

Abstract
Cutinases are powerful hydrolases that can cleave ester bonds of polyesters such as poly(ethyleneterephthalate) (PET), opening up new options for enzymatic routes for polymer recycling and surface modification reactions. Cutinase from Apsergillus oryzae (AoC) is promising owing to the presence of an extended groove near the catalytic triad which is important for the orientation of polymeric chains. However, the catalytic efficiency of AoC on rigid polymers like PET is limited by its low thermostability; as it is essential to work at or over the glass transition temperature (Tg) of PET i.e. 70 °C. Consequently, in this study we worked towards the thermostabilization of AoC. Use of Rosetta computational protein design software in conjunction with rational design led to a 6 °C improvement in the thermal unfolding temperature (Tm) and a 10-fold increase in the half-life of the enzyme activity at 60 °C. Surprisingly, thermostabilization did not improve the rate or temperature optimum of enzyme activity. Three notable findings are presented as steps toward designing more thermophilic cutinase: (a) surface salt bridge optimization produced enthalpic stabilization, (b) mutations to proline reduced the entropy loss upon folding and (c) the lack of a correlative increase in the temperature optimum of catalytic activity with thermodynamic stability suggests that the active site is locally denatured at a temperature below the Tm of the global structure.
Introduction
Enzyme catalysts provide “green” routes for chemical synthesis and modification reactions that avoid chemically intensive processes such as those requiring high temperature-pressure, protection-deprotection steps, and which generate toxic by-products (1, 2). Exciting opportunities exist to develop enzyme-catalyzed routes that enable polymer surface modification as well as polymer recycling. Polymer recycling generally requires energetically and chemically intense methods (e.g. mechanical, thermal and chemical recycling) 3,4. Similarly, surface modification of polymers often requires costly and specialize reagents and equipment (e.g. plasma modification as well as harsh reaction conditions 5–7. In contrast, inherent advantages of enzymatic surface chemistry includes specificity, reduced energy consumption, reactions in aqueous media and which occur exclusively at material surfaces since proteins are immiscible with most polymeric materials 8. Avoiding degradation well below the surface of polymeric materials avoids losses in mechanical properties. Furthermore, enzymatic material hydrolysis can continue from the surface, creating new surfaces that eventually lead to complete hydrolysis of a polymeric material (see examples below).
Cutinases are esterases produced by various phytopathogenic organisms to breakdown cutin, the biopolyester coating found on leaves and fruit 9 They also are powerful hydrolases that can cleave ester bonds of aliphatic polyesters such as poly(ε-caprolactone), PCL 10, poly(butylene succinate), PBS 11 and poly(3-hydroxybutyric acid, P3BH 12. In addition, cutinases catalyze the hydrolysis of relatively rigid aliphatic-aromatic polyesters such as poly(ethylene terephthalate), PET 13–16. This opens new options for recycling PET that is used as a packaging material in a wide range of bottles and containers that hold products such as beverages, detergents and cosmetics. Indeed, PET products have a high recycling value 17. Furthermore, cutinases can be used for surface modification of polyesters including PET to increase surface hydrophilicity (17, 18). In addition, cutinases hydrolyze ester side chains. Important examples include cutinase catalyzed deacetylation of polyvinyl acetate that can used for enzymatic removal of adhesives from paper during repulping 20, cellulose acetate deacetylation to improve the cellulose acetate fiber hydrophilicity for dyeing 21 or for its complete degradation by combining cutinases with cellulases 22.
Cutinases from several microbial sources have been identified and characterized for their activity on polymeric substrates e.g. Fusarium solani pisi (FsC) 23, Aspergillus niger 24, Aspergillus oryzae (AoC) 10, Pseudozyma Antarctica 11, Thielavia terrestris 12, Thermobifida fusca 25, Thermobifida alba 26 among others. However, for practical applications, the stability and catalytic efficiency of cutinases must be improved. Recently, Chen et al. reviewed strategies employed to engineer cutinase for improved activity on different substrates 27. Frequently reoccurring optimization rationales are as follows: i) create more accessible space for the active site 13, ii) increase the hydrophobicity near the active site 28 and iii) improve substrate binding 29.
Cutinase activity for polyester hydrolysis is highly dependent on whether the enzyme is functioning below or above the substrate glass transition temperature, Tg (the temperature at which the mobility of chains increases such that amorphous materials become more rubber-like), and the degree of crystallinity of semi-crystalline polymers. Enzymatic hydrolysis below Tg (or in locally crystalline domains) is generally slower because of decreased chain mobility and, therefore, poorer access of enzyme active sites to labile groups 30–32. Wild type cutinases have evolved to act on cutin, an amorphous polymer that has a fairly low Tg (e.g 22-25 °C for tomato fruit cutin depending on the age after anthesis) 33. However, aliphatic-aromatic copolyester such as poly(butylene terephthalate), PBT, and PET have Tg values of about 66 and 75 °C, respectively, that are well above that of cutin. Most of the known cutinases are stable from 45-50 °C with the exception of a few produced by thermophilic microorganisms, which are stable in the range of 55-70 °C e.g. H. insolens (Tm=63 °C) 34, T. fusca (Tm =70 °C) 35, T. Alba (Tm =60 °C) (25, 35) and a mesophilic fungi A. oryzae (Tm = 62 °C) 10. Recently a cutinase identified from leaf branch compost using a metagenomics approach was reported to be highly thermostable with the Tm of 83 °C, however the enzyme suffers lower kinetic stability at higher temperature (t1/2 of 40 minutes at 70 °C) at the same time loses activity at temperatures higher than 50 °C 36. Given the commercial importance of catalysis at or exceeding Tg for polymers such as PBT and PET to allow efficient surface modification and mild recycling to monomers, this paper is focused on thermostabilization of the cutinase from Aspergillus oryzae (AoC).
Thus far, the majority of studies to improve cutinase performance have been focused on enhancing activity. The following summarizes published research on cutinase thermostabilization. Chin et al. achieved a 2.5 fold increase in the half-life of Glomerella cingulata cutinase at 50 °C with a single mutation (N177D). They also reported an increase in the activity optimum temperature 37. Kawai et al. reported an increase in the Tm by approximately 4 °C (to final Tm of 59 °C) of a putative cutinase from Saccharomonospora viridis AHK190 by a single mutation (S226P-proline insertion) 38. While these studies using rational design strategies are promising, higher degrees of thermostabilization are desired as discussed above using PET recycling as a prominent example.
Thermostabilization strategies include random mutagenesis 39 and directed evolution 40. However rational design, as an optimization strategy, ideally saves significant experimental time 41 and can make use of multiple stabilization mechanisms observed in naturally evolved thermostable proteins 42. The software platform Rosetta has been a central tool for computer aided rational protein design (42, 43), and has been used in isolation and as a component of larger pipelines to improve bothenzyme activity and protein thermostability45–49
The cutinase from Aspergillus oryzae (AoC) was shown to have better activity on longer chain-length aliphatic ester substrates owing to its extended hydrophobic groove near the active site. The enzyme has also shown good thermostability (Tm of 62 °C at pH 8) owing to a third disulfide bond 10. In this work, we used a rational design approach to further improve the thermostability of AoC with the goal of enhancing activity towards polymeric substrates. We used a fixed backbone design protocol to identify sets of variants for experimental testing, which yielded two improved variants. The mutations in the better variants were combined or retained for the next set of the designs. After two stages of designing an overall 6 °C improvement in the Tm and 10 fold increase in the t1/2 at 60 °C was obtained. However, surprisingly, this did not result in an improvement in the enzyme activity optimum temperature. Furthermore, the enzyme activity was lower than the wild-type. The results were rationalized to gain insight into the structure-activity and stability relationships of the cutinases.
Materials and Methods
Chemicals
Substrate 4-nitrophenyl butyrate (pNPB) and salts monosodium phosphate and disodium phosphate were purchased from Sigama Aldrich. Zeocine was purchased from Invitrogen. All the media components used 10 were also procured from Sigma Aldrich. The polymeric substrates PCL (Tone grade 787, Mw = 80 000,) was purchased from Dow Corning. Polybutylene succinate adipate (PBSA) (Bionelle 3001MD, Mn=23,300,) was a kind gift from Showa Denko. The polymeric films were prepared by compression molding the crystallinity of the films was determined using Differential scanning calorimetric analysis and was found to be 50-55% and 30-35% for PCL and PBSA respectively.
