

Abstract
Reactivated androgen receptor (AR) signaling drives castration-resistant prostate cancer (CRPC). Novel AR targeting drugs abiraterone and enzalutamide have improved survival of CRPC patients. However, resistance to these agents develops and patients ultimately succumb to CRPC. Potential mechanisms of resistance include the following: 1) Expression of AR splice variants such as the AR-V7 isoform which lacks the ligand-binding domain, 2) AR missense mutations in the ligand-binding domain, such as F876L and T877A, and 3) Mutation or overexpression of androgen biosynthetic enzymes or glucocorticoid receptor. Several novel agents may overcome resistance mechanisms. Galeterone acts through multiple mechanisms that include degradation of AR protein and is being evaluated in CRPC patients positive for AR-V7. EPI-001 and related compounds inhibit AR splice variants by targeting the N-terminal transactivation domain of AR. Promising therapies and novel biomarkers, such as AR-V7, may lead to improved outcomes for CRPC patients.
Keywords: castration-resistant prostate cancer, androgen receptor, resistance, enzalutamide, abiraterone, galeterone, EPI-001
Introduction
In the United States, prostate cancer is the most commonly diagnosed malignancy, and is the second leading cause of cancer-related death among men. It is estimated that 220,800 new cases will be diagnosed, and 27,540 deaths caused by prostate cancer will occur in 2015.1 For over seven decades, surgical or medical androgen deprivation therapy (ADT) has been the primary treatment paradigm for men with advanced prostate cancer (PC).2 Surgical ADT is achieved through bilateral orchiectomy, while medical ADT may be achieved through the use of luteinizing hormone-releasing hormone (LH-RH) agonists or LH-RH antagonists, and both surgical and medical ADT have been shown to successfully palliate symptoms associated with metastatic PC.
While surgical and medical ADT is initially effective at stabilizing or causing disease regression in most patients, their effects are transient and virtually all patients develop disease progression to a stage referred to as castration-resistant PC (CRPC). Ultimately, metastatic CRPC (mCRPC) remains incurable, and traditionally patients had been resigned to receive traditional cytotoxic chemotherapeutics, such as mitoxantrone or docetaxel. However, during the past decade the number and types of viable treatment options for patients with mCRPC have expanded to include the immunotherapeutic sipuleucel-T for asymptomatic or minimally symptomatic mCRPC patients,3 the semi-synthetic taxane-derivative cabazitaxel designed to overcome docetaxel resistance,4 the α-emitting radiopharmaceutical radium-223 targeting bone metastases,5 and finally the bone-modifying agent denosumab that prevents or delays clinical sequelae associated with bone metastases.6, 7
Historically, CRPC had been considered “androgen-independent” or “hormone-refractory”; however, recent preclinical and clinical data have elucidated that CRPC remains highly dependent on the AR signaling axis in the castrate host.8, 9 The development and subsequent approval of two second generation AR axis targeting agents, abiraterone and enzalutamide, by the U.S. Food and Drug Administration (FDA) have confirmed the central role of the AR signaling axis on CRPC pathophysiology.10-13 While the treatment landscape for patients with CRPC has dramatically changed and now includes two approved agents that effectively target AR signaling, acquired resistance to these two agents limits treatment durability, and eventually the disease will become lethal to virtually all mCRPC patients.
The review will discuss the two approved second generation AR-targeting agents, highlight the most plausible mechanisms that lead to drug resistance in these two AR-targeting medications, and review the latest literature regarding novel agents in development that may overcome these resistance mechanisms related to the AR signaling axis (e.g. galeterone and EPI-001).
The Role of Androgen Receptor in Prostate Cancer
The human androgen receptor (AR) is 110 kD protein comprised of approximately 919 amino acids that is encoded by the AR gene (AR). AR is more than 90 kb long, is comprised of eight exons, and is located on the X chromosome at Xq11-12. The AR protein contains several functional domains that include the N-terminal transactivation domain (NTD) that is critical for engaging the cellular transcription complex, the DNA-binding domain (DBD) that directs the binding of AR protein to specific DNA sequences, the hinge region encoding the nuclear translocation signal, and the ligand-binding domain (LBD) that binds the androgen ligands. The NTD is encoded in exon 1, the DBD is encoded in exons 2 and 3, the hinge region is encoded in exon 4, and the LBD is encoded in exons 5-8 (Figure 1).14
Figure 1. The full-length androgen receptor compared with the AR-V7 splice variant.
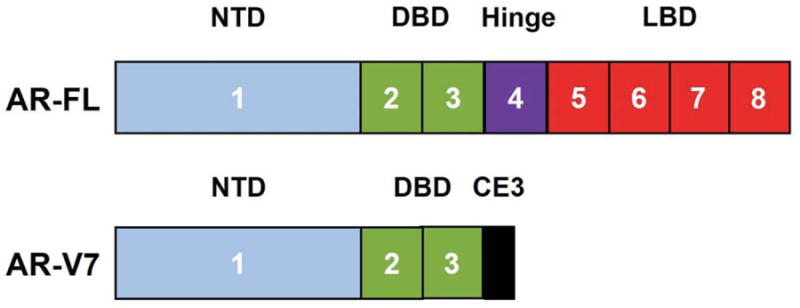
The AR gene is comprised of nine exons. The full length AR protein contains the N-terminal transactivation domain (encoded in exon 1) that is critical for engaging the cellular transcription complex, the DNA binding domain (encoded in exons 2-3) that directs the binding of AR protein to specific DNA sequences, the hinge region (encoded in exon 4) encoding the nuclear translocation signal, and the ligand-binding domain (encoded in exons 5-8) that binds the androgen ligands. The AR-V7 splice variant is produced by alternate splicing of the AR gene that leads to the addition of cryptic exon 3. This leads to premature termination of the AR protein, which results in the loss of the hinge region and LBD and the formation of truncated androgen receptor. AR-V7 is constitutively localized to the nucleus and binds DNA and promotes transcription of target genes without the need for androgen ligands. Therefore, AR-V7 is not inhibited by agents such as abiraterone or enzalutamide that targets the ligand-binding domain of AR. Each number represents the corresponding exon in the AR. Abbreviations: AR-FL, full length androgen receptor; AR-V7, androgen receptor splice variant V7; CE3, cryptic exon 3; DBD, DNA binding domain; LBD, ligand-binding domain; NTD, N-terminal transactivation domain.
