

Abstract
Background
In Ts65Dn, a mouse model of Down syndrome (DS), brain and craniofacial abnormalities that parallel those in people with DS are linked to an attenuated cellular response to sonic hedgehog (SHH) signaling. If a similarly reduced response to SHH occurs in all trisomic cells, then chronic up-regulation of the pathway might have a positive effect on development in trisomic mice, resulting in amelioration of the craniofacial anomalies.
Results
We crossed Ts65Dn with Ptch1tm1Mps/+ mice and quantified the craniofacial morphology of Ts65Dn;Ptch+/− offspring to assess whether a chronic up-regulation of the SHH pathway rescued DS-related anomalies. Ts65Dn;Ptch1+/− mice experience a chronic increase in SHH in SHH-receptive cells due to haploinsufficiency of the pathway suppressor, Ptch1. Chronic up-regulation had minimal effect on craniofacial shape and did not correct facial abnormalities in Ts65Dn;Ptch+/− mice. We further compared effects of this chronic up-regulation of SHH to acute pathway stimulation in mice treated on the day of birth with a SHH pathway agonist, SAG. We found that SHH affects facial morphology differently based on chronic vs. acute postnatal pathway up-regulation.
Conclusions
Our findings have implications for understanding the function of SHH in craniofacial development and for the potential use of SHH-based agonists to treat DS-related abnormalities.
Keywords: Patched, Down syndrome, development, Ts65Dn, geometric morphometrics
INTRODUCTION
Certain brain and craniofacial abnormalities found in individuals with DS, a developmental disorder caused by trisomy of human chromosome 21 (Hsa21), have been attributed to disruptions in SHH signaling (Roper et al., 2006; Roper et al., 2009; Trazzi et al., 2011; Currier et al., 2012; Das et al., 2013; Dutka et al., 2015). Mouse models of DS, such as Ts65Dn, have been used to investigate the mechanisms underlying the disabilities associated with DS (Davisson et al., 1993; Das et al., 2011). The Ts65Dn mouse is trisomic for orthologs of approximately half of the conserved genes on Hsa21, plus a number of genes that are not orthologous to Hsa21 that are included in a freely segregating chromosome (Duchon et al., 2011; Reinholdt et al., 2011), and is the most widely reported model for DS research.
Mouse models of DS have revealed that some trisomic cells exhibit a reduced mitogenic response to SHH signaling. This change in the receptivity to SHH signaling was found to affect the cellularity and size of the cerebellum, along with learning behaviors associated with the hippocampus (Dahmane et al., 1999; Baxter et al., 2000; Roper et al., 2006; Das et al., 2013). An acute increase of SHH signaling via an injection of a SHH pathway agonist, SAG 1.1 (SAG) (Chen et al., 2002) in newborn trisomic mice was able to normalize cerebellar hypoplasia and rescue some hippocampal function in adult Ts65Dn mice (Roper et al., 2006; Das et al., 2013). SAG up-regulates the canonical SHH pathway by binding and activating Smoothened, the regulator of the canonical HH pathway (Chen et al., 2002).
Skeletal hypoplasia of the mid-face of Ts65Dn mice (Richtsmeier et al., 2000) can be traced to defects in delamination, migration and mitosis of neural crest cells (Roper et al., 2009), which give rise to the facial skeleton and the complex network of facial connective tissue (Hall, 1999; Bhatt et al., 2013). Trisomic mice present alterations in the facial skeleton corresponding to those found in humans with DS (Allanson et al., 1993; Richtsmeier et al., 2000; Richtsmeier et al., 2002; Guihard-Costa et al., 2006). Among its many roles, SHH signaling is important for survival of cranial neural crest cells (CNCC) during the early stages of embryonic development (~E8.5) and for promoting cell proliferation in the later stages (E10.5) to mediate the size of the facial primordia (Ahlgren and Bronner-Fraser, 1999; Hu and Helms, 1999; Jeong et al., 2004). Clinical anomalies in the facial skeleton of individuals with DS are predominately in the mid-facial region including reduction in maxillary (and mandibular) dimensions and inter-orbital distance, but brachycephalic cranial shape and a smaller overall head size are also characteristic of this syndrome (Richtsmeier et al., 2000; Silva and Valladares-Neto, 2013).
Much of the data regarding the role of SHH in facial development comes from experimental work on murine and avian models (Marcucio et al., 2005; Hu and Marcucio, 2009a). This evidence implicates a signaling zone, the Frontonasal Ectodermal Zone or FEZ, located in the cephalic ectoderm of the upper jaw in mammals and avians (Hu et al., 2003). FEZ is defined as the boundary between Shh and Fibroblast growth factor 8 (Fgf8) (Marcucio et al., 2011). FEZ induces skeletogenesis and patterning of the underlying mesenchymal cells to control morphogenesis of the upper jaw region in mammals and birds (Hu et al., 2003; Hu and Marcucio, 2009b). SHH signaling within the forebrain induces Shh in the FEZ and changes in the expression of Shh cause morphological variation in mice and chicks (Hu and Marcucio, 2009b). The importance of SHH signaling within CNCC of the face has been demonstrated by several studies (Ahlgren and Bronner-Fraser,1999; Brito et al., 2006; Jeong et al., 2004; Welsh and O’Brien, 2009), and disruption in the ability of developing CNCC to transduce Hedgehog (HH) signaling inhibits proliferation of the mesenchymal cells and produces malformations of the upper jaw (Jeong et al., 2004). Thus there is a vital link between SHH signaling, CNCC proliferation, and formation of craniofacial structures.
