

Abstract
We assessed structural brain damage in obstructive sleep apnea syndrome (OSA) patients (21 males) and the effects of long‐term continuous positive airway pressure (CPAP) treatment (18.2 ± 12.4 months; 8–44 months) on brain structures and investigated the relationship between severity of OSA and effects of treatment. Using deformation‐based morphometry to measure local volume changes, we identified widespread neocortical and cerebellar atrophy in untreated patients compared to controls (59 males; Cohen's D = 0.6; FDR < 0.05). Analysis of longitudinally scanned magnetic resonance imaging (MRI) scans both before and after treatment showed increased brain volume following treatment (FDR < 0.05). Volume increase was correlated with longer treatment in the cortical areas that largely overlapped with the initial atrophy. The areas overlying the hippocampal dentate gyrus and the cerebellar dentate nucleus displayed a volume increase after treatment. Patients with very severe OSA (AHI > 64) presented with prefrontal atrophy and displayed an additional volume increase in this area following treatment. Higher impairment of working memory in patients prior to treatment correlated with prefrontal volume increase after treatment. The large overlap between the initial brain damage and the extent of recovery after treatment suggests partial recovery of nonpermanent structural damage. Volume increases in the dentate gyrus and the dentate nucleus possibly likely indicate compensatory neurogenesis in response to diminishing oxidative stress. Such changes in other brain structures may explain gliosis, dendritic volume increase, or inflammation. This study provides neuroimaging evidence that revealed the positive effects of long‐term CPAP treatment in patients with OSA. Hum Brain Mapp 37:395–409, 2016. © 2015 Wiley Periodicals, Inc.
Keywords: obstructive sleep apnea syndrome, continuous positive airway pressure, deformation‐based morphometry, anatomical MRI, intermittent hypoxemia
INTRODUCTION
Obstructive sleep apnea (OSA) is a common sleep disorder that is characterized by recurrent episodes of upper airway collapse, resulting in intermittent hypoxemia and recurrent arousals from sleep [Young et al., 2002]. An insufficient oxygen supply to the brain tissue due to hypoxemic events in OSA is thought to be a factor for deficits in cognitive functions in patients with such a condition [Beebe et al., 2003].
Structural and functional neuroimaging studies have supported the presence of OSA‐related brain damage, identifying regional gray matter (GM) loss [Canessa et al., 2011; Joo et al., 2010; Torelli et al., 2011], inflammatory changes [Chen et al., 2015], neural functional disruption [Jones et al., 2014], and white matter (WM) integrity decrease [Kumar et al., 2012; Macey et al., 2008]. Animal models have also shown that chronic exposure to intermittent hypoxia causes neurodegenerative changes, particularly increases in programmed cell death in the hippocampus, which subsequently result in impaired spatial working memory [Gozal et al., 2001a].
Continuous positive airway pressure (CPAP) has been the most effective and widely used treatment in OSA [Vennelle et al., 2010]. The therapeutic effects of CPAP on the brain include improved daytime functioning and cognitive performance [Ferini‐Strambi et al., 2003; Lau et al., 2010; Lim and Pack, 2014; Olaithe and Bucks, 2013], especially executive functioning associated with frontal lobe processes [Lau et al., 2010; Olaithe and Bucks, 2013]. It has yet to be determined if this enhanced functional improvement is due to the recovery of initial structural damages or whether CPAP treatment leads to such a reversal. While the effects of short‐term treatment (<6 months) on brain structures have been debated [Canessa et al., 2011; O'Donoghue et al., 2005], longer treatments (<12 months) in OSA patients change metabolite levels and WM water diffusion in multiple brain regions closer to the levels seen in good sleepers [Castronovo et al., 2014; O'Donoghue et al., 2012], indicating that both acute and mild brain damage can be reversed with effective treatments.
T1‐weighted MRI provides high‐resolution biometrics related to brain morphological changes. Deformation‐based morphometry (DBM) has been one of the best methodological frames for longitudinal studies [Chung et al., 2001] as it does not rely on “error‐prone” tissue classification but only on the performance of the nonlinear registration [Tosun et al., 2011].
Diffeomorphic nonlinear registration methods, such as DARTEL, which is part of the SPM8 package, have demonstrated their superior sensitivity to cortical volume changes by minimizing misregistration [Shigemoto et al., 2013].
Our study aimed to assess brain structural damage in untreated OSA patients and to investigate the effects of CPAP treatment on the brain structures of these patients. Furthermore, we explored whether the severity of the disease or of cognitive functional impairment in untreated OSA was associated with the degree of brain structural changes after CPAP treatment. The strength of the study lies in the following methodological considerations: (1) inclusion of patients with long‐term CPAP treatments (average: 18.2 months; range: up to 44 months); (2) assessment of volume changes using DBM with an optimal design for longitudinal analyses; and (3) evaluation of the pattern of post‐CPAP brain recovery and its comparison to the pattern of pre‐CPAP brain damage.
METHODS
We studied 21 male patients (age = 49.2 ± 7.7, range = 31–61 years old) with OSA who were recruited from the Sleep Clinic at Samsung Medical Center in Seoul, South Korea. Inclusion criteria were an apnea‐hypopnea index (AHI) >15; 20 patients presented with severe OSA (AHI ≥ 30), whereas only one patient exhibited moderate symptoms (AHI = 25.3). All patients were scanned on a 1.5 Tesla MRI scanner within 1 week prior to initiating CPAP treatment (pre‐CPAP scan). They were scanned once more after treatment (post‐CPAP scan, duration: 18.2 ± 12.4 months, range: 8–44 months). The mean use time (average nightly use of CPAP) was measured based on downloads of usage tracked during the treatment period with memory cards from ResMed machines (ResMed, San Diego, CA).
We also recruited 59 healthy males whose age was in a similar range as that of patients (age = 44.3 ± 10.1, range = 25–76 years old; P = 0.08). They were recruited using an advertisement in the local community. Each candidate underwent a detailed clinical interview and overnight polysomnography (PSG) and also completed sleep questionnaires.
Exclusion criteria were as follows: (1) controls with an average nightly sleep time of <7 h over the most recent 2 weeks based on sleep diary entries; (2) obstructive sleep apnea syndrome (OSA, apnea‐hypopnea index > 5/h); (3) moderate‐to‐severe periodic limb movement during sleep (PLMS, total PLM index > 25/h); (4) circadian rhythm sleep disorder determined by sleep‐wake cycles of sleep diaries; (5) history of medication for hypertension, diabetes, heart, or respiratory diseases; (6) history of cerebrovascular disease; (7) diagnosis of other neurological (neurodegenerative diseases, epilepsy, or head injury) or psychiatric diseases (psychosis, current depression); (8) alcohol or illicit drug abuse or current intake of psychoactive medications; and (9) a structural lesion on brain MRI. Five patients who showed diffuse brain atrophy on brain MRI were excluded from the initial patient group (n = 26). Informed consent was obtained from all participants, and the Institutional Review Board of Samsung Seoul Hospital authorized the study protocol and design.
Overnight Polysomnography
Participants were asked not to consume any alcohol or caffeinated beverages 24 h prior to the sleep study. Sleep studies were recorded using a Remlogic (Embla Systems, Denver, CO). We have previously provided a detailed description of the test procedures [Joo et al., 2013].
Neuropsychological Assessments
Participants underwent a battery of neuropsychological tests and a standardized intelligence test. Neuropsychological tests assessed six broad domains: working memory (the Digit Span Test from the Wechsler Memory Scale‐Revised and the Corsi Block Tapping Tests, forward and backward); executive functioning (Trail Making Tests A and B and the Stroop Test), verbal information processing (Digit Symbol Test), verbal memory (Korean California Verbal Test), visual memory (Rey Complex Figure Test), and verbal fluency (controlled oral word association test). Information regarding the protocol of the neuropsychological assessments can be found elsewhere [Noh et al., 2012].