The AR is a ligand-dependent transcription factor that is part of the nuclear receptor superfamily. Its primary role is to respond to androgenic steroid hormones, such as testosterone and dihydrotestosterone (DHT). In the absence of one of these androgenic ligands, the AR is sequestered in the cytoplasm bound to chaperone proteins (e.g. HSP90) where it is inactive, yet in a conformation that possesses high affinity for ligand binding.15 Upon androgen binding to the LBD of the AR, the receptor disassociates from the chaperone complex, and then translocates into the nucleus where it dimerizes with a second AR and binds to androgen response elements in cis-regulatory regions to regulate transcription of androgen-dependent target genes (e.g. KLK3, which encodes for prostate specific antigen [PSA]) (Figure 2).15, 16 Transcriptional regulation of these target genes, through persistent AR signaling, contributes to PC proliferation and survival.
Figure 2. Schematic of inhibition of AR signaling axis by AR targeting agents.
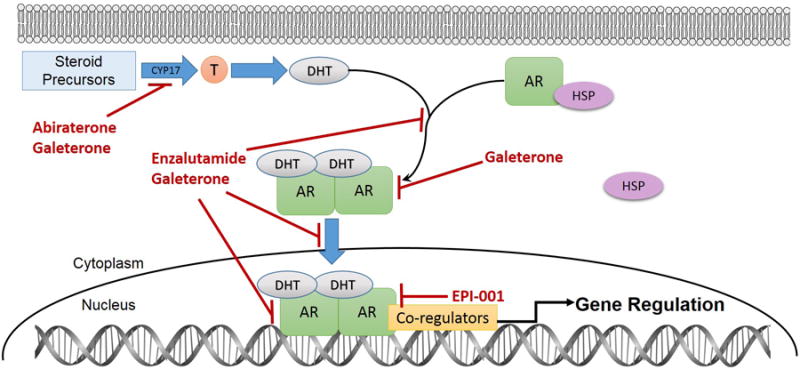
In CRPC tumor cells, the CYP17A1 enzyme is required for production of testosterone and DHT from precursors. Abiraterone is a selective and irreversible inhibitor of intratumoral androgen biosynthesis by potently blocking CYP17A1-mediated production of testosterone and DHT, which limits the amount of available ligand for AR axis signaling. Enzalutamide is a second generation antiandrogen that binds to the AR LBD and more potently antagonizes the AR than first generation antiandrogens. It also inhibits AR complex-mediated transcription by preventing AR translocation into the cell nucleus and binding to DNA. Galeterone is a novel agent with tri-modal mechanism of action. It is similar to abiraterone because it inhibits CYP17A1 to prevent intratumoral androgen synthesis, but is also similar to enzalutamide because it is an AR antagonist. Additionally, it also degrades the AR and decreases AR protein levels. EPI-001 is a novel AR-targeting agent that reduces AR transcriptional activity. It inhibits transactivation of the AR NTD through inhibition of protein-protein interactions of AR and co-regulators, thereby blocking induction of androgen target genes Abbreviations: AR, androgen receptor; CYP17A1, cytochrome P450 c17; DHT, dihydrotestosterone; DNA, deoxyribonucleic acid; HSP, heat shock protein; LBD, ligand-binding domain; NTD, N-terminal transactivation domain; T, testosterone.
ADT is initially effective in the majority of PC patients through suppression of gonadal testosterone production. Reduction of circulating serum testosterone to castrate levels ultimately renders the AR transcriptionally inactive. As a result, the AR no longer activates androgen-dependent target genes that drive the PC viability and proliferation. However, in the context of metastatic PC, the positive effects of ADT on AR signaling are temporary, and patients progress on ADT within approximately 18-30 months.17 Reactivation of the AR ultimately leads to a CRPC phenotype in virtually all patients where serum PSA levels rise and/or there is evidence of disease progression, despite effective suppression of testosterone below castrate levels (≤50 ng/dL). Several mechanisms have been proposed to explain how the AR is reactivated and leads to CRPC. These mechanisms include: 1) AR gene overexpression (with or without gene amplification) that results in the increased protein level and hypersensitization to low concentrations of androgens, 2) AR point mutations that lead to promiscuous activation of AR in response to atypical ligands such as adrenal androgens, other steroid hormones, or antiandrogen drugs, 3) de novo intratumoral synthesis of androgens, and 4) expression of constitutively active AR splice variants that lack the LBD.15, 18, 19
Next Generation Androgen Receptor Targeting Agents
Antiandrogen agents have been developed to inhibit DHT and testosterone binding to the AR, thus diminishing the ability of the AR to exert transcriptional control over target genes responsible for PC viability and proliferation. First generation antiandrogen medications (e.g. bicalutamide, flutamide, and nilutamide) competitively inhibit androgenic ligands (e.g. testosterone and DHT) from binding to the AR. In the context of advanced CRPC, these agents provide only modest, temporary clinical benefit.20 Bicalutamide (the most commonly used first generation antiandrogen) monotherapy is inferior to ADT,21 and as part of a combined androgen blockade (CAB) paradigm, a meta-analysis of CAB trials revealed only a modest survival benefit (approximately 2% at 5 years).22 Moreover, at the molecular level, first generation antiandrogens have actually been shown to have AR agonist activity in CRPC cells where AR protein has been overexpressed.23, 24 In response to the knowledge that CRPC remains dependent on androgens and AR signaling, as well as the shortcomings of first generation antiandrogens, two second generation AR-targeting agents have been recently approved by the FDA for the treatment of mCRPC patients.
