

Abstract
Persistence of Mycobacterium tuberculosis results from bacterial strategies that manipulate host adaptive immune responses. Infected dendritic cells (DCs) transport M. tuberculosis to local lymph nodes but activate CD4 T cells poorly, suggesting bacterial manipulation of antigen presentation. However, M. tuberculosis antigens are also exported from infected DCs and taken up and presented by uninfected DCs, possibly overcoming this blockade of antigen presentation by infected cells. Here we show that the first stage of this antigen transfer, antigen export, benefits M. tuberculosis by diverting bacterial proteins from the antigen presentation pathway. Kinesin-2 is required for antigen export and depletion of this microtubule-based motor increases activation of antigen-specific CD4 T cells by infected cells and improves control of intracellular infection. Thus, although antigen transfer enables presentation by bystander cells, it does not compensate for reduced antigen presentation by infected cells and represents a bacterial strategy for CD4 T cell evasion.
Graphical abstract

Antigen export: In M. tuberculosis-infecfed cells, bacterial antigens are exported by kinesin 2-dependent vesicular transport, which reduces the quantity of antigen routed to the MHC class II antigen presentation pathway. Sub-optimal antigen presentation limits CD4 T ceil activation by infected cells.
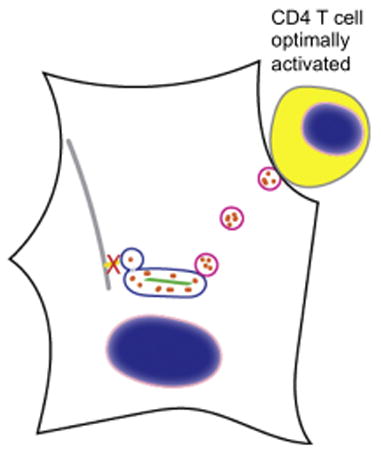
Blockade of antigen export: Depletion of kinesin 2 from M. tuberculosis-infected cells decreases antigen export. More antigen is available to the MHC class II antigen presentation pathway, resulting in more effective CD4 T cell activation by infected cells.
Introduction
Infection with Mycobacterium tuberculosis is a major global health problem, due to its ease of transmission by the aerosol route, the lack of an efficacious vaccine, and increasing emergence of bacterial drug resistance (Philips and Ernst, 2012). Even though the HIV pandemic has amplified the global problem of tuberculosis (TB), most people with TB are immunocompetent, indicating that M. tuberculosis possesses effective mechanisms for evasion of innate and adaptive immunity. Although the onset of adaptive immune responses is delayed after M. tuberculosis infection of humans (Poulsen, 1950) or mice (Chackerian et al., 2002; Gallegos et al., 2008; Reiley et al., 2008; Wolf et al., 2008), most infected individuals and experimental animals develop antigen-specific CD4 and CD8 T cell responses, and the resulting T cells have appropriate effector functions as assessed by ex vivo restimulation (Ernst, 2012). However, M. tuberculosis persists despite measurable T cell responses, suggesting that the bacteria manipulate the host to prevent effector T cells from exerting their functions at the site of infection (Urdahl et al., 2011).
Since M. tuberculosis resides in macrophages and DC in vivo (Tailleux et al., 2003; Wolf et al., 2007), the suboptimal efficacy of effector T cells may not be due to an intrinsic property of the antigen-specific effector T cells themselves, but instead may be secondary to bacterial manipulation of the infected antigen presenting cells. Indeed, multiple studies have reported that mycobacterial infection of antigen presenting cells interferes with MHC class II antigen presentation in vitro, although the mechanisms that interfere with antigen presentation are poorly understood (reviewed in (Baena and Porcelli, 2009)). Likewise, it is unclear whether the phenomenon observed in vitro contributes to suboptimal CD4 T cell efficacy in vivo (Ernst, 2012).
We recently reported that M. tuberculosis-infected host cells export bacterial antigens for uptake and presentation by uninfected cells, and we and Pamer and colleagues found that antigen transfer to uninfected DC in the lymph node allows priming of CD4 T cells in vivo (Samstein et al., 2013; Srivastava and Ernst, 2014), implying that this phenomenon helps to compensate for impaired antigen presentation by M. tuberculosis-infected cells (Hudrisier and Neyrolles, 2014). Because of the potential importance of antigen transfer for immunity to TB, we sought to further understand its underlying mechanisms and significance. Here we report the unexpected finding that the first stage of antigen transfer, antigen export, benefits the pathogen by shunting bacterial antigens away from the MHC class II antigen presentation pathway, thereby minimizing recognition of infected cells by antigen-specific CD4 T cells. We also discovered that the second stage of antigen transfer, antigen uptake and presentation by uninfected cells, only partially compensates for the impaired antigen presentation capacity of infected cells. Our results indicate that antigen export is a potent and effective mechanism of immune evasion that promotes persistence M. tuberculosis despite development of antigen-specific T cell responses.
Results
Evidence for antigen export and transfer to uninfected lung cells in vivo
We first confirmed and extended our finding that M. tuberculosis antigens can be acquired by uninfected cells in vivo in a process of antigen export from infected cells followed by uptake and processing by uninfected bystander cells. In addition to the earlier finding of antigen transfer in lymph nodes after aerosol infection (Samstein et al., 2013; Srivastava and Ernst, 2014), we found that antigen transfer occurs in the lungs. After infecting mice with GFP-expressing M. tuberculosis, we isolated and sorted live lung leukocytes on the basis of their phenotype and presence (GFP+) or absence (GFP-) of intracellular bacteria, and used the sorted cells to stimulate M. tuberculosis Ag85B-specific TCR transgenic (P25TCR-Tg) CD4 T cells. We found that the distinct subsets of infected cells (lung DC, recruited interstitial macrophages, and monocytes) differed in their capacity to activate CD4 T cells in vitro (Figure S1A). Notably, uninfected cells in each of the three subsets also activated CD4 T cells in this assay. Indeed, in all three subsets, the uninfected cells were superior to the infected cells, indicating that they had acquired bacterial antigen for processing and presentation to CD4 T cells.
To assure that antigen acquisition by uninfected myeloid cells in the lungs was not an artifact of cell isolation and sorting, we stained frozen lung sections from mice that had been infected with GFP-expressing M. tuberculosis. This demonstrated that both infected (GFP+) and uninfected (GFP-) CD11c+ cells contain Ag85, and that the uninfected cells that had acquired Ag85 were in proximity to infected cells, implying that they had acquired antigen released from infected cells (Figure S1B).