Design of variants
The crystal structure of wt-AoC (PDB code - 3GBS) was used for the rational design. We sought to identify primarily local ‘cliques’ of mutations that may confer thermostabilization, and which could later be recombined in future rounds should they prove to have stabilizing effects. Rosetta3 was used to generate 32 fixed backbone redesigns 44. During the design simulation, key active site residues, disulfides, and prolines were held fixed in the native rotamer conformation (namely residues 37, 113, 63, 76, 177, 184, 181, 194, 126, 90, 36, 39, 51, 60, 81, 88, 93, 116, 169, 192, and 193). Additionally, several surface arginines (162, 82, and 65) were only allowed to change to alternate rotamer conformations. All other positions were allowed to mutate to any type other than cysteine and histidine. See supplemental material for the Rosetta command line used for the design of the variants. The designed sequences were analyzed for favorable cliques of mutations via visual inspection and sequence comparison to cutinases from Aspergillus flavus and Neosartorya fischeri (as exemplars to identify conserved residues). We selected 9 variants for experimental evaluation (Table I). The number of mutated residues for each AoC variant in the initial round of 9 constructs ranged from 2-7. After analysis of the first set of variants, a second set was designed. In two of these new variants (10 and 11) the mutations of the two best variants from the first round (3 and 5) were combined (variant 11 added one additional presumed stabilizing mutation). In the other new variant (12), residues near the mutations of the variants 3 were redesigned with Rosetta.
Table I. AoC Variants studied: Mutations and the predicated rationale for thermostabilization.
| Variant | Mutation | Rationale |
|---|---|---|
| First stage variants designed using Rosetta | ||
| 1 | V150I, I136V | Minor core repacking |
| 2 | L26D, G28E, D30R, K67R, | Replace a glycine, add surface salt bridges |
| 3 | A102D, Q105R, G106E | Replace a mid-helix glycine, add surface salt bridges |
| 4 | Q110W, K114W | Add hydrophobic contacts |
| 5 | N133A, S140P, E161T, A166P | Add two prolines, shorter side chains |
| 6 | A178P, V179P | Add consecutive prolines |
| 7 | R46P | Add one proline |
| 8 | K174R, Y176F, A178E, D200R, G202E, D203E, D206R | Surface salt bridge network |
| 9 | T84R, D86L, A99E, A100S | Surface salt bridge network |
| Second stage designs | ||
| 10 | A102D, Q105R, G106E, N133A, S140P, E161T, A166P | Variant 3 plus Variant 5 |
| 11 | A102D, Q105R, G106E, N133A, S140P, E161T, A166P, K137E | Variant 10 plus charge stabilization |
| 12 | A102D, Q105R, G106E, Q98N, A99D, E109Q | Variant 3 plus new salt bridges involving A99D. Q98N removes one methylene of non-polar |
Gene synthesis and transformation
The wt-AoC and variants were heterologously expressed in Pichia pastoris using the strong methanol-induced AOX I promoter. The variant genes were synthesized using site directed mutagenesis with optimized codon usage at DNA2.0. The protein was expressed with the Saccharomycec cerevisiae alpha factor tag on the N terminal for extra- cellular secretion and the six histidine tag on the C-terminus for ease of purification using affinity chromatography. The gene was cloned in PJ912 express vector with Pichia pastoris AOX I promoter homologous sequence. Multiple copies of the vector were generated in E.coli DH10α cells. The vector was purified and subsequently linearized using the Sac I restriction enzyme which has the site at the AOX I promoter homologous sequence of the plasmid. The linearized plasmid was then transformed into electro-competent Pichia cells by electroporation. The selection of the transformants was performed on a YPD agar plate with 1mg/ml zeocine. The best expressing colony was picked from 10 prominent colonies on YPD Zeocine 1mg/ml plates based on a small scale expression test. Where, the transformants were grown in 96 well plates at 1 ml scale and the protein secreted upon induction was analyzed using Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) analysis of concentrated cell free supernatant.
Protein expression and purification
The bulk production of proteins was performed using a DASGIP (Germany) parallel fermentation system where 4 proteins can be produced simultaneously at the scale of 0.5-1L. Protein expression and purification was performed using a previously published protocol 10 with slight modification. During fermentations, dissolved oxygen was maintained at 30% during the feeding of 50% glycerol by varying the feed rate apart from the agitation cascade control. Protein purification was performed by metal ion affinity chromatography using a Poros MC20 column (Life technologies) with a GE AKTA Pure FPLC system (GE AKTA Pure 25) at 4 °C. It is essential to perform the purification at cold temperatures (4-15 °C). SDS PAGE analysis of the purified proteins was performed to confirm their purity.
Protein analysis
The protein concentrations of wt-AoC and variants used for various analyses were determined using a standard Bicinchonic acid assay (BCA) analysis. The standard curve was prepared using pure wt-AoC.
Enzyme activity measurements
PNPB hydrolysis assay
A standard p-nitrophenyl butyrate (PNPB) hydrolysis assay was used to determine esterase activity. The assay was performed at 3ml scale in a 3.5ml cuvette in Molecular devices Spectrophotometer M2. The assay was initiated by addition of 75 μl PNPB solution (40mM in methanol) to the diluted enzyme solution (5-10nM) in 20 mM phosphate buffer pH 8 containing 1% glycerol. The absorbance at 405 nm was monitored for 1.5 min. One unit of enzyme activity was determined as one μM of PNP (p-nitrophenol) formed per min per ml enzyme solution using 0.00127 M-1 as the extinction coefficient determined experimentally under assay conditions.
Polymer hydrolysis assay
Cutinase activity for polyester hydrolysis was assayed using a pH-stat apparatus (Titrando 842, Metrohm) equipped with Tiamo 1.1 software. All reactions were performed in the assay buffer (0.5 mM Tris solution with 10% glycerol). The assay was performed on polymeric substrates in film form, cut into squares (0.5 × 0.5 cm). A total film surface area of 2 cm2/mL was transferred into 7.5 ml assay buffer and 40 °C. The hydrolysis was initiated by the addition of enzyme solution to the final concentration of 100nM. Polyester hydrolysis leads to a decrease in pH owing to release of the acid. The pH was maintained at a desired value by automated addition of 100 mM NaOH. Control experiments (without enzyme) were performed to determine background chemical hydrolysis which was subtracted from total hydrolysis in the presence of enzyme. The polyester hydrolysis activity was calculated as the micromoles of NaOH per hour per unit reaction volume from the rate of addition of the NaOH. The variants were compared based on their activity for PCL hydrolysis (Table II) and PCL was also used to determine the pH optimum of variant and wt-AoC (Fig. S2- Supplemental material). To determine the temperature optimum, the assay was carried out at varying temperature and polybutylene succinate adipate (PBSA) was used as the substrate exactly as was described above for assays with PCL (Fig 6).
Table II. Thermostability and activity analysis of AoC variants in comparison to Wt-AoC.
| Variant | Tm (°C) | ΔG (thermal) at 20°C (kJ/mol) | ΔΔG vs wt at 20°C | ΔS (kJ/mol/°K) | ΔG (urea) at 20°C, 0M urea (kJ/mol) | ΔΔG vs wt at 0M urea (kJ/mol) | equilibrium m-value (kJ/mol/mol/L urea) | t1/2 at 60°C (Hrs.) | % of wt Activity: PNPB | % of wt Activity: PCL | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| First stage designs | |||||||||||
| wt-AoC | 62.3±0.3 | -74.3 | 0 | -1.76 | -22.3 | 0 | 5.7 | 4.6±0.25 | 100±5.6 | 100±6.2 | |
| 1 | 60±1 | - | - | - | - | - | - | 1.6±.3 | 80.9±2. | 84±5.2 | |
| 2 | 63±1 | - | - | - | - | - | - | 1.8±0.15 | 79.1±2.9 | 27±9.1 | |
| 3 | 65.7±0.2 | -72.5 | 1.8 | -1.59 | -39.6 | -17.3 | 9.1 | 13.5±0.5 | 90.9±7.9 | 93±7 | |
| 4 | No expression | ||||||||||
| 5 | 64.0±0.3 | -49.8 | 24.5 | -1.13 | -19.2 | 3.1 | 5.7 | 9.6±0.5 | 78.2±8.5 | 73±9 | |
| 6 | 58±1 | - | - | - | - | - | - | 1.7±0.2 | 52.7±7.2 | 25±7.8 | |
| 7 | 59±1 | - | - | - | - | - | - | 18.2±9.1 | 4.5±10.3 | ||
| 8 | 60±1 | - | - | - | - | - | - | 6±0.5 | 89.2±8.6 | 78±8.1 | |
| 9 | 52±1 | - | - | - | - | - | - | 1.7±0.3 | 38.2±6.8 | 9±11.1 | |
| Second stage designs | |||||||||||
| 10 | 68.0±0.5 | -55.5 | 18.8 | -1.16 | -23.7 | -1.4 | 5.4 | 59.2±2.5 | 64.2±4.6 | 68.1±5.6 | |
| 11 | 68.3±0.2 | -66.2 | 8.1 | -1.37 | -37.7 | -15.4 | 8.5 | 48.6±1.5 | 57±6.2 | 54±2.4 | |
| 12 | 67.1±0.4 | -81 | -6.7 | -1.72 | - | - | - | 11.2±0.5 | 86±3.2 | 94±5.1 | |
Figure 6.