MRI Acquisition
MRI scanning of patients and controls was performed with a GE Signa 1.5‐Tesla scanner (GE Medical Systems, Milwaukee, WI). T1‐weighted coronal spoiled gradient recalled (SPGR) MRI was performed with the following scanning variables: 1.6‐mm thickness, no gap, 124 slices, TR/TE = 30/7 ms, flip angle = 45°, NEX = 1, matrix size = 256 × 192, and field of view = 22 × 22 cm2. The voxel dimensions in the SPGR MRI images were 0.86 × 0.86 × 1.6 mm3.
Voxel‐wise Deformation‐based Morphometry of the Entire Brain Structure
Each image underwent automated correction for intensity non‐uniformity [Sled et al., 1998] prior to subsequent analyses. (1) Measuring volumes in OSA patients prior to CPAP treatment and in controls. The pre‐CPAP scans were first linearly and then nonlinearly registered to the MNI‐ICBM 152 template using DARTEL, a diffeomorphic anatomical registration [Ashburner, 2007] included in the statistical parametric mapping toolkit (SPM8; http://www.fil.ion.ucl.ac.uk/spm/). While linear registration normalized intra‐cranial volume across subjects, nonlinear registration yielded voxel‐wise deformation fields between the pre‐CPAP scans and the template. To examine the local volume change in each subject, the Jacobian determinant (Jacobian henceforth) in the deformation field was computed at every voxel [Chung et al., 2001]. This metric provides a simple and direct means of determining local volume changes, such as voxel‐wise expansion (Jacobian > 1) or compression (0 < Jacobian < 1), relative to the reference space (= MNI‐ICBM template). We also applied these steps to determine volume variations in healthy individuals. (2) Measuring volume changes in patients during the CPAP treatment. Post‐CPAP scans were first linearly registered to the stereotaxic space using the transformation parameters of the pre‐CPAP scan. These images were first linearly and then finally non‐linearly registered to their pre‐CPAP scans in stereotaxic space. From the deformation field, the individual Jacobian was extracted to quantify volume changes from the pre‐CPAP scan to the post‐CPAP scan. To allow for inter‐subject point correspondence, the measured Jacobians were mapped on the MNI‐ICBM template using the nonlinear transformation obtained by the previous registration of the pre‐CPAP scans to the template. All Jacobian maps were smoothed using an 8‐mm full‐width at half‐maximum isotropic Gaussian kernel.
Statistical Analysis
Analysis was performed using SurfStat (http://www.math.mcgill.ca/keith/surfstat/) [Worsley et al., 2009]. The up‐to‐date version that was used in this study incorporated a module performing voxel‐based analysis, such as DBM or voxel‐based morphometry [Chung et al., 2010; Worsley et al., 2009]. To compare demographics, mood questionnaires, PSG parameters, and neuropsychological scores between patients and controls, a one‐way analysis of variance was used.
Age and depressive mood scores were included as covariates in general linear models for all the subsequent DBM analyses. We assessed the volume decrease at the pre‐CPAP scan of OSA patients compared to healthy controls using voxel‐wise Student's t tests on the Jacobian. In the following tests where the effects of CPAP were assessed, we included the mean arterial pressure (MAP) that was calculated using a standardized equation (MAP = [(2 × diastolic) + systolic blood pressure]/3) as an additional covariate in order to control for possible confounding effects due to BP changes after CPAP treatment (Table 1). We also included the frequency (night/duration %) and average time (hours per day) of CPAP use as covariates because the CPAP adherence may also influence brain volume changes. We assessed volume increases induced by CPAP treatment using voxel‐wise paired t tests on the post‐CPAP Jacobian (measured from pre‐ to post‐CPAP scans). Effects of CPAP duration on brain changes (i.e., treatment of a longer duration associated with brain volume) were assessed using a linear model that included a term of inter‐scan intervals. We also investigated whether the severity of pre‐CPAP OSA symptoms changed the effect of the treatment on brain volume recovery by correlating the PSG parameters with the post‐CPAP Jacobian at each voxel. To assess the association between the degree of impairment in pre‐CPAP cognitive function and volume changes after treatment, we correlated neuropsychological scores with Jacobian. To investigate whether brain morphological changes after CPAP treatment can be attributed to normal aging, we also examined cross‐sectional aging effects in controls using a linear model that correlated age with Jacobian at each voxel. We visually compared patterns of normal aging to changes that emerged in patients after treatment.
Table 1.
Characteristics of untreated patients with OSA and controls
| Patients (n = 21) | Controls (n = 59) | P value | |
|---|---|---|---|
| Mean age (years) | 49.8 ± 7.7 | 44.3 ± 10.1 | 0.08 |
| Body mass index (kg m−2) | 27.4 ± 3.8 | 26.3 ± 1.3 | 0.1 |
| Mean arterial pressure (mm Hg) | 108.3 ± 12.1 | 88.7 ± 9.3 | <0.001a |
| Overnight polysomnography | |||
| Total sleep time (min) | 294 ± 91.2 (175.5–486.0) | 380.9 ± 40.0 (312.0–488.0) | <0.001a |
| Mean sleep latency (min) | 15.2 ± 20.1 (2.0–91.5) | 9.1 ± 7.1 (1.0–24.5) | 0.003a |
| Mean REM latency (min) | 122.6 ± 50.1 (56.5–245.0) | 90.4 ± 33.9 (4.5–158) | 0.001a |
| Sleep efficiency (%) | 79.6 ± 11.4 (57.1–92.2) | 88.1 ± 5.8 (76.3‐97.8) | <0.001a |
| AHI, per hour | 60.4 ± 23.5 (25.3–92.3) | 3.8 ± 2.4 (0–4.8) | <0.001a |
| Mean arousal index, per hour | 56.1 ±19.2 (23.9–90.8) | 12.3 ± 4.2 (5.9–22) | <0.001a |
| Respiratory arousal (%) | 78.0 ± 16.5 (41.5–98.3) | 23.5 ± 25.6 (0–52.4) | <0.001a |
| NREM sleep 1 (%) | 42.7 ± 16.5 (20.7–76.5) | 11.9 ± 6.3 (4–15.8) | <0.001a |
| NREM sleep 2 (%) | 38.9 ± 14.0 (7.8–56.7) | 53.4 ± 8.4 (34.3–78.8) | <0.001a |
| NREM sleep 3 (%) | 3.1 ± 4.8 (0–17.2) | 10.5 ± 8.2 (0–32.5) | <0.001a |
| REM sleep (%) | 15.3 ± 6.2 (0.5–25.9) | 24.2 ± 4.8 (11.2–35.5) | <0.001a |
| % Total sleep time < 90% | 17.2 ± 11.5 (10–65) | 0.10 ± 0.15 (0–0.4) | <0.001a |
| Apnea maximum duration (s) | 57.0 ± 22.8 (26.0–106.5) | 9.3 ± 11.4 (0‐15.3) | <0.001a |
| Mean ESS | 13.9 ± 4.8 (3–21) | 3.0 ± 2.4 (0–8) | <0.001a |
| Mean SSS | 3.7 ± 2.1 (1–7) | 1.7 ± 1.0 (0–3) | 0.02a |
| Beck depression index | 12.5 ± 4.3 (5–20) | 5.2 ± 3.7 (3–8) | 0.01a |
Independent t test, P < 0.05.
OSA; obstructive sleep apnea syndrome, REM; rapid eye movement, NREM; non‐REM, AHI, apnea‐hypopnea index, ESS; Epworth sleepiness scale, SSS; Stanford sleepiness scale, mean arterial pressure= [(2 × diastolic) + systolic blood pressure]/3. All values are expressed as mean ± standard deviation, and the range of values is in parentheses.
To better localize findings with respect to the neocortex, we projected the voxel‐wise significances to their nearest points on the cortical surface extracted from the MNI‐ICBM template. Findings in the hippocampus were separately visualized by interpolating the volumetric P value maps mapped on the MNI‐ICBM 152 nonlinear template on to the closest point of the hippocampal surface that was extracted from the template. We interpreted the hippocampal volume changes with respect to a previously published subfield atlas [Joo et al., 2014]). For other subcortical structures as well as the cerebellum, we localized findings on the MNI‐ICBM volume template.