M. tuberculosis Ag85 is present in extraphagosomal vesicles in infected cells
To further understand the significance of antigen export, we first sought to identify cellular mechanistic steps required for antigen export from infected cells. Since our earlier studies indicated that M. tuberculosis antigen export from infected cells did not involve apoptosis or exosome shedding, and released undegraded bacterial proteins, we used immunofluorescence staining and confocal microscopy to localize M. tuberculosis antigen 85 (Ag85) in infected cells. Ag85, which consists of the closely-related proteins Ag85A, Ag85B, and Ag85C, is secreted by M. tuberculosis, and is a component of multiple TB vaccine candidates currently in development (Kaufmann et al., 2014). We found Ag85 (principally Ag85B) staining in a punctate pattern in DC infected with wild-type, but not with Ag85B-null M. tuberculosis (Figure 1A). Punctate staining of Ag85 was not concentrated in the perinuclear region, and Ag85+ puncta were present in the periphery of the infected cells. Since the punctate pattern suggested that Ag85 is contained in vesicles, we used immunogold staining and electron microscopy of cryosectioned M. tuberculosis-infected bone marrow-derived DC to further characterize the location of extraphagosomal intracellular Ag85. This revealed Ag85 in bacteria-containing phagosomes and in small membrane-bound vesicles devoid of bacteria or electron-dense bacterial fragments (Figure 1B). This finding resembles the reported location of Ag85 in M. tuberculosis-infected human monocytes (Harth et al., 1996) and murine macrophages (Beatty and Russell, 2000), determined using a different antibody to Ag85. While the fixation conditions compatible with immunogold staining are not optimal for preservation of membrane ultrastructure, the Ag85-containing vesicles detected were bounded by a single bilayer (Figure 1B, inset). We refer to these vesicles as antigen export vesicles, or AEV.
Figure 1. M. tuberculosis Ag85 is located in extraphagosomal vesicles distinct from classical recycling endosomes.

(A) Confocal microscopy of DC infected with GFP-expressing M. tuberculosis (pseudo-colored blue) and stained for surface CD45.2 (pseudo-colored green), fixed, permeabilized, and stained with an antibody to Ag85B (red). Top row: DC infected with wild-type M. tuberculosis H37Rv; bottom row: DC infected with H37Rv:Δ85B. Although the antibody to Ag85B cross reacts with Ag85A and Ag85C on immunoblots, the predominant signal detected by immunofluorescence is from Ag85B. Scale bars 10 μm. (B) Immunoelectron microscopy of Ag85 in cryosections of DC infected with wild-type M. tuberculosis H37Rv. Ag85 staining associated with bacteria (arrow heads), and membrane-bound small vesicles (arrows with long tail; enlarged inset image). Scale bar: 500 nm. (C) DC infected with M. tuberculosis H37Rv expressing cyan fluorescent protein (CFP) (blue) and analysed for transferrin receptor-containing recycling endosomes. Transferrin receptor (green) and Ag85B (red). Scale bars 10 μm. (D) DC infected with M. tuberculosis H37Rv expressing CFP (blue), stained for MHC-II (green) and Ag85B (red). Scale bars 10 μm. Data in panel A, C and D represent 2-4 independent staining experiments. Panel B is representative of 20 images.
Phagosomes containing live mycobacteria, including M. tuberculosis, have been reported to interact with recycling endosomes containing transferrin and transferrin receptors (Clemens and Horwitz, 1996; Sturgill-Koszycki et al., 1996). We therefore hypothesized that AEV might be equivalent to recycling endosomes that acquired bacterial proteins present from the lumenal space of phagosomes, trafficked to the cell surface, and released their cargo to the extracellular space. However, immunofluorescence microscopy revealed lack of colocalization of extraphagosomal Ag85 and transferrin receptor-bearing endosomes (Figures 1C and 2D). Similar findings were obtained when recycling endosomes were labeled using fluorescently-labeled transferrin (not shown). These findings indicate that AEV are not classical recycling endosomes that have intersected with M. tuberculosis-containing phagosomes. They also imply that an additional population of intracellular vesicles interacts with mycobacterial phagosomes and is involved in transport of bacterial products from phagosomes to the extracellular space. We also found that most, but not all of the AEV are segregated from MHC class II-containing vesicles in infected cells (Figure 1D) suggesting that a limited fraction of Ag85 is present in the MHC-II pathway; AEV also did not resemble early endosomes, as they did not colocalize with EEA1 (not shown).
Figure 2. Ag85B export from infected cells depends on microtubules.
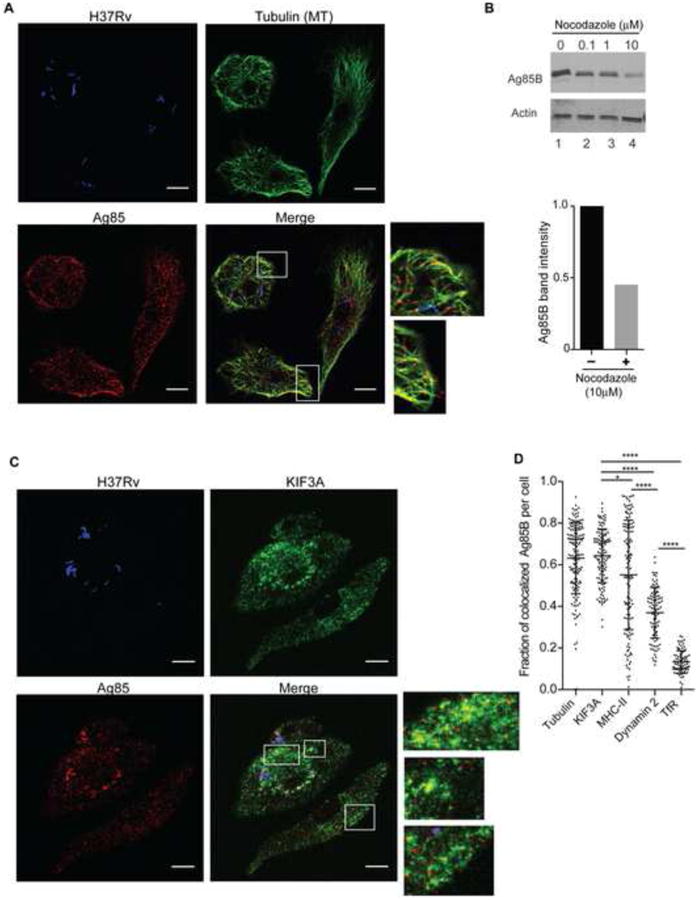
(A) Antigen export vesicles (AEV) colocalize with tubulin. DC infected with CFP-expressing M. tuberculosis H37Rv (blue) were stained for tubulin (green) and Ag85B (red). Scale bars 10 μm. (B) Inhibition of tubulin polymerization decreases Ag85B export. Upper panel: immunoblot of Ag85 in conditioned medium (CM) collected from DC that had been infected and cultured in the presence of the designated concentrations of nocodazole for 20 h. The same DC that were used to generate CM were lysed and probed for actin. Lower panel: quantitation of Ag85B band intensity by Image J of conditioned medium of M. tuberculosis-infected cells cultured in the absence or presence of 10 μM nocodazole, determined using the immunoblot in the upper panel. (C) Colocalization of M. tuberculosis Ag85 and kinesin-2 component KIF3A. DC infected with CFP-expressing M. tuberculosis (blue), were stained for KIF3A (pseudo-colored green) and Ag85 (red). Scale bars 10 μm. (D) Quantitative analysis of the fraction of AEV that colocalize with tubulin, KIF3A, dynamin 2 or transferrin receptor (TfR) in a given cell. Confocal z-stack images were acquired and analysed using the Manders' overlap coefficient (Dunn et al.) analysis module of the Volocity image analysis software. Data in panels A-D represent 2-4 independent experiments where 100-300 cells were analysed to obtain the Manders' colocalization coefficient. A similar trend with respect to colocalization was confirmed by analysis with the JACoP (Just another colocalization plugin) of the ImageJ software (Costes et al., 2004).
