

Abstract
Heritable mutations in the BAP1 tumor suppressor gene predispose individuals to mesothelioma and other cancers. However, a large-scale assessment of germline BAP1 mutation incidence and associated clinical features in mesothelioma patients with a family history of cancer has not been reported. Therefore, we examined the germline BAP1 mutation status of 150 mesothelioma patients with a family history of cancer, 50 asbestos-exposed control individuals with a family history of cancers other than mesothelioma, and 153 asbestos-exposed individuals without familial cancer. No BAP1 alterations were found in control cohorts, but were identified in 9 of 150 mesothelioma cases (6%) with a family history of cancer. Alterations among these cases were characterized by both missense and frameshift mutations, and enzymatic activity of BAP1 missense mutants was decreased compared to wild-type BAP1. Furthermore, BAP1 mutation carriers developed mesothelioma at an earlier age that was more often peritoneal than pleural (5 of 9), and exhibited improved long-term survival compared to mesothelioma patients without BAP1 mutations. Moreover, many tumors harboring BAP1 germline mutations were associated with BAP1 syndrome, including mesothelioma and ocular/cutaneous melanomas, as well as renal, breast, lung, gastric, and basal cell carcinomas. Collectively, these findings suggest that mesothelioma patients presenting with a family history of cancer should be considered for BAP1 genetic testing to identify those individuals who might benefit from further screening and routine monitoring for the purpose of early detection and intervention.
Keywords: mesothelioma, asbestos, familial cancer, cancer predisposition, BAP1
Introduction
A history of asbestos exposure has been documented in about 80% of individuals diagnosed with malignant mesothelioma (MM) (1). However, MM has been shown to develop in only about 5% of heavily exposed asbestos miners who were followed for 45 years (2), and just 3,200 new cases of MM are diagnosed each year (3) among the nearly 27 million workers in the US who were exposed to asbestos between 1940 and 1979 (4). Additionally, there is no dose-response relationship between asbestos exposure and MM (1). Collectively, these data have led investigators to conclude that asbestos exposure appears to be necessary, but not sufficient, for the development of MM (5).
Intriguingly, MM has been shown to cluster in certain families, i.e., those with so-called familial MM. Furthermore, a history of other common cancers is frequent among individuals who develop both familial and sporadic MM, and the enhanced susceptibility to common cancers appears to extend to other members of such families, as well (6–8). In a study of familial MM, clustering was not found to be due to similar exposures to asbestos, leading investigators to conclude that genetic factors must be involved (7). Thus, genetic susceptibility likely has a significant role in the etiology of MM (8, 9).
In 2011, germline mutations of the BAP1 tumor suppressor gene were reported in two families with multiple MMs and/or uveal melanomas (UMs) as well as other tumors such as kidney cancer (8). In addition, germline BAP1 mutations were identified in 2 of 26 sporadic MMs, and both individuals with mutant BAP1 were previously diagnosed with UM. Concurrently, Weisner et al. described germline BAP1 mutations in two families with atypical melanocytic tumors as well as cutaneous melanoma (CM) and UM (10). To investigate the potential contribution of germline BAP1 mutations in UM patients with possible predisposition to hereditary cancer, Abdel-Rahman et al. identified a patient with a germline truncating mutation of BAP1, which segregated in several family members with UM, lung adenocarcinoma, and meningioma (11). Moreover, several other members of this family had MM. A number of other papers have confirmed these findings and/or extended the disease phenotype to other cancer types, including renal cell carcinoma (RCC) (12). Collectively, these findings have led to the identification of a BAP1 tumor predisposition syndrome characterized by MM, CM, UM, RCC and potentially other tumors due to heterozygous germline mutations of BAP1 (13, 14).
Based on these studies, we hypothesized that germline mutations in BAP1 may contribute to susceptibility to MM in asbestos-exposed individuals through a mechanism that involves a gene-environment interaction. We further postulated that germline BAP1 mutations would more likely occur if an MM index case had a family history of various cancers, consistent with a genetic predisposition to cancer. We recently reported in vivo genetic evidence that germline heterozygous mutation of Bap1 accelerates development of asbestos-induced MM (15). No spontaneous MMs were seen in unexposed Bap1-mutant mice, suggesting that high penetrance of MM requires environmental exposure to asbestos of other carcinogenic fibers. To further investigate its role in asbestos-induced MM, we determined the prevalence of germline BAP1 mutations in a population of asbestos-exposed MM cases and controls. The MM cases were selected for a past personal or family history of cancer and compared with asbestos-exposed controls either with or without a personal or family cancer history. While no alterations of BAP1 were found in controls, BAP1 mutations were identified in a 9 of 150 MM cases with a personal or family history of cancer. The findings presented here imply that patients presenting with a MM and a family history of cancer should be considered for genetic testing to identify families who might benefit from screening and regular monitoring, with the goal of early cancer detection and intervention.
Materials and Methods
Study Populations and Data Acquisition
All MM cases had a history of asbestos exposure and were identified through one or more of the following sources: 1) their health care providers; 2) a support organization (Mesothelioma Applied Research Foundation, MARF); or 3) independent medical evaluations for medical legal purposes. All study participants gave informed consent for their participation, and the protocol was approved by the Institutional Review Board at Wake Forest School of Medicine. Additionally, MM cases were selected based on a past personal or family history of one or more of the cancers previously reported in BAP1 syndrome families, as above. Study entry criteria consisted of a pathology report, including immunohistochemical staining confirming the diagnosis of MM, from a CLIA-certified hospital laboratory within the United States or Canada. All cases had a personal interview with the examining physician or study coordinator to clarify questionnaire responses, and most subjects also underwent a complete physical examination and physician review of the full medical record. Legal cases frequently had independent pathologic review in addition to the clinical pathologic diagnostic workup noted above. Demographic data, such as gender, age, age at first exposure to asbestos, occupational history, and personal and family health history, were collected and archived from the questionnaire/interview.