In all analyses, significances were thresholded using the false discovery rate procedure [Benjamini and Hochberg, 1995], with FDR ≤ 0.05.
RESULTS
Clinical Characteristics
Patients and healthy controls were all right‐handed and mostly middle‐aged. The mean duration of CPAP therapy was 18.2 ± 12.4 months (8–44 months), and the mean use time was 6.3 ± 1.9 h day−1. The average CPAP pressure of patients was 9.5 ± 2.0 mm H2O. Detailed PSG findings in untreated OSA patients and healthy controls are summarized in Table 1. Untreated OSA patients had significantly higher blood pressure readings (Table 1) and lower scores in working memory, verbal information processing, and verbal memory (FDR ≤ 0.05) compared to controls (Table 2).
Table 2.
Neuropsychological analyses of patients with OSA and controls
| Patients (n = 21) | Good sleepers (n = 59) | Possible range of scores | P value | |
|---|---|---|---|---|
| Working memory composite score | −0.7 ± 1.4 | 0.0 ± 1.0 | <0.05a | |
| Digit span forward | 9.0 ± 2.2 | 9.5 ± 2.0 | 0–16 | |
| Digit span backward | 7.2 ± 1.6 | 8.6 ± 2.1 | 0–16 | |
| Corsi block forward | 8.5 ± 1.9 | 10.7 ± 1.8 | 0–16 | |
| Corsi block backward | 9.3 ± 2.9 | 9.9 ± 0.9 | 0–14 | |
| Trail making test A | 38.6 ± 13.9 | 34.8 ± 10.4 | N/A | |
| Trail making test B | 104.5 ± 45.3 | 82.0 ± 30.1 | N/A | |
| Executive functioning composite score | 0.1 ± 0.5 | 0.0 ± 1.0 | 0.6 | |
| Stroop word correct responses | 112.0 ± 1.0 | 106.0 ± 14.9 | 0–112 | |
| Stroop word correct responses time | 106.8 ± 10 | 108.4 ± 12 | N/A | |
| Verbal information processing composite score | −0.9 ± 0.3 | 0.0 ± 1.0 | <0.001a | |
| Digit symbol test | 54.6 ± 3.6 | 65.2 ± 12.1 | 0–135 | |
| Verbal memory composite score | −0.6 ± 1.08 | 0.0 ± 1.0 | <0.01a | |
| Korean California verbal test | ||||
| Total | 49.7 ± 10.0 | 57.2 ± 7.4 | 0–48 | |
| Short delay free recall | 10.3 ± 3.0 | 12.5 ± 3.0 | 0–16 | |
| Long delay free recall | 11.3 ± 2.8 | 12.7 ± 2.7 | 0–16 | |
| Recognition | 14.9 ± 1.2 | 15.1 ± 1.3 | 0–16 | |
| Verbal fluency composite score | 0.0 ± 0.8 | 0.0 ± 1.0 | 0.9 | |
| Controlled oral word association test | ||||
| Phonetic word fluency | 35.6 ± 9.3 | 32.1 ± 12.7 | N/A | |
| Semantic word fluency | 34.7 ± 6.0 | 36.2 ± 7.7 | N/A | |
| Visual memory composite score | −0.1 ± 0.9 | 0.0 ± 1.0 | 0.8 | |
| Rey complex figure test | ||||
| Copy | 34.6 ± 1.6 | 34.6 ± 2.5 | 0–36 | |
| Immediate recall | 21.8 ± 6.6 | 21.6 ± 7.3 | 0–36 | |
| Delayed recall | 21.0 ± 6.8 | 20.8 ± 7.1 | 0–36 | |
| Recognition | 20.0 ± 2.4 | 20.8 ± 1.9 | 0–36 |
Significant after Bonferroni adjustment for multiple comparisons. – Units of Neuropsychological Tests: Trail Making Test A & B = response time in seconds; Stroop word correct responses time = response time in seconds; controlled oral word association test, phonetic and semantic word fluency = number of responses during given time period.
All our patients had a good CPAP use (frequency = 89 ± 12, 74–100%: ≥70% of nights; average time = 6.0 ± 0.1, 5–7 h: ≥4 h/night) and only a small leak of mask. After the course of CPAP therapy, daytime sleepiness improved significantly (ESS 13.9 vs. 4.5 from pre‐ to post‐treatment, FDR = 0.005). Blood pressure was definitely decreased after CPAP; decreased MAP (108.3 ± 12.1 → 97.8 ± 7.1 m mHg), systolic BP (140.0 ± 15.4 → 126.8 ± 9.0 mm Hg), and diastolic BP (92.5 ± 11.4 → 83.3 ± 7.1 mm Hg) (FDR ≤ 0.005). However, a reduction in post‐CPAP blood pressure level was not associated with duration of treatment (FDR ≥ 0.3).
Pre‐CPAP Brain Damage in OSA Patients Relative to Controls
We found brain cortical atrophy in patients compared to controls (z‐score: −0.8 ± 0.9; t = 2.7; FDR < 0.05; Fig. 1). Cortical atrophy was localized bilaterally in the lateral prefrontal, central, and anterior/posterior cingulate cortices and unilaterally in the left medial prefrontal, right orbitofrontal, left superior temporal, right middle/inferior temporal, right insular, left hippocampal, right parahippocampal, and right lateral occipital cortices, and the left cuneus (t > 2.7; FDR < 0.05, Table 3). Atrophy was also found in the thalamus (latero‐posterior division) and the cerebellum (posterior lobe, lobular IV, VII) in OSA patients compared to controls (FDR < 0.05). No volume increases were identified in patients (FDR > 0.2).
Figure 1.
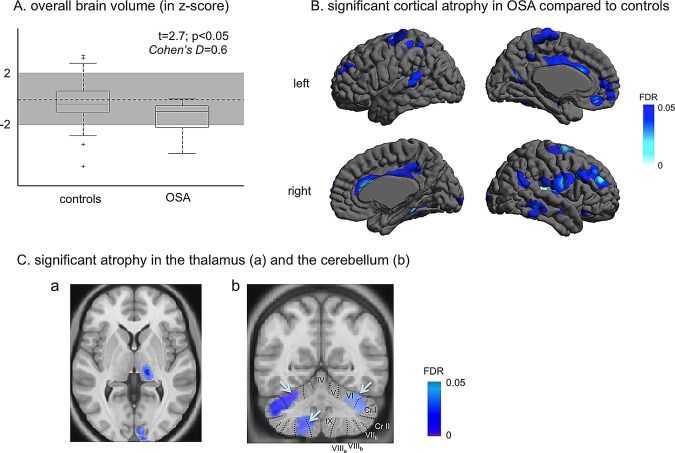
Comparison of brain volumes between pre‐CPAP OSA patients and controls. Decreases in overall brain volume (A); in local cortical volume (B); in the left thalamus and bilateral cerebelli (C). Significance values are thresholded at FDR < 0.05. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Table 3.