Following the premise that if all trisomic cells have a similarly attenuated response to SHH signaling, up-regulation of the HH pathway might normalize the developmental, including craniofacial, anomalies associated with DS, Dutka et al. (2015) crossed Ptch1tm1Mps/+ with Ts65Dn mice to yield Ts;Ptch+/− mice (Table 1). The resulting increase of SHH signaling in Ts;Ptch+/− mice improved nest building activities and corrected structural defects in the cerebellum compared to Ts65Dn (Dutka et al., 2015), partly replicating the results previously reported for trisomic mice acutely exposed to the SHH agonist, SAG (Roper et al., 2006; Das et al., 2013). However, in contrast to trisomic mice that were treated with SAG, the Ts;Ptch+/− mice did not show improvement in behavioral activities mediated by the hippocampus (Das et al., 2013; Dutka et al., 2015). Moreover, despite the beneficial effects of SAG on cerebellar morphology of Ts65Dn mice, further examination of SAG-treated euploid mice (controls) revealed that acute up-regulation of the pathway caused dysmorphology in the midline structures of the face in some treated mice (Singh et al., 2015).
Table 1.
Sample used in the study
| Ploidy | Genotype | Groups | Abbreviation | Sample size |
|---|---|---|---|---|
|
| ||||
| Euploid | Wildtype | Euploid;Wildtype | Eu;WT | 13 |
| Euploid | Ptch1tm1Mps/+ | Euploid; Ptch1tm1Mps/+ | Eu; Ptch+/− | 8 |
| Trisomic | Ts65Dn | Ts65Dn;Wildtype | Ts;WT | 9 |
| Trisomic | Ptch1tm1Mps/+ | Ts65Dn;Ptch1tm1Mps/+ | Ts;Ptch+/− | 9 |
| Total | 39 | |||
|
| ||||
| Euploid | 20μg/g SAG-treated | Euploid | Eu;SAG | 32 |
| Euploid | Vehicle treated | Euploid | Eu;Veh | 23 |
| Trisomic | 20μg/g SAG-treated | Ts65Dn | Ts;SAG | 4 |
| Trisomic | Vehicle treated | Ts65Dn | Ts;Veh | 7 |
| Total | 66 | |||
Here we test whether chronic, low-level pathway up-regulation might have ameliorative effects on the development of the craniofacial skeleton. Correction of developmental consequences of down-regulated SHH signaling in trisomic mice might suggest therapeutic strategies for ameliorating these phenotypes in humans with DS.
RESULTS
Does chronic up-regulation of SHH rescue cranial phenotypes in trisomic mice?
We crossed Ts65Dn and Ptch1tm1Mps/+ mice to generate four genotypes: Ts65Dn mice (Ts;WT); Ts65Dn mice haploinsufficient for Ptch1 (Ts;Ptch+/−); euploid control (Eu;WT); and euploid haploinsufficient for Ptch1 allele (Eu;Ptch+/−) (Table 1) (Dutka et al., 2015). Three-dimensional coordinates of biological landmarks measured on the skulls of adult mice were collected from micro-CT images and analyzed (Fig. 1). Principal component analysis (PCA) of the Procrustes shape coordinates separated the euploid mice from the trisomic mice along PC1 (summarizing 31.1% of the total variance in the sample) (Fig. 2). PC2 (15.5%) captured differences between the Ptch+/− mice and their WT littermates, Ts;WT and Ts;Ptch+/− showing more distinction in shape along this axis than Eu;WT and Eu;Ptch+/− (Fig. 2). Shape changes between the euploid and trisomic groups along PC1 were primarily in the neurocranium and snout (Fig. 2). Compared to euploid mice, the neurocranium of trisomic mice was rounded and raised and the snout slightly retracted and reduced dorsoventrally; this particular neurocranial shape is more evident in the Ts;Ptch+/− mice on the far positive end of PC1 relative to the Ts;WT. PC2 mainly accounted for changes in the position of the snout relative to the neurocranium and width of the overall cranium (not shown).
Fig 1.

Forty landmarks used in the study. Definitions provided in Table 2. A) Oblique supero-lateral view; B) Anterior view; C) Inferior view.
Fig 2.

Principal component (PC) analysis of forty 3D cranial landmarks. PC1 primarily captures the differences between the euploid and trisomic morphology, with some distinction between the Ptch+/− (dashed convex hulls) and wildtype (solid convex hulls) individuals in the respective euploid and triosmic groups. The shape changes along the PCs are represented by surface reconstructions computed from the grand mean of the total dataset and warped according to the variation defined along the respective PC scores. PC1 shows shape changes consistent with ploidy in the neurocranium, which is markedly more rounded and raised in the Ts;Ptch+/− and Ts;WT compared to the Eu;Ptch+/− and Eu;WT. PC2 mainly accounts for the within-group variation in all four groups, more so than in PC1, and also shape changes between the Ts;WT and Ts;Ptch+/− . The morphological changes on PC2 relate to the orientation of the snout and width of neucrocranium (not shown).
To further examine within-ploidy differences between Ptch1+/− and WT mice, we conducted separate Euclidean Distance Matrix analyses (EDMA) between Eu;Ptch+/− and Eu;WT and between Ts;Ptch+/− and Ts;WT. The separate within-ploidy comparisons showed similar, though subtle patterns of morphological change relative to mice with normal Ptch function, especially in the cranial vault. That is, the way in which the Ptch+/− mice differed from their respective WTs was similar, regardless of ploidy. Additionally, between-group mean shape comparisons across the four genotypes revealed that Ts;Ptch+/− mice had the most rounded and supero-inferiorly raised cranial vault, followed by Eu; Ptch+/−, Ts;WT and Eu;WT.