We developed a scoring system to prioritize the intensity of this “cancer signal.” As part of this system, the following four conditions were identified: 1) personal history of cancer; 2) parent, 3) sibling, or 4) child with history of cancer. To ascertain individual case scores the following criteria were used. Initially, the population was narrowed by identifying MM cases that had either a personal history of cancer or a first degree relative with a cancer. In order to help prioritize which of the MM subjects had a higher cancer signal, we developed a “Cancer Epi” scoring system. As part of this system, the following four conditions were identified: personal history of cancer, parent with history of cancer, sibling with history of cancer, and child with history of cancer. Scores were assigned as follows: 4 conditions, 4 points; 3 conditions, 3 points, 2 conditions, 2 points; and 1 condition only, 1 point. In addition, MM cases were given an additional point for each of the following two situations that were thought to indicate a higher cancer signal in the family: sib ratio ≥ 0.5 (at least half of the subject’s siblings must have cancer), and a child with cancer. Finally, subjects were also given an additional point for having a personal or family history of one of the following known or potential BAP1 syndrome-related cancers: CM, UM, RCC and breast cancer, and/or MM in another family member. Altogether, we identified 150 MM cases with known asbestos exposure and a family history of cancer (i.e., high cancer signal).
Like the MM case cohort, two control cohorts were identified through screening for litigation. One control cohort consisted of 50 individuals chosen using the same scoring criteria as for our MM cases (i.e., asbestos exposed and with a high family cancer signal), except that MM was not observed in the index cases or their families. Nineteen of the index cases in this control cohort had been diagnosed with a cancer other than MM, and the remaining 31 had no personal history of cancer. A second control group consisted of 153 subjects without a familial cancer history but with a known asbestos exposure history; these individuals were diagnosed with mild asbestosis and/or pleural plaques. This group was selected specifically to minimize cancer signal and to maximize latency (the time elapsed from date of first asbestos exposure to diagnosis of asbestos related diseases, other than MM). The second control cohort therefore shared the following characteristics: 1) long latency; 2) older age; and 3) absence of a personal or family history of cancer.
Sequence Analysis
Nine PCR products encompassing the entire BAP1 coding exons, adjacent intron splice sites, and 5′ and 3′ untranslated regions were amplified for sequencing. PCR primer pairs used to amplify the BAP1 gene were previously described (16). PFU Turbo (Stratagene/Agilent; Santa Clara, CA) was used following the manufacturer’s suggested protocol and the following cycling program: 95°C 2 min; 45 cycles of (95°C 30 sec, 65°C 30 sec, 72°C 2 min); 72°C 10 min. PCR products were gel purified and Sanger sequenced using the following primers: preT7, gggaggtctatataagcaga; and T3, attaaccctcactaaaggga. Variants found were subjected to analysis through dbSNP to find previously described polymorphisms. BAP1 mutations were described using cDNA and protein mutation nomenclature standardized by the Human Genome Variation Society (HGVS, http://www.hgvs.org/mutnomen) and cDNA accession # NM_004656 and protein accession # NP_004647 as references.
Ub-AMC Assay
293T cells were transfected with Myc-tagged wild-type (WT) or mutant BAP constructs. Whole cell lysates were extracted with NP40 lysis buffer. Bap1 antibody was pre-coated on protein A/G PLUS-Agarose beads in immunoprecipitation (IP) binding buffer and A/G resin and rotated overnight at 4°C. Then 0.5 mg cell lysate in 0.5 ml of IP binding buffer was mixed with 100 μl of BAP(rAb) conjugated A/G beads, followed by rotation at 4°C for 2 h and then washed several times in washing buffer and then in Ub-AMC Reaction Buffer. The resin was resuspended in 40 μl of 1× Reaction Buffer for Ub-AMC assay. For each reaction, 15 nM of Ub-AMC (3 μl) was diluted into 12 μl of 1× Reaction Buffer, and then 15 μl of Ub-AMC substrate was added into each of IP-BAP1 reaction. Purified BAP1 protein (1 nM) was used as positive control for the Ub-AMC assay. Ub-AMC activity was assayed at 25°C in 20 mM HEPES buffer (pH 7.5), 100 mM NaCl, 1 mM EDTA, 5 mM DTT, and 0.05% (w/v) Tween-20. Assays were performed in 384-well plates (PerkinElmer), and fluorescence was measured at 5-min intervals at excitation and emission wavelengths of 355 nm and 460 nm, respectively. The hydrolysis rate was linear for 40~60 min. and corrected for background signal (no enzyme). BAP1 [A/G bead conjugated BAP (rAb)] (~40 μl) was mixed with Ub-AMC (~15 μl) and then rotated at room temperature for 5, 10 or 15 min. The resin was spun down and 25 μl of supernatant was used for each activity reading. After completing the Ub-AMC assay, immunoblotting was performed to determine BAP1 protein expression level using anti-BAP1 monoclonal antibody (Oncogene) (1:1000). For each sample, Ub-AMC activity was normalized based on its BAP1 protein level.
Splicing Assay
The splicing assay was performed using mini-gene expression constructs. A PCR-based strategy was used to clone genomic BAP1 sequences encompassing exons and intervening introns flanking the regions of analysis (i.e., splice site mutation sites), using genomic DNA from samples MC7039 and MC7058 as templates. The primers incorporated a XhoI restriction site at the 5′ end and an EcoRI restriction site at the 3′ end of the PCR product for directional cloning into the pcDNA 3.1(−) plasmid (Invitrogen, Carlsbad, CA). Individual clones were sequenced to identify WT as well as the splice site mutant clones. The plasmids were then transfected, individually, into 293T cells (Lipofectamine 2000, Invitrogen) following the manufacturer’s suggested protocol. Two days later, total RNA (Trizol, Invitrogen) was isolated and converted into first-strand cDNA (Superscript II, Invitrogen). T7 and BGH-rev primers were used to PCR amplify the ectopically expressed BAP1 cDNA from each sample for analysis by gel electrophoresis and Sanger sequencing.