Significant brain volume reduction in untreated OSA patients
| Peak location | Side | MNI coordinates (mm) | T value | P value corrected by FDR | ||
|---|---|---|---|---|---|---|
| x | y | Z | ||||
| Lateral prefrontal cortex | L | −29 | 60 | 4 | 4.0 | <0.01 |
| Lateral prefrontal cortex | R | 42 | 43 | 27 | 6.2 | <0.001 |
| Central cortex | L | −41 | −17 | 65 | 4.3 | <0.005 |
| Central cortex | R | 28 | −6 | 67 | 5.5 | <0.001 |
| Cingulate cortex | L | −3 | 19 | 25 | 3.4 | <0.01 |
| Cingulate cortex | R | 5 | 37 | 18 | 3.9 | <0.01 |
| Medial prefrontal cortex | L | −4 | 45 | −20 | 3.3 | <0.05 |
| Orbitofrontal cortex | R | 17 | 36 | −25 | 3.3 | <0.05 |
| Superior temporal cortex | L | −67 | −41 | 10 | 3.4 | <0.01 |
| Middle temporal cortex | R | 65 | −40 | −12 | 3.6 | <0.01 |
| Insula | R | 35 | −22 | 13 | 5.8 | <0.001 |
| Hippocampus | L | −28 | −12 | −23 | 3.1 | <0.05 |
| Parahippocampal cortex | R | 19 | −34 | 16 | 3.9 | <0.01 |
| Lateral occipital cortex | R | 11 | −101 | 1 | 3.3 | <0.05 |
| Cuneus | L | −15 | −68 | 24 | 3.1 | <0.05 |
| Thalamus | R | 14 | −27 | 3 | 4.2 | <0.005 |
| Cerebellum | L | −23 | −57 | −20 | 5.2 | <0.001 |
MNI coordinates: Coordinates in MNI‐ICBM 152 template space; L/R: left/right.
Overall Effects of CPAP Treatment on Brain Structures in OSA Patients
After CPAP treatment, patients presented a trend of an increase in the overall volume of the cerebral cortex (t = 2.7; FDR = 0.09; uncorrected P = 0.01; Fig. 2A). Regionally, we found a significant volume increase in numerous cortical areas including the bilaterally superior frontal, orbitofrontal, central, and precuneus cortices, as well as the unilaterally left temporal pole and the left medial prefrontal, right anterior cingulate, left hippocampal, right lateral posterior temporal, and right lateral parietal cortices (FDR < 0.05, Table 4).
Figure 2.
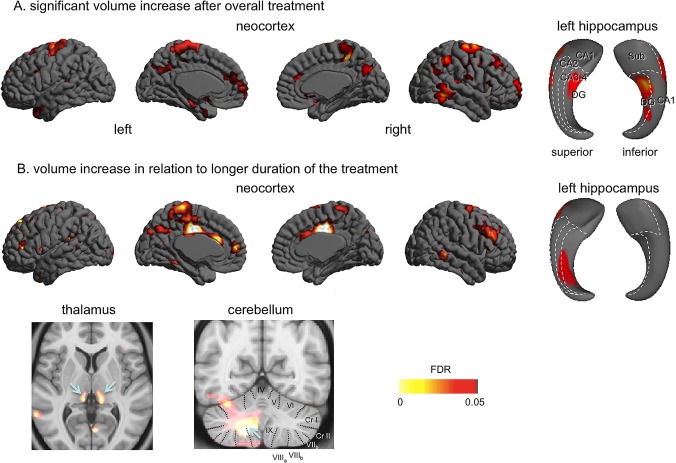
Effects of CPAP treatment on brain structural volume in OSA patients. A significant volume increase after treatment (no changes in thalamic or cerebellar volume) (A); Volume increase in relation to a longer duration of treatment (B). Arrows indicate locations with the most significant volume increase: the bilateral thalamic lateroposterior nuclei and deep left cerebellar structures. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Table 4.
Volume increases in OSA patients after CPAP treatment
| Peak location | Side | MNI coordinates (mm) | T value | P value corrected by FDR | ||
|---|---|---|---|---|---|---|
| x | y | Z | ||||
| Superior frontal cortex | L | −26 | 57 | 25 | 3.2 | <0.05 |
| Superior frontal cortex | R | 23 | 40 | 45 | 3.8 | <0.01 |
| Orbitofrontal cortex | L | −9 | 13 | −20 | 3.3 | <0.05 |
| Orbitofrontal cortex | R | 10 | 28 | −25 | 3.1 | <0.05 |
| Central cortex | L | −21 | −10 | 68 | 4.6 | <0.005 |
| Central cortex | R | 22 | −16 | 72 | 5.2 | <0.001 |
| Precuneus | L | −3 | −66 | −34 | 3.3 | <0.05 |
| Precuneus | R | 3 | −64 | 24 | 3.1 | <0.05 |
| Anterior temporal pole | L | −49 | 13 | −38 | 3.4 | <0.01 |
| Medial prefrontal cortex | L | −4 | 64 | −12 | 3.5 | <0.01 |
| Anterior cingulate cortex | R | 5 | 47 | 12 | 3.1 | <0.05 |
| Hippocampus | L | −27 | −16 | −18 | 3.4 | <0.01 |
| Posterior temporal cortex | R | 59 | −48 | −13 | 4.5 | <0.005 |
| Lateral parietal cortex | R | 48 | −50 | 51 | 3.4 | <0.05 |
Surface mapping of the left hippocampal volume increases after treatment localized more specifically the areas corresponding to the anterior dentate gyrus (FDR < 0.05). The pattern of changes in the right hippocampus was similar but did not reach statistical significance (FDR > 0.1).
No volume decrease was shown in any brain structures after the treatment (FDR > 0.15). No volume increase was found in deep GM structures or the cerebellum (FDR > 0.2). No volume increase was found in relation to the frequency or average time of CPAP use (FDR ≥ 0.1).
Relationships Between Duration of Treatment and Brain Structures in OSA Patients
Among the areas demonstrating a volume increase after CPAP, the medial prefrontal, superior frontal, central, precuneus, and posterior temporal cortices presented a larger volume increase with a longer duration of treatment (FDR < 0.05, Table 5). As seen in Figure 3A, patients who were treated for a longer amount of time presented with volume increases that extended to broad neighboring areas, including the bilateral cingulate, bilateral dorsolateral prefrontal cortices, and left parahippocampal gyrus (FDR < 0.05).
Table 5.
Volume increases associated with longer‐term CPAP treatment
| Peak location | Side | MNI coordinates (mm) | T value | P value corrected by FDR | ||
|---|---|---|---|---|---|---|
| x | y | z | ||||
| Lateral prefrontal cortex | L | −34 | 41 | −5 | 4.4 | <0.005 |
| Lateral prefrontal cortex | R | 46 | 41 | 22 | 4.6 | <0.005 |
| Cingulate cortex | L | −3 | 23 | 36 | 6.4 | <0.001 |
| Cingulate cortex | R | 3 | −24 | 41 | 6.8 | <0.001 |
| Medial prefrontal cortex | L | −4 | 48 | −4 | 4.8 | <0.001 |
| Superior frontal cortex | L | −22 | 47 | 42 | 4.3 | <0.005 |
| Posterior temporal cortex | R | 66 | −52 | −1 | 4.1 | <0.005 |
| Central cortex | L | −2 | −30 | 69 | 4.9 | <0.001 |
| Central cortex | R | 3 | −38 | 70 | 3.1 | <0.05 |
| Precuneus | L | −4 | −66 | −40 | 3.2 | <0.05 |
| Hippocampus | L | −30 | −30 | −9 | 3.1 | <0.05 |
| Thalamus | L | −11 | −28 | 3 | 3.9 | <0.005 |
| Thalamus | R | 12 | −28 | 5 | 4.3 | <0.005 |
| Cerebellum | L | −25 | −58 | −40 | 6.3 | <0.001 |
Figure 3.
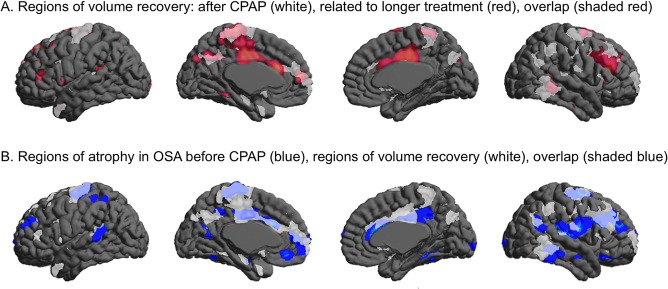
Qualitative assessment of the pattern of cortical volume changes in patients with OSA. Regions of volume recovery after overall treatment (white), after long‐term treatment (red), and their overlap (shaded red) (A). Regions of atrophy in OSA before treatment (blue), regions of volume recovery after treatment (white), and their overlap (shaded blue) (B). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Regions that combined areas displaying a volume increase after the overall CPAP treatment and a volume increase related to a longer treatment duration overlapped largely with regions that had displayed atrophy before the treatment (Fig. 3B).
