

SUMMARY
Long noncoding RNAs (lncRNAs) have emerged as regulators of diverse biological processes. Here we describe the initial functional analysis of a poorly characterized human lncRNA (LINC00657) that is induced after DNA damage, which we termed Noncoding RNA Activated by DNA Damage or NORAD. NORAD is highly conserved and abundant, with expression levels of approximately 500–1,000 copies per cell. Remarkably, inactivation of NORAD triggers dramatic aneuploidy in previously karyotypically-stable cell lines. NORAD maintains genomic stability by sequestering PUMILIO proteins, which repress the stability and translation of messenger RNAs to which they bind. In the absence of NORAD, PUMILIO proteins drive chromosomal instability by hyperactively repressing mitotic, DNA repair, and DNA replication factors. These findings introduce a mechanism that regulates the activity of a deeply conserved and highly dosage-sensitive family of RNA binding proteins and reveal unanticipated roles for a lncRNA and PUMILIO proteins in the maintenance of genomic stability.
Graphical Abstract
INTRODUCTION
Long noncoding RNAs, or lncRNAs, have recently attracted significant attention due to their emerging functions in development and disease (Fatica and Bozzoni, 2014; Li and Chang, 2014). lncRNAs represent a heterogeneous family of RNAs that are defined by a length of greater than 200 nucleotides and by the lack of any detectable open reading frame (ORF). The exact number of lncRNAs encoded in the human genome is a matter of debate, but most estimates place the number in the tens of thousands (Iyer et al., 2015; Ulitsky and Bartel, 2013). The biological roles and molecular functions of the overwhelming majority of these transcripts remain unexplored or elusive. Compared to other known noncoding RNA classes, lncRNAs stand out due to their enormous diversity with respect to evolutionary conservation, expression level, molecular function, and cellular localization (Ulitsky and Bartel, 2013). In the nucleus, lncRNAs have been shown to regulate transcription in cis or trans, organize subnuclear structure, and mediate chromosomal interactions (Clemson et al., 2009; Hacisuleyman et al., 2014; Rinn and Chang, 2012). Cytoplasmic lncRNAs are known to modulate the activity or abundance of interacting proteins or mRNAs (Faghihi et al., 2008; Gong and Maquat, 2011; Kino et al., 2010; Kretz et al., 2013; Liu et al., 2015). Nevertheless, studies of lncRNA function are still at an early stage. Due to their generally low abundance and modest evolutionary conservation relative to protein-coding genes (Cabili et al., 2011; Ulitsky and Bartel, 2013), it has been suggested that a large fraction of lncRNAs represent products of promiscuous transcription rather than independently functional RNAs (Struhl, 2007). To resolve this issue, detailed functional studies are needed to establish the biological role and molecular activity of lncRNAs of interest.
Pumilio-Fem3-binding factor (PUF) proteins represent a deeply conserved family of RNA binding proteins that act as negative regulators of gene expression (Wickens et al., 2002). PUF proteins bind with high specificity to sequences in the 3′ UTRs of target mRNAs through their PUMILIO homology domains (Zamore et al., 1997) and stimulate deadenylation and decapping, resulting in accelerated turnover and decreased translation (Miller and Olivas, 2011). There are two human and mouse PUF proteins, PUMILIO1 (PUM1) and PUMILIO2 (PUM2), that bind to target transcripts containing an eight nucleotide sequence (UGUANAUA), referred to as the PUMILIO response element (PRE). Many mammalian PUM targets have been identified using high-throughput approaches (Chen et al., 2012; Galgano et al., 2008; Hafner et al., 2010; Morris et al., 2008), revealing diverse functions for these proteins in germline homeostasis (Chen et al., 2012; Spassov and Jurecic, 2003), cell cycle control (Kedde et al., 2010; Miles et al., 2012) and neuronal activity and function (Driscoll et al., 2013; Vessey et al., 2010). Notably, Pum1 haploinsufficiency in mice has recently been reported to result in neurodegeneration (Gennarino et al., 2015), demonstrating that PUM dosage must be precisely controlled in vivo to avoid significant pathologic consequences. Nevertheless, the mechanisms through which PUM activity is regulated remain unknown.
Here we describe the unexpected finding that a poorly characterized mammalian lncRNA, which we termed NORAD, functions as a major regulator of PUM activity in human cells. This lncRNA initially came to our attention due to its induction after DNA damage, its strong evolutionary conservation, and its ubiquitous, abundant expression in human tissues and cell lines. Surprisingly, inactivation of NORAD resulted in chromosomal instability in previously stably diploid human cell lines. Identification of NORAD-interacting proteins revealed that this lncRNA functions as a multivalent binding platform for PUM proteins, with the capacity to sequester a significant fraction of the total cellular pool of PUM1 and PUM2. We further showed that PUM proteins regulate a large set of target transcripts that play a critical role in maintaining the fidelity of chromosome transmission and whose excessive repression in the absence of NORAD results in genomic instability. These findings have revealed a lncRNA-dependent mechanism that regulates a highly dosage-sensitive family of RNA binding proteins, uncovering a post-transcriptional regulatory axis that maintains genomic stability in mammalian cells.
RESULTS
Characterization of NORAD, an abundant, conserved human lncRNA
This study was initiated in an attempt to identify human lncRNAs that regulate the DNA damage response. To this end, we examined a set of previously identified mouse lncRNAs that are induced after doxorubicin treatment in a p53-dependent manner (Guttman et al., 2009). Among these transcripts, we noted a poorly-characterized 4.9 kilobase (kb) unspliced lncRNA, annotated as 2900097C17Rik, that exhibits a high degree of evolutionary conservation in mammals. A clear ortholog of this transcript, with 65% nucleotide identity to 2900097C17Rik, is expressed from the syntenic location in the human genome (Figure 1A). Annotated in RefSeq as LINC00657, this 5.3 kb lncRNA is broadly and abundantly expressed in human cell lines and tissues (Figure 1A, S1A). Like the mouse ortholog, the human transcript has features of an RNA polymerase II transcription unit, including an enrichment of H3K4me3-modified histones at the transcription start site (Figure 1A) and a canonical polyadenylation signal at the 3′ end, use of which was confirmed by 3′ rapid amplification of cDNA ends (RACE) (Figure S1B).
Figure 1. Characterization of NORAD, a highly abundant, conserved mammalian noncoding RNA induced by DNA damage.

(A) Schematic representation of NORAD (annotated in RefSeq as LINC00657) with associated UCSC Genome Browser tracks depicting mammalian conservation (PhastCons) as well as ENCODE RNA-seq and H3K4me3 ChIP-seq coverage in human cell lines (Rosenbloom et al., 2013).
(B) qRT-PCR analysis of NORAD expression relative to 18S rRNA in p53+/+ and p53−/− HCT116 cells (Bunz et al., 1998) with or without treatment with 1μM doxorubicin for 24 hours. For this and all subsequent qPCR figures, error bars represent standard deviations from 3 independent measurements.