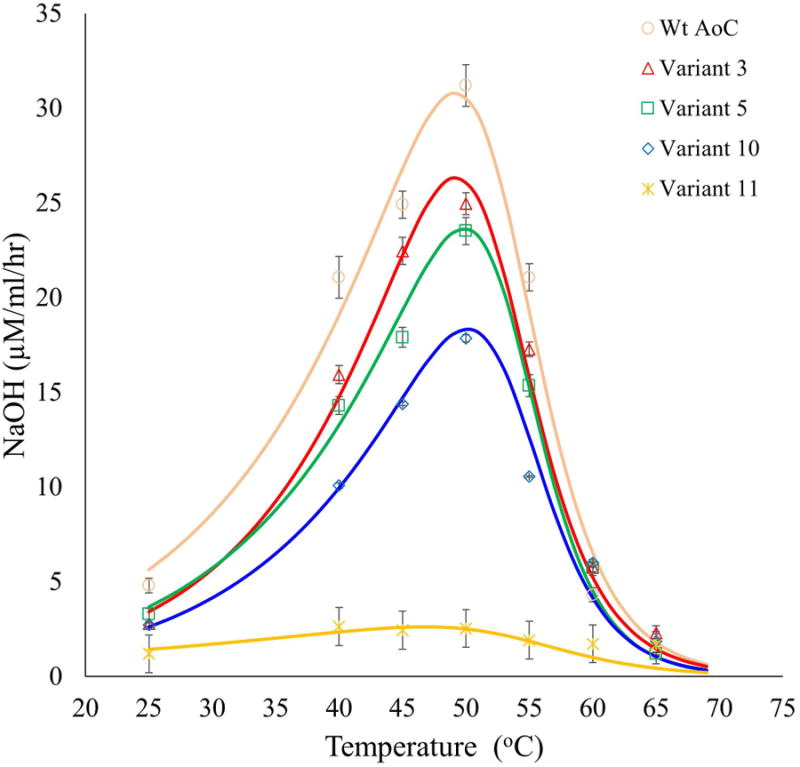
Analysis of the optimum temperature for PBSA hydrolytic activity for variants with higher thermostability than wt-AoC. Data were fit to the Arrhenius equation and two-state thermal equilibrium. There are no significant differences in the temperatures of inactivation or the temperature of optimum activity. Continuous lines represent fitted data.
Kinetic stability studies
The kinetic stability analysis of wt-AoC and AoC variants was performed using residual activity analysis. Protein solution (1 μM) in 20mM phosphate buffer pH 8 was incubated at 60 and 65 °C in an Eppendorf thermomixer with constant shaking at 300 rpm. Sample aliquots were withdrawn from incubations at specific time intervals, rapidly cooled on ice for 5 min and then kept at room temperature for 10 min before activity measurements were recorded using the PNPB assay. The data obtained were fit to first order deactivation kinetics and the half-life times were determined (Except for variant 5 and 10 where the two step deactivation pathway was observed and the half life time was reported as the approximate time when the 50% of the enzyme activity is lost)
Circular dichorism (CD) measurements
CD spectra were recorded using a JASCO J-815 Spectropolarimeter equipped with a Jasco Peltier-type temperature controller. The secondary structure and thermostability comparison of the variants was performed at pH 8 (20mM phosphate buffer) using 10 μM protein solutions. The data for wavelength scans were collected at 1nm intervals from 190-250nM. The temperature scan was performed at 1 °C/min from 20-75 °C. Observed ellipticities were fit to the Gibbs equation with a linear baseline correction, giving Tm, enthalpy, and entropy of folding. At 1 °C/min heating rate the equilibrium at each temperature would be rapidly attained. However thermal unfolding is reversible if cooled quickly after heating, but not if misfolding or aggregation occurs upon longer incubation. Hence these data cannot be considered to be at equilibrium. Instead, the resulting fit parameters should be interpreted qualitatively.
Denaturation with urea
Protein solutions (10 μM) were prepared in pH 8 phosphate buffer containing varying concentrations of urea (0M- 8M) and incubated for 24 hours at 25 °C. After incubations, the CD wavelength scan was performed from 240-200nm. The averaged ellipticity at 222nm (CD222) was plotted against urea concentration to track the folded to unfolded transition. The fraction unfolded (U) was calculated as U = [CD222(x) - CD222(0M)]/[CD222(8M) - CD222(0M)], where x is the urea concentration.
The value of U was fit to a two-state or three-state model assuming a linear relationship between urea concentration and the free energy of folding. That is, the fraction unfolded U was fit to ef/(1 + ef), where f = (ΔG0 - m[urea])/RT, giving the equilibrium free energy of folding in pure water ΔG0, and the equilibrium m-value. A three state fit was used if the data deviated significantly from the best two-state fit. For a three state fit, U was fit to Aef/(1 + ef) + (1-A)ef/(1 + ef), with three additional parameters including the relative amplitude of the two transitions. Explanations are proposed for the presence or absence of a second transition at low urea concentration.50
Results and Discussions
First set of variants
Design
The crystal structure of AoC (PDB id 3GBS) was subject to fixed backbone redesign with Rosetta3 as described in the methods section. From this analysis, 9 variants, mostly containing local cliques of mutations, were selected and experimentally screened for thermostabilization effects [Table I & Fig. 1a,1b]. The 9 variants can be divided to various strategies of stabilization. For example, Variant 1 (V150I, I136V) explores small changes in hydrophobic amino acids to improve core repacking. Variants 2, 3, 8, and 9 all introduce surface salt bridges. In addition, Variant 2 also replaces a native glycine with a negatively charged glutamate residue. Indeed, surface salt bridge formation is a proven strategy for thermostabilization 51. Variant 4 introduces two new tryptophan residues to enhance hydrophobic contacts. Variants 5, 6, and 7 introduce relatively rigid proline residues. Proline insertion is a well-established approach towards protein thermostabilization aimed at decreasing the unfolding entropy 52.
Figure 1.
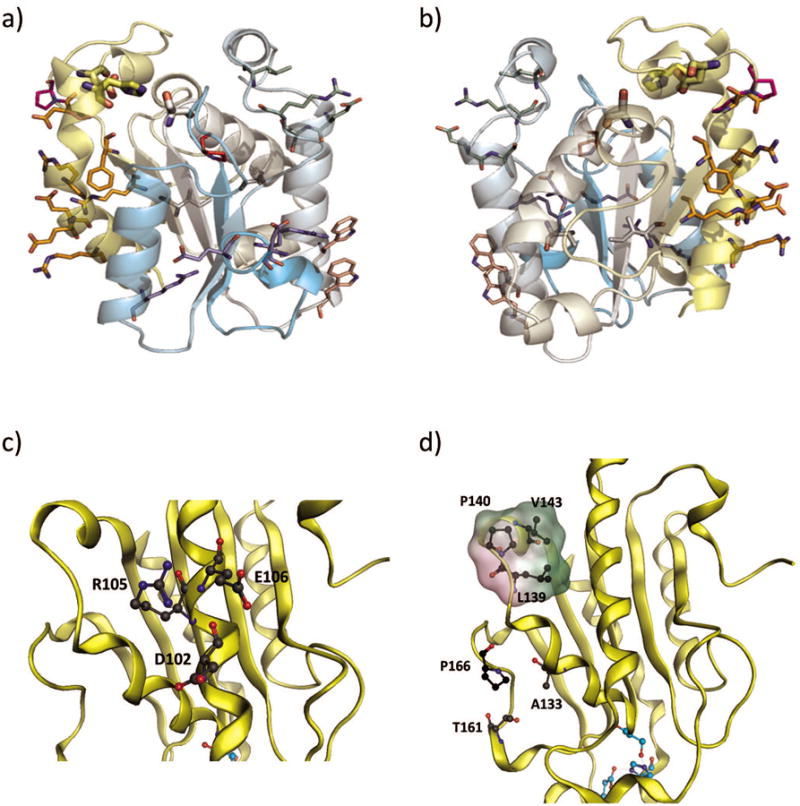
AoC variants used in this study. a) and b), two views of AoC showing mutations of Variant 1 (white), 2 (purple), 4 (salmon), 6 (magenta), 7 (red), 8 (orange), and 9 (pale green). The backbone is colored from cyan N-terminius to yellow C-terminus and the catalytic triad side chains are shown as large sticks. c) Variant 3 surface salt bridge network formation are shown by backbone color. d) Variant 4 proline insertion and the hydrophobic patch formed by (P140, L139, V143- surface rendered green for hydrophobicity)
Thermostability analysis of first round designs
Comparative thermostability of wt-AoC with the variants was analyzed by CD thermal scans to determine Tm (unfolding temperature), a measure of the thermodynamic stability. However, owing to the irreversible nature of the thermal unfolding of wt-AoC and variants Tm referred in this study is an apparent Tm. Concurrently, the half-life time (t1/2) or time for loss of 50% activity at 60 °C was determined as a measure of the kinetic stability. Successful expression in Pichia pastoris of 8 of the 9 AoC variants, designed by Rosetta for increased thermal stability, was achieved in volumetric yields ranging from 400 to 600 mg/l.