In the hippocampus, volumes of areas corresponding to the posterior dentate gyrus and Cornu Ammonis 3–4 were increased in relation to a longer duration of treatment. A thalamic volume increase was found in a similar location of pre‐CPAP atrophy, whereas the cerebellar volume increase was identified in the deep structures, including the dentate nucleus and WM, and did not closely overlap with pre‐CPAP atrophy (Fig. 3).
Relationships Between Severity of OSA or pre CPAP Cognitive Impairment, and Brain Structural Recovery After Treatment
We found that CPAP treatment increased the medial prefrontal cortical volume in patients with more severe OSA (AHI > 64, n = 10; Fig. 4) compared to those with a less severe type (n = 11, FDR ≤ 0.05; Fig. 4A). A retrospective analysis of pre‐CPAP scans showed that this area displayed a trend of more atrophy in untreated patients with more severe OSA (P ≤ 0.01, uncorrected).
Figure 4.
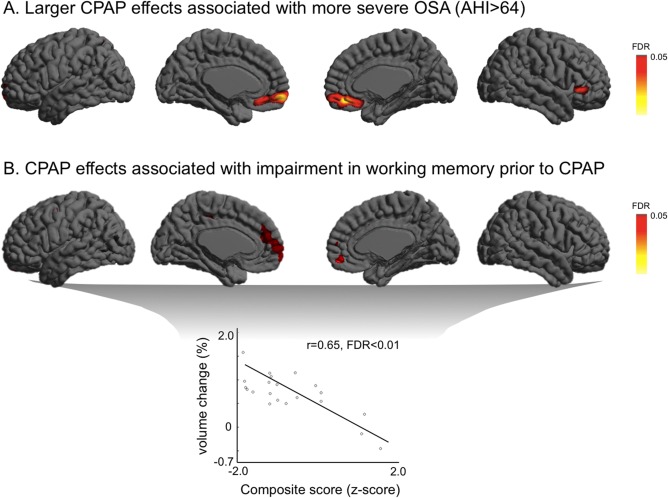
Various CPAP effects on brain volume in relation to clinical and neuropsychological conditions prior to treatment. (A). Bilateral medial prefrontal cortices display larger volume recovery in patients with severe OSA (AHI ≥ 64) after CPAP treatment compared to those with a less severe type (AHI ≤ 64). (B). More severe impairment in working memory before treatment induced larger volume increases in bilateral prefrontal and left posterior cingulate cortices after treatment. [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
Impairment in working memory in patients was associated with increase in volume of bilateral prefrontal cortices and a focal area of left posterior cingulate gyrus (r = 0.65; FDR ≤ 0.05; Fig. 4B). No association was found between other neuropsychological scores and post‐CPAP volume changes (FDR ≥ 0.2).
Aging in Healthy Controls
Aging affected negatively whole cortical volume (r = −0.45; FDR ≤ 0.01; Fig. 5). The cortical areas presenting volume decrease related to aging were mapped with scattered clusters in frontal, temporal, and parietal lobes (r = −0.65; FDR ≤ 0.005). In the subcortical area, bilateral thalamus showed volume decrease (r = −0.78; FDR ≤ 0.001). The size of lateral ventricles increased as subjects became old (r = 0.82; FDR ≤ 0.001). The pattern of brain volume decrease was not similar to that of atrophy in pre CPAP OSA. The direction of volume changes related to aging was opposite of that associated with CPAP treatment.
Figure 5.
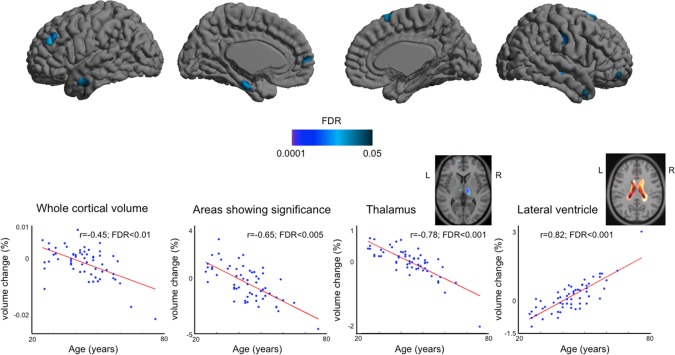
Aging effect on brain volume in healthy controls. Top: Cortical volume decrease in relation to Aging; Bottom: Plots showing volume change over ages (%/year) in whole cortical areas as well as other areas than the cortex. There were no volume increases found in cortical or subcortical GM (FDR ≥ 0.1). [Color figure can be viewed in the online issue, which is available at http://wileyonlinelibrary.com.]
DISCUSSION
The current study investigated brain volume changes in patients with untreated OSA and the effects of CPAP treatment on the brain structures of these patients using longitudinally designed deformation‐based morphometry (DBM). Continuing the treatment for a longer duration of time was associated with extensive brain volume increases in areas that largely overlapped with the areas of initial damage. Patients with more severe OSA presented with a greater amount of medial prefrontal atrophy and a larger volume increase after treatment. Working memory impairment in untreated OSA was correlated with increases in dorsolateral prefrontal and hippocampal volume after treatment.
Structural Damages in OSA: Findings and Possible Mechanisms
Our DBM analysis revealed decreased volume of the cerebral cortex and the cerebellum in untreated patients with OSA compared to healthy controls. The cortical areas displaying atrophy in OSA included the lateral/medial prefrontal, orbitofrontal, superior/middle temporal, insular, and anterior/posterior cingulate and sensorimotor cortices, as well as the hippocampus and parahippocampal gyrus. This finding is concordant with previous studies that have reported structural and functional abnormalities in OSA patients [Canessa et al., 2011; Jones et al., 2014; Joo et al., 2010, 2013; Kumar et al., 2012; Macey et al., 2008; Torelli et al., 2011]. In particular, a recent meta‐analysis summarizing previous VBM studies localized the hippocampus, parahippocampal cortex, lateral/medial frontal cortex, and middle temporal cortex as the areas most likely to display atrophy in OSA [Weng et al., 2014], confirming some areas of atrophy noted in our study. A potential mechanism for such neuronal damage is glutamate excitotoxicity induced by a chronic condition of hypoxia/ischemia [Banasiak et al., 2000; Gozal et al., 2001a]. Excessive activation of ionotropic N‐methyl‐d‐aspartate receptors through this mechanism may eventually increase programmed cell death, such as apoptosis [Gozal et al., 2001a; Kaindl et al., 2012].
The prefrontal cortex is known to be not only a site of neuronal reorganization in OSA but also that it may constitute a cerebral biomarker of disease severity in functional MRI studies [Prilipko et al, 2011; Zhang et al, 2013]. More frequent and more desaturating respiratory events are associated with more pronounced alterations of prefrontal activity, which provides the pathogenetic hypothesis of the brain structural changes by repetitive hypoxia in OSA.
Hypoxia‐induced apoptosis has been investigated and is well‐reproduced through N‐methyl‐d‐aspartate receptor‐expressing cells in the hippocampus [Gozal et al., 2001a] and the neocortex [Gozal et al., 2001b]. Oxidative stress induced by chronic intermittent hypoxia is another possible factor that contributes to cellular damage of the brain in addition to cardiovascular complications due to microvascular endothelial dysfunction [Buchner et al., 2011]. Although most neuroimaging studies report brain atrophy in OSA, a recent study that analyzed patients with a large spectrum of severity controversially also found bilateral enlargement of the hippocampus [Rosenzweig et al., 2013], suggesting the role of hippocampal neurogenesis as a response to ischemic/hypoxic preconditioning in the neuropathology of OSA. On the other hand, the patients analyzed in our study were clinically indicated to have severe OSA.