Export of M. tuberculosis Ag85B from infected cells involves the motor protein kinesin-2
Although the mechanisms of exocytosis and protein secretion from macrophages and DC are incompletely characterized, microtubule-dependent vesicle transport promotes secretory vesicle transport and exocytosis in diverse cell types, from neurons to cytotoxic T lymphocytes (Kuhn and Poenie, 2002; Stinchcombe et al., 2006; Vale et al., 1985). Immunofluorescence confocal microscopy revealed a high frequency of localization of AEV with microtubules in M. tuberculosis-infected cells (Figures 2A and D). AEV were closely aligned with microtubules, including at the periphery of infected cells (Figure 2A). AEV (red) frequently colocalized with tubulin (green), as indicated by yellow fluorescence; in addition, AEV were frequently in close proximity to microtubules. To determine the functional significance of colocalization of AEV and microtubules, we inhibited tubulin polymerization using nocodazole and observed a dose-dependent decrease in the extracellular release of Ag85 (Figure 2B). The maximum tolerated concentration of nocodazole decreased Ag85B export by approximately 50%. These results are consistent with microtubule-dependent vesicular transport as an essential step in export of an M. tuberculosis antigen from infected cells.
Microtubule-dependent organelle transport toward the cell periphery depends on one or more members of the kinesin family of motor proteins (reviewed in (Hirokawa et al., 2009)). We examined kinesin-2, since it is implicated in transport of vesicular organelles; its subunits, KIF3A, KIF3B, and KIF-associated protein 3 (KAP3 or KIFAP3) are expressed in macrophages and myeloid DC (Gautier et al., 2012; Miller et al., 2012) including CD11bhi lung macrophages and CD103-CD11b+ lung DC (http://www.immgen.org); and its KIF3A subunit has been implicated in virus release from HIV-1-infected human macrophages (Gaudin et al., 2012). Immunofluorescence confocal microscopy revealed that AEV colocalized with the kinesin-2 component KIF3A in infected cells (Figure 2C). We used automated image analysis to quantitate the fraction of AEV that colocalized with microtubules and/or with KIF3A in 100-300 infected cells and found that in the majority of infected cells, >60% of AEV colocalized with tubulin or KIF3A (Figure 2D), suggesting that microtubule associated kinesin-2 contributes to trafficking of AEV. In contrast, less than 20% of Ag85+ AEV colocalized with transferrin receptor-containing vesicles in any infected cell (Figure 2D).
Since members of the dynamin protein family mediate vesicle budding and membrane scission in diverse intracellular processes (Ferguson and De Camilli, 2012; Praefcke and McMahon, 2004), we hypothesized that dynamin-2 (the form expressed in myeloid cells) might contribute to generation and/or maturation of AEV, and found that in the majority of cells, 40-60% of Ag85+ AEV colocalized with dynamin-2 (Figure 2D) consistent with a partial or transient association of AEV and dynamin 2.
Kinesin-2 depletion blocks antigen export from infected cells
Since disruption of microtubule polymerization decreased export of Ag85 from infected cells, and AEV colocalized with microtubules and with KIF3A, we examined the functional importance of kinesin-2 in antigen export. Kinesin-2 typically functions as a heterotrimer of KIF3A, KIF3B, and KIF3 adaptor protein (KAP3 or KIFAP3) (Cole et al., 1993; Hirokawa et al., 2009), so we used siRNAs targeting KIF3A and KAP3 to deplete kinesin-2. siRNA treatment of infected cells resulted in marked depletion of both KIF3A and KAP3 as indicated by immunoblots of cell lysates (Figures 3A and S2A). Depletion of these components of kinesin-2 decreased export of Ag85B from infected cells by approximately 70%, as assayed by transferring conditioned medium to naive uninfected DC which were then used to activate Th1-polarized Ag85B-specific TCR transgenic (P25TCR-Tg) CD4 T cells (Figures 3B and S2B). Kinesin-2 depletion decreased the concentration of Ag85B protein in conditioned medium by a similar amount as detected by immunoblotting, without a difference in the number of bacteria in kinesin-2 depleted and control cells (Figures 3C and S2C-D). Kinesin-2 depletion also decreased export of at least three other M. tuberculosis antigenic proteins, ESAT-6, CFP-10, and EsxH (Figures 3C and S2C-D), indicating that the export pathway of these proteins from M. tuberculosis-infected cells resembles that of Ag85B. A similar decrease of exported bacterial antigens was observed when infected macrophages were used instead of DC (Figures 3D and E). The finding that export of M. tuberculosis antigens from infected cells depends on kinesin-2 is consistent with the results that implicated an intact microtubule cytoskeleton in antigen export. Moreover, the finding that kinesin-2 is required for export of at least four M. tuberculosis proteins, which are secreted by the bacteria using three distinct secretion systems (Ag85: SecA1; ESAT-6 and CFP-10: the type VII secretion system, Esx-1; and EsxH: Esx-3) indicates that AEV export multiple bacterial proteins that are targets of T cell recognition.
Figure 3. Kinesin 2 is required for antigen export from M. tuberculosis-infected DC and macrophages.

(A) Efficacy of siRNA knockdown of kinesin-2 components KIF3A and KAP3 in DC. (B) Kinesin-2 depletion decreases export of Ag85B antigenic activity from infected DC. Conditioned medium (CM) from infected DC treated with either a non-targeting siRNA (Control) or siRNAs targeting KIF3A and KAP3 (Kinesin-2 depleted) was transferred to fresh uninfected DC, which were used to stimulate Ag85B-specific P25TCR-Tg CD4 T cells. CD4 T cell activation was quantitated as IFNδ secretion. (C) Kinesin-2 depletion blocks export of Ag85B, CFP-10, ESAT-6, and EsxH. (D) Kinesin-2 (KIF3A and KAP3) depletion by siRNA treatment of bone marrow-derived macrophages (MØ) decreases secretion of Ag85B protein. Upper panels show efficacy of siRNA depletion of KIF3A and KAP3; the lower panel shows Ag85B in CM generated from macrophages from the same experiment. (E) Kinesin-2 depletion blocks export of Ag85B antigenic activity from macrophages. Methods were identical to those used in panel B, using bone marrow-derived macrophages instead of DC. (F) Dynasore treatment of infected DC decreases Ag85B export to CM as assayed by immunoblot or T cell activation assay (upper and lower left panels); addition of recombinant Ag85B to CM from dynasore-treated DC restored T cell activation (right panel). Data represent 4-10 independent experiments *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t-test).
