. doi: 10.1159/000265539. [DOI] [PubMed] [Google Scholar]
- 8.Asero R, Mistrello G, Roncarolo D, Amato S, Caldironi G, Barocci F, et al. Immunological cross-reactivity between lipid transfer proteins from botanically unrelated plant-derived foods: a clinical study. Allergy. 2002;57:900–6. doi: 10.1034/j.1398-9995.2002.t01-1-23541.x. [DOI] [PubMed] [Google Scholar]
- 9.Pascal M, Munoz-Cano R, Reina Z, Palacin A, Vilella R, Picado C, et al. Lipid transfer protein syndrome: clinical pattern, cofactor effect and profile of molecular sensitization to plant-foods and pollens. Clin Exp Allergy. 2012;42:1529–39. doi: 10.1111/j.1365-2222.2012.04071.x. [DOI] [PubMed] [Google Scholar]
- 10.Romano A, Scala E, Rumi G, Gaeta F, Caruso C, Alonzi C, et al. Lipid transfer proteins: the most frequent sensitizer in Italian subjects with food-dependent exercise-induced anaphylaxis. Clinical and Experimental Allergy. 2012;42:1643–53. doi: 10.1111/cea.12011. [DOI] [PubMed] [Google Scholar]
- 11.Cardona V, Luengo O, Garriga T, Labrador-Horrillo M, Sala-Cunill A, Izquierdo A, et al. Co-factor-enhanced food allergy. Allergy. 2012;67:1316–8. doi: 10.1111/j.1398-9995.2012.02877.x.. [DOI] [PubMed] [Google Scholar]
- 12.Niggemann B, Beyer K. Factors augmenting allergic reactions. Allergy. 2014;69:1582–7. doi: 10.1111/all.12532. [DOI] [PubMed] [Google Scholar]
- 13.Moneret-Vautrin DA, Morisset M. Adult food allergy. Curr Allergy Asthma Rep. 2005;5:80–5. doi: 10.1007/s11882-005-0060-6. [DOI] [PubMed] [Google Scholar]
- 14.Vidal C, Bartolome B, Gonzalez-Quintela A, Rodriguez V, Armisen M. Prawns, barnacles, and nonsteroidal anti-inflammatory drugs: effect modifiers or diagnostic confounders [corrected] J Investig Allergol Clin Immunol. 2007;17:113–8. [PubMed] [Google Scholar]
- 15.Paul E, Gall HM, Muller I, Moller R. Dramatic augmentation of a food allergy by acetylsalicylic acid. J Allergy Clin Immunol. 2000;105:844. doi: 10.1067/mai.2000.105011. [DOI] [PubMed] [Google Scholar]
- 16.Matsukura S, Aihara M, Sugawara M, Kunimi Y, Matsuki M, Inoue Y, et al. Two cases of wheat-dependent anaphylaxis induced by aspirin administration but not by exercise. Clin Exp Dermatol. 2010;35:233–7. doi: 10.1111/j.1365-2230.2009.03709.x. [DOI] [PubMed] [Google Scholar]
- 17.Matsuo H, Morimoto K, Akaki T, Kaneko S, Kusatake K, Kuroda T, et al. Exercise and aspirin increase levels of circulating gliadin peptides in patients with wheat-dependent exercise-induced anaphylaxis. Clin Exp Allergy. 2005;35:461–6. doi: 10.1111/j.1365-2222.2005.02213.x. [DOI] [PubMed] [Google Scholar]
- 18.Bol-Schoenmakers M, Bleumink R, Marcondes Rezende M, Mouser E, Hassing I, Ludwig I, et al. Diclofenac enhances allergic responses in a mouse peanut allergy model. Clin Exp Allergy. 2011;41:424–33. doi: 10.1111/j.1365-2222.2010.03594.x. [DOI] [PubMed] [Google Scholar]
- 19.Stone SF, Bosco A, Jones A, Cotterell CL, van Eeden PE, Arendts G, et al. Genomic responses during acute human anaphylaxis are characterized by upregulation of innate inflammatory gene networks. PLoS One. 2014;9:e101409. doi: 10.1371/journal.pone.0101409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Casadevall J, Ventura PJ, Mullol J, Picado C. Intranasal challenge with aspirin in the diagnosis of aspirin intolerant asthma: evaluation of nasal response by acoustic rhinometry. Thorax. 2000;55:921–4. doi: 10.1136/thorax.55.11.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kohane IS, Valtchinov VI. Quantifying the white blood cell transcriptome as an accessible window to the multiorgan transcriptome. Bioinformatics. 2012;28:538–45. doi: 10.1093/bioinformatics/btr713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pascual V, Chaussabel D, Banchereau J. A genomic approach to human autoimmune diseases. Annu Rev Immunol. 2010;28:535–71. doi: 10.1146/annurev-immunol-030409-101221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaussabel D. Assessment of immune status using blood transcriptomics and potential implications for global health. Semin Immunol. 2015 doi: 10.1016/j.smim.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Fasano A. Zonulin, regulation of tight junctions, and autoimmune diseases. Ann N Y Acad Sci. 2012;1258:25–33. doi: 10.1111/j.1749-6632.2012.06538.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–63. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diesner SC, Olivera A, Dillahunt S, Schultz C, Watzlawek T, Forster-Waldl E, et al. Sphingosine-kinase 1 and 2 contribute to oral sensitization and effector phase in a mouse model of food allergy. Immunol Lett. 2012;141:210–9. doi: 10.1016/j.imlet.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olivera A, Rivera J. An emerging role for the lipid mediator sphingosine-1-phosphate in mast cell effector function and allergic disease. Adv Exp Med Biol. 2011;716:123–42. doi: 10.1007/978-1-4419-9533-9_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 29.Kim SH, Nam EJ, Kim YK, Ye YM, Park HS. Functional variability of the adenosine A3 receptor (ADORA3) gene polymorphism in aspirin-induced urticaria. Br J Dermatol. 2010;163:977–85. doi: 10.1111/j.1365-2133.2010.09983.x. [DOI] [PubMed] [Google Scholar]
- 30.Miyahara JT, Karler R. Effect of salicylate on oxidative phosphorylation and respiration of mitochondrial fragments. Biochem J. 1965;97:194–8. doi: 10.1042/bj0970194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cronstein BN, Vandestouwe M, Druska L, Levin RI, Weissmann G. Nonsteroidal Antiinflammatory Agents Inhibit Stimulated Neutrophil Adhesion to Endothelium - Adenosine-Dependent and Independent Mechanisms. Inflammation. 1994;18:323–35. doi: 10.1007/BF01534273. [DOI] [PubMed] [Google Scholar]
- 32.Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–53. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- 33.Perrier C, Thierry AC, Mercenier A, Corthesy B. Allergen-specific antibody and cytokine responses, mast cell reactivity and intestinal permeability upon oral challenge of sensitized and tolerized mice. Clin Exp Allergy. 2010;40:153–62. doi: 10.1111/j.1365-2222.2009.03329.x. [DOI] [PubMed] [Google Scholar]
- 34.Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev. 2011;91:151–75. doi: 10.1152/physrev.00003.2008. [DOI] [PubMed] [Google Scholar]
- 35.Mancardi DA, Iannascoli B, Hoos S, England P, Daeron M, Bruhns P. FcgammaRIV is a mouse IgE receptor that resembles macrophage FcepsilonRI in humans and promotes IgE-induced lung inflammation. J Clin Invest. 2008;118:3738–50. doi: 10.1172/JCI36452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van der Poel CE, Spaapen RM, van de Winkel JG, Leusen JH. Functional characteristics of the high affinity IgG receptor, FcgammaRI. J Immunol. 2011;186:2699–704. doi: 10.4049/jimmunol.1003526. [DOI] [PubMed] [Google Scholar]
- 37.Mancardi DA, Albanesi M, Jonsson F, Iannascoli B, Van Rooijen N, Kang X, et al. The high-affinity human IgG receptor FcgammaRI (CD64) promotes IgG-mediated inflammation, anaphylaxis, and antitumor immunotherapy. Blood. 2013;121:1563–73. doi: 10.1182/blood-2012-07-442541. [DOI] [PubMed] [Google Scholar]
- 38.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, et al. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 39.Rispens T, Derksen NI, Commins SP, Platts-Mills TA, Aalberse RC. IgE production to alpha-gal is accompanied by elevated levels of specific IgG1 antibodies and low amounts of IgE to blood group B. PLoS One. 2013;8:e55566. doi: 10.1371/journal.pone.0055566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khodoun MV, Strait R, Armstrong L, Yanase N, Finkelman FD. Identification of markers that distinguish IgE- from IgG-mediated anaphylaxis. Proc Natl Acad Sci U S A. 2011;108:12413–8. doi: 10.1073/pnas.1105695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vadas P, Perelman B, Liss G. Platelet-activating factor, histamine, and tryptase levels in human anaphylaxis. J Allergy Clin Immunol. 2013;131:144–9. doi: 10.1016/j.jaci.2012.08.016. [DOI] [PubMed] [Google Scholar]
- 42.Hansen G, Berry G, DeKruyff RH, Umetsu DT. Allergen-specific Th1 cells fail to counterbalance Th2 cell-induced airway hyperreactivity but cause severe airway inflammation. J Clin Invest. 1999;103:175–83. doi: 10.1172/JCI5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu M, Eckart MR, Morgan AA, Mukai K, Butte AJ, Tsai M, et al. Identification of an IFN-gamma/mast cell axis in a mouse model of chronic asthma. J Clin Invest. 2011;121:3133–43. doi: 10.1172/JCI43598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okayama Y, Kirshenbaum AS, Metcalfe DD. Expression of a functional high-affinity IgG receptor, Fc gamma RI, on human mast cells: Up-regulation by IFN-gamma. J Immunol. 2000;164:4332–9. doi: 10.4049/jimmunol.164.8.4332. [DOI] [PubMed] [Google Scholar]