The primary sensorimotor and orbitofrontal (or ventromedial prefrontal) cortices and the cerebellum play a major role in modifying respiratory patterning or altering upper airway or diaphragmatic muscle tone [Macey et al., 2002; Marks et al., 1987]. Furthermore, the insular, cingulate, and orbitofrontal cortices are associated with autonomic regulation [Brannan et al., 2001]. A previous study showed that cingulate cortical thinning was correlated with increased respiratory arousals [Joo et al., 2013].
The current finding of structural damage in the aforementioned regions might therefore indicate early injury that predisposes an individual to OSA. The cerebellar atrophy in our study and its pattern (i.e., maximal change in the left lobe VIII and near XI) correspond well to findings in a previous study that analyzed a relatively large sample of OSA patients [Morrell et al., 2010]. Noticeably, cerebellar damage resulting from hypoxic conditions is often found in neonates, and such early injury is frequently accompanied by reduced muscle tone and difficult breathing [Huang and Castillo, 2008].
Effects of Treatment on OSA‐induced Hypoxic Brain Injury: Recovery, Compensation, or Inflammation?
Controversy exists regarding the reversibility of hypoxemic brain injury in OSA. However, the latest neuroimaging studies using structural T1‐weighted MRI, diffusion‐tensor imaging and single‐photon emission computed tomography [Canessa et al., 2011; Castronovo et al., 2014; Shiota et al., 2014] have provided evidence that brain structures can recover following treatment. In particular, multisite brain recovery has been found after long‐term CPAP treatments due to improved subcortical WM integrity [Castronovo et al., 2014] and cortical functional metabolism [O'Donoghue et al., 2012].
This investigation also qualitatively confirmed that brain volume increases related to CPAP treatment are not likely driven by the natural aging process. The current study revealed a brain volume increase after the initiation of treatment, which is probably not a result of the natural aging process; our analysis found only a negative effect of aging on brain volume. This conclusion might further indicate that some of the cortical atrophy observed in OSA can be reversed, as shown by the broad overlap between the areas of initial cortical atrophy and the volume increase after treatment. The mechanism underlying the recovery of the hypoxemic cortical damages remains unclear. Excessive glutamate release led by intermittent hypoxemic events affects not only the distribution of neurons, but also neuropils, including intra‐cortical glial cells [Choi and Rothman, 1990], and myelin‐supporting oligodendrocytes cells [Dewar et al., 2003]. As neocortical neurogenesis is rarely observed in older adults, the cortical volume increase induced by treatment might reflect an alteration of neuronal contents, particularly in the presence of an increased size of the neuropil. Proliferation or hypertrophy of several types of neuropil is indeed considered a responsive process to neurodegenerative diseases [Miller et al., 2004] and to intermittent hypoxia [Baronio et al., 2013].
The dentate gyrus of the hippocampus (DGH) and the dentate nucleus of the cerebellum (DNC) have been of particular interest in human adults as granule cells in these regions are generated continuously in the adult mammalian brain [Ming and Song, 2011] and play an important role in controlling motor function, regulating cognition, and storing new memories [Galliano et al., 2010; Kitamura et al., 2009]. Neurogenesis of granule cells in the DGH and DNC can be suppressed under stress conditions [Mirescu and Gould, 2006], which can be induced by sleep fragmentation [Guzman‐Marin et al., 2008]. We found that volumes corresponding to increases in DGH and DNC occurred with longer treatment of OSA despite the dissimilar pattern of initial atrophy in the hippocampus and the cerebellum. This finding might therefore indicate a secondary compensatory neurogenesis process in response to diminishing oxidative stress within neural tissue exposed to hypoxic events.
Questions have arisen regarding whether an inflammatory response to excessive changes in oxygen level induced by CPAP also reflects an increase in cortical volume. However, there is evidence that effective CPAP therapy reduces the molecules that govern the processes of inflammation, leukocyte activation, and leukocyte‐endothelial interaction [Jelic and Le Jemtel, 2008]. Human in vitro studies have shown that CPAP decreased the levels of soluble adhesion molecules and TNF‐α and reduced monocyte adhesion capacity in cultured endothelial cells [Chin et al., 2000; Phillips et al., 2007; Yokoe et al., 2003]. Such molecular changes are associated with a decreased level of inflammation. However, any positive effect of long‐term CPAP on inflammation in OSA remains to be assessed.
Our untreated patients presented with a high level of blood pressure (BP) that disappeared after treatment. Although the CPAP helped relieve high BP in our study, the duration of the treatment itself was not associated with a decrease in BP level. To control for a possible confounding effect, we included BP changes as a covariate in our statistical analyses. Thus, the reversibility of brain volume seen in our data is likely not due to any normalization of the initial high BP.
Our study did not fully support the reversibility of brain structural injury in OSA after treatment, as the areas of atrophy and volume increases partially overlapped, suggesting no significant recovery in some regions, while other regions showed increases in areas that had not initially been affected. Furthermore, it is unclear whether the volume increases fully reversed the initial lower volume; the group analysis of pre‐CPAP damage and the longitudinal analysis of CPAP effects employed different uses of the reference/baseline with which the Jacobian was computed.
Does More Severe Memory Impairment Prompts More Positive Effects of CPAP on Related Brain Structures?
In the current study, untreated OSA patients showed cognitive impairment in working memory and verbal memory and processing compared to controls; these results were consistent with the findings of previous studies [Ferini‐Strambi et al., 2003; Lau et al., 2010; Lim and Pack, 2014; Olaithe and Bucks, 2013]. Intriguingly, impairment in working memory for patients with untreated status was associated with a volume increase in the prefrontal cortex (PFC) after CPAP treatment. The PFC is known as a hub that processes working memory and executive function [Monsell, 2003]. It has been hypothesized that intermittent hypoxemia due to sleep‐disordered breathing putatively induces cellular and biochemical stresses and subsequently alters the efficacy of memory restorative processes during sleep [Harrison and Horne, 2000; Olaithe and Bucks, 2013]. When sleep deprivation occurs, the PFC, especially the lateral PFC, appears to be overrun during a complex memory task [Chee and Choo, 2004] in order to compensate for the dysfunction of the related areas given a difficult‐to‐solve task. Inversely, CPAP normalizes both sleep disruption and oxidative stress in OSA patients, likely stabilizing the over‐usage of PFC and its related memory circuits. This structural recovery process might further result in the restoration of impaired working memory in OSA after treatment [Kingshott et al., 2000; Olaithe and Bucks, 2013]. This association is supported by a recent observation of simultaneous recovery in prefrontal WM integrity and improvement of working memory after long‐term treatment [Castronovo et al., 2014].
Technical Consideration
DBM can directly quantify voxel‐wise volume changes using the Jacobian determinant [Chung et al., 2001]. Voxel‐based morphometry can also assess volume changes by incorporating the Jacobian with the tissue concentration map [Good et al., 2001]. This method, however, provides a relative rather than a quantitative measurement. The strength of DBM is its ability to detect subtle changes in longitudinal studies, as misregistration between within‐subject serial scans is minimal while avoiding “error‐prone” tissue segmentation [Tosun et al., 2011].
Restoration of impaired daytime (cognitive) function in OSA after treatment has been previously reported [Kingshott et al., 2000; Olaithe and Bucks, 2013]. Our study raised the question of whether this functional restoration is driven by the brain structural recovery achieved through treatment. Even though the initial severity of functional impairment was related to the effect of CPAP on brain volume, the absence of repeated neuropsychological testing after treatment in the current study limits our investigation to the direct causality between brain structural recovery and brain function.
Because of the difficulty in forcing patients to undergo long‐term treatment after moderation of major OSA symptoms, only a relatively small group of patients participated in the current study. We should admit that it is extremely difficult to acquire longitudinal scans of controls, which might be a limitation of this study. Instead, we cross‐sectionally assessed the aging effect in a large sample controls and found decrease in volume in multiple focal areas, which did not correspond to the broad areas of atrophy found in OSA and areas of volume increase after CPAP treatment.