Results
BAP1 mutations
Among the 150 MM cases with a family history of cancer, germline mutations of BAP1 were identified in 9 index cases, of which 8 tumors (88.9%) were of the epithelioid subtype, and 1 was sarcomatous. Of the 141 MM cases without BAP1 mutations, the histopathological subtype was available in 126 cases, of which 94 (74.6%) were epithelioid, 13 (10.3%) were biphasic, and 19 (15.1%) were sarcomatous. Of the 9 index cases with a germline BAP1 mutation, 5 MMs (55.6%) were peritoneal and the 4 (44.4%) were thoracic. Of the MM cases without a germline BAP1 mutation, 116 (82.3%) were thoracic tumors and 25 (17.7%) were peritoneal neoplasms. No mutations of BAP1 were found in either of the two control cohorts, i.e., 50 cases from families with a history of cancers other than MM and 153 asbestos-exposed cases without a familial history of cancer. The demographic data on these cohorts are summarized in Table 1.
Table 1.
Demographics of asbestos-exposed MM cases and controls.
| No. | Mean age at Dx* | Latency | Histology (%)** | MM location (%)*** | Gender (% M vs. F) | Cancer score | |
|---|---|---|---|---|---|---|---|
| BAP1-mutant MM cases | 9 | 57.9 ± 10.7 | 44.5 ± 9.9 | 88.9/11.1/0/0 | 44.4/55.6 | 77.8/22.2 | 3.00 ± 1.22 |
| WT BAP1 MM cases | 141 | 68.2 ± 10.9 | 50.9 ± 11.2 | 73.8/9.9/13.5/2.8 | 82.3/17.7 | 75.2/24.8 | 2.4 ± 0.95 |
| Significance: BAP1-mutant vs. WT BAP1 MM controls | p < 0.01 | NS | NS | p < 0.05 | NS | NS | |
| Controls: asbestosis +/− pleural plaques | 153 | 74.6 ± 4.9 | 54.4 ± 3.7 | 92.2/7.8 | |||
| Controls: High cancer score without MM | 50 | 72.8 ± 9.8 | 86.0/14.0 | 4.34 ± 1.46 |
For controls, age at time that blood sample was collected.
Epithelioid/biphasic/sarcomatous/unknown
% pleural/% peritoneal
Key: WT, wild-type (normal) BAP1 status; Dx, diagnosis; M, male; F, female
Of the 9 cases found to harbor germline mutations affecting the coding sequence of the BAP1 protein, 3 were missense mutations, 2 were splice site mutations, and 4 were insertions/deletions (in/dels) (Table 2). Analysis of the missense mutations was done using three tools: Protein Variation Effect Analyzer (PROVEAN), Sorting Intolerant from Tolerant (SIFT), and PolyPhen-2 (Fig. 1A). Generally, there was no consensus among the programs in predicting the effect of the missense mutations on protein function. However, two missense mutations sites, Asn78 and Arg383, are highly conserved across different vertebrate species (Fig. 1B), implying the importance of the amino acids at these sites. In addition, the Arg383Cys mutation is not a conservative change, as a basic, positively-charged arginine is changed to a non-basic, uncharged cysteine and, thus, could potentially affect BAP1 protein structure or function. In addition, the introduction of a cysteine residue could alter disulfide bonds within the protein. The third missense mutation (p.Ser585Leu), involves a serine that is not as conserved across species (Fig. 1B), explaining how two of the three software tools predicted the mutation to be ‘neutral’ or ‘benign’. However, several studies have shown that BAP1 Ser583 is phosphorylated in human cells after UV light exposure, providing functional significance of the serine at position 583 (17, 18).
Table 2.
Germline BAP1 mutations observed in MM cases.
| Sample ID | DNA Mutation Change | Predicted Protein Change | Mutation Type |
|---|---|---|---|
| ABS2570 | c.233A>G | p.78 Asn>Ser | Missense |
| ABS3023 | c.1147C>T(rs201844078) | p.383 Arg>Cys | Missense |
| ABS3313 | c.1748C>T | p.583 Ser>Leu | Missense |
| ABS2573 | c.1695insT | p.Glu566fs*1 | In/del, frameshift |
| ABS2778 | c.1717delC | p.Leu573fs*3 | In/del, frameshift |
| ABS3428 | c.1882_1885delTCAC | p.Ser628fs*8 | In/del, frameshift |
| ABS3554 | c.1717delC | p.Leu573fs*3 | In/del, frameshift |
| MC7039 | c.1729+1G>A | Unknown | Splice site |
| MC7058 | c.1891-1G>A | Unknown | Splice site |
Figure 1.

Analysis of the missense BAP1 mutations identified in a population of asbestos-exposed malignant mesothelioma (MM) cases. A) Analysis performed using Protein Variation Effect Analyzer (PROVEAN), Sorting Intolerant from Tolerant (SIFT), and PolyPhen-2 software tools. B) Assessment of the degree of conservation across different vertebrate species at sites of three BAP1 missense mutations identified among MM cases. Note that two missense mutations sites, Asn78 and Arg383, are highly conserved, indicative of the importance of the amino acids at these sites.
The Ub-AMC assay revealed that the enzymatic activity of each of the three BAP1 missense mutants was reproducibly decreased compared to that of WT BAP1 protein (Fig. 2). However, as expected, these BAP1 mutant proteins could still bind ASXL2 substrate, which was expected given that the missense mutations do not affect the carboxy-terminal ASXL1/2 binding domain of BAP1. As a control, we used a BAP1 mutant construct corresponding to the S628fs*8 mutation seen in family ABS3428. This mutation results in a predicted BAP1 protein truncation with loss of its ASXL1/2 binding motif. As expected, this protein was unable to bind ASXL2, based on IP experiments in which BAP1 and ASXL2 were co-expressed in 293T cells (data not shown). Location of missense mutations and the S628fs*8 mutation are shown in Fig. 2A, and representative Ub-AMC experiment of exogenously expressed wild-type and mutant BAP1 proteins are depicted in Fig. 2B.