Thermostability analysis revealed significant improvement for three variants. Specifically, Variant 2, 3 and 5 showed an improvement in Tm of 0.7, 3.4 and 1.7 °C, respectively. Furthermore, Variants 3 and 5 showed increased half-life at 60 °C relative to the wt-AoC of approximately 3 and 2-fold, respectively. A decrease in thermostability relative to the wild-type enzyme was observed for Variants 6, 7 and 9, whereas, the remaining variants did not show a significant change in thermostability (Table II). Detailed structural analysis coupled with the thermodynamic analysis of the protein unfolding data was performed to rationalize the results of the first stage variants.
Variant 1 was the only variant which directly altered packing of the hydrophobic core (V150I, I136V). These substitutions are conservative, constituting an exchange of a methylene unit between adjacent residues. They did not provide any improvement in stability.
Variant 2 introduces a pair of salt bridges (between L26D and D30R, G28E and K67R) which are > 20 Å from the catalytic site. The more modest effect of these mutations on thermostability may be due to the proximity of this clique to the N-terminus (L26 is the most N-terminal residue resolved in 3GBS). If this region is more flexible, the formation of salt bridges there may be less effective for thermostabilization.
Variant 3 introduces a surface charged cluster (A102D, Q105R, G106E) to a location that is distant from the active site (A102 is > 20 Å from the catalytic His) (Fig. 1c). Of the 4 variants that introduce new surface charge networks (2, 3, 8, and 9), three (2, 3, and 8) had improved stability relative to wt-AoC (Variant 8 showed an increased kinetic stability). These results suggest surface charge modulations are a productive optimization strategy to explore more deeply. Stability of variant 3 is also supported by G106E, which removes an unfavorable glycine from the middle of a helix. Because variant 3 incorporated ionizable residues, its pH sensitivity was determined and compared to wt-AoC. However, both for activity and stability, the corresponding pH optima for variant 3 and wt-AoC were identical within error [Fig S1, Supplemental Material].
Variant 4 (Q110W, K114W), designed for improved hydrophobic contacts, did not express. In retrospect, the two mutations (Q110W, K114W) alter a sequence pattern for a helix cap motif 53 and, in doing so, may have interfered with folding leading to difficulties in secretion. Similarly, the secretion of the variant (G82A, A85F, V184I, A185L, L189F) of Fusarium solani cutinase (FsC), designed to improve hydrophobic contacts, was found to be affected in Saccharomyces cerevisiae 54. This enzyme was retained in the endoplasmic reticulum whereas wt-FsC was secreted.
Variant 5 introduces two new prolines and two compensating mutations to alleviate steric clashes. The mutations are not proximal to the active site. Proline mutations contribute to themostabilization by decreasing the entropy lost upon folding (Table II), Furthermore, enhanced thermostabilization results by introducing new hydrophobic interactions between 140P, 143V and 139L (Fig 1d). However, A166P and S166P were also responsible for the loss of one backbone hydrogen bond each, leading to a decreased enthalpy of folding.
Variant 6 introduces two sequential prolines that result in a net loss of stability and activity. This is attributed to the loss of one backbone hydrogen bond and deleterious steric clashes with Y197.
Variant 7, in which a single proline mutation (R46P) was introduced, has a Tm that is 3 °C below that of wt-AoC. The position of this proline mutation is at the N-terminal base of a loop with the amino acid sequence RASTE that is adjacent to the catalytic triad. Although Rosetta finds a proline to be highly favorable given the residue's crystallographic backbone conformation, it appears that the energetic contribution of the R46 side chain in coordinating four backbone oxygens was underestimated.
Variant 8 has 7 mutations, the largest number of amino acid substitutions in the first design set (Table I). Furthermore, mutations in this variant include those close to the active site (A178E is within 7 Å of the catalytic His). The clique also includes the Y176F mutation that affects the boundary of the hydrophobic core. Given the large number of alterations relative to the wt-AoC, rationalizing the contributions of all individual interactions with the experiential data is challenging.
Variant 9 introduced salt bridges that destabilize the protein. The introduction of surface hydrophobicity at D89L may have led to poor folding kinetics. The mutation was intended to pack against T84R and compensate for the loss of the native hydrogen bond between T84 and D89. Additionally, the introduction of A99E and A100S creates a helix N-cap sequence pattern that would extend the helix by one residue, but this is incompatible with the native structure 55.
Thermostability analysis of second round designs
Based on the results of the first set of designs, substitutions in Variants 3 and 5 were deemed most promising and worthy of further examination. Three new variants were designed including the combination variant of Variant 5 and 3; Variant 10 (Fig 2). Given the spacing of substitutions in Variant 5 and 3, we expected the energetic changes to be additive. Variant 11 differs from Variant 10 by one extra mutation, K137E, which was incorporated to stabilize the positive charge cluster in that region (Fig 2). To create Variant 12 (Fig 2), Rosetta simulations were run to design clusters of residues around the mutations of Variant 3. Of the nine selected residues (97, 98, 99, 101, 107, 109, 110, 135, 139), Rosetta identified 4 stabilizing mutations: Q98N, A99D, E109Q and Q110L. Q110L was rejected since a leucine on the enzyme surface would stabilize the unfolded state. The mutation Q98N removes a methylene's worth of hydrophobic surface area. An aspartic acid in place of alanine at 99 (A99D) was predicted to form a stabilizing salt bridge with R82. E109Q was predicted to interact with Q105R and G106E. Furthermore, Q109 is conserved in A. Flavous cutinase.
Figure 2.
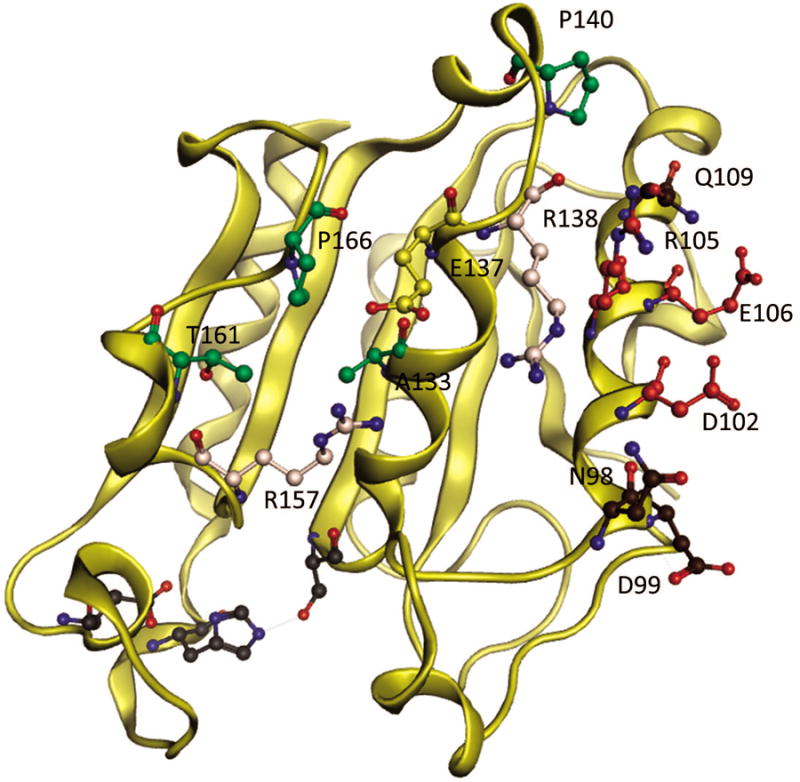
Aspergillus oryzae cutinase second stage designs, Variant 11- combination variant of Variant 5 (residues in green) and Variant 3 (residues in red) with an extra mutation of K137E (in yellow) as compared to Variant 10. K137E stabilizing positive charge cluster formed by native residues R157, R138 in peach, Variant 12 – residues in brown are mutations in addition to those derived from Variant 3. Residues in grey represent the active site.
Analysis by CD temperature scans revealed an improvement in the thermostability of Variants 10 and 11 relative to the wt-AoC by 5.7 and 6.0 °C, respectively. In contrast to these modest improvements, the kinetic stability of Variants 10 and Variant 11 at 60 °C revealed large improvements. While the t1/2 at 60 °C for wt-AoC is 4.6 h, Variant 10 and 11 have t1/2 values that exceed 40 h (Fig 3 and 4, Table II). At 65 °C, wt-AoC has a t1/2 of 10 min and is nearly 100% unfolded in 25 min. In contrast, at 65 °C, variants 10 and 11 have t1/2 values of 4 and 5 h, respectively. The higher stabilities of Variants 10 and 11 relative to 3 and 5 confirmed the additive effect of stabilization introduced for individual mutations in Variant 3 and 5. Variant 10 has the same low folding entropy as Variant 5, and the free energy of folding falls in between that of Variants 3 and 5 as is expected for an additive effect. Variant 11 has a slightly higher folding entropy but also a higher enthalpy, probably due to the formation of a network of salt bridges. The melting temperature of Variant 11 is not significantly different from that of Variant 10. However, the deactivation kinetics analysis of Variant 10 and 11 at 60 °C showed important differences. Variant 10 has a two phase deactivation profile (Fig 4) while Variant 11 fits a single phase. The two phase deactivation was also observed for Variant 5 but wasn't clear owing to the faster deactivation. The two phase deactivation may be attributed to the relatively fast formation of a partially unfolded state that retains a lower level of activity, or to aggregation as explained by Baptista et al in case of wt-FsC. However, formation of the hypothesized intermediate partially unfolded state or aggregate was apparently rescued by the mutation K137E in Variant 11. K137E, as explained earlier, was designed to stabilize positive charge, which accumulated by the combination of mutations in Variants 3 and 5. The observed rescue by this single mutation in Variant 11 points towards the significance of the region in thermal unfolding.