To explore specific changes in the face, we performed a PCA using only the facial landmarks (Table 2; Fig. 3). Similar to the overall result (Fig. 2), the biggest separation on PC1 (33.1%) was between the trisomic and euploid groups, with little distinction between Eu;Ptch1+/− and control Eu;WT individuals along this axis (Fig. 3). PC2 (18.2%) showed shape differences between the Ptch+/− and WT groups. These morphological differences due to decrease in Ptch1 function are more prominent between the Ts;Ptch+/− and Ts;WT than between the two euploid genotypes, as indicated by the respective group scatters (Fig. 3).
Table 2.
Definition of the forty landmarks used in the study. Landmark numbers correspond to
| Landmark # | Landmark definition |
|---|---|
| 1 | Anterior nasal spine is the most anterior point of interpremaxiallary suture at base of nasal aperture, midline |
| 2 | Nasale is the intersection of nasal bones at rostral point, midline |
| 3 | Nasion is the intersection of nasal bones at caudal point, midline |
| 4 | Bregma is the intersection of frontal bones and parietal bones at midline |
| 5 | The intersection of parietal bones with anterior aspect of interparietal bone, midline |
| 6 | The intersection of interparietal bone with squamous portion of occipital bone, midline |
| 7 | Opisthion is the midsaggital point on the posterior margin of the foramen magnum, midline |
| 8 | Basion is the midsaggital point on the anterior margin of the foramen magnum, midline |
| 9&10 | Antero-superior most tip on the nasal bone |
| 11 & 12 | Anterior-most point at intersection of premaxilla and nasal bones |
| 13 & 14 | Anterior notch on frontal process lateral to infraorbital fissure |
| 15 & 16 | Intersection of frontal process of maxilla with frontal and lacrimal bones |
| 17 & 18 | Point left of nasion intersection of nasal bone and premaxilla |
| 19 & 20 | Intersection of the coronal suture and the temporal crest |
| 21 & 24 | Intersection of zygomatic process of maxilla with zygoma (jugal), superior surface |
| 22 & 25 | Posterior point at the joining of the squamosal body to zygomatic process of squamosal |
| 23 & 26 | Intersection of parietal temporal and occipital bones |
| 27 & 28 | Postero-lateral most point on the tympanic bulla |
| 29 & 30 | Medial most notch on the tympanic bulla, basicapsular fissure |
| 31 & 32 | Most antero-lateral point on corner of the basioccipital |
| 33 & 34 | Medial most tip of the tympanic bulla |
| 35 & 36 | The posterior-most point on the central anteriorposterior axis of the left molar alveolus |
| 37 & 38 | Most posterior point of the anterior palatine foramen |
| 39 & 40 | Most anterior point of the anterior palatine foramen |
Fig 3.

Principal component analysis of twenty-three facial landmarks. PC1 separates the euploid and trisomic groups, showing little difference between the Ptch+/− (dashed convex hulls) and their wildtype (solid convex hulls) individuals along this axis. The wireframe diagrams illustrate the shape changes (in black) from the negative to the positive end along each PC relative to the grand mean shape (in gray) computed from all the specimens in the sample. PC1 captures mediolateral expansion and contraction of the face (narrow and elongated in euploids and laterally expanded in both the trisomic groups), marked by the position of the orbital margin. PC2 accounts for changes between the Ptch+/− and WT mice, particularly between the Ts;Ptch+/− and Ts;WT. Shape changes along PC2 captures differences in the relative length of the snout, being shorter and retracted in the Ptch+/− mice compared to their WT counterparts.
Given our previous demonstration of cranial mean size differences between euploid and trisomic mice (Richtsmeier et al., 2002; Hill et al., 2007), we conducted a multivariate regression analysis of shape on centroid size to examine allometric variation (i.e., size related shape variation) among the groups (Fig. 4). The majority of euploid mice were considerably larger than the trisomic, with the trisomic mice occupying the lower left corner on the plot (Fig. 4). The Eu;Ptch+/− mice were larger than the Eu;WT while the distribution of Eu;WT mice overlaps with both trisomic groups and with Eu;Ptch+/−. Overall, the trisomic mice showed more variation in aspects of shape than size while the euploid mice showed relatively more variation in size.
Fig 4.
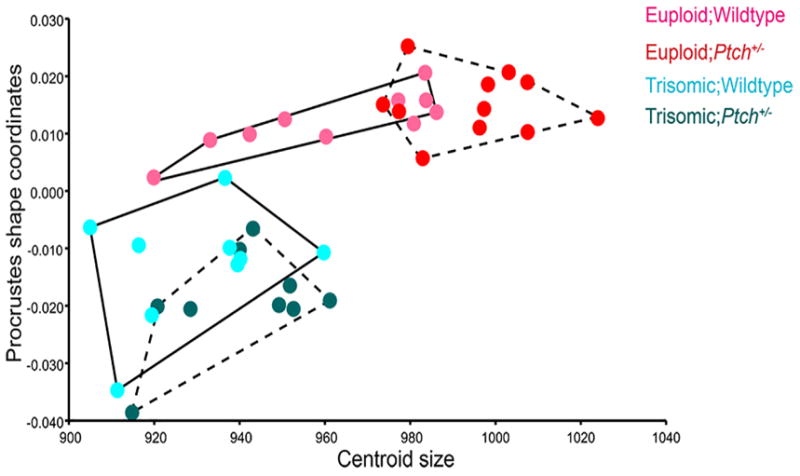
Multivariate regression analysis of all forty Procrustes shape coordinates on centroid size. The plot illustrates differences in allometric variation between the euploid and trisomic mice. Both Ts;Ptch+/− (dashed convex hull) and Ts;WT (solid convex hull) mice are considerably smaller than the euploids, and show more variation in aspects of shape than size. The euploids are distinct from the trisomic mice in shape and are larger in size, with the Eu;Ptch+/− (dashed convex hull) being slightly larger in size than the Eu;WT (solid convex hull).