Figure 2.
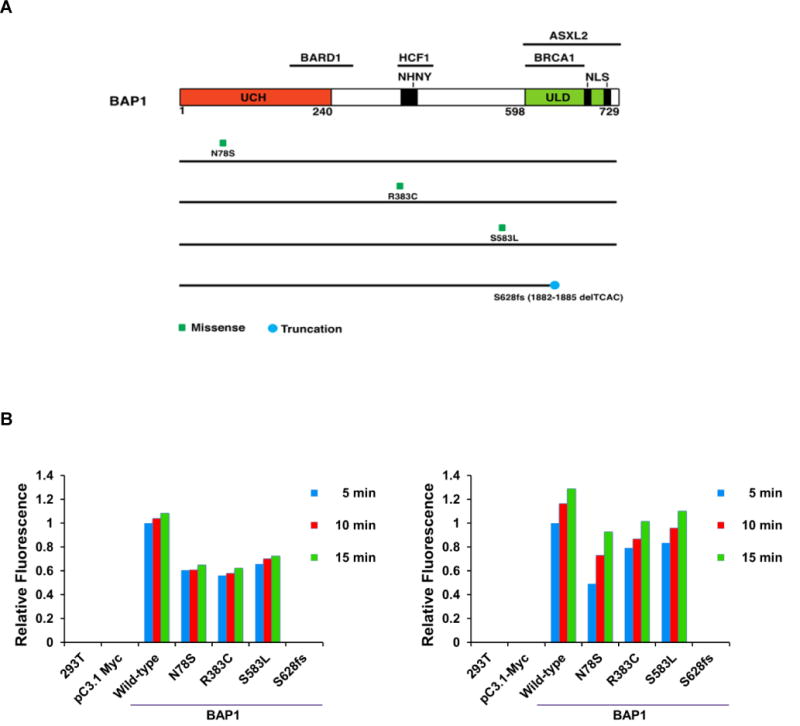
Representative results of Ub-AMC assay depicting decreased activity of three exogenously expressed BAP1 missense mutant proteins (N78S, R383C, and S583L – corresponding to mutations seen in families ABS2570, ABS3023 and ABS3313, respectively) compared to the activity of the wild-type BAP1 protein. Exogenous expression of a BAP1 mutant construct corresponding to the S628fs*8 mutation (case ABS3428) showed no BAP1 activity. 293T cells were transfected with individual BAP1 expression constructs, and then protein levels for each construct were normalized prior to Ub-AMC assay. BAP1 and Ub-substrate were incubated for 15 min, and then fluorescence indicative of activity was assessed after 5, 10 and 15 min. A) Location of each BAP1 missense mutation and S628fs*8 mutation. B) Results of fluorescence activity observed in two representative Ub-AMC experiments to assay activity of exogenously expressed wild-type and mutant BAP1 proteins. The experiment was repeated a total of three times with similar results.
Two cases had germline BAP1 mutations at consensus splice sites: cases MC7039 and MC7058. These mutations affect intron splice sites that are highly conserved and required for proper intron/exon splicing. We utilized a mini-gene expression assay to functionally test the implications of these mutations. The mutation in sample MC7039 was found to lead to an mRNA message lacking exon 13 (Fig. 3A). On the other hand, the mutation in case MC7058 resulted in the retention of a portion of intron 14 in the mRNA (Fig. 3B). Based on sequence analysis, the latter may have resulted from the use of an alternative, cryptic splice site within intron 14. In both of these MM cases, the loss of an exon and the retention of part of an intron were each predicted to lead to a frameshift and premature truncation of the translated BAP1 protein.
Figure 3.
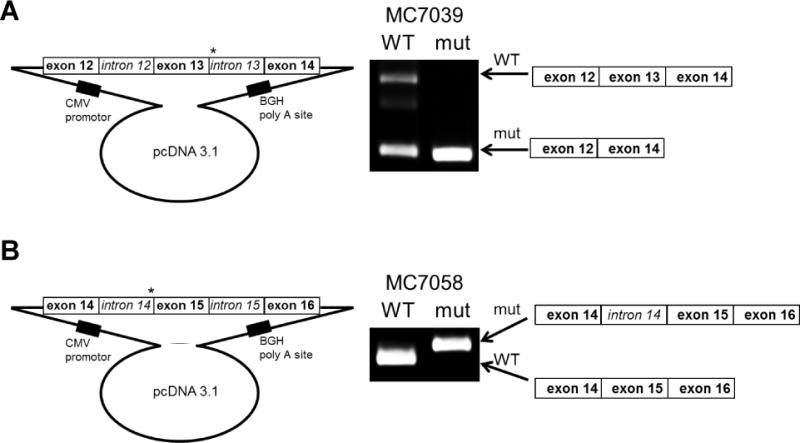
Splicing assay using mini-gene expression constructs. Genomic DNA encompassing exons 12 through 14 or exons 14 through 16 of wild-type and splice site-mutant BAP1 were cloned into pcDNA 3.1 expression constructs. The plasmids were then transfected into 293T cells. A) RT-PCR analysis of BAP1 expression amplified two strong bands in cells transfected with the wild-type construct but only the smaller band in the cells transfected with a mutant construct containing the mutation found in case MC7039. Sequencing of the PCR products (not shown) revealed that the higher molecular weight band consists of the correctly spliced exons 12, 13 and 14, whereas the smaller band containing only exons 12 and 14. B) RT-PCR analysis of BAP1 expression amplified a smaller band in cells transfected with the wild-type construct, whereas a larger band was observed in cells transfected with the mutant construct containing the mutation found in case MC7058. Sequencing of the PCR products (not shown) revealed that the higher molecular weight band retained sequences from part of intron 14. *, location of splice site mutations.