(C) Northern blot analysis of NORAD expression in total RNA in HCT116 cells.
(D) Absolute quantification of NORAD transcript copy number per cell, determined by qRT-PCR, in various human cell lines with or without treatment with 1μM doxorubicin for 24 hours.
(E) Maximum CSF scores of NORAD as well as other known coding and noncoding RNAs determined by analysis with PhyloCSF (Lin et al., 2011).
See also Figure S1.
As in mouse, the human lncRNA is induced after DNA damage in a p53-dependent manner in the colon cancer cell line HCT116 (Figure 1B). We therefore named this lncRNA Noncoding RNA Activated by DNA Damage, or NORAD. Despite its p53-dependent induction, we were unable to identify an obvious p53 binding site in the vicinity of the NORAD promoter nor was one identified in a recent p53 ChIP-seq study performed in this cell line (Sanchez et al., 2014), likely indicating indirect regulation of NORAD by p53.
NORAD is easily detectable as a discrete transcript of the expected size by northern blotting (Figure 1C). Absolute copy number analysis in a panel of human cell lines with or without doxorubicin treatment revealed that NORAD is present at ~300–1400 copies per cell, similar in abundance to highly expressed mRNA transcripts such as ACTB (Islam et al., 2011) (Figure 1D).
Because lncRNAs may encode conserved peptides (Anderson et al., 2015; Bazzini et al., 2014), we examined the coding potential of NORAD using PhyloCSF, an algorithm that discriminates protein coding from noncoding transcripts based on their evolutionary signatures (Lin et al., 2011). This analysis confirmed the low coding potential of NORAD, which received a maximum codon substitution frequency (CSF) value similar to other well-characterized lncRNAs (Figure 1E). NORAD also lacks the potential to encode any recognizable protein domains, based on a BLASTX analysis of all possible reading frames. Based on these findings that established NORAD as a highly conserved, ubiquitously expressed, abundant lncRNA, we set out to investigate its functions in human cells.
NORAD loss-of-function results in chromosomal instability
To elucidate potential functions of NORAD, we designed 3 pairs of transcription activator-like effector nucleases (TALENs) that target within the first 300 nucleotides of the lncRNA to facilitate the homology-directed insertion of a transcriptional stop element and puromycin-resistance cassette flanked by loxP sites (Figure 2A). Initially, this approach was used to inactivate NORAD in HCT116 cells, a stably diploid human cell line that has been extensively used to study the p53 pathway and the human DNA damage response (Bunz et al., 1998; Jallepalli et al., 2001). All 3 TALEN pairs produced correctly targeted subclones with high efficiency after puromycin selection, allowing the derivation of a large number of NORAD−/− lines (15/147 clones with homozygous insertions). Correct targeting was confirmed by Southern blotting (Figure S1C) and resulted in the expected loss of NORAD expression (Figures 2B, S1D).
Figure 2. Genetic inactivation of NORAD results in chromosomal instability in human cells.

(A) NORAD was inactivated in human cell lines using custom TALEN pairs (represented as scissors) that cleave within the first 300 nucleotides of the gene, thereby stimulating the insertion of a puromycin resistance cassette (PuroR) followed by tandem polyadenylation signals (STOP). Green triangles represent loxP sites.
(B) Northern blot analysis of NORAD in HCT116 clones of the indicated genotypes.
(C) Flow cytometry histograms showing DNA content, as measured by propidium iodide staining, in representative diploid and tetraploid NORAD−/− HCT116 clones.
(D) Metaphase spreads of wild-type HCT116 cells and representative tetraploid and diploid NORAD−/− clones. The number in the lower right corner of each image shows the number of chromosomes present. Abnormal chromosome numbers indicated in red.
(E) Representative images of chromosome 7 and 20 FISH in NORAD+/+ and NORAD−/− HCT116 cells. White arrowheads highlight cells with chromosome loss or gain.
(F) NORAD−/− cells exhibit significantly elevated levels of aneuploidy. At least 100 interphase nuclei in each of 3 independent knockout clones were assayed for chromosome 7 and 20 using DNA FISH and the frequency of cells exhibiting a non-modal chromosome number was scored. **p<0.005, chi-square test.
(G) Representative time-lapse images of mitoses in NORAD+/+ and NORAD−/− HCT116 cells. Time stamp indicates minutes elapsed.
(H, I) Quantification of the percentage of mitoses exhibiting the indicated mitotic errors in time-lapse imaging experiments. Values represent the average of 3 independent experiments with 39–100 mitoses imaged per genotype per experiment. Error bars represent standard deviations. *p<0.05; **p<0.01, Student’s t-test.
See also Figures S1, S2, and S3.
Despite the induction of NORAD after DNA damage, we observed no consistent defect in the p53-dependent G1 or G2 checkpoints in NORAD−/− cells (Figure S2A–C), indicating that NORAD is not required for these aspects of the DNA damage response. In the course of these experiments, however, we unexpectedly observed that 2/15 NORAD−/− clones appeared to have stably tetraploid DNA content (Figure 2C). These findings were confirmed by examining metaphase chromosome spreads. With rare exception, wild-type HCT116 cells had 45 chromosomes, consistent with the reported karyotype (Masramon et al., 2000) (Figure 2D). In contrast, tetraploid NORAD−/− cells had variable chromosome numbers, with DNA content approaching 4N. As described below, our subsequent experiments have demonstrated that the spontaneous generation of tetraploid HCT116 subclones is exceedingly rare and we have never observed stable tetraploidization of these cells without NORAD inactivation.
Even apparently diploid NORAD−/− clones displayed a range of chromosome numbers (Figure 2D), suggesting that this karyotypically-stable cell line had adopted a chromosomal instability (CIN) phenotype, defined as the frequent loss or gain of whole chromosomes (Geigl et al., 2008). Human cancer cells frequently exhibit CIN, which is believed to be an important driver of tumorigenesis (Rajagopalan et al., 2003), yet the the role of lncRNAs in regulating this phenotype is poorly understood. Therefore, to more quantitatively assess whether loss of NORAD induces CIN, we employed an established fluorescent in situ hybridization (FISH) assay in which marker chromosomes are labeled and scored in hundreds of interphase cells (Jallepalli et al., 2001). Assaying chromosomes 7 and 20 with this approach verified that wild-type HCT116 cells exhibit a low frequency of chromosomal gain or loss (Figure 2E–F). In contrast, up to 25% of NORAD−/− cells displayed gain or loss of one of these chromosomes, confirming the presence of a CIN phenotype. Importantly, since only 2 chromosomes were assayed in these experiments, these measurements likely represent a significant underestimate of the frequency of aneuploidy in NORAD−/− cells. In addition, live cell imaging documented a high rate of mitotic errors, including anaphase bridges and mitotic slippage, in NORAD−/− clones (Figure 2G–I). Finally, karyotyping of representative NORAD−/− clones revealed the presence of non-recurrent de novo structural chromosomal rearrangements (Figure S2D). These findings were documented in multiple NORAD−/− clones generated with distinct TALEN pairs, strongly suggesting that this CIN phenotype is not caused by an off-target effect of TALEN-mediated genome editing.