To further characterize AEV and the process of antigen export, we examined the functional role of dynamin 2, which mediates budding and scission of diverse populations of intracellular vesicles (Ferguson and De Camilli, 2012), and since immunofluorescence microscopy revealed evidence for association of AEV and dynamin 2. We first depleted dynamin 2 from DC using siRNA prior to infection, but this reduced uptake of the bacteria, consistent with the previously-reported role of dynamin 2 in phagocytosis (Gold et al., 1999). As an alternative, we took advantage of the rapid kinetics of dynasore, a cell-permeable chemical inhibitor of dynamin GTPase activity (Macia et al., 2006), and added it to DC after infection with M. tuberculosis. Dynamin inhibition reduced export of Ag85B by ∼60%, as reflected by immunoblotting of CM and by activation of P25TCR-Tg Th1 cells after transfer of CM to fresh DC (Figure 3F; left panel). The decreased responses of P25TCR-Tg cells to CM from dynasore-treated infected DC were due to decreased availability of antigen in CM and not due to dynasore carried over into the T cell response assay, since P25TCR-Tg cells were able to respond when recombinant Ag85B was added to CM from dynasore-treated DC (Figure 3F; right panel). Together, the findings that dynamin 2 associates with AEV and that dynamin inhibition blocks antigen export imply a role for dynamin 2 in one or more vesicle membrane budding/scission events in the antigen export pathway.
Blocking antigen export decreases CD4 T cell priming by bystander cells and increases T cell priming by infected cells in vivo
To determine whether kinesin-2 dependent antigen export impacts activation of Ag85B-specific CD4 T cells in vivo, we used intratracheal transfer of M. tuberculosis-infected bone marrow-derived DC, followed by assay of P25TCR-Tg CD4 T cell proliferation in the mediastinal lymph node of recipient mice (Srivastava and Ernst, 2014). Transfer of M. tuberculosis-infected kinesin-2-depleted MHC II-/- DC (that are impaired in antigen export and also cannot directly activate CD4 T cells) to naïve MHC II+/+ recipients resulted in less proliferation of antigen-specific CD4 T cells than observed when infected kinesin-2 replete MHC II-/- DC were transferred (Figures 4 A-D). These results support our in vitro observations that kinesin-2 depletion in infected DC decreases export of bacterial antigens and decreases availability of antigen that can be acquired and presented by bystander cells.
Figure 4. Blockade of antigen export decreases CD4 T cell priming by bystander cells and increases priming by infected cells in vivo.

(A-D): Kinesin-2 depletion in infected donor MHC-II-/- DC blocks antigen transfer to resident MHC-II+/+ cells reflected as decreased CD4 T cell priming in vivo. (A) Frequency of Ag85B-specific P25TCR-Tg CD4+ T cells in lymph nodes of MHC II+/+ recipients following transfer of infected MHC-II-/- control (left panel) or kinesin-2-depleted (right panel) DC. (B) Quantitation of P25TCR-Tg cells in lymph nodes of the groups of mice in A. (C) Proportion of P25TCR-Tg cells that underwent ≥1 cycle of proliferation (CFSEdim) in lymph nodes of groups of mice shown in A. (D) Quantitation of P25TCR-Tg cells that underwent ≥1 cycle of proliferation (CFSEdim) in lymph nodes of groups of mice in C. (E-H): Kinesin-2 depletion in infected donor MHC-II+/+ DC increases their ability to directly prime antigen-specific CD4 T cells in vivo in MHC-II-/- mice. (E) Frequency of P25TCR-Tg cells in lymph nodes of MHC II -/- recipients following transfer of infected MHC II+/+ control (left panel) or kinesin-2-depleted (right panel) DC. (F) Quantitation of P25TCR-Tg cells in lymph nodes of groups of mice in E. (G) Proportion of P25TCR-Tg cells that underwent ≥1 cycle of proliferation (CFSEdim) in lymph nodes of mice shown in E. (H) Quantitation of P25TCR-Tg cells that underwent ≥1 cycle of proliferation (CFSEdim) in lymph nodes of groups of mice in G. Data are mean ± SEM of three pools of mice (n=6) per experimental group and represent 2 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t-test).
To determine the impact of blocking antigen export on the ability of infected cells to directly present antigen and activate antigen-specific CD4 T cells, we transferred M. tuberculosis-infected kinesin-2 depleted or kinesin-2 replete MHC class II+/+ DC into MHC class II-/- recipients and assayed proliferation of adoptively-transferred naive P25TCR-Tg CD4 T cells. This yielded the unexpected result that kinesin-2-depleted DC (which are impaired in antigen export) stimulated more T cell proliferation than did DC that were treated with a control siRNA (Figures 4E-H); this was not due to differences in the number of bacteria in the transferred DC nor to enhanced migration of kinesin-2 depleted DC to the local lymph node (Figures S3A-C). Since the only cells that can activate CD4 T cells under these conditions are the infected, transferred DC, this result indicates that disrupting antigen export increases the antigen presenting efficacy of M. tuberculosis-infected cells. These results indicate that antigen export is a mechanism that decreases MHC class II antigen presentation by M. tuberculosis-infected cells.
Antigen export limits overall CD4 T cell activation in vivo
Our previously-published finding that M. tuberculosis-infected cells export antigen for uptake and presentation by uninfected bystander cells implied that antigen transfer compensates for impaired antigen presentation by infected cells (Srivastava and Ernst, 2014). In the context of those results, our finding that blocking antigen export could increase antigen presentation by infected cells raised the question whether antigen transfer to bystander cells fully compensates for impaired antigen presentation by infected cells. To address this question, we transferred wild-type M. tuberculosis-infected kinesin-2 depleted (impaired for antigen transfer) or control (competent for antigen transfer) DC into wild-type mice and quantitated proliferation of adoptively-transferred naïve P25TCR-Tg CD4 T cells. This revealed quantitatively greater proliferation of antigen-specific CD4 T cells in recipients of kinesin-2 depleted DC compared with that in recipients of control DC (Figures 5A-D); this was not due to a difference in the number of antigen-producing bacteria in the two groups of cells or a difference in their ability to migrate to the local lymph node (Figures S4A-D). These results indicate that blocking antigen export enhances CD4 T cell activation to a greater extent than can be accomplished by antigen export followed by antigen uptake and presentation by bystander cells. These results also indicate that antigen export is a quantitatively significant mechanism that reduces MHC class II antigen presentation by M. tuberculosis-infected cells.