The four cases with in/dels consisted of three different mutations. Interestingly, two cases (ABS2778 and ABS3554) had the same germline mutation, c.1717delC, and the pedigrees of these two cases did not indicate any known relationship, although they may have a common ancestor. Interestingly, this mutation is also identical to our previously identified germline mutation in a sporadic MM case who also had UM (SP-002) (8).
Four intronic SNPs in BAP1 (rs116234404, rs141765555, rs187797012 and rs190356708), were found in ≤ 4 cases and in ≤ 1 control. To determine if these SNPs had any potential functional relevance, we examined regulatory data from a number of sources, including ENCODE histone modifications and transcription factor binding sites, RegulomeDB (19), Haploreg (20), as well the eQTL browser (21). None of these four SNPs were in obvious regulatory regions.
Demographics
By design, controls without familial cancers were older and had a longer latency than cases. Compared with MM cases without a BAP1 mutation, the cases harboring germline mutations that could affect BAP1 protein coding sequence were diagnosed at an earlier age and were more likely to have peritoneal rather than pleural MM (Table 1). There was no significant difference in cancer signal score, gender or latency between cases with and without a BAP1 mutation. Both families with and without mutations of BAP1 exhibited a significant number of malignancies that have been repeatedly associated with the BAP1 syndrome, i.e., MM, CM, UM and RCC, although the relative proportion of these neoplasms appeared to be greater in families of MM cases with a BAP1 mutation. A total of 11 such tumors (4 RCC, 3 MMs, 2 CMS, and 2 UMs) were identified in the 9 families in which the MM proband has a germline BAP1 mutation. Among the 141 MM cases without a BAP1 mutation, 27 such tumors (7 RCCs, 9 MMs, 10 CMs, and 1 ocular tumor) were documented. Atypical cutaneous melanocytic tumors are also associated with the BAP1 syndrome (10), and we cannot rule out the existence of such lesions in some members of the 9 families with a germline BAP1 mutation, but detailed dermatological assessments of these families were not available. Among the families of cases with BAP1 mutations, 4 MM index cases had a second primary tumor, including 3 with RCC and 1 with UM. Of the cases without a BAP1 mutation, 5 MM index cases had a second primary tumor (2 with RCC and 3 with CM). One MM case with a BAP1 mutation (ABS3554) had two siblings who also developed MM; similarly, two MM cases without a BAP1 mutation each had two siblings who developed MM. Thus, the existence of multiple MMs in a given family need not be due to inheritance of a BAP1 mutation.
Among the three cases (ABS2570, ABS3023, ABS3313) with missense mutations, multiple malignancies were observed in their past personal and family histories. The cancers observed in families of these cases were similar to those identified in the six families with frameshift or splice site mutations, suggesting that families with missense BAP1 mutations also belong to the BAP1 syndrome. For example, case ABS3023 had a daughter with breast cancer, and case ABS3313 had a daughter with cutaneous melanoma (Fig. 4A). Moreover, case ABS2570 had a kidney cancer diagnosed 2 years prior to his MM diagnosis, and his father and three brothers were reported to have gastric cancer.
Figure 4.
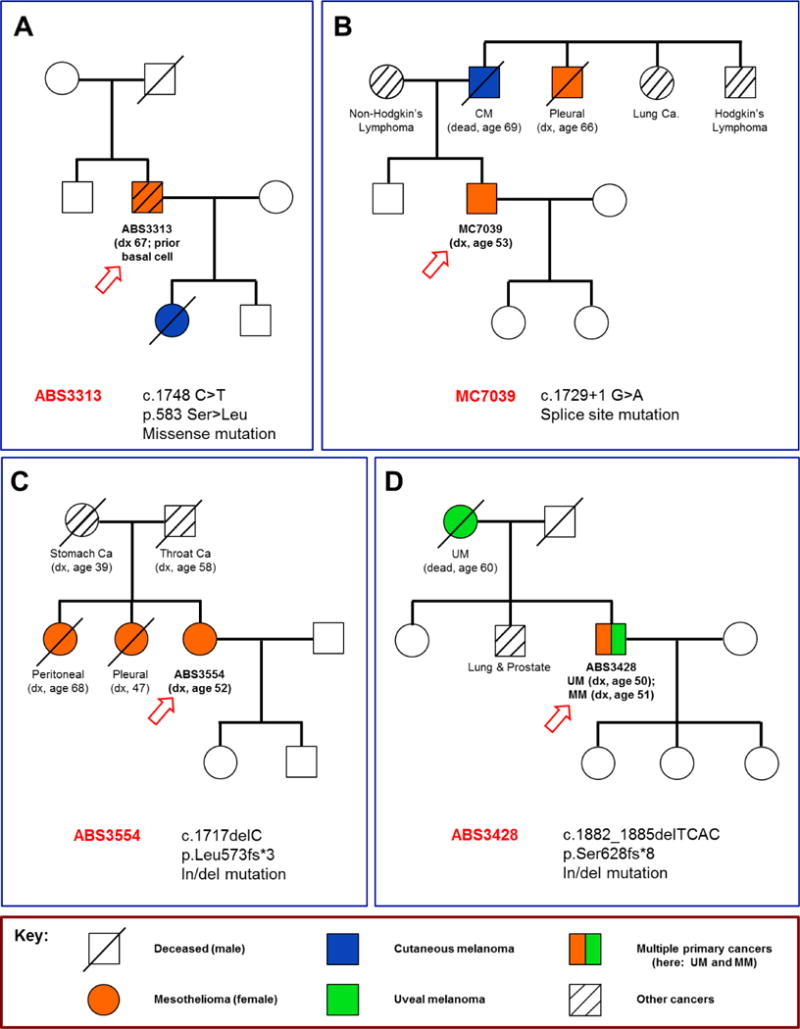
Pedigrees of four families with MM and family history of cancer, with each index case (arrow) and BAP1 mutation shown. A) Pedigree of family of MM case ABS3313, whose germline DNA contained a missense mutation of BAP1, c.1748 C>T. The index case has had both MM and basal cell carcinoma, and his daughter died of cutaneous melanoma (CM). B) Pedigree of family of MM case MC7039, whose germline DNA contained a splice site mutation of BAP1, c.1729+1 G>A. The family of the index case (MC7039) has numerous malignancies, including the proband’s father, who died of metastatic CM, as well as a paternal uncle, who had MM. C) Pedigree of family of MM case ABS3554, who had a germline in/del mutation of BAP1, c.1717delC. In addition to the index case, two sisters had MM, one with peritoneal and the other with pleural disease. D) Pedigree of family of MM case ABS3428, who was found to have a germline in/del mutation of BAP1, c.1882_1885delTCAC. Case ABS3428 was diagnosed with both peritoneal MM and UM, which is depicted as a split-colored box symbol. His mother was diagnosed with UM, and a brother had two distinct primary malignancies, prostate and lung cancer.