To confirm that this phenotype is not unique to HCT116 cells, we introduced the transcriptional stop cassette into the NORAD locus in BJ-5ta cells, a telomerase-immortalized non-transformed diploid fibroblast cell line (Figure S3A–B). Although NORAD−/− BJ-5ta cells were grossly diploid by flow cytometric analysis of DNA content (data not shown), they exhibited significantly elevated levels of aneuploidy, as determined by quantification of chromosomes 7 and 20 by FISH (Figure S3C).
Chromosomal instability is specifically due to NORAD loss-of-function
We next performed a series of experiments to confirm that the CIN phenotype that we observed in NORAD−/− cells was specifically due to loss of this lncRNA rather than a consequence of genome manipulation with TALENs. First, we used a published TALEN pair to insert a puromycin resistance cassette at the AAVS1/PPP1R12C locus (Sanjana et al., 2012). Quantification of chromosomes 7 and 20 documented normal chromosome numbers in homozygously targeted HCT116 and BJ-5ta cells (Figures 3A, S3C). Live cell imaging confirmed a low rate of mitotic errors (Figure 2G–I). Furthermore, analysis of 70 subclones of HCT116 cells transfected with these TALENs revealed that all retained diploid DNA content (data not shown). Thus, neither CIN nor tetraploidy is a general property of cells that have undergone TALEN-mediated genome editing.
Figure 3. Chromosomal instability is specifically due to NORAD inactivation.

(A) Insertion of a puromycin resistance cassette at the AAVS1/PPP1R12C locus was performed using a published TALEN pair (Hockemeyer et al., 2009; Sanjana et al., 2012) and the frequency of aneuploidy in homozygous targeted HCT116 clones was assessed using DNA FISH as in Figure 2E–F. n.s., not significant (chi-square test).
(B) qRT-PCR analysis of NORAD expression, relative to 18S rRNA, in HCT116 cells 48 hours after transfection with control (siNT) or NORAD-targeting siRNAs.
(C) Aneuploidy in siRNA-transfected HCT116 cells 12 days after siRNA transfection, assayed as in Figure 2E–F. At least 200 nuclei were scored per condition. P value calculated by chi-square test.
(D) Flow cytometry histograms showing DNA content, as measured by propidium iodide staining, in representative HCT116 subclones generated after transfection with the indicated siRNAs.
(E) qRT-PCR analysis of NORAD expression in NORAD+/+ and NORAD−/− HCT116 cells with or without adenovirus-Cre infection.
(F) Subclones generated from untreated or adenovirus-Cre infected NORAD−/− HCT116 cells were scored for aneuploidy as in Figure 2E–F. P value calculated by Student’s t-test.
Next, we depleted the NORAD transcript using multiple siRNAs and assessed chromosome content by FISH after 12 days of subsequent growth (Figure 3B, C). As observed following TALEN-mediated inactivation of NORAD, knockdown of this transcript resulted in significant aneuploidy. Subclones of control or NORAD knockdown cells were then produced, revealing infrequent but reproducible de novo generation of tetraploid lines derived specifically from cells transfected with NORAD-targeting siRNAs (Figure 3D).
Lastly, we used Cre recombinase to excise the transcriptional stop cassette in NORAD−/− cells, resulting in restored NORAD expression (Figure 3E). Rescued subclones exhibited significantly lower levels of aneuploidy than subclones derived from control-treated cells (Figure 3F). Based on these findings, we conclude that NORAD is essential for the maintenance of genomic stability in human cells.
NORAD directly regulates both ploidy and chromosomal stability
It has been proposed that CIN can result from whole genome duplication events that produce a transient tetraploid state that subsequently resolves into an unstable pseudo-diploid state (Ganem et al., 2007). Therefore, since we recovered both tetraploid and diploid NORAD−/− clones that each exhibited CIN, it was unclear whether loss of NORAD primarily causes tetraploidization, which then results in CIN, or whether NORAD directly regulates both ploidy and chromosomal stability. The fact that CIN can be rescued by NORAD reactivation in diploid knockout cells (Figure 3E, F) supports the latter possibility. If CIN were due to a prior, now resolved, tetraploid state, restoration of NORAD should no longer have the capacity to revert genomic instability in diploid cells. Furthermore, if the CIN phenotype of NORAD−/− cells is solely a secondary consequence of polyploidization, tetraploid knockout cells should revert to a diploid state at a measureable frequency. However, 32/32 analyzed subclones derived from tetraploid NORAD−/− cells retained tetraploid DNA content. In contrast, approximately 10% of subclones of diploid NORAD−/− cells gained tetraploid DNA content (data not shown). These results support a primary role for NORAD in regulating both ploidy and chromosomal stability in diploid cells.
NORAD is a cytoplasmic multivalent PUMILIO binding platform
Alignment of the NORAD sequence to itself using the BLAST algorithm uncovered a repetitive ~400 nucleotide domain that recurs 5 times in the transcript (Figure S4A–B). We termed this sequence the NORAD domain (ND1-ND5). Notably, a large fraction of the conserved sequence within NORAD is encompassed within these repetitive regions. Furthermore, subcellular fractionation (Figure S4D) and single molecule RNA FISH (Figure S4E) demonstrated a nearly exclusively cytoplasmic localization of the NORAD RNA. Based on these results, we hypothesized that the NORAD domain represents a binding platform through which this lncRNA is able to assemble a multivalent cytoplasmic ribonucleoprotein (RNP) complex.
To identify components of this putative NORAD RNP, we synthesized 7 biotinylated RNA fragments encompassing each NORAD domain as well as the 5′ and 3′ segments of the transcript (Figure S4C). Mass spectrometry was used to identify proteins that bind to these fragments, and corresponding antisense controls, in HCT116 lysates (Figure S5A, Table S1). Candidate interactors were filtered for those that were detectable above background in all five NORAD domain pull downs with at least 5-fold enrichment compared to each corresponding antisense pull down. Only a single protein, PUMILIO 2 (PUM2), fulfilled these criteria (Figure S5B). We confirmed the binding of PUM2 to all five NORAD domains as well as the 5′ end of NORAD using western blotting (Figure 4A). Western blotting also revealed detectable interaction of NORAD with the related protein PUMILIO 1 (PUM1). Additionally, immunoprecipition of endogenous PUM proteins resulted in highly significant enrichment of endogenous NORAD (Figure S5C). Consistent with these data, both NORAD (Figure S4D, E) as well as PUM1/PUM2 (Morris et al., 2008; Narita et al., 2014; Ponten et al., 2008) are predominantly localized to the cytoplasm.
Figure 4. NORAD interacts with PUMILIO proteins.

(A) Western blot analysis of PUM1 and PUM2 in sense (S) and antisense (AS) NORAD fragment pull-downs. GAPDH served as a negative control.