Figure 3.
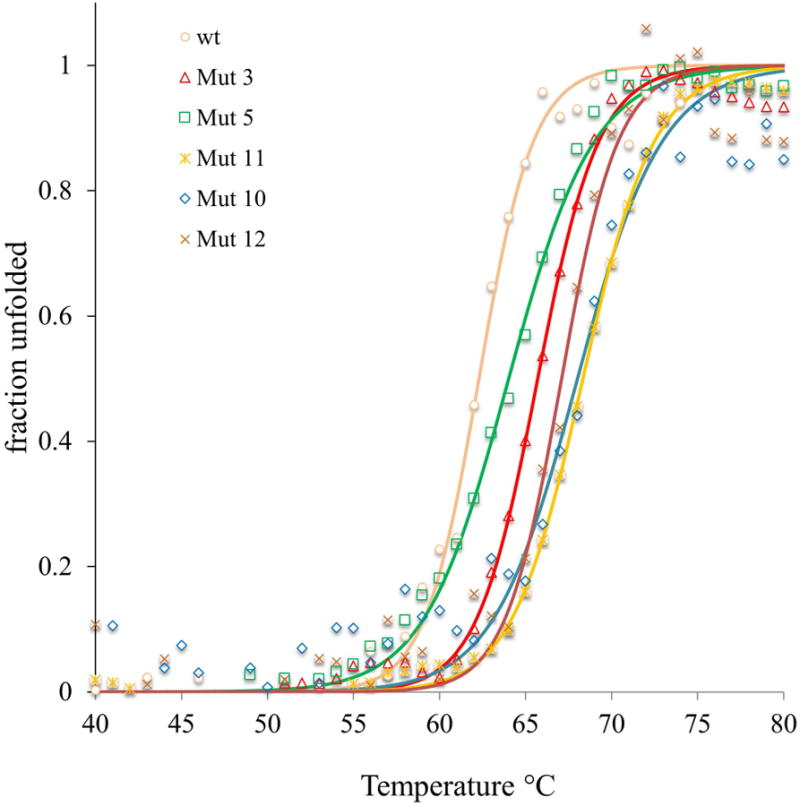
CD temperature scans at 222 nm for variants with higher thermostability than wt-AoC. Continuous lines represent fitted data.
Figure 4.
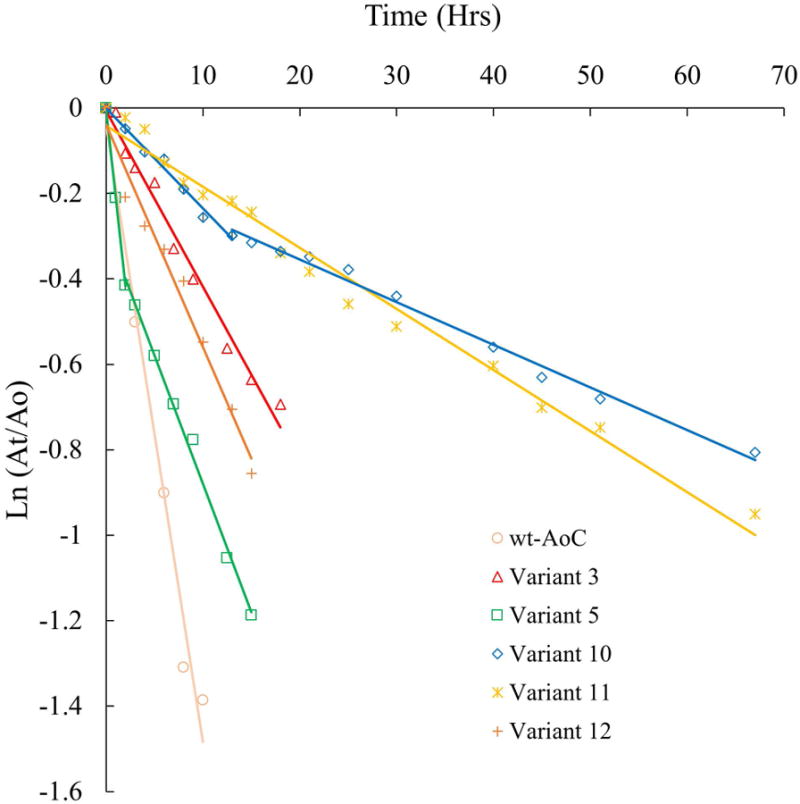
Kinetic stability analysis at 60 °C for variants with higher thermostability than wt-AoC. Continuous lines represent fitted data
Variant 12 was found to be the thermodynamically most stable variant, improving on wt-AoC by 6.7 kJ/mol. The thermodynamic stabilization can be attributed to the formation of a salt bridge network along the central 97-116 helix. Removal of one glycine, combined with the addition of longer side chains, resulted in no net change in the entropy of folding. However, Variant 12, which has a Tm 1.4 °C higher than Variant 3, has a t1/2 at 60 °C of 11.2 h, 2.9 h less than Variant 3, showing the important contribution of lower the folding entropy towards longer half-life. While Variant 12 is more stable at 25 °C, Variants 10 and 11, with proline mutations that decrease the entropy loss upon folding, are more stable at 60 °C. The G106E mutation was intended to decrease the entropy loss on folding. This decrease in folding entropy was observed in Variant 3, but this effect was lost in Variant 12.
Stabilization against urea denaturation
Thermal stabilization may lead to increased stabilization against urea denaturation and/or chemical denaturation. Thermal stabilization depends more on the entropy of folding, while stability to chemical denaturation depends more on the enthalpic contributions and the amount of buried surface area 50. The stabilities of wt AoC and Variants 3, 5, 10 and 11 were further evaluated by the equilibrium urea denaturation (24 h incubation) experiment. Unlike thermal denaturation urea denaturation was found to be completely reversible for wt-AoC and variants studied, based on the retention of the total activity upon dialysis to remove urea (Data not shown). The fraction unfolded was determined by CD at points ranging from 0 to 8M urea, and these were fit to 2 or 3-state equilbrium models, depending on the data. The deviation of the 1 h and 24 h (equilibrium) measurements was viewed as a crude assessment of kinetic stability. The equilibrium m-value of folding, a measure of exposed surface area in the unfolded state, was obtained from the fits. As expected, no significant change in the m-value was observed, signifying that the mutations did not affect the structure of the unfolded state.
Variant 3 is more resistant to urea than wt-AoC, but Variant 5 is significantly less resistant. This is surprising since Variant 5 is more thermostable than the wt-AoC. However, the lower resistance of Variant 5 to urea is consistent with the enthalpically destabilizing effect of the two proline substitutions and the concomitant loss of hydrogen bonds. The additive effect towards thermostabilization observed upon combining mutations in Variants 3 and 5 wasn't observed in case of urea denaturation for Variants 10 and 11. Both Variant 10 and Variant 11 were found to have similar resistance as Variant 3 against urea denaturation i.e 3.5-4M urea.
For Variants 5, 10 and wt-AoC, the unfolding traces were best fit to a 3-state model, with some observed unfolding at lower concentrations of urea. Variant 3 clearly fit a 2-state model, implying a locally increased resistance to urea. Variant 11, with one additional mutation, lost the early unfolding state but did not affect the main unfolding transition. The structural nature of the low-urea state is not clear, but that unfolding step must involve the region around the K137E mutation, which is the only difference between Variants 10 and 11.K137E introduces a partially-buried salt-bridge between E137 and R157, which crosses the deep active site cleft. The early loss of structure, and perhaps the loss of activity at temperatures below the melting point, may be due to the localized unfolding of the active site region, approximately residues 154-194.
AoC structure-activity relationships
Activity analysis of the variants in comparison with the wt-AOC
The activity of variants were evaluated on soluble (PNPB) and insoluble (poly[ε-caprolactone], PCL) substrates. The first set of variants was designed by Rosetta keeping the active site residues fixed. Nevertheless, the activities of this variant set showed large fluctuations. For example, by the PNPB assay, the activity of Variant 7 and 9 decreased by 80 and 60% relative to wt-AoC (Table II). In contrast, the activity of Variant 3 was nearly identical to wt-AoC. Furthermore, inspection of CD wavelength scans for the variant set shows only small variations in the corresponding spectra [Fig SI 1 Supplemental material]. Hence, AoC activity was highly sensitive to minor changes in protein structure that, based on CD, did not cause major changes in protein conformation. In addition to PNPB activity, Table II also lists AoC variant activities for PCL hydrolysis. Even though the two substrates used to determine activity are extraordinarily different, generally, the magnitude of activity loss for variants was similar for both assays. The largest deviation in activity loss for the two assays was observed for Variant 2 (79 vs 27%).