Figure 5. Blockade of antigen export from infected DC enhances overall antigen-specific CD4 T cell activation in vivo.

Comparison of Ag85B-specific CD4 T cell proliferation in MHC II+/+ recipients following transfer of infected kinesin-2 depleted or control MHC II+/+ DC. (A) Frequency of Ag85B-specific P25TCR-Tg CD4+ T cells in lymph nodes. (B) Quantitation of total P25TCR-Tg CD4+ T cells in lymph nodes of groups of mice shown in A. (C) Proportion of P25TCR-Tg cells that underwent ≥1 cycle of proliferation (CFSEdim) in lymph nodes of groups of mice shown in A. (D) Quantitation of P25TCR-Tg cells that underwent ≥1 cycle of proliferation (CFSEdim) in lymph nodes of groups of mice in C. Data are mean ± SEM of four pools of mice (n=8) per experimental group and represent 2 independent experiments. *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t-test).
Blocking antigen export enhances activation of antigen-specific Th1 effector T cells and T cell-dependent antimycobacterial activity
The unexpected finding that blocking antigen transfer in vivo enhances priming and proliferation of naive CD4 T cells suggested that blocking antigen transfer might also enhance activation of CD4 effector T cells. When varying numbers of kinesin-2-depleted or control DC infected with M. tuberculosis were cultured with a constant number of Ag85B-specific Th1-polarized P25TCR-Tg CD4 T cells, kinesin-2 depleted DC were superior to kinesin-2-replete DC at all DC:T cell ratios examined, with 3-fold greater T cell activation by kinesin-2-depleted cells at the lowest DC:T cell ratio examined (Figure 6A). Enhanced activation of antigen-specific CD4 effector cells was due to enhanced presentation of antigen produced by intracellular bacteria and not due to increased surface expression of MHC II or costimulatory molecules, differential apoptosis, or cytokine secretion; depletion of kinesin-2 in DC did not affect activation of Ag85B-specific CD4 T cells when antigen was provided as recombinant Ag85B or as peptide 25 (Figures S5A-E). At a constant ratio of DC:effector T cells, enhanced activation of CD4 effector cells by kinesin-2-depleted DC depended on the dose of siRNA used; as little as 25 nM siRNA caused a significant increase in T cell secretion of IFNγ. (Figure 6B). Kinesin-2 depletion did not affect uptake of M. tuberculosis, as reflected by the number of viable bacteria present immediately following infection, prior to addition of P25TCR-Tg Th1 cells (Figure S5F).
Figure 6. Blockade of antigen export by kinesin 2 depletion increases activation of Ag85B-specific CD4 T cells and enhances control of intracellular M. tuberculosis.

(A): Kinesin-2 depletion in DC increases their ability to activate P25TCR-Tg Th1 CD4 effector T cells (IFNγ secretion) at a range of DC: T cell ratios. (B) Increased activation of P25TCR-Tg Th1 CD4 effector T cells by kinesin-2 depleted cells is siRNA dose-dependent. (C) Increased activation of P25TCR-Tg Th1 CD4 effector T cells by kinesin-2 depleted DC results in enhanced control of intracellular bacteria. (D) Increased activation of P25TCR-Tg Th1 CD4 effector T cells by kinesin-2 depleted DC depends on MHC class II and bacterial antigen. (E) Enhanced control of intracellular M. tuberculosis in kinesin 2-depleted DC by P25TCR-Tg Th1 CD4 effector T cells requires MHC II and bacterial antigen. Data represent 2-4 independent experiments *p < 0.05; **p < 0.01; ***p < 0.001 (Student's t-test).
The evidence that blocking antigen export from M. tuberculosis-infected cells caused increased CD4 effector T cell activation suggested that blocking antigen export might also enhance T cell-dependent control of intracellular M. tuberculosis infection. When Ag85B-specific P25TCR-Tg Th1 cells were cocultured with M. tuberculosis-infected bone marrow DC that were depleted of kinesin-2 by treatment with KIF3A and KAP3 siRNAs, there was an siRNA dose-dependent decrease in the number of viable bacteria recovered after 20 hours, compared with that in export-competent control cells (Figure 6C). The decreased recovery of viable bacteria was not a consequence of decreased DC viability or bacterial uptake (Figure S5F), indicating that inhibition of antigen export not only increased activation of antigen-specific CD4 T cells, but also improved T cell-dependent control of intracellular infection. The apparent threshold for improved bacterial control was higher than that required for increased CD4 T cell activation: treatment of DC with 25 nM siRNA was sufficient to increase activation of antigen-specific CD4 T cells, while 50 nM was required to enhance control of intracellular M. tuberculosis. In these experiments, CD4 T cell activation and bacterial control required both MHC class II and specific antigen, as MHC-II-/- DC infected with wild-type M. tuberculosis (H37Rv) and MHC-II+/+ DC infected with Ag85B-null M. tuberculosis (H37Rv:Δ85B) did not activate P25TCR-Tg Th1 cells, and did not enhance control of bacterial growth (Figures 6D, 6E and S5G). These results indicate that antigen export from M. tuberculosis-infected cells contributes to survival of the bacteria in the presence of antigen-specific Th1 cells.
Discussion
The studies reported here reveal that DC and macrophages infected with Mycobacterium tuberculosis divert mycobacterial antigens away from the MHC class II antigen presentation pathway and to the extracellular space, and by doing so reduce their activation of CD4 T cells, leading to improved survival of the intracellular bacteria. Although we and others have previously reported that antigen transfer from infected to uninfected DC after aerosol infection provides a mechanism to compensate for impaired antigen presentation by infected cells (Samstein et al., 2013; Srivastava and Ernst, 2014), our present results provide two major insights. They reveal that the first stage of antigen transfer, antigen export from infected cells, clearly benefits the pathogen, by reducing antigen presentation and CD4 T cell activation by infected cells. They also reveal that the second stage, antigen uptake and presentation by uninfected cells, provides incomplete compensation for the decreased antigen presentation by infected cells, indicating that the net effect of antigen export is to diminish activation of antigen-specific CD4 T cells at the site of infection.