(B, C) Histogram of the total number of CLIP reads per PUM2 target transcript in PAR-CLIP data generated with FLAG-PUM2 (Hafner et al., 2010) (B) or endogenous PUM2
(C). Number of NORAD CLIP reads shown in red text in parentheses.
(D) Location, sequence, and conservation of PREs in NORAD. ND, NORAD domain.
(E) Location and read depth of endogenous PUM2 (upper) or FLAG-PUM2 (lower) PAR-CLIP clusters mapped to NORAD. Black bars, clusters overlapping PREs; gray bars, non-PRE clusters.
See also Figures S4 and S5 and Tables S1 and S2.
Since RNAs and proteins are known to reassociate after cell lysis (Mili and Steitz, 2004), we took advantage of a previously generated photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP) dataset generated with human FLAG-tagged PUM2 (Hafner et al., 2010) that detects specific PUM2:RNA binding interactions that occur in intact cells. 7,523 PUM2 binding sites, occurring in ~3,000 transcripts, were identified in this study. Five PUM2 binding sites within NORAD were reported, with a site in ND4 representing the 11th most frequently crosslinked site transcriptome-wide (http://www.mirz.unibas.ch/restricted/clipdata/RESULTS/PUM2/PUM2.html). These results further document direct PUM2:NORAD interactions.
In the course of these studies, we noted the presence of a large number of sequences with homology to NORAD distributed throughout the human genome, with at least 43 genomic loci that exhibit 84–98% identity to NORAD over at least a 500 bp span. Many of these homologous sequences have features of processed pseudogenes, including target site duplications and terminal poly(A) sequences (Kazazian, 2014). Analysis of Illumina BodyMap 2.0 RNA-seq data revealed little evidence of transcription of most of these loci (data not shown), with the notable exception of a nearly full-length NORAD-related sequence on chromosome 6, which is annotated in Refseq as HCG11. However, HCG11 has an average FPKM of 2.0 in BodyMap data compared to an average FPKM of 31.8 for NORAD. Accordingly, use of sequence-specific Taqman assays demonstrated that HCG11 abundance is >200-fold lower than NORAD abundance in HCT116 cells (data not shown). Thus, at present, there is no evidence that any of these NORAD-related sequences are functional in human cells, although it remains possible that some may be transcribed at biologically relevant levels in specific tissues or cell types. Importantly, our Southern blot strategy (Figure S1C) confirmed that these NORAD-related sequences did not confound our genome editing approach since all analyzed NORAD−/− clones had single copy insertions of the lox-STOP-lox cassette at the desired site.
The presence of numerous NORAD pseudogenes likely confounded the mapping of sequencing reads in the prior PAR-CLIP study (Hafner et al., 2010), since reads that mapped to multiple genomic locations were excluded from further analyses. We therefore reanalyzed these data, first extracting all reads that map to NORAD prior to transcriptome-wide mapping. Remarkably, this revealed that NORAD was the most highly represented PUM2 CLIP target by a large margin (Figure 4B). To complement these data, we performed PAR-CLIP on endogenous PUM2 in NORAD+/+ and NORAD−/− HCT116 cells. Recovery of PUM2 was less efficient in this experiment than the prior study, which used heterologous expression of epitope-tagged PUM2, resulting in less comprehensive transcriptome-wide PUM2 target identification (Table S2). Nevertheless, NORAD was again the most highly represented target of endogenous PUM2 and, as expected, was not detected in NORAD−/− cells (Figure 4C, Table S2), demonstrating that the NORAD pseudogenes do not confound our modified mapping approach.
PUM1 and PUM2 exhibit a strong preference to bind to the PUMILIO response element (PRE) sequence UGUANAUA (Galgano et al., 2008; Wang et al., 2002). Indeed, the PRE is the most highly represented sequence motif within CLIP clusters identified using endogenous PUM2 (data not shown) or heterologously-expressed PUM2 (Hafner et al., 2010). The PRE is expected to occur once in approximately 16 kb of sequence by chance. Strikingly, there are 15 conserved sequences perfectly matching the PRE in the 5.3 kb NORAD transcript, with the large majority clustering in or near the NORAD domains (Figure 4D). This is in stark contrast to other PUM-bound transcripts, 90% of which have 2 or fewer PREs (Galgano et al., 2008). Analysis of CLIP cluster distribution on NORAD confirmed the binding of endogenous PUM2 to 7/15 PREs and heterologously-expressed PUM2 to 15/15 PREs (Figure 4E). Together, these data provide compelling evidence demonstrating multivalent interaction of PUMILIO proteins with NORAD and indicate that NORAD is the preferred PUM2 target transcript in human cells.
NORAD acts as a negative regulator of PUMILIO activity
With 15 PREs per transcript, at an expression level of ~500–1,000 copies per cell (Figure 1D), NORAD has the capacity to bind ~7,500–15,000 PUMILIO protein molecules per cell in HCT116. Quantitative western blotting documented an average of ~15,000 PUM1 and ~2,000 PUM2 proteins per cell (Figure S6). Based on these estimates, we hypothesized that NORAD sequesters a large fraction of the pool of PUMILIO proteins, thus negatively regulating their ability to repress target mRNAs. This model invokes at least 3 key predictions: First, in NORAD−/− cells, PUM1/2 should be hyperactive resulting in relative repression of PUM1/2 targets; second, PUM1 and/or PUM2 overexpression should phenocopy NORAD loss-of-function; and third, depletion of PUM1/2 should suppress the NORAD loss-of-function phenotype.
To test these predictions, we first performed RNA-seq on NORAD+/+ and NORAD−/− HCT116 cells (Table S3). Consistent with PUMILIO hyperactivity, PUM2 CLIP targets were statistically-significantly downregulated in NORAD−/− cells (Figure 5A). Significant downregulation of these targets was also confirmed by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) (Figure S7A).
Figure 5. Chromosomal instability in NORAD−/− cells is mediated through PUMILIO hyperactivity.

(A–C) Cumulative distribution plots depicting behavior of PUM2 CLIP targets, as defined in Hafner et al. and this study, versus non-PUM2-targets in the indicated RNA-seq experiments. P value calculated by Kolmogorov–Smirnov test demonstrates significant repression of PUM2 targets in all tested datasets.
(D) PUM1 and PUM2 overexpressing clones were assayed for aneuploidy using chromosome 7/20 FISH as in Figure 2E–F. At least 200 nuclei were scored per clone. n.s., not significant; *p<0.05; **p<0.005; ***p<0.0005, chi-square test.
(E, F) Cells of the indicated genotypes were assayed for aneuploidy as in (D). *p<0.05, Student’s t-test, comparing NORAD−/−; PUM1+/+; PUM2+/+ to NORAD−/−; PUM1−/−; PUM2+/+ or NORAD−/−; PUM1+/+; PUM2−/−.