Abiraterone acetate (the prodrug of abiraterone) is a selective, irreversible inhibitor of intratumoral androgen biosynthesis by potently blocking the cytochrome P450 c17 (CYP17A1). CYP17A1 is an enzyme with 17α-hydroxylase and C17,20-lyase activity central to androgen biosynthesis, and is key in the conversion of pregnenolone to dehydroepiandrosterone (DHEA) (Figure 2).25 DHEA is an important upstream precursor of DHT and testosterone, and thus inhibiting its production correspondingly reduces the amount of ligand available to stimulate AR signaling. Preclinically, abiraterone has been shown to be a potent inhibitor of both 17α-hydroxylase and C17,20-lyase.26 Results of an open-label observational study of 57 mCRPC patients revealed that abiraterone is capable of achieving sustained suppression of both circulating testosterone and testosterone in bone marrow aspirates infiltrated with metastatic tumor cells.27 Results from several Phase I/II trials demonstrated that abiraterone is effective and safe with or without corticosteroids (although symptoms associated with secondary mineralocorticoid excess were higher in patients not concomitantly administered a corticosteroid), that there was a ≥50% PSA decline in the majority of patients, and that abiraterone was efficacious in men with mCRPC who had prior exposure to ketoconazole (a weak CYP17A1 inhibitor).28-31 Based on results from these phase I/II trials, the double-blinded, placebo-controlled phase III COU-AA-301 trial was conducted in men with mCRPC who had previously been treated with chemotherapy (n=1195).10 The primary endpoint for COU-AA-301 was overall survival (OS). In the abiraterone-treated patients, there was a 35% reduction in the risk of death (hazard ratio [HR], 0.65; 95% confidence interval [CI], 0.54 to 0.77; p<0.001) with a 3.9 month increased median OS, when compared to placebo (14.8 versus 10.9 months). Abiraterone was also shown to be superior to placebo for all secondary endpoints, including: radiographic progression free survival (PFS), time to PSA progression, and PSA response rate. Mineralocorticoid-related adverse events (e.g. fluid retention, hypertension, and hypokalemia) were more common among patients treated with abiraterone. Based on the COU-AA-301 results, in 2011 the U.S. FDA approved abiraterone as a second-line treatment option for mCRPC patients after traditional cytotoxic chemotherapy with docetaxel. A second randomized, double-blinded, placebo-controlled phase III trial (COU-AA-302) was conducted in chemotherapy-naive mCRPC patients (n=1088).11 The co-primary endpoints for COU-AA-302 were OS and radiographic PFS. In the abiraterone-treated patients, there was a 25% reduction in the risk of death (HR, 0.75; 95% CI, 0.61 to 0.93; p=0.01) and a 47% reduced risk of progression in patients treated with abiraterone (HR, 0.53; 95% CI, 0.45 to 0.62; p<0.001), with an 8.3 month increased median PFS (16.5 versus 8.2 months), when compared to placebo. Abiraterone also showed superiority among all secondary endpoints, including: time to initiation of cytotoxic chemotherapy, opiate use for cancer-related pain, PSA progression, and decline in performance status. Again, mineralocorticoid-mediated toxicities were significantly more common among the patients treated with abiraterone. In the updated final analysis of COU-AA-302, the abiraterone-treated patients demonstrated a statistically significant improvement in OS (34.7 versus 30.3 months in the placebo group).32 Based on the COU-AA-302 results, in 2012 the U.S. FDA approved abiraterone as a first-line treatment option for mCRPC.
Enzalutamide (formerly known as MDV3100) was developed to overcome the resistance to first generation antiandrogens in CRPC. Unlike first generation antiandrogens, enzalutamide has been shown to be a pure antagonist without also possessing agonist characteristics in prostate cancer cells with overexpressed AR.33 Enzalutamide binds to the AR LBD and more potently antagonizes the receptor than first generation antiandrogens. It also inhibits AR complex-mediated transcription by preventing AR translocation into the cell nucleus, recruitment of AR cofactors, and binding to DNA (Figure 2). Results from a phase I/II trial of patients with progressive mCRPC (n=140) demonstrated several anti-tumor effects at all doses investigated.34 Investigators noted a ≥50% PSA decline and stabilized bone disease in 56% of patients, decreased circulating tumor cells (CTCs) in 40%, and responses in soft-tissue disease in 22% of patients. PSA declines were dose dependent from 30-150 mg, but plateaued between 150-240 mg.34 Additionally, another phase II trial of enzalutamide-treated mCRPC patients (n=60) provided the first clinical data supporting the hypothesis that the therapeutic benefit of enzalutamide can be attributed to AR inhibition manifested by re-localization of the nuclear AR to the cytoplasm.35 Based on results from the original phase I/II trial, the double-blinded, placebo-controlled phase III AFFIRM trial was conducted in men with mCRPC who had previously been treated with chemotherapy (n=1199).12 The primary end point of AFFIRM was OS. For patients treated with enzalutamide, median OS was 4.8 months longer (18.4 versus 13.6 months), and the risk of death was decreased by 37%, when compared to placebo (HR, 0.63; 95% CI, 0.53 to 0.75; p<0.001). Enzalutamide was also shown to be superior with regards to all secondary endpoints including: radiographic PFS time, the proportion of patients with a ≥50% PSA reduction, time to PSA progression, soft-tissue and quality of life response rates, and time to first skeletal-related event (SRE). Based on the AFFIRM results, in 2012 the U.S. FDA approved enzalutamide as a second-line treatment option for mCRPC patients after they received docetaxel. A second randomized, double-blinded, placebo-controlled phase III trial was conducted in chemotherapy-naıve mCRPC patients (n=1717).13 The co-primary endpoints for the PREVAIL study were OS and radiographic PFS. In the enzalutamide-treated patients, there was a 29% reduction in the risk of death (HR, 0.71; 95% CI, 0.60 to 0.84; p<0.001) with a 2.2 month increased median OS, when compared to placebo (32.4 versus 30.2 months). The rate of radiographic PFS at 12 months was also higher in enzalutamide-treated patients, when compared to placebo (65% versus 14%), with an 81% reduced risk of progression (HR, 0.19; 95% CI 0.15 to 0.23; p<0.001). Enzalutamide also showed superiority among all secondary endpoints including: time to initiation of cytotoxic chemotherapy, time to first SRE, rate of a complete or partial soft-tissue response, time to PSA progression, and a rate of decline of at least 50% in PSA. Based on the PREVAIL results, in 2014 the U.S. FDA approved enzalutamide as a first-line treatment option for mCRPC.