To reduce the effects of size differences between trisomic and euploid mice, we conducted a PCA on the regression residuals to explore non-allometric shape variation among the groups (Fig. 5). PC1 (22.7%) accounted for slight differences among all four genotypes, also showing similarities between the Eu;Ptch+/− and Ts;Ptch+/− groups, which clustered closer together on the positive end of PC1. PC2 (15.8%) captured more of the within-group variation displayed by each group (Fig. 5).
Fig 5.
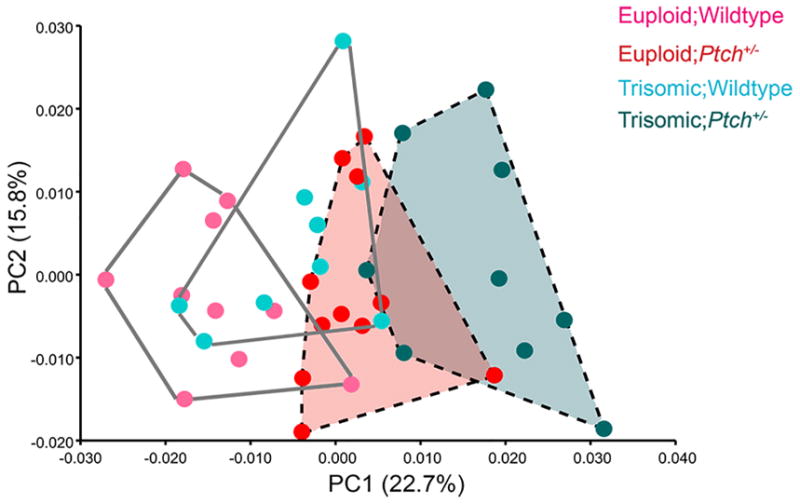
Principal component analysis on the multivariate regression residuals. The euploid and trisomic mice are less distinct from one another after the effects of size have been removed from the analysis. The Eu;Ptch+/− and Ts;Ptch+/− (dashed convex hulls) both occupy the positive end of PC1, showing slight overlap and subtle similarities along this axis.
To summarize, our overall results show that chronic up-regulation of the SHH pathway does not correct midfacial dysmorphology in the Ts;Ptch+/−. In fact, haploinsufficiency of Ptch1 affects cranial shape of trisomic and eupoid mice (Ts;Ptch+/− and Eu;Ptch+/−) in similar ways.
Do chronic and acute up-regulation of the SHH pathway have similar effects?
Injection of SAG results in acute, high-level up-regulation of the canonical SHH pathway. In newborn mice this results in dosage sensitive penetrance, i.e., only a subset of injected mice are affected and higher doses increase the number of animals affected (Singh et al., 2015). We compared the craniofacial phenotypes of the mice that were injected with an acute dose of SAG or vehicle on the day of birth, to the craniofacial skeleton of mice haploinsufficient for Ptch1. PCA results revealed marked differences between the affected SAG-treated euploid mice and all the other groups in the sample (Table 1), driving the shape change along PC1 (27.4%) (Fig. 6). The subset of nine SAG treated euploid mice (orange dots on the positive end of PC1) shared a snout morphology distinct from all the other mice. These nine animals had retracted and depressed nasal bones, medio-laterally expanded snouts and prominent ridging along the lateral aspects of the fronto-nasal-premaxillary junction (Singh et al., 2015); none of the Ptch+/− mice (euploid or trisomic) showed shape deformations similar to the changes exhibited by the affected SAG-treated mice. PC2 (18.9%) separated all the trisomic mice, regardless of genotype, from all the euploid mice (Fig. 6). The Ptch1+/− mice clustered with mice of similar ploidy and showed no distinction from their WT counterparts. PC2 also captured increased variation within the affected SAG-treated euploid mice (the same subset of SAG-treated euploid mice that separated on the positive end of PC1).
Fig 6.

Principal component analysis of all Ptch+/− mice (the Eu;Ptch+/− and Ts;Ptch+/− groups are indicated by the dashed convex hulls) and their wildtype littermates (solid convex hulls), and the SAG and Vehicle (Veh) treated euploid and trisomic mice (solid convex hulls). The plot illustrates the difference between an acute and chronic up-regulation of SHH on craniofacial morphology. The shape changes (in black) along the respective PCs are represented by wireframe diagrams of the extreme shapes at the end of each axis relative to the grand mean shape (in gray) computed from all the specimens in the dataset. PC1 separates a subset of affected SAG-treated mice from all the other groups in the sample. PC2 distinguishes all the trisomic mice, irrespective of gentoype, from all the euploid mice in the sample.