(G, H) Quantification of the percentage of mitoses exhibiting the indicated mitotic errors in time-lapse imaging experiments after transfection with control siRNA (siNT) or two distinct sets of siRNAs targeting PUM1 and PUM2. Values represent the average of 3 independent experiments with 85–200 mitoses imaged per condition per experiment. Error bars represent standard deviations. *p<0.05; **p<0.01, Student’s t-test.
See also Figures S6 and S7 and Table S3.
We next generated HCT116 cell lines with stable overexpression of PUM1 or PUM2 (Figure S7B). Importantly, NORAD expression was unchanged in these cells (Figure S7C). RNA-seq confirmed the expected downregulation of PUM2 PAR-CLIP targets (Figure 5B, C, Table S3). Furthermore, PUM1 or PUM2 overexpression produced a gene expression signature that was similar to that observed upon NORAD inactivation, with genes that were down- or upregulated in NORAD−/− cells showing a similar pattern of expression in PUM1/2 overexpressing cells (Figure S7D). Accordingly, PUM2 and, to a lesser extent, PUM1 overexpression was sufficient to induce significant levels of aneuploidy (Figure 5D). Thus, PUMILIO overexpression phenocopies both the molecular and phenotypic consequences of NORAD inactivation.
Lastly, we used two approaches to deplete PUM1/2 in NORAD−/− cells. First, CRISPR/Cas9-mediated genome editing was used to inactivate PUM1, PUM2, or both (Figure S7E), followed by TALEN-mediated insertion of the transcriptional stop cassette at the NORAD locus. Individual knockout of PUM1 or PUM2 resulted in partial suppression of CIN in NORAD−/− cells, consistent with functional redundancy of these proteins (Figure 5E). Unexpectedly, double knockout of PUM1 and PUM2 led to measureable aneuploidy (Figure 5F). Together with our finding that PUM1 or PUM2 overexpression also results in aneuploidy (Figure 5D), these results suggest that precise control of PUM1/2 levels is necessary to maintain genomic stability. Importantly, knockout of NORAD in the PUM1−/−; PUM2−/− background did not result in a further increase in CIN (Figure 5F). Finally, we demonstrated that siRNA-mediated depletion of PUM1/2 in NORAD−/− cells (Figure S7F) significantly reduced the frequency of mitotic errors, as documented by time-lapse imaging (Figure 5G, H). These data establish a critical role for PUMILIO proteins downstream of NORAD in the maintenance of genomic stability.
PUMILIO proteins repress key mitotic, DNA repair, and DNA replication factors
Finally, to determine why PUMILIO hyperactivity results in CIN, we examined the expression of PUM2 CLIP targets in RNA-seq data from NORAD−/− cells. Among the 1,303 genes that were statistically-significantly downregulated in NORAD−/− cells were 193 PUM2 targets (Figure 6A, Table S3) that were significantly enriched for regulators of the cell cycle, mitosis, DNA repair, and DNA replication (Figure 6B). Notably, individual knockout or knockdown of many of these targets has previously been shown to be sufficient to induce genomic instability, including components of the cohesin complex (e.g. SMC1A, SMC3, and ESCO2), centromere components (e.g. CENPJ), and key factors necessary for DNA repair and replication (e.g. PARP1, PARP2, EXO1, BARD1, MCM4, and MCM8) (summarized in Table S4). We validated the downregulation of a large set of these transcripts with qRT-PCR in NORAD−/− cells, as well as in cells that overexpress PUM1 or PUM2 (Figures 6C, D, S7G). This coordinated downregulation of a broad set of targets that are necessary to maintain genomic stability would be expected to strongly impair accurate chromosome transmission.
Figure 6. Genes required for the maintenance of chromosomal stability are repressed in NORAD−/− and PUM1/2-overexpressing cells.

(A) Venn diagram showing overlap of genes that are significantly downregulated in NORAD−/− HCT116 cells (adjusted p value ≤ 0.05; see Table S3) and PUM2 PAR-CLIP targets identified in Hafner et al. and this study.
(B) Gene ontology analysis of the 193 PUM2 PAR-CLIP targets that are downregulated in NORAD−/− cells, demonstrating enrichment of genes involved in mitosis, the cell cycle, DNA replication, and DNA repair.
(C) qRT-PCR validation of PUM2 PAR-CLIP targets that have a known role in the maintenance of genomic stability (see Table S4) and were downregulated in NORAD−/− cells according to RNA-seq. Gene expression was normalized to 18S rRNA. All genes shown were significantly downregulated in NORAD−/− cells (p≤0.05, Student’s t-test).
(D) qRT-PCR demonstrating expression of genes from panel C that are significantly downregulated in both PUM1- and PUM2-overexpressing HCT116 cells (p≤0.05, Student’s t-test) (see also Figure S7G).
DISCUSSION
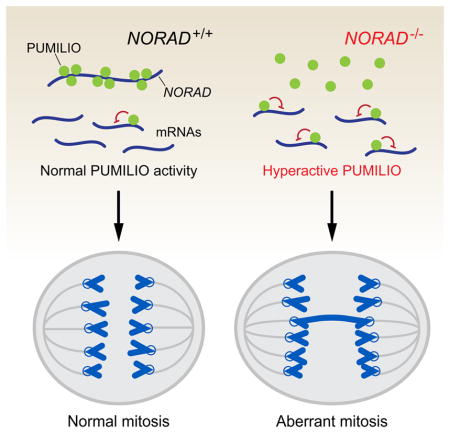
Here we report the initial functional characterization of a highly conserved lncRNA that we termed NORAD, which is broadly and abundantly expressed in mammalian cells and tissues. Our studies of this lncRNA have yielded several important and unexpected findings. First, inactivation of NORAD is sufficient to produce a chromosomal instability (CIN) phenotype in previously karyotypically-stable cell lines, revealing an essential role for a lncRNA in the maintenance of chromosomal stability in mammalian cells. Second, we show that NORAD preserves genomic stability by acting as a multivalent binding platform for the PUMILIO family of RNA binding proteins. Indeed, NORAD appears to be the preferred PUMILIO target transcript in human cells. Due to its high abundance and multitude of PUMILIO binding sites, NORAD is able to sequester a significant fraction of the total cellular pool of PUMILIO proteins, thereby limiting their ability to repress target mRNAs. Among PUMILIO targets are a large set of factors that are critical for mitosis, DNA repair, and DNA replication whose excessive repression in the absence of NORAD perturbs accurate chromosome segregation and can induce tetraploidization (Figure 7). The elucidation of this lncRNA:PUMILIO regulatory interaction has expanded our understanding of lncRNA functions and has uncovered a heretofore-unknown role for PUMILIO proteins in the regulation of genomic stability in mammals.
Figure 7. A NORAD-PUMILIO axis that regulates genomic stability.

Due to its abundance and multitude of PUMILIO binding sites, NORAD acts as a potent negative regulator of PUMLIO activity. In the absence of this lncRNA, PUMILIO is released to hyperactively repress a program of genes necessary to maintain chromosomal stability and a euploid state, including key factors required for mitosis, DNA replication, and DNA repair.