In an attempt to explain how certain variants had larger effects on enzyme activity, correlation between the distance from the active site and the decrease in activity was investigated. Hydrophobic core amino acid substitutions (V150I, I136V), located about ∼18 Å from the catalytic triad, result in about a 20% decrease in both PNPB and PCL hydrolytic activity. Variant 2 (L26D, G28E, D30R, K67R) includes building in new surface salt bridges (between E28 and R67 and between R30, D34, and D26) that are distant from the active site (about 20 Å). The mutations built into Variant 2, result in a small decrease in PNPB hydrolysis (about 20%) but PCL hydrolysis activity decreased by about 70%. The discrepancy between Variant 2 activity losses for the soluble and insoluble substrates is likely explained by loss of the surface hydrophobic L26 that can affect enzyme adsorption to the substrate. For both substrates, the activity of Variant 3 was within experimental error of wt-AoC. Furthermore, for Variant 3 in which a charged mutation was introduced, there was no observed effect on the pH-activity relationship relative to wt-AoC (Fig S2 Supplemental material). This potential effect was considered since charged mutations can alter the protonation state at the active site.
Variant 5 (N133A, S140P, E161T, A166P) displays ∼20-30% decrease in both PNBP and PCL activity indicating that the mutations introduced did not substantially alter enzyme adsorption to PCL. However, P155 and T161 introduced in Variant 5 may reduce favorable modes of protein flexibility as these residues reside in a loop (residues 157-171) that abuts the ‘binding loop’ (approximately residues 180-190) which forms one lip of the hydrophobic groove just C-terminal to the catalytic His194. The amino acid substitutions introduced into Variant 6 (A178P, V179P) are highly detrimental to enzyme activity (50% and 75% loss for PNPB and PCL, respectively). These two mutations that reside at the N-terminal base of the binding loop must cause a change in loop structure or dynamics that is harmful to enzyme activity.
Variant 7 (R46P) showed a remarkable decrease (∼80%) in enzyme activity. Thus, while Rosetta predicts that the introduction of P at 46 is favorable given the residue's crystallographic backbone conformation, mutation to a proline may alter important dynamic motions or induce a subtle conformational shift of the loop that causes reduced activity. Furthermore, R46P was recommended along with the compensating mutation of Y83D by Rosetta. However, the native Y83 was retained in the mutant as it was considered to provide key hydrophobic packing contacts but this may have caused steric clashes between P46 and Y83 leading to loss of activity. Also, the mutation of R46, which is in close proximity to the active site and in coordination with four backbone oxygen's, would create a hole in the region perturbing enzyme activity.
Variant 8 (K174R, Y176F, A178E, D200R, G202E, D203E, D206R) explores the effects of several mutations across a large surface patch of AoC. The relatively small (10-20%) reduction in activity for both substrates suggests that substitutions within this surface region have little influence on either active site confirmation or binding to hydrophobic surfaces. Large losses in PNPB (∼70%) and PCL (∼90%) activities for Variant 9 (T84R, D86L, A99E, A100S) are attributed to adverse changes to the ‘helical flap’ (approximately residues 80-87) that pairs with the binding loop to enclose the active site.
Comparison of variants in Table II provides insight into how changes in activity of first round Variants are manifested in second stage variant designs. Variant 10 combines amino acid substitutions in Variant 3 and 5. If the decreases in activity found for Variant 10 results from the additive contributions of Variant 3 and 5, the decrease in activity for PNPB and PCL hydrolysis would both be about 30% relative to wt-AoC. Indeed, Variant 10 losses about 35 and 32% activity that, within experimental error, is consistent with this simplistic model. Variant 11, which is identical to Variant 10 except for one additional change (K137E), shows a small decrease in PCL and PNPB hydrolysis activity relative to Variant 10. However, Variant 11 has about an order of magnitude decrease in hydrolytic activity for polybutylene succinate adipate (PBSA) (Fig 6). This decrease can be attributed to loss of a critical interaction with the substrate, which could be a requirement for productive binding or the orientation of the enzyme to PBSA. Variant 12 showed activity similar to Variant 3 as it retained mutations in 3 and, the new variations were in the same region as that of 3.
Temperature optima
Thermophilic enzymes are known to be active at higher temperature but, often, their specific activity is comparable with that of their mesophilic counterparts at the respective optimum temperatures 56. While thermostabilization is governed by the rigidity of the structure, molecular flexibility is essential for both substrate binding and catalysis 57. Hence, thermostabilization often leads to loss of activity at lower temperature but, increased activities can be realized by its retained activity at elevated temperatures. Hence, the influence of thermostabilization of Variants 3, 5, 10, 11 and wt-AoC on the temperature at which optimal activity is observed was investigated (Fig 6). PNPB and PCL hydrolysis assays could not be used to explore hydrolytic activity at temperatures above 55 °C. This is due to high background activity caused by chemical hydrolysis of PNPB at elevated temperatures and the fact that PCL undergoes a melting transition (Tm) at about 55 °C which will influence activity measurements below and above this temperature. To determine the activity as a function of temperature from 25 to 70 °C, film of polybutylene succinate, PBSA, was selected since, within this temperature range, PBSA does not undergo either a glass (Tg) or melting transition (Tm for PBSA is 90 °C). The optimum temperature for wt-AoC activityis 50 °C, which is just below the onset of protein unfolding based on the CD analysis. This explains why an increase in the reaction temperature to 55 °C and above results in a precipitous decrease in PBSA hydrolytic activity. Variants with enhanced thermostability relative to wt-AoC were expected to show increased activity to correspondingly higher temperatures. Based on CD thermal scans, Variants 3, 10 and 11 were expected to have higher temperature optimum activity values in the range of 55 to 60 °C. However, temperature optima for Variants 3, 10 and 11 did not significantly differ from that of wt-AoC. This behavior may be due to that, as the temperature is increased above 50 °C, small structural changes occur near the active site that result in reduced activity. However, these changes in structure at the active site are not reflected in CD spectra. This is explained by that the mutations improved the stability of wt-AoC at locations that are sufficiently distant from the active site such that CD spectra show that the overall structure of the protein is stabilized. Similar observations are reported for the LCC where the difference between the thermal unfolding temperature and the optimum temperature for activity is remarkably high i.e 36 °C which was again explained based on the lower active site stability as compared to the global structural stability 36.
Conclusions
Computational protein design coupled with intuitive rationalization was used to improve AoC stability without loss of enzyme activity. Through this work, important insights were obtained about AoC structure-stability relationships. Notably, surface charge modifications or formation of salt bridges (Variant 3) and Proline insertions (variant 5) at the right location are promising strategies which should be further investigated in order to increase AoC's thermostability. The known stabilization mechanisms, i.e enthalpic stabilization for former and entropic stabilization for latter, were consistent with the case of AoC. The entropic stabilization in the case of variant 5 was achieved at the cost of breaking the hydrogen bond contributing to the enthalpy. This effect becomes evident with urea denaturation analysis, indicating that such experiments can be useful to detect enthalpic losses associated with mutations to proline, even if the overall effect is thermostabilization.
The loss of enzyme activity at temperatures well below the thermal unfolding temperatures of both Wt-AoC and the improved variants confirmed the early loss of active site structure during thermal unfolding. In these studies, mutations near the active site were avoided, assuming implications on activity. However, considering the lowered stability of the active site region, mutations in these regions should be considered for future designs. Further intuitive rationalization should also be pursued, incorporating the predicted effects of these mutations on the activity. The activity analysis of the variants designed for thermostabilization revealed the remarkable effect of a small number of mutations on the activity regardless of their position with respect to the active site. Our rationalization of these results to understand the possible indirect effects of these mutations will be useful for future designing efforts. Insight gained into the structure and property relationships of AoC will also be useful in the design of other cutinase homologues (such as FsC and HiC), as they have high sequence and structural similarities to AoC.
Supplementary Material
Figure 5.