Combined, these studies provide evidence that both enzalutamide and abiraterone are clinically effective by suppressing the reactivated AR signaling axis, which is central to CRPC pathophysiology and progression.
Mechanisms of Resistance to Approved Androgen Receptor Directed Drugs
Although abiraterone and enzalutamide represent a major conceptual and clinical advancement in the treatment of mCRPC, approximately 20-40% of patients present with primary resistance to these agents (e.g. no initial PSA response).10-12, 34 But perhaps just as importantly, patients who experience an initial PSA response after treatment with either abiraterone or enzalutamide will eventually develop secondary resistance to the drug.36 Despite distinct mechanisms by which the AR signaling is inhibited, there may be cross-resistance between these two drugs.37, 38 This suggests a possibility that there may be a common mechanism of resistance and limits the clinical options for patients with progressive disease on abiraterone or enzalutamide. However, other negative prognostic features (e.g. more advanced disease, greater tumor burden, and more symptoms) were observed more frequently in enzalutamide-refractory patients who received abiraterone, when compared to the original phase III abiraterone trial, and could conceivably account for the modest response and a shorter PFS that was observed.10, 38 Therefore, cross-resistance mechanisms of abiraterone and enzalutamide require additional preclinical and clinical validation.
While multiple mechanisms of acquired resistance to enzalutamide and abiraterone have been proposed,39 substantial clinical evidence has emerged for the role of AR splice variants (AR-V's) as mediators of resistance to abiraterone and enzalutamide. AR-V's are commonly truncated versions of the AR, and have lost their C-terminal LBD due to alternative splicing of AR mRNA.40 Over 20 AR-V isoforms have been identified in vitro in cell lines and most AR-V mRNA species retain exons 1-3 encoding the AR NTD and DBD domains. In many AR-V's, aberrant splicing of the AR mRNA leads to the addition of one of small “cryptic exons” after exon 3 and premature termination of the AR protein. For example, AR splice variant-7 (AR-V7) contains cryptic exon 3 (Figure 1).41 As a result of the loss of the C-terminal LBD, AR-V's are constitutively active without androgenic ligands present and localize to the nucleus and promote transcription of target genes.42 AR-V7 has emerged as a putative variant that underlies clinical resistance to enzalutamide and abiraterone. In preclinical xenograft models, it was shown that abiraterone and enzalutamide induced AR-V7 expression.43, 44 A recent CTC assay, capable of detecting AR-V7 mRNA expression, was used to prospectively evaluate AR-V7 in mCRPC patients treated with either enzalutamide or abiraterone (n=62).36 In this study, 31% of the enzalutamide-treated patients and 19% of the abiraterone-treated patients had detectable AR-V7 in CTCs. Among the enzalutamide-treated patients, AR-V7-positive patients experienced significantly lower PSA response rates (0% versus 53%, p=0.004), and achieved significantly shorter OS (median 5.5 months versus not reached, p=0.002), PSA PFS (median 1.4 versus 6.0 months, p<0.001), and radiographic PFS (median 2.1 versus 6.1 months, p<0.001), when compared to AR-V7-negative patients. Similarly, among the abiraterone-treated patients, AR-V7-positive patients experienced significantly lower PSA response rates (0% versus 68%, p=0.004), and achieved significantly shorter OS (median 10.6 months versus not reached, p=0.006), PSA PFS (median 1.3 months versus not reached, p<0.001), and radiographic PFS (median 2.3 versus not reached, p<0.001), when compared to AR-V7-negative patients. These data suggest that for patients treated with enzalutamide and/or abiraterone, the AR-V7 is likely predictive of resistance to both agents and associated with negative clinical outcomes. While these findings require independent confirmation in a prospective cohort, they provide plausible mechanisms by which tumor cells escape from selective pressures of potent AR targeted therapy and why cross-resistance between abiraterone and enzalutamide exists. Interestingly, serial measurements of AR-V7 revealed that some patients converted from the AR-V7 negative to the AR-V7 positive status with both AR-targeted therapy and cytotoxic chemotherapy. However, loss of AR-V7 expression only occurred with taxane chemotherapy.45 Moreover, data from a separate study suggests that patients with detectable AR-V7 in CTCs may benefit more from taxane treatment, when compared to either abiraterone or enzalutamide treatment.46 Among the AR-V7 positive patients, PSA responses were higher in those treated with a taxane than in patients treated with an AR-targeting agent (41% vs 0%; p<0.001). In addition, PSA PFS (HR, 0.19; 95%CI, 0.07 to 0.52; p=0.001) and radiologic PFS (HR, 0.21; 95%CI, 0.07 to 0.59; p=0.003) were both significantly longer in patients treated with a taxane. Interestingly, there were no observed differences in treatment efficacy between the taxanes and AR-targeting agents among the patients with undetectable AR-V7; however, this was a small study and not sufficiently powered to detect differences in efficacy among the AR-V7 positive patients. These data suggest that the AR-V7 assay may have the potential to become a clinically useful biomarker in directing the choice of therapy for CRPC patients.