Even though DBM can assess WM and GM changes, our main results did not show WM volume changes, suggesting that macroscopic WM changes are unlikely. Assessment of WM‐related damage in OSA and its reversibility might be complemented using DTI, which can identify changes in the distribution of extra‐axonal water molecules [Castronovo et al., 2014].
CONCLUSION
Our findings emphasize the necessity of the long‐term use of CPAP in brain structural recovery; this type of treatment moderates general OSA symptoms, including impairment in daytime memory function. The large overlap between the initial brain damage and recovery at post‐treatment suggests the reversibility of non‐permanent structural damage. All our patients had a good CPAP use and variables of CPAP adherence did not correlate with volume changes. The results suggest that the CPAP treatment and its duration are the significant factors associated with brain structural recovery as long as the patients used CPAP properly. Volume increases in the dentate gyrus and dentate nucleus might indicate compensatory neurogenesis in response to diminishing oxidative stress. This study provided neuroimaging evidence that revealed the positive effects of long‐term treatment with CPAP in participants with OSA.
ACKNOWLEDGMENTS
The authors are grateful to Felix Carbonell Ph.D. at McGill University for his technical consulting. All authors report no biomedical financial interests or potential conflicts of interest.
Correction added on 2 November 2015, after first online publication.
REFERENCES
- Ashburner J (2007): A fast diffeomorphic image registration algorithm. Neuroimage 38:95–113. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Xia Y, Haddad GG (2000): Mechanisms underlying hypoxia‐induced neuronal apoptosis. Prog Neurobiol 62:215–249. [DOI] [PubMed] [Google Scholar]
- Baronio D, Martinez D, Fiori CZ, Bambini‐Junior V, Forgiarini LF, Pase da Rosa D, Kim LJ, Cerski MR (2013): Altered aquaporins in the brains of mice submitted to intermittent hypoxia model of sleep apnea. Respir Physiol Neurobiol 185:217–221. [DOI] [PubMed] [Google Scholar]
- Beebe DW, Groesz L, Wells C, Nichols A, McGee K (2003): The neuropsychological effects of obstructive sleep apnea: A meta‐analysis of norm‐referenced and case‐controlled data. Sleep 26:298–307. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y (1995): Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat Soc 57:289–300. [Google Scholar]
- Brannan S, Liotti M, Egan G, Shade R, Madden L, Robillard R, Abplanalp B, Stofer K, Denton D, Fox PT (2001): Neuroimaging of cerebral activations and deactivations associated with hypercapnia and hunger for air. Proc Natl Acad Sci USA 98:2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchner NJ, Quack I, Woznowski M, Stahle C, Wenzel U, Rump LC (2011): Microvascular endothelial dysfunction in obstructive sleep apnea is caused by oxidative stress and improved by continuous positive airway pressure therapy. Respiration 82:409–417. [DOI] [PubMed] [Google Scholar]
- Canessa N, Castronovo V, Cappa SF, Aloia MS, Marelli S, Falini A, Alemanno F, Ferini‐Strambi L (2011): Obstructive sleep apnea: Brain structural changes and neurocognitive function before and after treatment. Am J Respir Crit Care Med 183:1419–1426. [DOI] [PubMed] [Google Scholar]
- Castronovo V, Scifo P, Castellano A, Aloia MS, Iadanza A, Marelli S, Cappa SF, Strambi LF, Falini A (2014): White matter integrity in obstructive sleep apnea before and after treatment. Sleep 37:1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee MW, Choo WC (2004): Functional imaging of working memory after 24 hr of total sleep deprivation. J Neurosci 24:4560–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HL, Lu CH, Lin HC, Chen PC, Chou KH, Lin WM, Tsai NW, Su YJ, Friedman M Lin CP, others (2015): White matter damage and systemic inflammation in obstructive sleep apnea. Sleep 38:361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin K, Nakamura T, Shimizu K, Mishima M, Nakamura T, Miyasaka M, Ohi M (2000): Effects of nasal continuous positive airway pressure on soluble cell adhesion molecules in patients with obstructive sleep apnea syndrome. Am J Med 109:562–567. [DOI] [PubMed] [Google Scholar]
- Choi DW, Rothman SM (1990): The role of glutamate neurotoxicity in hypoxic‐ischemic neuronal death. Annu Rev Neurosci 13:171–182. [DOI] [PubMed] [Google Scholar]
- Chung MK, Worsley KJ, Paus T, Cherif C, Collins DL, Giedd JN, Rapoport JL, Evans AC (2001): A unified statistical approach to deformation‐based morphometry. Neuroimage 14:595–606. [DOI] [PubMed] [Google Scholar]
- Chung MK, Worsley KJ, Nacewicz BM, Dalton KM, Davidson RJ (2010): General multivariate linear modeling of surface shapes using SurfStat. Neuroimage 53:491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar D, Underhill SM, Goldberg MP (2003): Oligodendrocytes and ischemic brain injury. J Cereb Blood Flow Metab 23:263–274. [DOI] [PubMed] [Google Scholar]
- Ferini‐Strambi L, Baietto C, Di Gioia MR, Castaldi P, Castronovo C, Zucconi M, Cappa SF (2003): Cognitive dysfunction in patients with obstructive sleep apnea (OSA): Partial reversibility after continuous positive airway pressure (CPAP). Brain Res Bull 61:87–92. [DOI] [PubMed] [Google Scholar]
- Galliano E, Mazzarello P, D'Angelo E (2010): Discovery and rediscoveries of Golgi cells. J Physiol 588:3639–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good CD, Johnsrude IS, Ashburner J, Henson RN, Friston KJ, Frackowiak RS (2001): A voxel‐based morphometric study of ageing in 465 normal adult human brains. Neuroimage 14:21–36. [DOI] [PubMed] [Google Scholar]
- Gozal D, Daniel JM, Dohanich GP (2001a): Behavioral and anatomical correlates of chronic episodic hypoxia during sleep in the rat. J Neurosci 21:2442–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal E, Row BW, Schurr A, Gozal D (2001b): Developmental differences in cortical and hippocampal vulnerability to intermittent hypoxia in the rat. Neurosci Lett 305:197–201. [DOI] [PubMed] [Google Scholar]
- Guzman‐Marin R, Suntsova N, Bashir T, Nienhuis R, Szymusiak R, McGinty D (2008): Rapid eye movement sleep deprivation contributes to reduction of neurogenesis in the hippocampal dentate gyrus of the adult rat. Sleep 31:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison Y, Horne JA (2000): The impact of sleep deprivation on decision making: A review. J Exp Psychol Appl 6:236–249. [DOI] [PubMed] [Google Scholar]
- Huang BY, Castillo M (2008): Hypoxic‐ischemic brain injury: Imaging findings from birth to adulthood. Radiographics 28:417–439; quiz 617. [DOI] [PubMed] [Google Scholar]
- Jelic S, Le Jemtel TH (2008): Inflammation, oxidative stress, and the vascular endothelium in obstructive sleep apnea. Trends Cardiovasc Med 18:253–260. [DOI] [PubMed] [Google Scholar]
- Jones SG, Riedner BA, Smith RF, Ferrarelli F, Tononi G, Davidson RJ, Benca RM (2014): Regional reductions in sleep electroencephalography power in obstructive sleep apnea: A high‐density EEG study. Sleep 37:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo EY, Tae WS, Lee MJ, Kang JW, Park HS, Lee JY, Suh M, Hong SB (2010): Reduced brain gray matter concentration in patients with obstructive sleep apnea syndrome. Sleep 33:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo EY, Jeon S, Kim ST, Lee JM, Hong SB (2013): Localized cortical thinning in patients with obstructive sleep apnea syndrome. Sleep 36:1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo EY, Kim H, Suh S, Hong SB (2014): Hippocampal substructural vulnerability to sleep disturbance and cognitive impairment in patients with chronic primary insomnia: Magnetic resonance imaging morphometry. Sleep 37:1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, Le Charpentier T, Josserand J, Ali C Vivien D, GL Collingridge, A Lombet, L Issa, F Rene, JP Loeffler, A Kavelaars, C Verney, J Mantz, P Gressens. (2012): Activation of microglial N‐methyl‐d‐aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol 72:536–549. [DOI] [PubMed] [Google Scholar]