Antigen export is a quantitatively important route for trafficking of mycobacterial antigens produced by intraphagosomal bacteria, as indicated by the 3-fold increase in activation of Ag85B-specific CD4 T cells when antigen export was blocked by depletion of kinesin-2 (Figure 6A). This implies that more antigen enters the export pathway than enters the MHC class II pathway in M. tuberculosis-infected cells. Diverting antigen to the export pathway has functional significance for survival of the bacteria, as blockade of antigen export also improves CD4 T cell-dependent control of intracellular infection. This finding is especially relevant in light of our previous results using mixed (MHC II-/- and MCH II+/+) bone marrow chimeric mice that revealed that CD4 T cells must directly recognize infected lung DC and macrophages to control intracellular infection in vivo (Srivastava and Ernst, 2013). By exporting bacterial antigens, M. tuberculosis-infected cells reduce the availability of antigen for presentation on MHC class II, diminishing their ability to be recognized by and to activate CD4 effector cells, thereby promoting bacterial persistence. A limitation of the present work is that we have not yet quantitated the impact of antigen export on bacterial persistence in vivo; additional experiments are needed to reveal this important information.
In this and our previous report (Srivastava and Ernst, 2014), we found that all of the M. tuberculosis antigens that we examined are exported from infected cells, and share a kinesin-2-dependent export pathway. While this establishes that antigen export is not unique to Ag85B, the antigen we studied in most detail, it raises the question whether all mycobacterial antigens are diverted from the MHC class II pathway and exported to a similar extent. One common property of the antigens we have examined so far is that they are secreted by the bacteria. These proteins may accumulate to especially high concentrations in the intraphagosomal space and therefore might be particularly prone to export by intracellular vesicles that bud from the phagosome membrane and enter the export pathway. Further studies of the intracellular trafficking of antigens that are not secreted by the bacteria are warranted, as our findings imply that antigens that are not exported from infected cells might induce more productive CD4 T cell activation and thus might be especially useful TB vaccine antigens.
Our finding that antigen export provides a mechanism for evasion of CD4 T cell activation by infected cells may also help explain the observation that few, if any, human T cell epitopes of M. tuberculosis exhibit evidence of escape mutations (Comas et al., 2010; Copin et al., 2014; Coscolla et al., 2015). The efficiency of antigen export and evidence that it limits activation of CD4 T cells by infected cells implies that it may reduce selection pressure from T cell recognition so that epitope escape mutations are neither selected nor necessary for persistence and proliferation of M. tuberculosis.
The results reported here provide initial insight into key mechanistic elements of antigen export. We found that Ag85B is present in extraphagosomal membrane vesicles, that one or more steps in antigen export depends on the GTPase activity of dynamin-2, and that antigen export vesicles (AEV) are associated with microtubules and the motor protein, kinesin-2. We also determined that disruption of microtubules or depletion of kinesin-2 decreases antigen export. Notably, we found that decreasing antigen export by kinesin-2 depletion improved the ability of M. tuberculosis-infected cells to activate Ag85B-specific CD4 T cells and the ability of CD4 T cells to control infection. These findings provide a strong rationale for efforts to identify additional mechanistic steps of antigen export and their essential mediators, including mechanisms for AEV budding from phagosomes, AEV targeting through a nondegradative pathway, and AEV membrane fusion with the plasma membrane for extracellular delivery of bacterial antigens. Such efforts will also clarify the identity of AEV and their relationship to previously-characterized vesicular compartments involved in intracellular protein transport, and will provide insight into whether AEV are an existing vesicle population that is co-opted by M. tuberculosis, or whether AEV are generated in response to infection. Identification of specific steps and mediators in the antigen export pathway may guide pharmacological intervention to block antigen export and enhance the efficacy of CD4 T cell responses in control of TB. As the interactions of mycobacterial phagosomes with intracellular vesicle networks have been thought to be limited, the further characterization of AEV will also shed light on a fundamental interaction of intracellular M. tuberculosis with host cells.
Another important matter is whether antigen export or a similar process contributes to the pathogenesis of other infections. One related phenomenon is the export of typhoid toxin from epithelial cells infected with the agent of typhoid fever, Salmonella enterica serovar Typhi (S. Typhi)(Spano and Galan, 2008). Like M. tuberculosis, S. Typhi resides in vesicles, and exports a heterotrimeric toxin from infected cells to intoxicate neighboring uninfected cells. Typhoid toxin appears as intracellular puncta in infected cells; upon release to the extracellular media typhoid toxin is neutralized by an antibody, indicating that it is no longer contained in a membrane-bound compartment (Spano and Galan, 2008). Export of typhoid toxin requires assembly of the trimeric complex of CdtB, PltA, and PltB, indicating that structural features of PltA and/or PltB are required to target the complex to export vesicles. Our finding that multiple structurally unrelated M. tuberculosis antigens are exported suggests that the export pathway in M. tuberculosis-infected cells does not require specific structural features for a protein to enter and to be transported. Together, the findings with typhoid toxin and M. tuberculosis antigen export provide evidence for a mechanism beyond cytokine secretion, bacterial membrane vesicle shedding (Rath et al., 2013), exosome shedding (Bhatnagar et al., 2007), and apoptotic vesicles (Divangahi et al., 2010; Winau et al., 2006) for pathogen-infected cells to communicate with and affect the function of other cells, by release of soluble pathogen proteins through an intracellular nondegradative pathway. Further characterization of the antigen export pathway of M. tuberculosis-infected cells will provide opportunities and targets to enhance the efficacy of natural and vaccine-induced T cells in TB, and may also provide means of intervening in the pathogenesis of other infectious diseases.
Experimental Procedures
Mice
Mice were bred and housed in specific pathogen-free facilities. Intratracheal administration of infected bone marrow-derived DC was done as we recently described (Srivastava and Ernst, 2014) followed by harvest and processing of MDLN and lungs into single cell suspensions. The staining and quantitation of antigen-specific CD4 T cell proliferation and bacteria in tissues was performed as previously described (Wolf et al., 2008; Wolf et al., 2007). All procedures were approved by the NYU School of Medicine IACUC.
Bone-marrow derived DC and macrophages
CD11c+ DC were sorted following culture of fresh or cryopreserved bone marrow in BMDC medium (RPMI 1640 with 10% heat-inactivated FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1× β-ME, 10 mM HEPES) containing 10 ng/ml recombinant murine GM-CSF for 7 days. For siRNA treatments, CD11c+ DC were sorted on day 5. Macrophages were generated by culture of bone marrow cells in BMDM medium (DMEM with 10% heat-inactivated FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 1× β-ME, 10 mM HEPES, and 20 ng/ml recombinant murine M-CSF from Peprotech) for six days prior to use in the designated assays.
Immunofluorescence and Confocal microscopy
BMDC on coverslips were stained with biotinylated anti-CD45.2 (Biolegend) followed by streptavidin Alexa-647 (Invitrogen) and fixed overnight in PFA. Fixed cells were permeabilized in cold PBS/0.5% Triton X-100 (5 min on ice), washed, and stained for Ag85, and transferrin receptor, tubulin, KIF3A, dynamin 2 or MHC-II (see Supplemental Experimental Procedures). Finally, coverslips were washed and mounted in Vectashield mounting medium (Vector Labs). An automated TCS SP5 II confocal microscope (Leica) with a 63×/1.4 oil immersion lens was used to acquire 3-D image stacks (0.3-0.5 μm) using appropriate laser combinations. Representative image stacks of average intensity from relevant channels were merged and converted to RGB format using Image J. All related groups were subjected to similar color settings for each color channel.