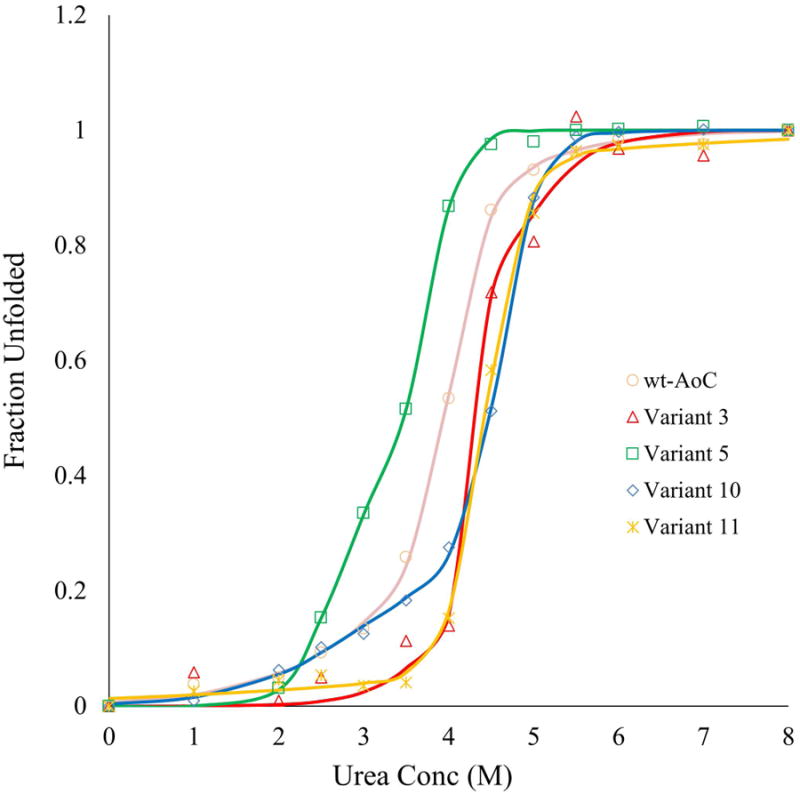
Fraction unfolded was measured by CD at varying concentrations of urea after 24 h equilibration at room temperature. For Variants 3 and 11, data were fit to a two-state equilibrium model. For wt-AoC and Variants 5 and 10, data were fit to a three-state equilibrium model. Continuous lines represent fitted data
Acknowledgments
This work was supported by the National Science Foundation Award # 1067415 to R.A.G. and NIH grant R01 GM099827 to C.B. We thank DNA2.0 for the gene synthesis and intellectual inputs to troubleshoot protein expression problems.
References
- 1.Clark J, Sheldon R, Raston C, Poliakoff M, Leitner W. 15 years of Green Chemistry. Green Chem. 2014;16:18. [Google Scholar]
- 2.Dubé Ma, Salehpour S. Applying the Principles of Green Chemistry to Polymer Production Technology. Macromol React Eng. 2014;8:7–28. [Google Scholar]
- 3.Al-Salem SM, Lettieri P, Baeyens J. Recycling and recovery routes of plastic solid waste (PSW): A review. Waste Manag. 2009;29:2625–2643. doi: 10.1016/j.wasman.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 4.Sasse BF, Emig G. Review Chemical Recycling of Polymer Materials. 1998;21:777–789. [Google Scholar]
- 5.Goddard JM, Hotchkiss JH. Polymer surface modification for the attachment of bioactive compounds. Prog Polym Sci. 2007;32:698–725. [Google Scholar]
- 6.Ratner BD. Surface modification of polymers: Chemical, biological and surface analytical challenges. Biosens Bioelectron. 1995;10:797–804. doi: 10.1016/0956-5663(95)99218-a. [DOI] [PubMed] [Google Scholar]
- 7.Biltresse P, Descamps D, Boxus T, Marchand-brynaert J. Attachment of Bis- (trifluoromethyl) aryl Labels onto the Chain Ends of Poly (ethylene terephthalate) (PET) Track- Etched Membranes and Films by Surface Wet Chemistry. 2000:3510–3520. [Google Scholar]
- 8.Guebitz GM, Cavaco-Paulo A. Enzymes go big: surface hydrolysis and functionalization of synthetic polymers. Trends Biotechnol. 2008;26:32–8. doi: 10.1016/j.tibtech.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 9.Purdy RE, Kolattukudy PE. Hydrolysis of plant cuticle by plant pathogens. Properties of cutinase I, cutinase II, and a nonspecific esterase isolated from Fusarium solani pisi. Biochemistry. 1975;14:2832–40. doi: 10.1021/bi00684a007. [DOI] [PubMed] [Google Scholar]
- 10.Liu Z, Gosser Y, Baker PJ, Ravee Y, Lu Z, Alemu G, Li H, Butterfoss GL, Kong XP, Gross R, et al. Structural and functional studies of Aspergillus oryzae cutinase: enhanced thermostability and hydrolytic activity of synthetic ester and polyester degradation. J Am Chem Soc. 2009;131:15711–6. doi: 10.1021/ja9046697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shinozaki Y, Morita T, Cao X, Yoshida S, Koitabashi M, Watanabe T, Suzuki K, Sameshima-Yamashita Y, Nakajima-Kambe T, Fujii T, et al. Biodegradable plastic-degrading enzyme from Pseudozyma antarctica: cloning, sequencing, and characterization. Appl Microbiol Biotechnol. 2013;97:2951–9. doi: 10.1007/s00253-012-4188-8. [DOI] [PubMed] [Google Scholar]
- 12.Yang S, Xu H, Yan Q, Liu Y, Zhou P, Jiang Z. A low molecular mass cutinase of Thielavia terrestris efficiently hydrolyzes poly(esters) J Ind Microbiol Biotechnol. 2013;40:217–26. doi: 10.1007/s10295-012-1222-x. [DOI] [PubMed] [Google Scholar]
- 13.Araújo R, Silva C, O'Neill A, Micaelo N, Guebitz G, Soares CM, Casal M, Cavaco-Paulo A. Tailoring cutinase activity towards polyethylene terephthalate and polyamide 6,6 fibers. J Biotechnol. 2007;128:849–57. doi: 10.1016/j.jbiotec.2006.12.028. [DOI] [PubMed] [Google Scholar]
- 14.Heumann S, Eberl A, Pobeheim H, Liebminger S, Fischer-Colbrie G, Almansa E, Cavaco-Paulo A, Gübitz GM. New model substrates for enzymes hydrolysing polyethyleneterephthalate and polyamide fibres. J Biochem Biophys Methods. 2006;69:89–99. doi: 10.1016/j.jbbm.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 15.Müller RJ, Schrader H, Profe J, Dresler K, Deckwer WD. Enzymatic Degradation of Poly(ethylene terephthalate): Rapid Hydrolyse using a Hydrolase from T. fusca. Macromol Rapid Commun. 2005;26:1400–1405. [Google Scholar]
- 16.Ronkvist ÅM, Xie W, Lu W, Gross Ra. Cutinase-Catalyzed Hydrolysis of Poly(ethylene terephthalate) Macromolecules. 2009;42:5128–5138. [Google Scholar]
- 17.Gallagher FG. Controlled degradation polyesters. Modern Polyesters: Chemistry and Technology of Polyesters and Copolyesters. 2003;89:591–698. [Google Scholar]
- 18.Guebitz GM, Cavaco-Paulo A. Enzymes go big: surface hydrolysis and functionalization of synthetic polymers. Trends Biotechnol. 2008;26:32–8. doi: 10.1016/j.tibtech.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 19.Vertommen MaME, Nierstrasz Va, Van Der Veer M, Warmoeskerken MMCG. Enzymatic surface modification of poly(ethylene terephthalate) J Biotechnol. 2005;120:376–86. doi: 10.1016/j.jbiotec.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 20.Ronkvist ÅM, Lu W, Feder D, Gross Ra. Cutinase-Catalyzed Deacetylation of Poly(vinyl acetate) Macromolecules. 2009;42:6086–6097. [Google Scholar]
- 21.Matamá T, Araújo R, Gübitz GM, Casal M, Cavaco-Paulo A. Functionalization of cellulose acetate fibers with engineered cutinases. Biotechnol Prog. 2009;26:636–43. doi: 10.1002/btpr.364. [DOI] [PubMed] [Google Scholar]
- 22.Altaner C, Saake B, Tenkanen M, Eyzaguirre J, Faulds C, Biely P, Viikari L, Siika-aho M, Puls J. Regioselective deacetylation of cellulose acetates by acetyl xylan esterases of different CE-families. J Biotechnol. 2003;105:95–104. doi: 10.1016/s0168-1656(03)00187-1. [DOI] [PubMed] [Google Scholar]
- 23.Martinez C, De Geus P, Lauwereys M, Matthyssens G, Cambillau C. Fusarium solani cutinase is a lipolytic enzyme with a catalytic serine accessible to solvent. Nature. 1992;356:615–618. doi: 10.1038/356615a0. [DOI] [PubMed] [Google Scholar]
- 24.Nyyssölä A, Pihlajaniemi V, Järvinen R, Mikander S, Kontkanen H, Kruus K, Kallio H, Buchert J. Enzyme and Microbial Technology Screening of microbes for novel acidic cutinases and cloning and expression of an acidic cutinase from Aspergillus niger CBS 513. 2013;8852:272–278. doi: 10.1016/j.enzmictec.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 25.Hegde K, Veeranki VD. Production optimization and characterization of recombinant cutinases from Thermobifida fusca sp. NRRL B-8184. Appl Biochem Biotechnol. 2013;170:654–75. doi: 10.1007/s12010-013-0219-x. [DOI] [PubMed] [Google Scholar]
- 26.Hu X, Thumarat U, Zhang X, Tang M, Kawai F. Diversity of polyester-degrading bacteria in compost and molecular analysis of a thermoactive esterase from Thermobifida alba AHK119. Appl Microbiol Biotechnol. 2010;87:771–9. doi: 10.1007/s00253-010-2555-x. [DOI] [PubMed] [Google Scholar]
- 27.Chen S, Su L, Chen J, Wu J. Cutinase: characteristics, preparation, and application. Biotechnol Adv. 2013;31:1754–67. doi: 10.1016/j.biotechadv.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 28.Kodama Y, Masaki K, Kondo H, Suzuki M, Tsuda S, Nagura T, Shimba N, Suzuki E, Iefuji H. Crystal structure and enhanced activity of a cutinase-like enzyme from Cryptococcus sp. strain S-2. Proteins. 2009;77:710–7. doi: 10.1002/prot.22484. [DOI] [PubMed] [Google Scholar]