Somatic point mutations in the AR gene have been implicated as etiologies underlying resistance to first generation AR-targeting medications.47 Missense mutations in the AR LBD can cause reduced specificity of binding between the AR and its ligands.9, 48 These somatic point mutations allow AR activation in response to other hormones (e.g. progesterone), and can affect coregulatory recruitment.48 One mutation, which causes a phenylalanine to leucine substitution at amino acid 876 (F876L) has been shown to convert enzalutamide into a partial agonist in prostate cancer cell lines.49-51 Moreover, one study also showed that this point mutation occurs spontaneously in cells treated with enzalutamide, which suggests that in vivo this could be an important mechanism that explains secondary resistance to enzalutamide.50 In support of this idea, the F876L mutation was detected in circulating tumor DNA of patients treated with enzalutamide or ARN-509, a novel AR antagonist similar to enzalutamide.49, 52 Another mutation, which causes a threonine to alanine substitution at amino acid 877 (T877A), is a gain of function mutation. T877A can be activated by antiandrogens (e.g. flutamide), but also by steroid hormones (e.g. progesterone) that are precursors of androgen synthesis. It was also shown that when CYP17A1 is effectively inhibited (e.g. by abiraterone), intracellular progesterone levels increase and drive transcription of target genes in cells expressing the AR T877A mutant. Ultimately, this results in clones able to overcome abiraterone inhibition.53 One study demonstrated that the T877A mutant (referred to as T878A in this publication) was detected in 3 of 18 CRPC cases after abiraterone treatment.54 Another study showed that AR point mutants H874Y and T877A were detected in circulating cell-free DNA of 7 out of 29 patients resistant to abiraterone. This study also documented the high frequency (53%) of AR amplification occurring in cell-free DNA of patients progressing on enzalutamide, in comparison to abiraterone or other treatments.52 Accumulating data point to the emergence of AR point mutations as a mechanism of resistance to enzalutamide and abiraterone, and it remains to be shown whether the spectrum of mutations may be different for enzalutamide versus abiraterone. Availability of noninvasive assays of CTCs or plasma DNA using high-throughput sequencing technology may lead to greater precision in determining the mechanism of resistance and allow for selection of more effective therapy for individual patients.
Preclinical studies of xenograft tumors have yielded additional hypotheses regarding mechanisms of resistance. Glucocorticoid receptor may bypass the need for AR, and in the context of potent AR inhibition, the glucocorticoid receptor is capable of activating a subset of AR target genes and promoting tumor progression.55, 56 A gain-of-function mutation (N367T) in the enzyme 3β-hydroxysteroid dehydrogenase 1, involved in androgen biosynthesis, may allow increased synthesis of DHT from precursors and this mutation was found in a subset of xenograft tumors after exposure to abiraterone.57 Increased expression of the enzyme 17β-hydroxysteroid dehydrogenase (AKR1C3) was found in enzalutamide-resistant xenograft tumors.58 These data raise the possibility that an alternate steroid receptor or increased androgen biosynthesis may reactivate the AR signaling axis, or downstream targets, after exposure to abiraterone and enzalutamide.
Agents in Development that Overcome Secondary AR Resistance
Despite the availability of enzalutamide and abiraterone for CRPC patients, secondary resistance mechanisms inevitably result in clinical progression. Galeterone (formerly VN/124-1 or TOK-001) and EPI-001 are two examples of novel compounds in development that target the AR and attempt to overcome issues related to secondary resistance to enzalutamide and/or abiraterone. Other antiandrogens in development (ARN-509 and ODM-201) have mechanisms of action similar to enzalutamide,59 and it is unclear as to how effectively these agents overcome resistance to abiraterone and/or enzalutamide.
Galeterone (3β-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16,-diene) was originally developed as a CYP17A1 inhibitor, and is set to enter phase III clinical trials for CRPC.60 Galeterone is a novel AR-targeting agent with a tri-modal mechanism of action. Similar to abiraterone, galeterone selectively and irreversibly inhibits CYP17A1 and prevents intratumoral androgen synthesis. However, galeterone is pharmacologically distinct from abiraterone in its mechanisms of CYP17A1inhibition. While abiraterone has been shown to be a potent inhibitor of 17α-hydroxylase and C17,20-lyase, galeterone only inhibits the latter. Inhibition of C17,20-lyase effectively blocks the production of androgens, whereas inhibition of 17α-hydroxylase can lead to the overproduction of progesterone and pregnenalone and cause a secondary mineralocorticoid excess. Symptoms of mineralocorticoid overproduction include hypokalemia, hypertension and fluid retention, all of which are abrogated by prednisone therapy.10 Clinically, this distinction is important because, as a selective inhibitor of C17,20-lyase, galeterone (and other selective C17,20-lyase inhibitors, such as VT-46461, 62) may be able to block the production of androgens without causing symptoms of secondary mineralocorticoid excess and thereby spare patients from concomitant corticosteroid therapy.