Quantitation of colocalization
Colocalization analysis was performed on raw images in Volocity image analysis software using the built-in Manders' overlap coefficient colocalization feature. Multiple confocal images were subjected to background subtraction followed by colocalization analysis on a per-cell basis. The data was presented as Manders' colocalization coefficient of Ag85 with respect to other proteins; that is, the fraction of Ag85 that colocalized with the other protein of interest in each analysed cell (Dunn et al., 2011). The data represent 2-3 independent experiments and 100-300 cells per experiment.
Cryo-Immunoelectron microscopy
BMDC infected with wild-type M. tuberculosis H37Rv or Ag85B-deficient M. tuberculosis (Wolf et al., 2008) were washed and fixed (PBS/2% PFA/0.2% glutaraldehyde) as pellets that were then processed for cryo-immunoelectron microscopy as described in Supplemental Experimental Procedures.
siRNA
ON-Target plus SMART pool Kif3a siRNA (L-042111-01), Kifap3 (KAP3) siRNA (L-047278-01) and ON-Target plus non-targeting control siRNA (D-001810-01) were obtained from GE Dharmacon (details in Table S1).
Generation of Conditioned Media (CM) and in vitro antigen transfer assay
Bone marrow-derived macrophages or CD11c+ DC were treated with control or targeting siRNAs beginning on day 5 of culture. After 48 hours of exposure to siRNAs, the cells were infected overnight with M. tuberculosis (see Supplemental Experimental Procedures for details). After overnight infection, cells were washed, treated with amikacin, washed again and cultured further in fresh BMDC or BMDM medium that was harvested after 20-24 hours as conditioned medium (CM). The CM was sterile filtered and added to uninfected BMDC in the presence of P25TCR-Tg CD4+ Th1 effector cells for 60 h, as described previously (Srivastava and Ernst, 2014). Culture supernatants were collected at 60 h and used for IFNγ quantitation by ELISA (BD Biosciences).
In vitro T cell activation by infected DC and bacterial quantitation
CD11c+ bone marrow-derived DC were cultured, treated with siRNAs, and infected with M. tuberculosis as in the preceding section. After amikacin treatment and washing, the DC were cultured further in fresh BMDC medium in the absence or presence of Th1 polarized P25-TCRTg CD4 T cells for 20-24 hours. After 20-24 hrs, plates were spun to pellet cells, and the supernatants were aspirated carefully, sterile filtered, and assayed for IFNγ by ELISA (BD Biosciences). The cells in the wells were lysed in PBS-0.5% Tween 80 at a cell density of 2 × 105/ml, and plated on 7H11 agar for bacterial quantitation (see Supplemental Experimental Procedures for details).
In vivo T cell activation following intratracheal transfer of infected, siRNA treated DC
CD11c+ bone marrow-derived DC were cultured and treated with siRNAs as for in vitro experiments, with the variation that siRNAs were used at a final concentration of 80 nM and siRNA treatment was for 3 days prior to overnight infection with M. tuberculosis (MOI, 1). After washing and amikacin treatment, the siRNA-treated infected DC were administered intratracheally (3-5 × 105 DC/mouse) one day after adoptive transfer of CFSE-labeled naive P25TCR-Tg CD4 cells (1-3 × 106/mouse). To assay the effect of blocking antigen export by transferred DC in the absence of direct T cell priming, MHC-II-/- DC were treated with relevant siRNAs, infected and administered intratracheally to MHC-II+/+ mice. To assay the effect of antigen export on direct T cell priming by transferred DC in the absence of antigen presentation by resident cells, MHC-II+/+ DC were treated with relevant siRNAs, infected, and administered intratracheally to MHC-II-/- mice. To assay the net effect of antigen export when antigen-specific CD4 T cells could be activated by direct antigen presentation by infected cells and by indirect activation after exported antigen is taken up and processed by resident uninfected cells, MHC-II+/+ DC were treated with relevant siRNAs, infected, and administered intratracheally to MHC-II+/+ mice. In all experiments, MDLN were harvested and processed sixty hours after BMDC transfer for bacterial quantitation and flow cytometry while lungs were processed for bacterial quantitation only.
Supplementary Material
Acknowledgments
We thank the NYU imaging core facility (Feng-Xia (Alice) Liang for cryo-immunoelectron microscopy, and Yan Deng for assistance with confocal microscopy (both supported by NCRR S10 RR024708)). We greatly appreciate early guidance by Sergio Grinstein, Ph.D., and are grateful to members of the Ernst Lab for helpful discussions and suggestions on early drafts of the manuscript. We also thank Jennifer Philips, M.D., Ph.D. and members of her laboratory for sharing their siRNA expertise with primary cells.
Supported by grants from the National Institutes of Health (AI 051242 and AI 084041).
Footnotes
Author contributions: S.S., P.S.G., and J.D.E. planned the experiments; S.S. and P.S.G. performed the experiments; S.S., P.S.G., and J.D.E. interpreted the results and wrote the manuscript.