DISCUSSION
Craniofacial structure of Ts65Dn;Ptch+/− mice differs from euploid morphology
Trisomic granule cell neuron precursors and cells of the first pharyngeal arch have a reduced response to SHH compared to their euploid counterparts (Currier et al., 2012). Here we tested whether that was the case for characteristic dysmorphologies of the trisomic facial skeleton. Our quantitative comparisons of Ts;Ptch+/− and Ts;WT are not consistent with our hypothesis as haploinsufficiency of Ptch1 failed to ameliorate craniofacial anomalies in Ts;Ptch+/− mice. In fact, the subtle shape changes between the Ts;Ptch+/− and Ts;WT mice indicate that the Ts;Ptch+/− mice express a more perturbed version of the Ts;WT morphological pattern, which is characterized by wider, more rounded neurocrania and shortened, retracted snouts compared to euploid mice (Richtsmeier et al., 2002). Eu;Ptch+/− mice also have slightly raised and rounded cranial vaults compared to the ‘flatter’ vaults of Eu;WT mice, suggesting that, while subtle, the cranial changes caused by the absence of one Ptch allele are similar in both the Eu;Ptch+/− and Ts;Ptch+/−. Given that substantial perturbations in SHH signaling can cause serious craniofacial defects (Hu and Helms 1999; Hu et al., 2003; Tapadia et al., 2005; Cordero et al., 2006; Singh et al., 2015), our results are surprising in that we did not find overt changes in the facial morphology of the Eu;Ptch1+/− mice compared to Eu;WT.
While SHH signaling plays a crucial role in the development of the skull, it also interacts with several other growth factors, and the reduced response to SHH signaling in trisomic cells might be just one of many contributors to the facial anomalies caused by trisomy. We can conclude from these results that a chronic increase in SHH signaling through haploinsufficiency of Ptch1 in trisomic mice is not sufficient to correct the skeletal craniofacial dysmorphology characteristic of trisomic mice. Further, it appears likely that not all trisomic cells have a similarly reduced response to SHH. Several related conclusions are certainly not eliminated, the most likely being that stoichiometry of SHH production and/or delivery to target cells and/or targeting of Ptch is different for different cell types so that the “one size fits all” up-regulation of the pathway in Ptch+/− mice results in the wrong level of pathway activation for craniofacial skeletal phenotypes.
The multivariate regression analysis (Fig. 4) does capture some size differences between the Eu;WT and Eu;Ptch+/− mice, with the latter showing a marked increase in size (on the higher end of the centroid size axis), suggesting that haploinsufficiency for Ptch1 does, to an extent, affect craniofacial size in the euploid mice (Dutka et al., 2015). This analysis also indicates that size related changes in cranial shape (i.e., allometry) are different between the euploid and trisomic mice. Specifically, the trisomic mice show large variation in shape that is not accompanied by corresponding changes in size, whereas the euploid mice show considerable size variation, but relatively little variation in shape. The result is that the effect of haploinsufficiency for Ptch1 on cranial size is stronger in the euploid sample than in our trisomic sample.
Chronic vs. Acute up-regulation of the SHH pathway
SAG is a short-lived pharmacological agonist of the SHH pathway that acts downstream of Ptch by binding to the pathway regulator, Smo (Chen et al., 2002). We showed previously that SAG, when injected at birth, affects postnatal craniofacial skeletal development in some euploid mice (i.e., it is incompletely penetrant) irrespective of any variation in adult age stage, and that this penetrance is dose dependent (Singh et al., 2015). Our comparison of the adult cranial morphology of SAG-treated vs. Ptch+/− mice defines a difference in the phenotypic effects of an acute vs. chronic up-regulation of the SHH pathway. Given the evidence from multiple studies (Hu and Helms 1999; Hu et al., 2003; Tapadia et al., 2005; Cordero et al., 2006; Young et al., 2010; Singh et al., 2015; Young et al., 2014) that an increase of SHH affects the width and outgrowth of the face both pre- and postnatally, particularly the midline structures, we expected the Eu;Ptch+/− to show craniofacial changes similar to those found in the affected SAG treated mice (Singh et al., 2015). However, our observations indicate that a chronic increase in SHH activity caused by a loss of function of one Ptch allele has markedly different morphological outcomes than an acute up-regulation of SHH signaling at birth. These differential effects also highlight the potentially different roles of Ptch and Smo as regulators of craniofacial development. Further investigation into the activity of these receptors might provide insight into cranial malformations caused by mutations in the HH pathway (Du et al., 2012). This information is important for the design of effective therapies for craniofacial malformation.
Our results indicate that stimulation of the SHH pathway does not function as a simple developmental toggle, but that developmental time (i.e. when the pathway is up-regulated), amount of increased SHH activity, and method of up-regulation can have varied and unpredictable effects on craniofacial morphology, targeting cells that form various tissues differently. Moreover, overexpression of SHH can cause an increase in expression of Indian hedgehog (IHH) inhibitor, PTHrP, altering normal growth of the cranial base in the later stages of development (Young et al., 2006; Pan et al., 2013). In this respect, our results cannot exclude either a potential effect of SAG on postnatal endochondral growth and patterning regulated by IHH or a potential prenatal effect of Ptch1 haploinsufficiency on early craniofacial cartilages (chondrocranium). Further investigation is needed to fully understand the extent of IHH involvement in facial development and whether there is an overlap in SHH and IHH function in prenatal and/or early postnatal craniofacial development.
Our studies reveal that euploid and abnormal craniofacial morphology resulting from effects of trisomy can be further affected by perturbations in SHH signaling during development. Knowledge of the relationship between shape and size of cranial structures during normal and abnormal development and the specific developmental time periods when significant craniofacial shape variation is generated is necessary not only for the development of treatment strategies for craniofacial dysmorphology in DS, but for developing treatments of any craniofacial anomalies in which this central signaling pathway is affected.