Galeterone is also similar to enzalutamide because it is an AR antagonist and blocks androgenic ligand binding. However, galeterone is distinct from either of its predecessors because it also has the ability to degrade the AR and decrease AR levels (Figure 2).60, 63, 64 Additionally, galeterone has also been shown to impair AR binding to chromatin.64 ARMOR1 was a phase I dose escalation study that tested the safety of galeterone in chemotherapy naïve CRPC patients with either metastatic or non-metastatic disease (n=49). This study revealed that galeterone was safe at all doses, and demonstrated activity in CRPC.65 Results from the phase II ARMOR2 trial (ClinicalTrials.gov Identifier: NCT01709734) (n=52) revealed that 82% of patients with treatment-naïve CRPC had a 30% reduction in PSA, and 75% had a 50% reduction in PSA. Among ARMOR2 patients with abiraterone-refractory disease, 27% had reductions in PSA, and 13% had a 30% reduction in PSA. Six of 11 patients with treatment naïve CRPC had high expression of AR splice variants containing the N-terminus of the AR and lacking the C-terminus. Five of these patients experienced at least a 50% reduction in PSA, suggesting that galeterone could still be effective in the treatment of CRPC clones positive for AR splice variants.66 Moreover, preclinical data has revealed that galeterone is able to effectively degrade AR-V7 splice variant receptors.60 Mechanisms by which galeterone (which presumably interacts with AR in the LBD region) induces AR-V7 degradation remain to be elucidated. In this context, it should be noted that CTCs expressing AR-V7 expressed high levels of full-length AR concomitantly.36 AR-V7 and full-length AR form a complex as a heterodimer.67 Galeterone may target the full-length AR/AR-V7 complex for degradation, thereby effectively inhibiting the transcriptional activity of AR-V7. Because galeterone directly degrades the AR, including the AR-V7 splice variant, it may prove to be more effective than abiraterone and/or enzalutamide for AR-V7 positive patients. A randomized phase III trial (ARMOR3-SV) is planned to compare galeterone and enzalutamide in abiraterone or enzalutamide treatment-naïve mCRPC patients with AR-V7-positive circulating prostate cancer cells (ClinicalTrials.gov Identifier: NCT02438007). Upon completion of this trial, a separate trial of galeterone should be conducted in patients with detectable AR-V7 who have had previous exposure to an AR-targeting agent to determine the efficacy of galeterone in the context of secondary resistance. Preclinical research has also shown that galeterone is particularly effective against prostate cancer cells with the T877A AR mutation; however, this observation still requires further confirmation in human subjects.64
Unlike previous antiandrogen therapies, a novel compound EPI-001 targets AR axis signaling by blocking the AR NTD.68, 69 EPI-001 is a mixture of four stereoisomers and binds to the activation function-1 (AF-1) region of the AR NTD. Binding to the NTD is a unique mechanism in comparison to currently available AR-targeting agents because it can potentially bypass secondary resistance mechanisms associated with the loss of the AR LBD. EPI-001 has been shown to be an effective inhibitor of AR transcriptional activity and can inhibit transactivation of the AR NTD and block induction of androgen target genes. Through covalent binding of the NTD, EPI-001 reduced protein-protein interactions between AR and co-regulators p300/CBP, which are required for AR-mediated transactivation (Figure 2). Treatment with EPI-001 caused cytoreduction of CRPC xenografts dependent on AR for growth and survival without causing toxicity and reduced protein-protein interactions with the AR NTD.68, 69 EPI-001 has also been shown to have a specificity for blocking AR-dependent growth of prostate cancer cells, while having no effect on the proliferation of cells that are not dependent on AR signaling for growth.68 But most importantly, EPI-001 has been shown to inhibit the transcriptional activity of the ARV567es splice variant.68 The ARV567es (variant 5, 6, 7 exon skipped) splice variant, like AR-V7, does not possess the AR LBD due to loss of exon 5-7.70 Therefore, it stands to reason that EPI-001 (or agents with similar mechanisms of action, such as EPI-506) would be effective in inhibiting the transcriptional activity of all AR-V's, including AR-V7; however, preclinical and clinical studies are necessary to test this hypothesis. EPI-506 will be tested in a phase I/II trial of CRPC patients that is expected to be initiated in 2015.
Conclusions
Reactivation of the AR axis signaling, after initial ADT treatment, underlies progression to a CRPC phenotype for virtually all men diagnosed with PC. Better understanding of CRPC tumor biology, such as AR amplification/overexpression/alteration and intratumoral androgen synthesis, led to the introduction of two AR-targeting agents (abiraterone and enzalutamide) associated with increased survival and clinical benefit. However, the gain in survival is modest (3-4 months) and CRPC remains a terminal disease with a uniformly fatal outcome. Preclinical and clinical studies have revealed that several acquired resistance mechanisms result in AR pathway activation, including: AR splice variants lacking the LBD, missense point mutations in the AR, overexpression or mutation in androgen biosynthetic enzymes, and a glucocorticoid receptor that may possess the ability to bypass the AR. Clinical observation of increasing serum PSA levels as the early sign of treatment failure after abiraterone and enzalutamide is consistent with the idea that CRPC remains a disease driven by AR signaling. Therefore, efforts are underway to develop novel AR-targeting drugs that can overcome the development of resistance.
The AR-V7 splice variant may be the first predictive biomarker for patients with mCRPC that can be used to help inform clinicians regarding resistance and treatment.71 However, additional preclinical and clinical validation must be performed before AR-V7 can be implemented. First, independent and more robust clinical studies need to be performed to provide large-scale validation of previous clinical findings.36 Also, mechanistic data that provide the role of AR-V7 and full-length AR in drug resistance to abiraterone and/or enzalutamide are still needed. Collectively, these additional studies should help delineate whether AR-V7 is truly a predictive biomarker, or simply a marker of prognosis. The ARMOR3-SV trial undoubtedly will help elucidate the role of AR-V7 in primary resistance, but a second trial, which enrolls mCRPC patients who have progressed on abiraterone and/or enzalutamide, is necessary to test AR-V7 as a predictive biomarker.
Furthermore, prospective clinical validation of missense point mutations in the AR is essential towards elucidating their role in mCRPC, in acquired resistance to abiraterone and enzalutamide, and in the effectiveness of novel AR-targeting agents in development (e.g. galeterone and EPI-001/506). Moreover, clinical validation of these potentially predictive biomarkers may also aid in treatment selection for the subset of patients who will benefit from a particular novel AR-targeting agents once acquired resistance to abiraterone and/or enzalutamide has occurred. Finally, the elucidation of additional mechanisms of acquired resistance to AR targeting drugs should be explored, both preclinically and clinically, to identify and validate additional informative predictive biomarkers. Biomarkers interrogating enzymes involved in androgen biosynthesis (e.g. 3β-hydroxysteroid dehydrogenase 1) and glucocorticoid receptor-mediated expression of AR target genes in the presence of AR inhibition could conceivably provide clinicians with an even greater number of predictive biomarkers that could be used during the treatment selection process.