- 45.Marchi LF, Sesti-Costa R, Ignacchiti MD, Chedraoui-Silva S, Mantovani B. In vitro activation of mouse neutrophils by recombinant human interferon-gamma: increased phagocytosis and release of reactive oxygen species and pro-inflammatory cytokines. Int Immunopharmacol. 2014;18:228–35. doi: 10.1016/j.intimp.2013.12.010. [DOI] [PubMed] [Google Scholar]
- 46.Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discov Today. 2012;17:359–66. doi: 10.1016/j.drudis.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mabley J, Soriano F, Pacher P, Hasko G, Marton A, Wallace R, et al. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–9. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- 48.Cohen S, Barer F, Bar-Yehuda S, AP IJ, Jacobson KA, Fishman P. A(3) adenosine receptor allosteric modulator induces an anti-inflammatory effect: in vivo studies and molecular mechanism of action. Mediators Inflamm. 2014;2014:708746. doi: 10.1155/2014/708746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leung CT, Li A, Banerjee J, Gao ZG, Kambayashi T, Jacobson KA, et al. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal. 2014;10:465–75. doi: 10.1007/s11302-014-9409-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhong H, Shlykov SG, Molina JG, Sanborn BM, Jacobson MA, Tilley SL, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol. 2003;171:338–45. doi: 10.4049/jimmunol.171.1.338. [DOI] [PubMed] [Google Scholar]
- 51.Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest. 1997;100:2849–57. doi: 10.1172/JCI119833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Newby AC, Holmquist CA, Illingworth J, Pearson JD. The control of adenosine concentration in polymorphonuclear leucocytes, cultured heart cells and isolated perfused heart from the rat. Biochem J. 1983;214:317–23. doi: 10.1042/bj2140317. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure E1- Zonulin levels in serum are increased in NSAID-LTP-A and LTP-A patients. Zonulin levels were measured by specific ELISA. Statistical analysis was performed using non-parametric tests. *p<.05, ***p<.001 compared to HV unless otherwise indicated.
Supplemental figure E2- Increased expression of enzymes in adenosine metabolism and adenosine type 3 receptor in NSAID-LTP-A patients Quantitative PCR measurements of ADORA3 (A) SCL29 (nucleoside transporter or Solute carrier family 29, a transporter for adenosine) and NTSE (5-Nucleotidase, ecto (CD73), that converts AMP into Adenosine) (B) were obtained from whole blood RNA. Values were corrected for GAPDH expression for each sample and expressed as a fold change (compared to HV) using ΔΔCt method. Statistical analysis was performed using non-parametric tests. *p<0.05 compared to HV unless otherwise indicated. C) Two polymorphisms at the promoter of ADORA3 (-1050G/T and -564T/C) previously associated to NSAID-induced urticaria were evaluated. The variants for each site are indicated in red. No statistical differences were found among the groups.
Supplemental Table E1- Pollen and food sensitization profile in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A) and LTP-anaphylaxis (LTP-A) patients.
Supplemental Table E2- Counts of blood cells in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A), LTP-anaphylaxis (LTP-A) patients and healthy volunteers (HV).
Supplemental Table E3- Top up- or down regulated gene transcripts in NSAID-related LTP-induced anaphylaxis (NSAID-LTP-A) and LTP-anaphylaxis (LTP-A) patients compared to healthy volunteers (HV) as identified by Ingenuity Pathway Analysis (IPA).
Supplemental Table E4- List of canonical pathways related to immune cell functions significantly affected in LTP-A patients compared to HV. Up-regulated and down-regulated gene transcripts (p<0.05; ≤2 or ≥2) were used for analysis using Ingenuity Pathway Analysis (IPA). Canonical pathways were sorted according to their p values.
Supplemental Table E5. Diseases or Function Annotations under the category of “Infectious, Immunological and Inflammatory Diseases” associated with upregulated gene transcripts in LTP-anaphylaxis patients (LTP-A).
Supplemental Table E6. ADORA3 PCR and Sequencing Primers