- Kingshott RN, Vennelle M, Hoy CJ, Engleman HM, Deary IJ, Douglas NJ (2000): Predictors of improvements in daytime function outcomes with CPAP therapy. Am J Respir Crit Care Med 161:866–871. [DOI] [PubMed] [Google Scholar]
- Kitamura T, Saitoh Y, Takashima N, Murayama A, Niibori Y, Ageta H, Sekiguchi M, Sugiyama H, Inokuchi K (2009): Adult neurogenesis modulates the hippocampus‐dependent period of associative fear memory. Cell 139:814–827. [DOI] [PubMed] [Google Scholar]
- Kumar R, Chavez AS, Macey PM, Woo MA, Yan‐Go FL, Harper RM (2012): Altered global and regional brain mean diffusivity in patients with obstructive sleep apnea. J Neurosci Res 90:2043–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau EY, Eskes GA, Morrison DL, Rajda M, Spurr KF (2010): Executive function in patients with obstructive sleep apnea treated with continuous positive airway pressure. J Int Neuropsychol Soc 16:1077–1088. [DOI] [PubMed] [Google Scholar]
- Lim DC, Pack AI (2014): Obstructive sleep apnea and cognitive impairment: Addressing the blood‐brain barrier. Sleep Med Rev 18:35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macey PM, Henderson LA, Macey KE, Alger JR, Frysinger RC, Woo MA, Harper RK, Yan‐Go FL, Harper RM (2002): Brain morphology associated with obstructive sleep apnea. Am J Respir Crit Care Med 166:1382–1387. [DOI] [PubMed] [Google Scholar]
- Macey PM, Kumar R, Woo MA, Valladares EM, Yan‐Go FL, Harper RM (2008): Brain structural changes in obstructive sleep apnea. Sleep 31:967–977. [PMC free article] [PubMed] [Google Scholar]
- Marks JD, Frysinger RC, Harper RM (1987): State‐dependent respiratory depression elicited by stimulation of the orbital frontal cortex. Exp Neurol 95:714–729. [DOI] [PubMed] [Google Scholar]
- Miller DW, Cookson MR, Dickson DW (2004): Glial cell inclusions and the pathogenesis of neurodegenerative diseases. Neuron Glia Biol 1:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming GL, Song H (2011): Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 70:687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirescu C, Gould E (2006): Stress and adult neurogenesis. Hippocampus 16:233–238. [DOI] [PubMed] [Google Scholar]
- Monsell S (2003): Task switching. Trends Cogn Sci 7:134–140. [DOI] [PubMed] [Google Scholar]
- Morrell MJ, Jackson ML, Twigg GL, Ghiassi R, McRobbie DW, Quest RA, Pardoe H, Pell GS, Abbott DF Rochford PD, GD Jackson, RJ Pierce, FJ O'Donoghue, DR Corfield. (2010): Changes in brain morphology in patients with obstructive sleep apnoea. Thorax 65:908–914. [DOI] [PubMed] [Google Scholar]
- Noh HJ, Joo EY, Kim ST, Yoon SM, Koo DL, Kim D, Lee GH, Hong SB (2012): The relationship between hippocampal volume and cognition in patients with chronic primary insomnia. J Clin Neurol 8:130–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donoghue FJ, Briellmann RS, Rochford PD, Abbott DF, Pell GS, Chan CH, Tarquinio N, Jackson GD, Pierce RJ (2005): Cerebral structural changes in severe obstructive sleep apnea. Am J Respir Crit Care Med 171:1185–1190. [DOI] [PubMed] [Google Scholar]
- O'Donoghue FJ, Wellard RM, Rochford PD, Dawson A, Barnes M, Ruehland WR, Jackson ML, Howard ME, Pierce RJ, Jackson GD (2012): Magnetic resonance spectroscopy and neurocognitive dysfunction in obstructive sleep apnea before and after CPAP treatment. Sleep 35:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olaithe M, Bucks RS (2013): Executive dysfunction in OSA before and after treatment: A meta‐analysis. Sleep 36:1297–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips CL, Yang Q, Williams A, Roth M, Yee BJ, Hedner JA, Berend N, Grunstein RR (2007): The effect of short‐term withdrawal from continuous positive airway pressure therapy on sympathetic activity and markers of vascular inflammation in subjects with obstructive sleep apnoea. J Sleep Res 16:217–225. [DOI] [PubMed] [Google Scholar]
- Prilipko O, Huynh N, Schwartz S, Tantrakul V, Kim JH, Peralta AR, Kushida C, Paiva T, Guilleminault C (2011): Task positive and default mode networks during a parametric working memory task in obstructive sleep apnea patients and healthy controls. Sleep 34:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig I, Kempton MJ, Crum WR, Glasser M, Milosevic M, Beniczky S, Corfield DR, Williams SC, Morrell MJ (2013): Hippocampal hypertrophy and sleep apnea: A role for the ischemic preconditioning? PLoS One 8:e83173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto Y, Matsuda H, Kamiya K, Maikusa N, Nakata Y, Ito K, Ota M, Matsunaga N, Sato N (2013): In vivo evaluation of gray and white matter volume loss in the Parkinsonian variant of multiple system atrophy using SPM8 plus DARTEL for VBM. Neuroimage Clin 2:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota S, Inoue Y, Takekawa H, Kotajima M, Nakajyo M, Usui C, Yoshioka Y, Koga T, Takahashi K (2014): Effect of continuous positive airway pressure on regional cerebral blood flow during wakefulness in obstructive sleep apnea. Sleep Breath 18:289–295. [DOI] [PubMed] [Google Scholar]
- Sled JG, Zijdenbos AP, Evans AC (1998): A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 17:87–97. [DOI] [PubMed] [Google Scholar]
- Torelli F, Moscufo N, Garreffa G, Placidi F, Romigi A, Zannino S, Bozzali M, Fasano F, Giulietti G, Djonlagic I, et al. (2011): Cognitive profile and brain morphological changes in obstructive sleep apnea. Neuroimage 54:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosun D, Dabbs K, Caplan R, Siddarth P, Toga A, Seidenberg M, Hermann B (2011): Deformation‐based morphometry of prospective neurodevelopmental changes in new onset paediatric epilepsy. Brain 134:1003–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vennelle M, White S, Riha RL, Mackay TW, Engleman HM, Douglas NJ (2010): Randomized controlled trial of variable‐pressure versus fixed‐pressure continuous positive airway pressure (CPAP) treatment for patients with obstructive sleep apnea/hypopnea syndrome (OSAHS). Sleep 33:267–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng HH, Tsai YH, Chen CF, Lin YC, Yang CT, Tsai YH, Yang CY (2014): Mapping gray matter reductions in obstructive sleep apnea: An activation likelihood estimation meta‐analysis. Sleep 37:167–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsley K, Taylor JE, Carbonell F, Chung MK, Duerden EG, Bernhardt B, Lyttelton O, Boucher M, Evans AC (2009): SurfStat: A Matlab toolbox for the statistical analysis of univariate and multivariate surface and volumetric data using linear mixed effects models and random field theory. Neuroimage 47:451 SA‐AM. 19409500 [Google Scholar]
- Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, Hirano T, Adachi M (2003): Elevated levels of C‐reactive protein and interleukin‐6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation 107:1129–1134. [DOI] [PubMed] [Google Scholar]
- Young T, Peppard PE, Gottlieb DJ (2002): Epidemiology of obstructive sleep apnea: A population health perspective. Am J Respir Crit Care Med 165:1217–1239. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang D, Qin W, Li Q, Chen B, Zhang Y, Yu C (2013): Altered restingstate brain activity in obstructive sleep apnea. Sleep 36:651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]