EXPERIMENTAL PROCEDURES
Ts65Dn mice were acquired from the Jackson laboratory and maintained at the Reeves’ laboratory as a colony of C57BL/6JxC3H/HeJ(B6xC3H) advanced intercross. The B6;129-Ptch1tm1Mps/J mice were also obtained from the Jackson Laboratory, backcrossed for five generations onto a B6 background, and then crossed with C3H mice to create an F1 generation of B6C3H mice. The Ptch1tm1Mps/+ mice were maintained in the Reeves’ laboratory colony as a B6 × C3H advanced intercross (Dutka et al., 2015). The B6C3H SAG and Vehicle treated euploid mice were also maintained in the Reeves’ laboratory and alternate litters were injected with either 20μg/g of SAG or Veh, subcutaneously at the back of the skull within hours after birth (Das et al., 2013; Singh et al., 2015) (Table 1). Between 10 and 18 weeks of age, all the mice in the dataset were anesthetized with isoflourane and euthanized by cervical dislocation or perfusion. Heads were removed and placed in 4% paraformaldehye (PFA) for at least 48hrs. The skulls were then washed and stored in phosphate buffered saline (PBS) at 4 °C until they were scanned by micro-computed tomography (μCT) for morphological analyses. All procedures were reviewed, approved, and carried out in compliance with animal welfare guidelines approved by the Johns Hopkins University and the Pennsylvania State University Animal Care and Use Committees.
μCT images of 39 adult Ptch+/− and their respective wildtype (WT) (Table 1) and 66 SAG and Vehicle-treated mouse (Table 1) crania were acquired from Johns Hopkins Medical Institutions through the Small Animal Resource Imaging Program at the Research Building Imaging Center, using a Gamma Medica X-SPECT/CT scanner (Northridge, CA, USA), with a resolution of 0.05mm long the x,y,z axes. The original image data were reduced from 16 to 8bit for image analyses. Isosurfaces of each specimen in the sample were reconstructed using the software package AVIZO 6.3 and 8.1.1 (Visualization Sciences Group, VSG) to visualize the cranial bones.
The three-dimensional coordinates of 40 anatomical landmarks were measured on the reconstructed isosurfaces of all the individuals used in this study and recorded for data analysis (Fig. 1; Table 2). The landmark configurations for each specimen were superimposed using generalized Procrustes analysis (GPA). This method extracts shape coordinates from the original 3D coordinates by superimposing the landmark data for all animals to the same orientation and scaling each specimen to unit centroid size. The resulting Procrustes shape coordinates were used in all subsequent PC analyses (Dryden and Mardia, 1998; Slice, 2005). We additionally conducted EDMA (Euclidean Distance Matrix Analysis) to statistically examine some of the small scale changes between Eu;Ptch+/− and Eu;WT and between the Ts;Ptch+/− and Ts;WT using the original coordinate data. EDMA is a coordinate system-free morphometric method that enables statistical testing of the morphological differences between groups. EDMA converts 3D landmark data into a matrix of all possible linear distances between unique landmark pairs and tests for statistical significance of differences between shapes using non-parametric confidence intervals (Lele and Richtsmeier, 2001).
Patterns of shape variation in the dataset were analyzed using principal component analyses (PCA). PCA is based on an eigenvalue decomposition of a covariance matrix, transforming Procrustes shape coordinates into scores along principal components (PCs) (Slice, 2005). To address our first objective of examining the craniofacial morphology of the 39 Ptch+/− and their WT mice, we first performed a PCA with all the 40 cranial landmarks and then another PCA with just the 23 facial landmarks (Fig. 1; Table 2). The latter was conducted to explore the effects of an up-regulation of SHH specifically on the face to assess whether the facial morphology of Ts;Ptch+/− mice was rescued as a result of increasing SHH signaling in trisomic cells. We additionally computed mean shapes for each of the four euploid and trisomic Ptch+/− and WT groups (Table 1) to directly compare between-group cranial shape differences. To explore size related shape changes and static allometric trajectories among the Ptch+/− mice, we conducted a multivariate regression analysis of all the Procrustes shape coordinates on centroid size. Subsequently, we conducted another PCA on the regression residuals to explore the overall shape variation in the sample without the effects of allometry, unlike the initial PCA. Lastly, to address our second objective of comparing the effects of chronic vs. acute up-regulation of SHH on craniofacial morphology, we conducted a second PCA with the combined datasets of all the 39 Ptch+/− and WT mice and the 66 SAG and Veh-treated groups.
The shape changes along the respective PC axes were visualized using surface scans and wireframe diagrams. The surface scans were generated in AVIZO 8.1.1 by warping the PC scores onto the grand mean shape of all the groups in the sample. The wireframe diagrams were constructed in MorphoJ (Klingenberg, 2011), and the shape changes along the respective PCs were depicted relative to the grand mean shape computed from all the specimens in the sample. All the analyses were performed in R programming software version 3.1 (The R FAQ; http://cran.r-project.org/doc/FAQ/R-FAQ.html) and MorphoJ.