The authors have no conflicts of interest relevant to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baena A, Porcelli SA. Evasion and subversion of antigen presentation by Mycobacterium tuberculosis. Tissue antigens. 2009;74:189–204. doi: 10.1111/j.1399-0039.2009.01301.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty WL, Russell DG. Identification of mycobacterial surface proteins released into subcellular compartments of infected macrophages. Infection and immunity. 2000;68:6997–7002. doi: 10.1128/iai.68.12.6997-7002.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood. 2007;110:3234–3244. doi: 10.1182/blood-2007-03-079152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chackerian AA, Alt JM, Perera TV, Dascher CC, Behar SM. Dissemination of Mycobacterium tuberculosis is influenced by host factors and precedes the initiation of T-cell immunity. Infection and immunity. 2002;70:4501–4509. doi: 10.1128/IAI.70.8.4501-4509.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Horwitz MA. The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. The Journal of experimental medicine. 1996;184:1349–1355. doi: 10.1084/jem.184.4.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole DG, Chinn SW, Wedaman KP, Hall K, Vuong T, Scholey JM. Novel heterotrimeric kinesin-related protein purified from sea urchin eggs. Nature. 1993;366:268–270. doi: 10.1038/366268a0. [DOI] [PubMed] [Google Scholar]
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nature genetics. 2010;42:498–503. doi: 10.1038/ng.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copin R, Coscolla M, Seiffert SN, Bothamley G, Sutherland J, Mbayo G, Gagneux S, Ernst JD. Sequence diversity in the pe_pgrs genes of Mycobacterium tuberculosis is independent of human T cell recognition. mBio. 2014;5:e00960–00913. doi: 10.1128/mBio.00960-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscolla M, Copin R, Sutherland J, Gehre F, de Jong B, Owolabi O, Mbayo G, Giardina F, Ernst JD, Gagneux S. M. tuberculosis T Cell Epitope Analysis Reveals Paucity of Antigenic Variation and Identifies Rare Variable TB Antigens. Cell host & microbe. 2015;18:538–548. doi: 10.1016/j.chom.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophysical journal. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nature immunology. 2010;11:751–758. doi: 10.1038/ni.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. American journal of physiology Cell physiology. 2011;300:C723–742. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst JD. The immunological life cycle of tuberculosis. Nature reviews Immunology. 2012;12:581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nature reviews Molecular cell biology. 2012;13:75–88. doi: 10.1038/nrm3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallegos AM, Pamer EG, Glickman MS. Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. The Journal of experimental medicine. 2008;205:2359–2368. doi: 10.1084/jem.20080353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudin R, de Alencar BC, Jouve M, Berre S, Le Bouder E, Schindler M, Varthaman A, Gobert FX, Benaroch P. Critical role for the kinesin KIF3A in the HIV life cycle in primary human macrophages. The Journal of cell biology. 2012;199:467–479. doi: 10.1083/jcb.201201144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology. 2012;13:1118–1128. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold ES, Underhill DM, Morrissette NS, Guo J, McNiven MA, Aderem A. Dynamin 2 is required for phagocytosis in macrophages. The Journal of experimental medicine. 1999;190:1849–1856. doi: 10.1084/jem.190.12.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harth G, Lee BY, Wang J, Clemens DL, Horwitz MA. Novel insights into the genetics, biochemistry, and immunocytochemistry of the 30-kilodalton major extracellular protein of Mycobacterium tuberculosis. Infection and immunity. 1996;64:3038–3047. doi: 10.1128/iai.64.8.3038-3047.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nature reviews Molecular cell biology. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Hudrisier D, Neyrolles O. Antigen smuggling in tuberculosis. Cell host & microbe. 2014;15:657–659. doi: 10.1016/j.chom.2014.05.018. [DOI] [PubMed] [Google Scholar]
- Kaufmann SH, Lange C, Rao M, Balaji KN, Lotze M, Schito M, Zumla AI, Maeurer M. Progress in tuberculosis vaccine development and host-directed therapies--a state of the art review. Lancet Respir Med. 2014;2:301–320. doi: 10.1016/S2213-2600(14)70033-5. [DOI] [PubMed] [Google Scholar]
- Kuhn JR, Poenie M. Dynamic polarization of the microtubule cytoskeleton during CTL-mediated killing. Immunity. 2002;16:111–121. doi: 10.1016/s1074-7613(02)00262-5. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Developmental cell. 2006;10:839–850. doi: 10.1016/j.devcel.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Miller JC, Brown BD, Shay T, Gautier EL, Jojic V, Cohain A, Pandey G, Leboeuf M, Elpek KG, Helft J, et al. Deciphering the transcriptional network of the dendritic cell lineage. Nature immunology. 2012;13:888–899. doi: 10.1038/ni.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annual review of pathology. 2012;7:353–384. doi: 10.1146/annurev-pathol-011811-132458. [DOI] [PubMed] [Google Scholar]
- Poulsen A. Some clinical features of tuberculosis. 1. Incubation period. Acta tuberculosea Scandinavica. 1950;24:311–346. [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nature reviews Molecular cell biology. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- Rath P, Huang C, Wang T, Wang T, Li H, Prados-Rosales R, Elemento O, Casadevall A, Nathan CF. Genetic regulation of vesiculogenesis and immunomodulation in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4790–4797. doi: 10.1073/pnas.1320118110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiley WW, Calayag MD, Wittmer ST, Huntington JL, Pearl JE, Fountain JJ, Martino CA, Roberts AD, Cooper AM, Winslow GM, et al. ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:10961–10966. doi: 10.1073/pnas.0801496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samstein M, Schreiber HA, Leiner IM, Susac B, Glickman MS, Pamer EG. Essential yet limited role for CCR2(+) inflammatory monocytes during Mycobacterium tuberculosis-specific T cell priming. eLife. 2013;2:e01086. doi: 10.7554/eLife.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano S, Galan JE. A novel pathway for exotoxin delivery by an intracellular pathogen. Current opinion in microbiology. 2008;11:15–20. doi: 10.1016/j.mib.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ernst JD. Cutting edge: Direct recognition of infected cells by CD4 T cells is required for control of intracellular Mycobacterium tuberculosis in vivo. Journal of immunology. 2013;191:1016–1020. doi: 10.4049/jimmunol.1301236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S, Ernst JD. Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell host & microbe. 2014;15:741–752. doi: 10.1016/j.chom.2014.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stinchcombe JC, Majorovits E, Bossi G, Fuller S, Griffiths GM. Centrosome polarization delivers secretory granules to the immunological synapse. Nature. 2006;443:462–465. doi: 10.1038/nature05071. [DOI] [PubMed] [Google Scholar]
- Sturgill-Koszycki S, Schaible UE, Russell DG. Mycobacterium-containing phagosomes are accessible to early endosomes and reflect a transitional state in normal phagosome biogenesis. The EMBO journal. 1996;15:6960–6968. [PMC free article] [PubMed] [Google Scholar]
- Tailleux L, Schwartz O, Herrmann JL, Pivert E, Jackson M, Amara A, Legres L, Dreher D, Nicod LP, Gluckman JC, et al. DC-SIGN is the major Mycobacterium tuberculosis receptor on human dendritic cells. The Journal of experimental medicine. 2003;197:121–127. doi: 10.1084/jem.20021468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdahl KB, Shafiani S, Ernst JD. Initiation and regulation of T-cell responses in tuberculosis. Mucosal immunology. 2011;4:288–293. doi: 10.1038/mi.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale RD, Schnapp BJ, Mitchison T, Steuer E, Reese TS, Sheetz MP. Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell. 1985;43:623–632. doi: 10.1016/0092-8674(85)90234-x. [DOI] [PubMed] [Google Scholar]
- Winau F, Weber S, Sad S, de Diego J, Hoops SL, Breiden B, Sandhoff K, Brinkmann V, Kaufmann SH, Schaible UE. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity. 2006;24:105–117. doi: 10.1016/j.immuni.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Wolf AJ, Desvignes L, Linas B, Banaiee N, Tamura T, Takatsu K, Ernst JD. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. The Journal of experimental medicine. 2008;205:105–115. doi: 10.1084/jem.20071367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AJ, Linas B, Trevejo-Nunez GJ, Kincaid E, Tamura T, Takatsu K, Ernst JD. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. Journal of immunology. 2007;179:2509–2519. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.