Clearly, there is an urgent need for the continued development and the U.S. FDA approval of novel AR-targeting agents that can be effective in the setting mCRPC with secondary resistance to AR axis signaling. Galeterone possesses many of the same mechanisms as both abiraterone and enzalutamide, but is distinct from its predecessors by its ability to degrade the AR and reduce AR levels. Galeterone has also been shown preclinically to be effective against clones with AR splice variants and missense point mutations. EPI-001 is a novel agent that targets the AR NTD, and is therefore not reliant on ligand binding to exert its effects on the AR. EPI-506 is currently entering clinical development. Collectively, this provides rationale for the continued development of these agents to be used in mCRPC patients with demonstrated secondary resistance to abiraterone and/or enzalutamide. Moreover, additional clinical trials will help provide insights into timing, sequencing, and novel combinations for the treatment of mCRPC.
Acknowledgments
The authors would like to thank Dr. Lana Crona for her help in reviewing and editing this review. D.J.C. was supported by T32GM086330 from the National Institute of General Medicine Sciences (NIGMS).
Y.E.W. received research funding from Astellas and Janssen and Tokai. M.I.M. received research funding from Janssen.
Footnotes
Conflicts of Interest Disclosure: D.J.C. stated no conflicts of interest.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Hellerstedt BA, Pienta KJ. The current state of hormonal therapy for prostate cancer. CA Cancer J Clin. 2002;52:154–79. doi: 10.3322/canjclin.52.3.154. [DOI] [PubMed] [Google Scholar]
- 3.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 4.de Bono JS, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet. 2010;376:1147–54. doi: 10.1016/S0140-6736(10)61389-X. [DOI] [PubMed] [Google Scholar]
- 5.Parker C, et al. Alpha emitter radium-223 and survival in metastatic prostate cancer. N Engl J Med. 2013;369:213–23. doi: 10.1056/NEJMoa1213755. [DOI] [PubMed] [Google Scholar]
- 6.Fizazi K, et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377:813–22. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith MR, et al. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 2012;379:39–46. doi: 10.1016/S0140-6736(11)61226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nelson PS. Molecular states underlying androgen receptor activation: a framework for therapeutics targeting androgen signaling in prostate cancer. J Clin Oncol. 2012;30:644–6. doi: 10.1200/JCO.2011.39.1300. [DOI] [PubMed] [Google Scholar]
- 9.Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–24. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Bono JS, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ryan CJ, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. doi: 10.1056/NEJMoa1209096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scher HI, et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–97. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 13.Beer TM, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–33. doi: 10.1056/NEJMoa1405095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gelmann EP. Molecular biology of the androgen receptor. J Clin Oncol. 2002;20:3001–15. doi: 10.1200/JCO.2002.10.018. [DOI] [PubMed] [Google Scholar]
- 15.Jentzmik F, Azoitei A, Zengerling F, Damjanoski I, Cronauer MV. Androgen receptor aberrations in the era of abiraterone and enzalutamide. World J Urol. 2015 doi: 10.1007/s00345-015-1624-2. [DOI] [PubMed] [Google Scholar]
- 16.van Royen ME, van Cappellen WA, de Vos C, Houtsmuller AB, Trapman J. Stepwise androgen receptor dimerization. J Cell Sci. 2012;125:1970–9. doi: 10.1242/jcs.096792. [DOI] [PubMed] [Google Scholar]
- 17.Oudard S. Progress in emerging therapies for advanced prostate cancer. Cancer Treat Rev. 2013;39:275–89. doi: 10.1016/j.ctrv.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Mitsiades N. A road map to comprehensive androgen receptor axis targeting for castration-resistant prostate cancer. Cancer Res. 2013;73:4599–605. doi: 10.1158/0008-5472.CAN-12-4414. [DOI] [PubMed] [Google Scholar]
- 19.Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- 20.Agarwal N, et al. Six-month progression-free survival as the primary endpoint to evaluate the activity of new agents as second-line therapy for advanced urothelial carcinoma. Clin Genitourin Cancer. 2014;12:130–7. doi: 10.1016/j.clgc.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bales GT, Chodak GW. A controlled trial of bicalutamide versus castration in patients with advanced prostate cancer. Urology. 1996;47 (1A Suppl):38–43. doi: 10.1016/s0090-4295(96)80007-0. discussion 48-53. [DOI] [PubMed] [Google Scholar]
- 22.Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Prostate Cancer Trialists' Collaborative Group. Lancet. 2000;355:1491–8. [PubMed] [Google Scholar]
- 23.Chen CD, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–91. doi: 10.1016/S1470-2045(09)70229-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Attard G, Reid AH, Olmos D, de Bono JS. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009;69:4937–40. doi: 10.1158/0008-5472.CAN-08-4531. [DOI] [PubMed] [Google Scholar]
- 26.Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38:2463–71. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 27.Efstathiou E, et al. Effects of abiraterone acetate on androgen signaling in castrate-resistant prostate cancer in bone. J Clin Oncol. 2012;30:637–43. doi: 10.1200/JCO.2010.33.7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Attard G, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 29.Attard G, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8. doi: 10.1200/JCO.2008.20.0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryan CJ, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–8. doi: 10.1200/JCO.2009.24.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim W, et al. Sequential use of the androgen synthesis inhibitors ketoconazole and abiraterone acetate in castration-resistant prostate cancer and the predictive value of circulating androgens. Clin Cancer Res. 2014;20:6269–76. doi: 10.1158/1078-0432.CCR-14-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ryan CJ, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16:152–60. doi: 10.1016/S1470-2045(14)71205-7. [DOI] [PubMed] [Google Scholar]
- 33.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–90. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scher HI, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–46. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Efstathiou E, et al. Molecular characterization of enzalutamide-treated bone metastatic castration-resistant prostate cancer. Eur Urol. 2015;67:53–60. doi: 10.1016/j.eururo.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Antonarakis ES, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–38. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrader AJ, et al. Enzalutamide in castration-resistant prostate cancer patients progressing after docetaxel and abiraterone. Eur Urol. 2014;65:30–6. doi: 10.1016/j.eururo.2013.06.042. [DOI] [PubMed] [Google Scholar]