Our discovery that NORAD sequesters PUMILIO proteins contributes to an emerging concept that a major class of lncRNAs function as molecular decoys. For example, noncoding transcripts that sequester microRNAs (miRNAs), referred to as competing endogenous RNAs (ceRNAs), have been proposed to act as broad regulators of gene expression (Salmena et al., 2011). lncRNAs that inhibit proteins through competitive binding have also been reported, such as GAS5 and the glucocorticoid receptor (Kino et al., 2010), GADD7 and TDP-43 (Liu et al., 2012), and PANDA and NF-YA (Hung et al., 2011). Nevertheless, due to the generally low abundance of lncRNAs and the frequent promiscuity of protein-RNA interactions, the extent to which lncRNAs function through this mechanism has been heavily debated. Importantly, several features of NORAD distinguish it from the majority of lncRNAs and strongly support its function as a bona fide molecular decoy. First, NORAD is unusually abundant with expression in the range of ~500–1000 copies per cell in human cell lines, comparable to abundant housekeeping transcripts such as ACTB. Moreover, the presence of at least 15 PUMLIIO response elements (PREs) per NORAD transcript further amplifies, by more than an order of magnitude, the number of competitive binding sites provided by this lncRNA. Indeed, careful measurements of the number of PUM1 and PUM2 protein molecules per cell revealed that NORAD has the potential to sequester 50–100% of the total PUMILIO protein pool in HCT116 cells. Finally, it is noteworthy that unlike many RNA binding proteins that interact with loosely defined or very short consensus sequences, PUMILIO proteins are known for their exquisite specificity (Wang et al., 2002). Thus, NORAD provides an optimized binding platform that would be expected to efficiently assemble a multivalent PUMILIO RNP, thereby greatly reducing the availability of PUMILIO proteins to act upon mRNA targets.
These results also establish a role for PUMILIO proteins as important regulators of genomic stability. CIN, a phenotype characterized by the frequent gain or loss of chromosomes during mitosis, is a hallmark of cancer cells (Hanahan and Weinberg, 2011; Kops et al., 2005) and is a key mechanism that contributes to gain- and loss-of-function of oncogenes and tumor suppressors. Therefore, the mechanisms that give rise to genomic instability have been the subject of intensive research. Our finding that PUMILIO proteins repress a program of genes whose expression is necessary to maintain chromosomal stability reveals a previously unrecognized pathway to CIN. Prominent among PUMILIO targets are many genes that function in DNA replication and repair as well as key mitotic factors (summarized in Table S4). It is plausible that the coordinated dysregulation of these targets under conditions of PUMILIO hyper- or hypoactivity would produce a state of severe genomic instability, as observed upon NORAD loss-of-function, PUM1/2 overexpression, or PUM1/2 inactivation. Importantly, it is presently unclear whether dysregulated PUMILIO activity contributes to CIN in human cancer cells since abnormalities in PUMILIO or NORAD have not been reported in human tumors. Nevertheless, in light of our findings, a more thorough examination of this pathway in cancer is merited.
These findings contribute to a growing appreciation that the activity of PUMILIO proteins must be maintained within a narrow range to maintain homeostasis in mammals. For example, Pum1 haploinsufficiency results in neurodegeneration in mice due to upregulation of the PUMILIO target Ataxin1 (Gennarino et al., 2015). Our data further document that either increased or decreased PUM1/2 activity results in deleterious consequences. Nevertheless, little is known regarding how PUMILIO proteins are regulated. The emergence of NORAD in mammals provides a robust mechanism to buffer PUMILIO activity and maintain it within tolerable limits. A major unresolved question, however, is whether NORAD functions primarily as a static buffer or whether its levels are modulated in order to further titrate PUMILIO activity under certain conditions. Importantly, since each NORAD transcript has the capacity to bind a large number of PUMILIO protein molecules, even small changes in NORAD levels can profoundly influence PUMILIO availability. For example, NORAD initially came to our attention due to its modest induction after DNA damage (~2 fold; see Figure 1B, D). Yet this small increase generates ~7,000 additional PREs, representing sufficient binding sites to sequester nearly half of the total pool of PUMILIO proteins in HCT116 cells. Interestingly, it was recently reported that NORAD (LINC00657) is induced by hypoxia in human endothelial cells (Michalik et al., 2014), suggesting broader roles for NORAD in cellular stress responses. How NORAD influences the functional outputs of these and other stress response pathways, and the broader roles of NORAD in normal physiology and disease, represent important areas for future research.
EXPERIMENTAL PROCEDURES
TALENs and targeting constructs
3 pairs of TALENs targeting NORAD were designed using ZiFit Targeter v4.1 (Sander et al., 2010) and constructed using the Restriction Enzyme And Ligation (REAL) assembly method (Sander et al., 2011) with Addgene Kit #1000000017. Sequences of target genomic DNA (gDNA) and TALEN RVDs are provided in Table S5. To construct donor templates for homologous recombination (HR), homology arms were amplified from gDNA (primers in Table S5) and cloned into Lox-Stop-Lox TOPO (Addgene plasmid #11584) (Jackson et al., 2001) using the In-Fusion HD cloning Kit (Clontech). A previously described TALEN pair targeting the AAVS1/PPP1R12C locus (Sanjana et al., 2012) and an AAVS1/PPP1R12C targeting construct (Hockemeyer et al., 2009) were obtained from Addgene (hAAVS1 1L TALEN, Plasmid #35431; hAAVS1 1R TALEN, Plasmid #35432; AAVS1 hPGK-PuroR-pA donor, Addgene plasmid #22072).
Cell culture, transfection, and adenovirus transduction
HCT116 and BJ-5ta cells were obtained from ATCC and cultured in either McCoy’s 5a or a 4:1 mixture of DMEM and Medium199 respectively, supplemented with 10% FBS and 1X Antibiotic-Antimycotic (Life Technologies). HCT116 cells were transfected with Fugene HD (Promega). 10 μg DNA and 30 μL of the transfection reagent were used per 10 cm dish. For BJ-5ta, 4×106 cells were suspended in 100 μL nucleofector solution SE with 3 μg DNA and transferred to 100 μL cuvettes, followed by nucleofection using a 4D-Nucleofector System (Lonza) with program EN-150. For genome editing experiments, plasmids were mixed at molar ratio of Left-TALEN:Right-TALEN:HR-donor = 1:1:8. Transfected cells were then selected with 1 μg/mL puromycin for at least 7 days and surviving cells were plated in 96 well plates at single cell density. Genomic DNA was isolated from single-cell clones with the DNeasy kit (Qiagen) and genotyped by PCR with primers provided in Table S5. Ad-Cre was obtained from the UT Southwestern Vector Core and cells were transduced with an MOI of 200 for 2 days. siRNAs (sequences in Table S5) were transfected using DharmaFECT 2 (GE Healthcare).