Chronic up-regulation of the SHH pathway does not rescue trisomic facial morphology
Chronic up-regulation of the SHH pathway affects the craniofacial morphology of trisomic and euploid mice similarly
Effects of the canonical SHH pathway on skeletal morphology varies with chronic vs. acute postnatal up-regulation
Acknowledgments
Supported by Public Health Service Awards R01-HD038384-16 (RR and JTR), R01-DE018500, ARRA 3R01DE018500-S1, R01-DE022988, P01HD078233 (JTR); by an award from the Lumind-RDS Foundation (RR); and by the National Science Foundation BCS-0725227(JTR).
We thank J. Yu, G. Green, Y. Wang (Johns Hopkins Small Animal Imaging Resource Program) and Hongseok Kim (Dept. of Anthropology, Pennsylvania State University) for their assistance in obtaining and processing the micro-CT images. The authors declare that they have no competing interests. R.H.R. is a member of Science Advisory Boards of the Lumind Foundation, the Linda Crnic Institute, and the National Down Syndrome Society; none of these positions is remunerated. All authors contributed substantially to the work presented in this paper. R.H.R., J.T.R. and N.S. designed the study. N.S. and J.T.R. collected and analyzed the quantitative data. T.D. performed all mouse breeding and phenotyping for the Ptch+/− mice and generated μCT scans. N.S., J.T.R and R.H.R wrote the paper with input from T.D.
References
- Ahlgren SC, Bronner-Fraser M. Inhibition of Sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Curr Bio. 1999;9:1304–1314. doi: 10.1016/s0960-9822(00)80052-4. [DOI] [PubMed] [Google Scholar]
- Allanson JE, O'Hara P, Farkas LG, Nair RC. Anthropometric craniofacial pattern profiles in Down syndrome. Am J Med Genet. 1993;47:748–752. doi: 10.1002/ajmg.1320470530. [DOI] [PubMed] [Google Scholar]
- Baxter LL, Moran TH, Richtsmeier JT, Troncoso J, Reeves RH. Discovery and genetic localization of Down syndrome cerebellar phenotypes using the Ts65Dn mouse. Hum Mol Genet. 2000;9:195–202. doi: 10.1093/hmg/9.2.195. [DOI] [PubMed] [Google Scholar]
- Bhatt S, Diaz R, Trainor PA. Signals and switches in mammalian neural crest cell differentiation. Cold Spring Harb Perspect Biol. 2013;5:a008326. doi: 10.1101/cshperspect.a008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito JM, Teillet MA, Le Douarin NM. An early role for sonic hedgehog from foregut endoderm in jaw development: Ensuring neural crest cell survival. Proc Natl Acad Sci. 2006;103:11607–11612. doi: 10.1073/pnas.0604751103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero D, Tapadia M, Helms JA. The role of sonic hedgehog signaling in craniofacial development. In: Fisher CE, SEM, editors. Shh and Gli Signaling and Development. Austin, TX: Bioscience and Springer Science; 2006. pp. 44–57. [Google Scholar]
- Currier G, Polk RC, Reeves RH. A Sonic hedgehog (Shh) response deficit in trisomic cells may be a common denominator for multiple features of Down Syndrome. Prog in Brain Research. 2012;197:223–236. doi: 10.1016/B978-0-444-54299-1.00011-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Das I, Reeves RH. The use of mouse models to understand and improve cognitive deficits in Down syndrome. Dis Model Mech. 2011;4:596–606. doi: 10.1242/dmm.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Park JM, Shin JH, Jeon SK, Lorenzi H, Linden DJ, Worley PF, Reeves RH. Hedgehog agonist therapy corrects structural and cognitive deficits in a Down syndrome mouse model. Sci Transl Med. 2013;5:1–10. doi: 10.1126/scitranslmed.3005983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davisson M, Schmidt C, Reeves N, Irving E, Akeson E, Harris B, Bronson R. Segmental trisomy as a mouse model for Down Syndrome. Prog Clin Biol Res. 1993;384:117–133. [PubMed] [Google Scholar]
- Dryden IL, Mardia KV. Statistical Shape Analysis. Chichester: Wiley; 1998. [Google Scholar]
- Du J, Fan Z, Ma X, Wu Y, Liu S, Gao Y, Shen Y, Fan M, Wang S. Expression of smoothened in mouse embryonic maxillofacial development. Biotech Histochem. 2012;87:187–194. doi: 10.3109/10520295.2011.602645. [DOI] [PubMed] [Google Scholar]
- Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm Genome. 2011;22:674–684. doi: 10.1007/s00335-011-9356-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutka T, Hallberg D, Reeves RH. Chronic up-regulation of the SHH pathway normalizes some developmental effects of trisomy in Ts65Dn mice. Mech Dev. 2015;135:68–80. doi: 10.1016/j.mod.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guihard-Costa AM, Khung S, Delbecque K, Menez F, Delezoide AL. Biometry of face and brain in fetuses with trisomy 21. Pediatr Res. 2006;59:33–38. doi: 10.1203/01.pdr.0000190580.88391.9a. [DOI] [PubMed] [Google Scholar]
- Hall BK. The neural crest in development and evolution. Springer; New York, NY: Kluwer Academic Publishers; 1999. [Google Scholar]
- Hill CA, Reeves RH, Richtsmeier JT. Effects of aneuploidy on skull growth in a mouse model of Down syndrome. J Anat. 2007;210:394–405. doi: 10.1111/j.1469-7580.2007.00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Helms JA. The role of Sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development. 1999;126:4873–4884. doi: 10.1242/dev.126.21.4873. [DOI] [PubMed] [Google Scholar]