- 38.Noonan KL, North S, Bitting RL, Armstrong AJ, Ellard SL, Chi KN. Clinical activity of abiraterone acetate in patients with metastatic castration-resistant prostate cancer progressing after enzalutamide. Ann Oncol. 2013;24:1802–7. doi: 10.1093/annonc/mdt138. [DOI] [PubMed] [Google Scholar]
- 39.Chism DD, De Silva D, Whang YE. Mechanisms of acquired resistance to androgen receptor targeting drugs in castration-resistant prostate cancer. Expert Rev Anticancer Ther. 2014;14:1369–78. doi: 10.1586/14737140.2014.928594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ware KE, Garcia-Blanco MA, Armstrong AJ, Dehm SM. Biologic and clinical significance of androgen receptor variants in castration resistant prostate cancer. Endocr Relat Cancer. 2014;21:T87–T103. doi: 10.1530/ERC-13-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu R, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan SC, Dehm SM. Constitutive activity of the androgen receptor. Adv Pharmacol. 2014;70:327–66. doi: 10.1016/B978-0-12-417197-8.00011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mostaghel EA, et al. Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants. Clin Cancer Res. 2011;17:5913–25. doi: 10.1158/1078-0432.CCR-11-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu R, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–62. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakazawa M, et al. Serial Blood-Based Analysis of AR-V7 in Men with Advanced Prostate Cancer. Ann Oncol. 2015 doi: 10.1093/annonc/mdv282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antonarakis ES, et al. Androgen Receptor Splice Variant 7 and Efficacy of Taxane Chemotherapy in Patients With Metastatic Castration-Resistant Prostate Cancer. JAMA Oncol. 2015;1:582–91. doi: 10.1001/jamaoncol.2015.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Taplin ME, et al. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21:2673–8. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 48.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 49.Joseph JD, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013;3:1020–9. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- 50.Korpal M, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer Discov. 2013;3:1030–43. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- 51.Balbas MD, et al. Overcoming mutation-based resistance to antiandrogens with rational drug design. Elife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Azad AA, et al. Androgen Receptor Gene Aberrations in Circulating Cell-Free DNA: Biomarkers of Therapeutic Resistance in Castration-Resistant Prostate Cancer. Clin Cancer Res. 2015;21:2315–24. doi: 10.1158/1078-0432.CCR-14-2666. [DOI] [PubMed] [Google Scholar]
- 53.Cai C, et al. Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors. Cancer Res. 2011;71:6503–13. doi: 10.1158/0008-5472.CAN-11-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen EJ, et al. Abiraterone treatment in castration-resistant prostate cancer selects for progesterone responsive mutant androgen receptors. Clin Cancer Res. 2015;21:1273–80. doi: 10.1158/1078-0432.CCR-14-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arora VK, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–22. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Isikbay M, et al. Glucocorticoid receptor activity contributes to resistance to androgen-targeted therapy in prostate cancer. Horm Cancer. 2014;5:72–89. doi: 10.1007/s12672-014-0173-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang KH, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–84. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu C, et al. Intracrine Androgens and AKR1C3 Activation Confer Resistance to Enzalutamide in Prostate Cancer. Cancer Res. 2015;75:1413–22. doi: 10.1158/0008-5472.CAN-14-3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clegg NJ, et al. ARN-509: a novel antiandrogen for prostate cancer treatment. Cancer Res. 2012;72:1494–503. doi: 10.1158/0008-5472.CAN-11-3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Njar VC, Brodie AM. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J Med Chem. 2015;58:2077–87. doi: 10.1021/jm501239f. [DOI] [PubMed] [Google Scholar]
- 61.Rafferty SW, Eisner JR, Moore WR, Schotzinger RJ, Hoekstra WJ. Highly-selective 4-(1,2,3-triazole)-based P450c17a 17,20-lyase inhibitors. Bioorg Med Chem Lett. 2014;24:2444–7. doi: 10.1016/j.bmcl.2014.04.024. [DOI] [PubMed] [Google Scholar]
- 62.Yin L, Hu Q. CYP17 inhibitors--abiraterone, C17,20-lyase inhibitors and multi-targeting agents. Nat Rev Urol. 2014;11:32–42. doi: 10.1038/nrurol.2013.274. [DOI] [PubMed] [Google Scholar]
- 63.Vasaitis T, et al. Androgen receptor inactivation contributes to antitumor efficacy of 17{alpha}-hydroxylase/17,20-lyase inhibitor 3beta-hydroxy-17-(1H-benzimidazole-1-yl)androsta-5,16-diene in prostate cancer. Mol Cancer Ther. 2008;7:2348–57. doi: 10.1158/1535-7163.MCT-08-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yu Z, Cai C, Gao S, Simon NI, Shen HC, Balk SP. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin Cancer Res. 2014;20:4075–85. doi: 10.1158/1078-0432.CCR-14-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taplin ME, Chu F, Morrison JP, Pili R, Rettig RB, Stephenson J, Vogelzang NJ, Montgomery FB. Abstract CT-07: ARMOR1: Safety of galeterone (TOK-001) in a Phase 1 clinical trial in chemotherapy naïve patients with castration resistant prostate cancer (CRPC) Cancer Res. 2012;72 CT-07. [Google Scholar]
- 66.Yaqub F. Galeterone activity in castration-resistant prostate cancer. Lancet Oncol. 2015;16:e10. doi: 10.1016/S1470-2045(14)71165-9. [DOI] [PubMed] [Google Scholar]
- 67.Xu D, et al. Androgen receptor splice variants dimerize to transactivate target genes. Cancer Res. 2015 doi: 10.1158/0008-5472.CAN-15-0381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Myung JK, et al. An androgen receptor N-terminal domain antagonist for treating prostate cancer. J Clin Invest. 2013;123:2948–60. doi: 10.1172/JCI66398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Andersen RJ, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–46. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 70.Sun S, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Antonarakis ES. Predicting treatment response in castration-resistant prostate cancer: could androgen receptor variant-7 hold the key? Expert Rev Anticancer Ther. 2015;15:143–5. doi: 10.1586/14737140.2015.999044. [DOI] [PMC free article] [PubMed] [Google Scholar]