RNA isolation, qPCR, and northern blotting
Total RNA was extracted from cultured cells with Trizol (Invitrogen) or the RNeasy kit (Qiagen) and treated with RNase-free DNase (Qiagen). For qRT-PCR experiments, the Taqman One-Step RT-PCR Master Mix (Life Technologies) was used with a custom NORAD Taqman assay or a commercial 18S rRNA Taqman assay (Life Technologies). For all other qPCR assays, RNA was reverse-transcribed with SuperScript III (Invitrogen) and Power SYBR Green PCR Master Mix (Life Technologies) was used. Primers and probes used for qPCR are provided in Table S5. To measure NORAD copies per cell, NORAD was first amplified from HCT116 cDNA and cloned into pcDNA3.1. This plasmid was then used to generate a standard curve for absolute quantification of NORAD abundance in defined numbers of cells. For northern blotting, 20 μg total RNA was separated on a 0.7% denaturing agarose gel containing formaldehyde and transferred to Hybond N+ membranes. The NORAD probe was PCR amplified with primers provided in Table S5 and radiolabeled using the Random Primed DNA Labeling Kit (Roche).
DNA FISH and Karyotyping
Chromosome enumeration probes for chromosome 7 (CHR7-10-GR) and chromosome 20 (CHR20-10-RE) were purchased from Empire Genomics. For interphase DNA FISH, cells were harvested with trypsin, washed with PBS, and incubated in hypotonic solution (0.4% KCl) for 10 minutes. Cells were then resuspended in fixation buffer (3:1 mix of methanol:glacial acetic acid) and spread on slides pre-treated with 1M HCl for 24 hours, then 70% EtOH for 24 hours and stored in distilled water. For analyzing metaphase spreads, cells were treated with 1 μg/mL colcemid (Roche) for 30 minutes, harvested and fixed as described above, and spread on slides in a climate-controlled hood, set at 25°C and 40% humidity. DNA FISH hybridizations and karyotype analyses were performed by the Veripath Cytogenetics laboratory at UT Southwestern.
Accession Numbers
The accession number for RNA-seq data reported in this paper is GEO GSE75440. The accession number for PAR-CLIP data reported in this paper is GEO GSE75439.
Additional materials and methods are provided in Extended Experimental Procedures.
Author Contributions
S.L., F.K., T.-C.C., A.S., B.C., S.S., H.Y., Y.X., and J.T.M. designed the experiments and interpreted the results. S.L., F.K., T.-C.C., A.S., B.C., and S.S. performed the experiments. S.L., F.K., and J.T.M. wrote the manuscript.
Supplementary Material
Highlights.
NORAD is a broadly expressed, highly abundant, conserved mammalian lncRNA.
Inactivation of NORAD in human cells triggers dramatic aneuploidy.
NORAD functions as a potent molecular decoy for PUMILIO proteins (PUM1/PUM2).
PUM1/PUM2 repress a program of genes necessary to maintain genomic stability.
Acknowledgments
We thank Tyler Jacks, Rudolf Jaenisch, Keith Joung, David Root, Didier Trono, and Feng Zhang for plasmids; Kathleen Wilson, Sangeeta Patel, and Charles Tiongson in the Veripath Cytogenetics laboratory at UT Southwestern; Vanessa Schmid, Ashley Guzman, and Rachel Bruce in the McDermott Center Next Generation Sequencing Core; and Hamid Mirzaei and David Trudgian in the UT Southwestern Proteomics Core. Stephen Johnson provided valuable assistance with software implementation. We also thank Jose Cabrera for assistance with figure preparation and Nicholas Conrad, Kathryn O’Donnell, Eric Olson, Hao Zhu, and members of the Mendell laboratory for critical reading of the manuscript. This work was supported by grants from CPRIT (R1008 to J.T.M., RP101251 to Y.X., and RP120717 to H.Y.) and the NIH (R01CA120185 and R35CA197311 to J.T.M. and R01CA152301 to Y.X.). J.T.M. and H.Y. are investigators of the Howard Hughes Medical Institute, J.T.M. is a CPRIT Scholar in Cancer Research, and F.K. is supported by the Leopoldina Fellowship Program (LPDS 2014-12) from the German National Academy of Sciences Leopoldina.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160:595–606. doi: 10.1016/j.cell.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33:981–993. doi: 10.1002/embj.201488411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 2011;25:1915–1927. doi: 10.1101/gad.17446611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Zheng W, Lin A, Uyhazi K, Zhao H, Lin H. Pumilio 1 suppresses multiple activators of p53 to safeguard spermatogenesis. Curr Biol. 2012;22:420–425. doi: 10.1016/j.cub.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson CM, Hutchinson JN, Sara SA, Ensminger AW, Fox AH, Chess A, Lawrence JB. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–726. doi: 10.1016/j.molcel.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll HE, Muraro NI, He M, Baines RA. Pumilio-2 regulates translation of Nav1.6 to mediate homeostasis of membrane excitability. J Neurosci. 2013;33:9644–9654. doi: 10.1523/JNEUROSCI.0921-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St Laurent G, 3rd, Kenny PJ, Wahlestedt C. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med. 2008;14:723–730. doi: 10.1038/nm1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nat Rev Genet. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Galgano A, Forrer M, Jaskiewicz L, Kanitz A, Zavolan M, Gerber AP. Comparative analysis of mRNA targets for human PUF-family proteins suggests extensive interaction with the miRNA regulatory system. PLoS One. 2008;3:e3164. doi: 10.1371/journal.pone.0003164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Storchova Z, Pellman D. Tetraploidy, aneuploidy and cancer. Curr Opin Genet Dev. 2007;17:157–162. doi: 10.1016/j.gde.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Geigl JB, Obenauf AC, Schwarzbraun T, Speicher MR. Defining ‘chromosomal instability’. Trends Genet. 2008;24:64–69. doi: 10.1016/j.tig.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Gennarino VA, Singh RK, White JJ, De Maio A, Han K, Kim JY, Jafar-Nejad P, di Ronza A, Kang H, Sayegh LS, et al. Pumilio1 haploinsufficiency leads to SCA1-like neurodegeneration by increasing wild-type Ataxin1 levels. Cell. 2015;160:1087–1098. doi: 10.1016/j.cell.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong C, Maquat LE. lncRNAs transactivate STAU1-mediated mRNA decay by duplexing with 3′ UTRs via Alu elements. Nature. 2011;470:284–288. doi: 10.1038/nature09701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP, et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacisuleyman E, Goff LA, Trapnell C, Williams A, Henao-Mejia J, Sun L, McClanahan P, Hendrickson DG, Sauvageau M, Kelley DR, et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat Struct Mol Biol. 2014;21:198–206. doi: 10.1038/nsmb.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jr, Jungkamp AC, Munschauer M, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung T, Wang Y, Lin MF, Koegel AK, Kotake Y, Grant GD, Horlings HM, Shah N, Umbricht C, Wang P, et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat Genet. 