- Hu D, Marcucio R, Helms JA. A zone of frontonasal ectoderm regulates patterning and growth in the face. Development. 2003;130:1749–1758. doi: 10.1242/dev.00397. [DOI] [PubMed] [Google Scholar]
- Hu D, Marcucio RS. Unique organization of the frontonasal ectodermal zone in birds and mammals. Dev Biol. 2009a;325:200–210. doi: 10.1016/j.ydbio.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Marcucio RS. A SHH-responsive signaling center in the forebrain regulates craniofacial morphogenesis via the facial ectoderm. Development. 2009b;136:107–116. doi: 10.1242/dev.026583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J, Mao J, Tenzen T, Kottman AH, McMahon AP. Hedgehog signaling in the neural crest cells regulates the patterning and growth of the facial primordia. Genes Dev. 2004;18:937–951. doi: 10.1101/gad.1190304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klingenberg CP. MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resour. 2011;11:353–357. doi: 10.1111/j.1755-0998.2010.02924.x. [DOI] [PubMed] [Google Scholar]
- Lele S, Richtsmeier JT. An invariant approach to statistical analysis of shapes. London: Chapman and Hall-CRC Press; 2001. [Google Scholar]
- Marcucio RS, Cordero D, Helms JA. Molecular interactions coordinating the development of the forebrain and face. Dev Biol. 2005;284:48–61. doi: 10.1016/j.ydbio.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Marcucio RS, Young NM, Hu D, Hallgrimsson B. Mechanisms that underline co-variation of the brain and face. Genesis. 2011;49:177–189. doi: 10.1002/dvg.20710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan A, Chang L, Nguyen A, James AW. A review of hedgehog signalling in cranial bone development. Front Physiol. 2013;4:1–14. doi: 10.3389/fphys.2013.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinholdt LG, Ding Y, Gilbert GJ, Czechanski A, Solzak JP, Roper RJ, Johnson MT, Donahue LR, Lutz C, Davisson MT. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm Genome. 2011;22:685–691. doi: 10.1007/s00335-011-9357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richtsmeier JT, Baxter LL, Reeves RH. Parallels of craniofacial maldevelopment in Down syndrome and Ts65Dn mice. Dev Dyn. 2000;217:137–145. doi: 10.1002/(SICI)1097-0177(200002)217:2<137::AID-DVDY1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Richtsmeier JT, Zumwalt A, Carlson E, Epstein CJ, Reeves RH. Craniofacial phenotypes in segmentally trisomic mouse models for Down syndrome. Amer J Med Genet. 2002;107:317–324. doi: 10.1002/ajmg.10175. [DOI] [PubMed] [Google Scholar]
- Roper RJ, Baxter LL, Saran NG, Klinedinst DK, Beachy PA, Reeves RH. Defective cerebellar response to mitogenic Hedgehog signaling in Down [corrected] syndrome mice. Proc Natl Acad Sci. 2006;103:1452–1456. doi: 10.1073/pnas.0510750103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roper RJ, VanHorn JF, Cain CC, Reeves RH. A neural crest deficit in Down syndrome mice is associated with deficient mitotic response to Sonic hedgehog. Mech Dev. 2009;126:212–219. doi: 10.1016/j.mod.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva FA, Valladares-Neto J. Craniofacial morphological differences between Down syndrome and maxillary deficiency children. Eur J Orthod. 2013;35:124–130. doi: 10.1093/ejo/cjr105. [DOI] [PubMed] [Google Scholar]
- Singh N, Dutka T, Devenney B, Kawasaki K, Reeves RH, Richtsmeier JT. Acute upregulation of hedgehog signaling causes differential effects on cranial morphology. Dis Model Mech. 2015;8:271–279. doi: 10.1242/dmm.017889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slice DE. Modern Morphometrics in Physical Anthropology. New York: Kluwer Academic Publishers; 2005. [Google Scholar]
- Tapadia MD, Cordero DR, Helms JA. It’s all in your head: new insights into craniofacial development and deformation. J Anat. 2005;207:461–477. doi: 10.1111/j.1469-7580.2005.00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trazzi S, Mitrugno VM, Valli E, Fuchs C, Rizzi S, Guidi S, Perini G, Bartesaghi R, Ciani E. APP-dependent up-regulation of Ptch1 underlies proliferation impairment of neural precursors in Down syndrome. Hum Mol Genet. 2011;20:1560–1573. doi: 10.1093/hmg/ddr033. [DOI] [PubMed] [Google Scholar]
- Welsh IC, O’Brien TP. Signaling integration in the rugae growth zone directs sequential SHH signaling center formation during the rostral outgrowth of the palate. Dev Biol. 2009;336:53–67. doi: 10.1016/j.ydbio.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young B, Minugh-Purvis N, Shimo T, St-Jacques B, Iwamoto M, Enomoto-Iwamoto M, Koyama E, Pacifici M. Indian and sonic hedgehogs regulate synchondrosis growth plate and cranial base development and function. Dev Biol. 2006;299:272–282. doi: 10.1016/j.ydbio.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Young NM, Chong HJ, Hu D, Hallgrimsson B, Marcucio RS. Quantitative analyses link modulation of sonic hedgehog signaling to continuous variation in facial growth and shape. Development. 2010;137:3405–3409. doi: 10.1242/dev.052340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young NM, Hu D, Lainoff AJ, Smith FJ, Diaz R, Tucker AS, Trainor PA, Schneider RA, Hallgrímsson B, Marcucio RS. Embryonic bauplans and the developmental origins of facial diversity and constraint. Development. 2014;141:1059–63. doi: 10.1242/dev.099994. [DOI] [PMC free article] [PubMed] [Google Scholar]