Of the two splice site mutations, the father of MM case MC7058 had a non-Hodgkin’s lymphoma and his sister had thyroid cancer. Notably, all three were diagnosed with a malignancy at a relatively young age (MC7058: age 47; father: 52; sister: 34). The family history of case MC7039 is replete with malignancy (Fig. 4B). His father had metastatic cutaneous melanoma, and his mother had non-Hodgkin’s lymphoma. Moreover, his paternal uncle had MM, an aunt and cousin (not shown in figure) both had lung cancer, and an additional uncle had Hodgkin’s lymphoma.
As noted above, both ABS2778 and ABS3554 exhibited the same mutation c.1717delC, which is identical to that described in a sporadic MM (SP-002) we reported earlier (8). Notably, both ABS2778 and ABS3554 had MM simultaneously affecting both the pleura and peritoneum. Case ABS3554 had two sisters with MM, one with peritoneal and the other with pleural disease. Her mother, by self-report, had gastric cancer, and her father had throat cancer. A pedigree of the family of case ABS3554 is shown in Figure 4C. Reportedly, the father of case ABS2778 died from lung cancer. His mother and two siblings were unaffected by cancer. No family history of melanoma was evoked from either case ABS2778 or ABS3554.
The c.1882_1885delTCAC germline mutation observed in case ABS3428 was also previously described by us in a sporadic case (SP-008) (8). Case ABS3428 was diagnosed with peritoneal MM and reported a previous UM. His mother was diagnosed with UM, and a brother reportedly had two distinct primary malignancies, prostate and lung cancer (Fig. 4D). The fourth MM case with an in/del, ABS2573, had a past medical history of breast cancer and kidney cancer; thus, ABS2573 had three separate primary tumors. The kidney cancer was diagnosed 21 years prior to the MM, and the breast cancer was diagnosed the year following her diagnosis of MM. A daughter had RCC, a sister breast cancer, and a brother had a brain tumor. Her mother died at the age of 46 from a possible malignancy.
Discussion
Altogether, we found germline mutations of BAP1 in 9 of 150 (6%) cases with a personal or family history of cancer based on a high cancer signal, as defined above. Among the 9 families with BAP1 mutations, a total of 11 MMs were identified. The median age of these 11 MM patients was 54.5 years, and the mean was 58.5 years (57.9 among the 9 index cases). Among the 141 cases without a BAP1 mutation, the mean age of MM diagnosis was 68.2 years (Table 1). Since MM is generally regarded as a disease of the 60s and 70s (6), the age of onset in our BAP1 mutation carriers is younger than among the general population of sporadic MM cases. This would be expected based of the increased susceptibility associated with a tumor predisposition syndrome, in this case due to the existence of a heterozygous germline mutation of BAP1. However, it is noteworthy that while BAP1 mutation carriers tend to develop MM prior to the age of 60, two probands were diagnosed at 67 and 81 years of age (ABS3313 and ABS3023, respectively), and another (a sister of ABS3554) developed peritoneal MM at age 68. Thus, not all MM cases with BAP1 mutations develop the disease at an early age.
Among the 11 MM cases identified from the 9 families with germline BAP1 mutations, the 7 deceased patients had a median survival of 60 months after the initial diagnosis. In contrast, the median age of MM survival among individuals without a germline BAP1 mutation was 17 months. This 3.5-fold increased median survival rate among mutation carriers is similar to the improved long-term survival reported in a recent study in which a 60-month median survival was observed in MM patients with germline BAP1 mutations compared to a 9-month median survival among all MM cases recorded in the U.S. Surveillance, Epidemiology, and End Results (SEER) data from 1973 to 2010 (22). In contrast, somatic BAP1 mutations in uveal melanomas are strongly associated with a more metastatic phenotype (23). Some factors that may contribute to the prolonged survival observed in BAP1 mutation carriers include the high proportion of peritoneal and epithelioid tumors in this subgroup as well as the younger age of diagnosis compared to non-carriers.
The BAP1 syndrome is characterized by a high incidence of MM, UM, CM and benign melanocytic tumors, RCC, and potentially other tumors such as carcinomas of the lung and breast (13, 14). Among the nine families corresponding to cases with germline BAP1 mutations, two had a single case of cutaneous melanoma (father of MC7039; daughter of ABS3313), and one family had two UMs (case ABS3428 and his mother). Three of the families with germline BAP1 mutations exhibited RCCs: ABS2573 and her daughter, ABS2570 (who also had MM), and ABS2778 (who also had MM). The findings are similar to those reported by Popova et al., who determined that hereditary RCCs were associated with germline mutations of BAP1 mainly in families that also exhibited other tumors known to be associated with the BAP1 syndrome (12).