2011;43:621–629. doi: 10.1038/ng.848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam S, Kjallquist U, Moliner A, Zajac P, Fan JB, Lonnerberg P, Linnarsson S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011;21:1160–1167. doi: 10.1101/gr.110882.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer MK, Niknafs YS, Malik R, Singhal U, Sahu A, Hosono Y, Barrette TR, Prensner JR, Evans JR, Zhao S, et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet. 2015;47:199–208. doi: 10.1038/ng.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, Jacks T, Tuveson DA. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM, Kinzler KW, Vogelstein B, Lengauer C. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445–457. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Kedde M, van Kouwenhove M, Zwart W, Oude Vrielink JA, Elkon R, Agami R. A Pumilio-induced RNA structure switch in p27-3′ UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol. 2010;12:1014–1020. doi: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- Kino T, Hurt DE, Ichijo T, Nader N, Chrousos GP. Noncoding RNA gas5 is a growth arrest- and starvation-associated repressor of the glucocorticoid receptor. Sci Signal. 2010;3:ra8. doi: 10.1126/scisignal.2000568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- Kretz M, Siprashvili Z, Chu C, Webster DE, Zehnder A, Qu K, Lee CS, Flockhart RJ, Groff AF, Chow J, et al. Control of somatic tissue differentiation by the long non-coding RNA TINCR. Nature. 2013;493:231–235. doi: 10.1038/nature11661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Chang HY. Physiological roles of long noncoding RNAs: insight from knockout mice. Trends Cell Biol. 2014;24:594–602. doi: 10.1016/j.tcb.2014.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Jungreis I, Kellis M. PhyloCSF: a comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics. 2011;27:i275–282. doi: 10.1093/bioinformatics/btr209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Sun L, Liu Q, Gong C, Yao Y, Lv X, Lin L, Yao H, Su F, Li D, et al. A cytoplasmic NF-kappaB interacting long noncoding RNA blocks IkappaB phosphorylation and suppresses breast cancer metastasis. Cancer Cell. 2015;27:370–381. doi: 10.1016/j.ccell.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Liu X, Li D, Zhang W, Guo M, Zhan Q. Long non-coding RNA gadd7 interacts with TDP-43 and regulates Cdk6 mRNA decay. EMBO J. 2012;31:4415–4427. doi: 10.1038/emboj.2012.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masramon L, Ribas M, Cifuentes P, Arribas R, Garcia F, Egozcue J, Peinado MA, Miro R. Cytogenetic characterization of two colon cell lines by using conventional G-banding, comparative genomic hybridization, and whole chromosome painting. Cancer Genet Cytogenet. 2000;121:17–21. doi: 10.1016/s0165-4608(00)00219-3. [DOI] [PubMed] [Google Scholar]
- Michalik KM, You X, Manavski Y, Doddaballapur A, Zornig M, Braun T, John D, Ponomareva Y, Chen W, Uchida S, et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389–1397. doi: 10.1161/CIRCRESAHA.114.303265. [DOI] [PubMed] [Google Scholar]
- Miles WO, Tschop K, Herr A, Ji JY, Dyson NJ. Pumilio facilitates miRNA regulation of the E2F3 oncogene. Genes Dev. 2012;26:356–368. doi: 10.1101/gad.182568.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mili S, Steitz JA. Evidence for reassociation of RNA-binding proteins after cell lysis: implications for the interpretation of immunoprecipitation analyses. RNA. 2004;10:1692–1694. doi: 10.1261/rna.7151404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Olivas WM. Roles of Puf proteins in mRNA degradation and translation. Wiley Interdiscip Rev RNA. 2011;2:471–492. doi: 10.1002/wrna.69. [DOI] [PubMed] [Google Scholar]
- Morris AR, Mukherjee N, Keene JD. Ribonomic analysis of human Pum1 reveals cis-trans conservation across species despite evolution of diverse mRNA target sets. Mol Cell Biol. 2008;28:4093–4103. doi: 10.1128/MCB.00155-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita R, Takahasi K, Murakami E, Hirano E, Yamamoto SP, Yoneyama M, Kato H, Fujita T. A novel function of human Pumilio proteins in cytoplasmic sensing of viral infection. PLoS Pathog. 2014;10:e1004417. doi: 10.1371/journal.ppat.1004417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponten F, Jirstrom K, Uhlen M. The Human Protein Atlas - a tool for pathology. J Pathol. 2008;216:387–393. doi: 10.1002/path.2440. [DOI] [PubMed] [Google Scholar]
- Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145–166. doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res. 2013;41:D56–63. doi: 10.1093/nar/gks1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Segura V, Marin-Bejar O, Athie A, Marchese FP, Gonzalez J, Bujanda L, Guo S, Matheu A, Huarte M. Genome-wide analysis of the human p53 transcriptional network unveils a lncRNA tumour suppressor signature. Nat Commun. 2014;5:5812. doi: 10.1038/ncomms6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK, Yeh JR. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nat Biotechnol. 2011;29:697–698. doi: 10.1038/nbt.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Maeder ML, Reyon D, Voytas DF, Joung JK, Dobbs D. ZiFiT (Zinc Finger Targeter): an updated zinc finger engineering tool. Nucleic Acids Res. 2010;38:W462–468. doi: 10.1093/nar/gkq319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171–192. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spassov DS, Jurecic R. The PUF family of RNA-binding proteins: does evolutionarily conserved structure equal conserved function? IUBMB Life. 2003;55:359–366. doi: 10.1080/15216540310001603093. [DOI] [PubMed] [Google Scholar]
- Struhl K. Transcriptional noise and the fidelity of initiation by RNA polymerase II. Nat Struct Mol Biol. 2007;14:103–105. doi: 10.1038/nsmb0207-103. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I, Bartel DP. lincRNAs: genomics, evolution, and mechanisms. Cell. 2013;154:26–46. doi: 10.1016/j.cell.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vessey JP, Schoderboeck L, Gingl E, Luzi E, Riefler J, Di Leva F, Karra D, Thomas S, Kiebler MA, Macchi P. Mammalian Pumilio 2 regulates dendrite morphogenesis and synaptic function. Proc Natl Acad Sci U S A. 2010;107:3222–3227. doi: 10.1073/pnas.0907128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, McLachlan J, Zamore PD, Hall TM. Modular recognition of RNA by a human pumilio-homology domain. Cell. 2002;110:501–512. doi: 10.1016/s0092-8674(02)00873-5. [DOI] [PubMed] [Google Scholar]
- Wickens M, Bernstein DS, Kimble J, Parker R. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet. 2002;18:150–157. doi: 10.1016/s0168-9525(01)02616-6. [DOI] [PubMed] [Google Scholar]
- Zamore PD, Williamson JR, Lehmann R. The Pumilio protein binds RNA through a conserved domain that defines a new class of RNA-binding proteins. RNA. 1997;3:1421–1433. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.