- 29.Ribitsch D, Yebra AO, Zitzenbacher S, Wu J, Nowitsch S, Steinkellner G, Greimel K, Doliska A, Oberdorfer G, Gruber CC, et al. Fusion of binding domains to Thermobifida cellulosilytica cutinase to tune sorption characteristics and enhancing PET hydrolysis. Biomacromolecules. 2013;14:1769–76. doi: 10.1021/bm400140u. [DOI] [PubMed] [Google Scholar]
- 30.Marten E, Müller RJ, Deckwer WD. Studies on the enzymatic hydrolysis of polyesters I. Low molecular mass model esters and aliphatic polyesters. Polym Degrad Stab. 2003;80:485–501. [Google Scholar]
- 31.Nishida H, Tokiwa Y. (1993) Effects of higher-order structure of Poly(3-hydrolxybutyrate) on biodegradation. ll Effects of crystal structure on microbial degradation. J Environ Polym Degrad. 1993;1(1):65–80. 1:65–80. [Google Scholar]
- 32.Tokiwa Y, Calabia BP, Ugwu CU, Aiba S. Biodegradability of plastics. Int J Mol Sci. 2009;10:3722–42. doi: 10.3390/ijms10093722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matas AJ, Cuartero J, Heredia A. Phase transitions in the biopolyester cutin isolated from tomato fruit cuticles. Thermochim Acta. 2004;409:165–168. [Google Scholar]
- 34.Baker PJ, Poultney C, Liu Z, Gross R, Montclare JK. Identification and comparison of cutinases for synthetic polyester degradation. Appl Microbiol Biotechnol. 2012;93:229–40. doi: 10.1007/s00253-011-3402-4. [DOI] [PubMed] [Google Scholar]
- 35.Roth C, Wei R, Oeser T, Then J, Föllner C, Zimmermann W, Sträter N. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl Microbiol Biotechnol. 2014;98:7815–23. doi: 10.1007/s00253-014-5672-0. [DOI] [PubMed] [Google Scholar]
- 36.Sulaiman S, You DJ, Kanaya E, Koga Y, Kanaya S. Crystal structure and thermodynamic and kinetic stability of metagenome-derived LC-cutinase. Biochemistry. 2014;53:1858–69. doi: 10.1021/bi401561p. [DOI] [PubMed] [Google Scholar]
- 37.Chin IS, Abdul Murad AM, Mahadi NM, Nathan S, Abu Bakar FD. Thermal stability engineering of Glomerella cingulata cutinase. Protein Eng Des Sel. 2013;26:369–75. doi: 10.1093/protein/gzt007. [DOI] [PubMed] [Google Scholar]
- 38.Kawai F, Oda M, Tamashiro T, Waku T, Tanaka N, Yamamoto M, Mizushima H, Miyakawa T, Tanokura M. A novel Ca(2+)-activated, thermostabilized polyesterase capable of hydrolyzing polyethylene terephthalate from Saccharomonospora viridis AHK190. Appl Microbiol Biotechnol. 2014;98(24):10053–64. doi: 10.1007/s00253-014-5860-y. [DOI] [PubMed] [Google Scholar]
- 39.Minagawa H, Nakayama N, Nakamoto S. Thermostabilization of lactate oxidase by random mutagenesis. Biotechnol Lett. 1995;17:975–980. [Google Scholar]
- 40.Stemmer WP. Rapid evolution of a protein in vitro by DNA shuffling. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 41.Filikov AV, Hayes RJ, Luo P, Stark DM, Chan C, Kundu A, Dahiyat BI. Computational stabilization of human growth hormone. Protein Sci. 2002;11:1452–61. doi: 10.1110/ps.3500102. Available from. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vieille C, Zeikus GJ, Vieille C. Hyperthermophilic Enzymes: Sources, Uses, and Molecular Mechanisms for Thermostability Hyperthermophilic Enzymes: Sources, Uses, and Molecular Mechanisms for Thermostability. Microbilology Mol Biol Rev. 2001;65:1–43. doi: 10.1128/MMBR.65.1.1-43.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J. Practically useful: what the Rosetta protein modeling suite can do for you. Biochemistry. 2010;49:2987–98. doi: 10.1021/bi902153g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leaver-Fay A, Tyka M, Lewis SM, Lange OF, Thompson J, Jacak R, Kaufman K, Renfrew PD, Smith Ca, Sheffler W, et al. ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–74. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borgo B, Havranek JJ. Automated selection of stabilizing mutations in designed and natural proteins. Proc Natl Acad Sci U S A. 2012;109:1494–9. doi: 10.1073/pnas.1115172109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dantas G, Kuhlman B, Callender D, Wong M, Baker D. A Large Scale Test of Computational Protein Design: Folding and Stability of Nine Completely Redesigned Globular Proteins. J Mol Biol. 2003;332:449–460. doi: 10.1016/s0022-2836(03)00888-x. [DOI] [PubMed] [Google Scholar]
- 47.Floor RJ, Wijma HJ, Colpa DI, Ramos-Silva A, Jekel Pa, Szymański W, Feringa BL, Marrink SJ, Janssen DB. Computational library design for increasing haloalkane dehalogenase stability. Chembiochem. 2014;15:1660–72. doi: 10.1002/cbic.201402128. [DOI] [PubMed] [Google Scholar]
- 48.Wijma HJ, Floor RJ, Janssen DB. Structure- and sequence-analysis inspired engineering of proteins for enhanced thermostability. Curr Opin Struct Biol. 2013;23:588–94. doi: 10.1016/j.sbi.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 49.Wijma HJ, Floor RJ, Jekel Pa, Baker D, Marrink SJ, Janssen DB. Computationally designed libraries for rapid enzyme stabilization. Protein Eng Des Sel. 2014;27:49–58. doi: 10.1093/protein/gzt061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Makhatadze GI, Loladze Vv, Ermolenko DN, Chen X, Thomas ST. Contribution of Surface Salt Bridges to Protein Stability: Guidelines for Protein Engineering. J Mol Biol. 2003;327:1135–1148. doi: 10.1016/s0022-2836(03)00233-x. [DOI] [PubMed] [Google Scholar]
- 52.Querol E, Perez-Pons Ja, Mozo-Villarias a. Analysis of protein conformational characteristics related to thermostability. Protein Eng. 1996;9:265–71. doi: 10.1093/protein/9.3.265. [DOI] [PubMed] [Google Scholar]
- 53.Bystroff C, Baker D. Prediction of local structure in proteins using a library of sequence-structure motifs. J Mol Biol. 1998;281:565–77. doi: 10.1006/jmbi.1998.1943. [DOI] [PubMed] [Google Scholar]
- 54.Sagt CMJ, Müller WH, Van Der Heide L, Boonstra J, Verkleij J, Verrips CT, Verkleij AJ. Impaired Cutinase Secretion in Saccharomyces cerevisiae Induces Irregular Endoplasmic Reticulum (ER) Membrane Proliferation, Oxidative Stress, and ER-Associated Degradation Impaired Cutinase Secretion in Saccharomyces cerevisiae. Induces Irregular Endo. 2002;68(5):2155–2160. doi: 10.1128/AEM.68.5.2155-2160.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Doig AJ, MacArthur MW, Stapley BJ, Thornton JM. Structures of N-termini of helices in proteins. Protein Sci. 1997;6:147–55. doi: 10.1002/pro.5560060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Danson MJ, Hough DW, Russell RJM, Taylor GL, Pearl L. Enzyme thermostability and thermoactivity. 1996;9:629–630. doi: 10.1093/protein/9.8.629. [DOI] [PubMed] [Google Scholar]
- 57.Jaenicke R. Do ultrastable proteins from hyperthermophiles have high or low conformational rigidity? Proc Natl Acad Sci U S A. 2000;97:2962–2964. doi: 10.1073/pnas.97.7.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.