Carcinomas of the lung and breast have been previously reported in several BAP1 families. In our series, breast cancer was observed in three individuals (ABS2573 and her sister; daughter of MM case ABS3023). Lung carcinomas were present in two individuals (father of ABS2778; brother of ABS3428). However, because lung and breast cancers are so common, it is still too early to know if these cancer types are associated with the BAP1 syndrome. Interestingly, gastric cancers were identified in 5 individuals among the 9 affected families (3 brothers and father of case ABS2570; mother of ABS3554). However, histopathological assessments of the gastric tumors were not available to confirm the diagnosis in these tumors. Thus, it is uncertain whether the tumors were gastric carcinomas, gastrointestinal stromal tumors, peritoneal MM, or metastatic tumors of unknown origin. To date, at least one stomach cancer has been previously reported in a BAP1 mutation carrier (24).
Interestingly, 5 of the 11 MM patients (each from a different family) from our 9 BAP1 families were diagnosed with a second primary cancer: one with RCC and breast cancer (3 primary tumors); two with RCC; and two with basal cell carcinoma. We previously reported a family with a remarkable history of multiple cancers, including the proband’s father, who had both MM and CM, and a paternal uncle, who had UM, CM, and lung cancer. The fact that several members of the family manifested two or more different types of cancer suggests widespread BAP1-related tumor susceptibility involving multiple organs (16). Our finding of two basal cell carcinomas in MM index cases is of interest in relation to recent work showing that germline BAP1 mutations can predispose to multiple basal cell carcinomas (25). In this study, multiple basal cell carcinomas were identified in four patients from two independent BAP1 families, with all tumors located in chronic sun-exposed areas. Immunohistochemical analysis revealed complete or partial loss of BAP1 protein nuclear expression in all 19 basal cell carcinomas from these four patients, but not in a control set of 22 sporadic basal cell carcinomas. Based on their observations, the investigators suggested that BCCs are also part of the BAP1 cancer syndrome, perhaps in relation with chronic sun exposure. However, it should be noted that basal cell carcinoma is the most common of all malignant tumors (26), and this tumor was seen even in families of cases without BAP1 mutations. In fact, even among the families of the 141 MM cases without BAP1 mutations, 11 family members were diagnosed with basal cell carcinoma, including 10 that were found in MM index cases, i.e., non-BAP1 associated individuals who developed two primary cancers.
The BAP1 mutation sites observed in this study were spread widely within the gene, although most mutations occurred in the 3′ end of the BAP1 coding region, with 7 of 9 mutations distributed between c.1695 and c.1891 (exons 13–14), corresponding to the ASXL1/2 binding domain of BAP1). Although no BAP1 mutation hotspot has been identified to date, it was remarkable that two unrelated families (ABS2778 and ABS3554) had an identical mutation: c.1717delC, p.Leu573fs*3. Upon review of reported germline hereditary and sporadic cases, we found the very same in/del mutation in a previously reported sporadic case of MM (SP-002) that was obtained through MARF(8). In addition, the germline BAP1 mutation found in family ABS3428, i.e., c.1882_1885delTCAC, is identical to that seen in a previously reported “sporadic” MM case, SP-008, obtained through MARF (8). In retrospect, SP-008 appears to be a member of a BAP1 syndrome family, given that both this patient and his mother had UM.
The six BAP1 changes that involve frameshift or splice site mutations are predicted to result in loss of the carboxy-terminal region of the protein, which contains the nuclear localization signal. In contrast, the carboxy terminus is retained in the three BAP1 missense mutants, although they appeared to have decreased activity (Fig. 2B). The Ub-AMC assay demonstrated that the missense mutations still permit the BAP1 enzyme to be activated, but to a lesser extent than the WT protein.
We had hypothesized that germline mutations of BAP1 contribute to susceptibility to MM in asbestos-exposed individuals through a mechanism that involves gene x environment interaction. Our finding of an increased prevalence of inherited BAP1 mutations in asbestos-exposed MM cases versus asbestos-exposed controls appears to be consistent with this notion. The results from the cohort of 153 individuals with a significant exposure to asbestos but without MM or a familial cancer history did not, as expected, show any germline BAP1 mutations. The other control cohort, consisting of 50 asbestos-exposed individuals from non-MM families with a high cancer signal similar to that of the 150 MM case cohort, also did not show any germline BAP1 mutations. These findings thus mitigate possible concerns about a selection bias related to the family history scoring system that was used in this investigation. Consistent with our findings, Betti et al. recently reported a truncating germline mutation of BAP1 in one of five families with exposure to asbestos and multiple MMs (27). These families were selected, as in our MM case cohort, based on a familial history of cancer that included MM. Unlike our cohort, however, each of these five families had at least two cases of MM. In their single family with a germline BAP1 mutation, three individuals who had potential exposure to asbestos developed MM, whereas another family member who was not exposed developed a different tumor type, i.e., a mucoepidermoid carcinoma.
If genetic factors were inconsequential, one might expect a similar incidence of germline mutations of BAP1 in both cases and controls. Instead, the significantly higher incidence of BAP1 mutations observed in MM cases versus controls suggests that asbestos and genetic factors cooperate to enhance the onset of MM. Similarly, we have recently shown that mice carrying a heterozygous germline mutation of Bap1 have a significantly higher incidence, and accelerated rate of onset, of asbestos-induced MM than similarly-exposed WT littermates (8). Taken together, the findings presented here indicate that patients presenting with MM and a personal or family history of other cancers should be considered for genetic testing, with the goal of identifying those families that might benefit from BAP1 mutation screening and regular clinical monitoring of family members for the purpose of early detection of cancers and clinical intervention.
Acknowledgments
Grant Support: Work supported by NCI grants CA175691 (J.R. Testa and F.J. Rauscher) and CA-06927 (J.R. Testa), NIEHS grant P42 ES023720 (UPenn Superfund Research and Training Program Center), a grant from the Mesothelioma Applied Research Foundation – The Anderson Family Grant (M. Cheung and J.R. Testa), an appropriation from the Commonwealth of Pennsylvania, and a gift from the Local #14 Mesothelioma Fund of the International Association of Heat and Frost Insulators & Allied Workers.
BAP1-related work performed by JRT and his colleagues at Fox Chase Cancer Center is supported by NCI grant CA175691 and NIEHS grant P42 ES023720 (UPenn Superfund Research and Training Program Center). FJR and HP are also supported by grant CA175691. JRT and MC have a pending patent application on BAP1 mutation testing. JAO has served as a consultant and expert witness for both the plaintiff and defense related to her Wake Forest University Occupational Medicine work. JRT has been a genetics consultant.
Footnotes
Conflict of Interest: All other authors declare no potential conflict of interest.
References
- 1.Robinson BW, Lake RA. Advances in malignant mesothelioma. N Engl J Med. 2005;353:1591–1603. doi: 10.1056/NEJMra050152. [DOI] [PubMed] [Google Scholar]
- 2.Sluis-Cremer GK, Liddell FD, Logan WP, Bezuidenhout BN. The mortality of amphibole miners in South Africa, 1946–80. Br J Ind Med. 1992;49:566–575. doi: 10.1136/oem.49.8.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henley SJ, Larson TC, Wu M, Antao VC, Lewis M, Pinheiro GA, et al. Mesothelioma incidence in 50 states and the District of Columbia, United States, 2003–2008. Int J Occup Environ Health. 2013;19:1–10. doi: 10.1179/2049396712Y.0000000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tossavainen A. Asbestos, asbestosis, and cancer: the Helsinki criteria for diagnosis and attribution. Scand J Work Environ Health. 1997;23:311–316. [PubMed] [Google Scholar]
- 5.Bocchetta M, Di Resta I, Powers A, Fresco R, Tosolini A, Testa JR, et al. Human mesothelial cells are unusually susceptible to simian virus 40-mediated transformation and asbestos cocarcinogenicity. Proc Natl Acad Sci U S A. 2000;97:10214–10219. doi: 10.1073/pnas.170207097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohar JA, Ampleford EJ, Howard SE, Sterling DA. Identification of a mesothelioma phenotype. Respir Med. 2007;101:503–509. doi: 10.1016/j.rmed.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Ugolini D, Neri M, Ceppi M, Cesario A, Dianzani I, Filiberti R, et al. Genetic susceptibility to malignant mesothelioma and exposure to asbestos: the influence of the familial factor. Mutat Res. 2008;658:162–171. doi: 10.1016/j.mrrev.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Testa JR, Cheung M, Pei J, Below JE, Tan Y, Sementino E, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matullo G, Guarrera S, Betti M, Fiorito G, Ferrante D, Voglino F, et al. Genetic variants associated with increased risk of malignant pleural mesothelioma: a genome-wide association study. PLoS One. 2013;8:e61253. doi: 10.1371/journal.pone.0061253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdel-Rahman MH, Pilarski R, Cebulla CM, Massengill JB, Christopher BN, Boru G, et al. Germline BAP1 mutation predisposes to uveal melanoma, lung adenocarcinoma, meningioma, and other cancers. J Med Genet. 2011;48:856–859. doi: 10.1136/jmedgenet-2011-100156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Popova T, Hebert L, Jacquemin V, Gad S, Caux-Moncoutier V, Dubois-d’Enghien C, et al. Germline BAP1 mutations predispose to renal cell carcinomas. Am J Hum Genet. 2013;92:974–980. doi: 10.1016/j.ajhg.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13:153–159. doi: 10.1038/nrc3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Testa JR, Malkin D, Schiffman JD. Connecting molecular pathways to hereditary cancer risk syndromes. Am Soc Clin Oncol Educ Book. 2013:81–90. doi: 10.1200/EdBook_AM.2013.33.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu J, Kadariya Y, Cheung M, Pei J, Talarchek J, Sementino E, et al. Germline mutation of Bap1 accelerates development of asbestos-induced malignant mesothelioma. Cancer Res. 2014;74:4388–4397. doi: 10.1158/0008-5472.CAN-14-1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheung M, Talarchek J, Schindeler K, Saraiva E, Penney LS, Ludman M, et al. Further evidence for germline BAP1 mutations predisposing to melanoma and malignant mesothelioma. Cancer Genet. 2013;206:206–210. doi: 10.1016/j.cancergen.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 17.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 18.Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baumann F, Flores E, Napolitano A, Kanodia S, Taioli E, Pass H, et al. Mesothelioma patients with germline BAP1 mutations have 7-fold improved long-term survival. Carcinogenesis. 2015;36:76–81. doi: 10.1093/carcin/bgu227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, et al. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aoude LG, Wadt K, Bojesen A, Cruger D, Borg A, Trent JM, et al. A BAP1 mutation in a Danish family predisposes to uveal melanoma and other cancers. PLoS One. 2013;8:e72144. doi: 10.1371/journal.pone.0072144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de la Fouchardière A, Cabaret O, Savin L, Combemale P, Schvartz H, Penet C, et al. Germline BAP1 mutations predispose also to multiple basal cell carcinomas. Clin Genet. 2015;88:273–277. doi: 10.1111/cge.12472. [DOI] [PubMed] [Google Scholar]
- 26.Rubin AI, Chen EH, Ratner D. Basal-cell carcinoma. N Engl J Med. 2005;353:2262–2269. doi: 10.1056/NEJMra044151. [DOI] [PubMed] [Google Scholar]
- 27.Betti M, Casalone E, Ferrante D, Romanelli A, Grosso F, Guarrera S, et al. Inference on germline BAP1 mutations and asbestos exposure from the analysis of familial and sporadic mesothelioma in a high-risk area. Genes Chromosomes Cancer. 2015;54:51–62. doi: 10.1002/gcc.22218. [DOI] [PubMed] [Google Scholar]