

Abstract
Major depressive disorder (MDD) is a common and potentially life-threatening mood disorder. Identifying genetic markers for depression might provide reliable indicators of depression risk, which would, in turn, substantially improve detection, enabling earlier and more effective treatment. The aim of this study was to identify rare variants for depression, modeled as a continuous trait, using linkage and post-hoc association analysis. The sample comprised 1221 Mexican-American individuals from extended pedigrees. A single dimensional scale of MDD was derived using confirmatory factor analysis applied to all items from the Past Major Depressive Episode section of the Mini-International Neuropsychiatric Interview. Scores on this scale of depression were subjected to linkage analysis followed by QTL region-specific association analysis. Linkage analysis revealed a single genome-wide significant QTL (LOD = 3.43) on 10q26.13, QTL-specific association analysis conducted in the entire sample revealed a suggestive variant within an intron of the gene LHPP (rs11245316, p = 7.8×10-04; LD-adjusted Bonferroni-corrected p = 8.6×10-05). This region of the genome has previously been implicated in the etiology of MDD; the present study extends our understanding of the involvement of this region by highlighting a putative gene of interest (LHPP).
Introduction
Major depressive disorder (MDD) is a common and potentially life-threatening mood disorder 1. It affects 16.2% of individuals in the US during their lifetime 2, and incurs great economic cost ($83.1 billion per annum in the US) 3. The illness also places an immense burden on the sufferer, such that the impact of MDD on wellbeing and functioning is in line with that seen in other major chronic conditions (e.g., arthritis and diabetes mellitus) 4. Moreover, functional impairments remain even after the remission of a depressive episode 5. Unsurprisingly, the World Health Organization (WHO) cites MDD as a leading cause of disability worldwide 6. Current methods of diagnosing and treating MDD are symptom based, that is, diagnosis is made based on the presence of symptoms outlined in the DSM 7 and successful treatment is defined by the reduction and eventual remission of those symptoms 8. Relying on symptoms alone, without regard for the etiological roots of a disorder, makes for mediocre diagnostic reliability 9-11 and inadequate treatment 12. The effectiveness of anti-depressant pharmacotherapy is hampered by our limited understanding of the biological basis of MDD 8, 13; indeed the administration of anti-depressant medications results in remission in only one third of patients 14. Identifying risk variants for depression would enhance our understanding of the etiology of MDD which in turn would enable earlier and more reliable detection as well as, potentially, the development of new and more effective therapies 15, 16.
Heritability estimates of MDD vary around 0.37 1, 17, indicating a substantial influence of genes on MDD risk. However, attempts to isolate specific genes which mediate MDD risk have been met with difficulty 16, 18: meta-analysis suggests that many of the early candidate gene studies were false positives 19, 20, and numerous genome-wide association (GWA) studies, including the latest mega-analysis of over nine thousand depressed individuals from the Psychiatric Genetics Consortium, have struggled to attain genome-wide significant results 20-30. As a consequence it has been suggested that even larger sample sizes are necessary for the identification of risk variants for MDD 30. However, linkage, a method that ostensibly measures rare in addition to common variation, has isolated numerous genome-wide significant loci in relatively small samples 31-34. Thus, while intuitively the assertion that greater sample sizes are needed to isolate genes for MDD makes good sense (particularly given that increasing sample size has worked for other disorders e.g. schizophrenia 35), it is also possible that the degree of genetic (and also phenotypic) heterogeneity is greater for MDD than for other disorders 36 and as a consequence increasing sample sizes might only serve to compound the problem. Therefore a complementary approach would be to focus on reducing genetic heterogeneity using, for example, a family-based approach when searching for MDD risk genes.
In order to effectively account for the phenotypic heterogeneity associated with MDD it is critical to develop optimal MDD phenotypes 30. MDD is typically treated as a categorical trait, it is assumed that MDD reflects the tail end of an underlying normal distribution of mood, and that diagnosis occurs when a threshold for liability is crossed. It seems plausible that the genes which moderate behavior at the tail end of the distribution are the same as those that underlie the regulation of normal mood37 and by dichotomizing the MDD distribution, we ignore a substantial proportion of variance that would contribute to gene-finding efforts. Conceptualizing MDD as a continuous dimension would capture this important information which would confer greater sensitivity and power to detect genes 38.
Thus, the present study, we report on univariate linkage and association analysis of a dimensional scale of depression derived from the Past Major Depressive Episode section of the Mini-International Neuropsychiatric Interview (MINI) within extended-pedigree data.
Methods
Participants
The sample comprised 1221 Mexican American individuals from extended pedigrees (132 families, average size 9.32 people, range = 1-129). The sample was 63% female and had a mean age of 46.01 (SD = 15.10; range = 18-97). Individuals in this San Antonio Family Study cohort have actively participated in research for over 18 years and were randomly selected from the community with the constraints that they are of Mexican American ancestry, part of a large family, and live within the San Antonio region (see (Olvera et al., 2011) for recruitment details). All participants provided written informed consent on forms approved by the institutional review board at the University of Texas Health Science Center of San Antonio.
Diagnostic Assessment
All participants received the Mini-International Neuropsychiatric Interview (MINI) 39, which is a semi-structured interview augmented to include items on lifetime diagnostic history. Masters-and doctorate-level research staff, with established reliability (κ ≥ .85) for affective disorders, conducted all interviews. All subjects with possible psychopathology were discussed in case conferences that included licensed psychologists or psychiatrists. Lifetime consensus diagnoses were determined based on available medical records, the MINI interview, and the interviewer's narrative.
Data Analysis
Depression Modelling: Confirmatory Factor Analysis
All items from the Past Major Depressive Episode (A3a-g) section of the MINI were modeled using a single factor score; Table 1 outlines each of these items. This enabled the categorical outcomes associated with the A3a-g MINI items to be modeled as a unitary quantitative trait. It is important to note that because the factor model included all items from the past major depressive episode section, the resultant score should be thought of as a lifetime rating of depression not a reflection of current symptom severity. the Specifically, a single-factor model was built using confirmatory factor analysis in Mplus (Figure 1). Family structure was taken into account using the cluster command, under the cluster command in Mplus standard errors in the model are adjusted in accordance with non-independence in the data, in this way family ID is treated as a nuisance covariate. Because the questionnaire items have categorical rather than continuous outcomes, factor analysis was applied to tetrachoric correlations derived from the raw phenotypic data. The resultant factor score (mean = 0.15, standard deviation = 0.59) was subjected to an inverse normalization to ensure normality.
Table 1.
Descriptive statistics for each item for the past major depressive episode section of the MINI.
| When you felt depressed or uninterested: | % Responded No | h2 | SE |
|---|---|---|---|
| Was your appetite decreased or increased nearly every day? Did your weight decrease or increase without trying intentionally? | .663 | .270 | .074 |
| Did you have trouble sleeping nearly every night? | .659 | .203 | .087 |
| Did you talk or move more slowly than normal or were you fidgety, restless or having trouble sitting still almost every day? | .725 | .177 | .243 |
| Did you feel tired or without energy almost every day? | .691 | .290 | .074 |
| Did you feel worthless or guilty almost every day? | .730 | .193 | .224 |
| Did you have daily difficulty concentrating or making decisions? | .744 | .245 | .100 |
| Did you repeatedly consider hurting yourself, feel suicidal, or wish that you were dead? | .840 | .339 | .153 |
Figure 1.
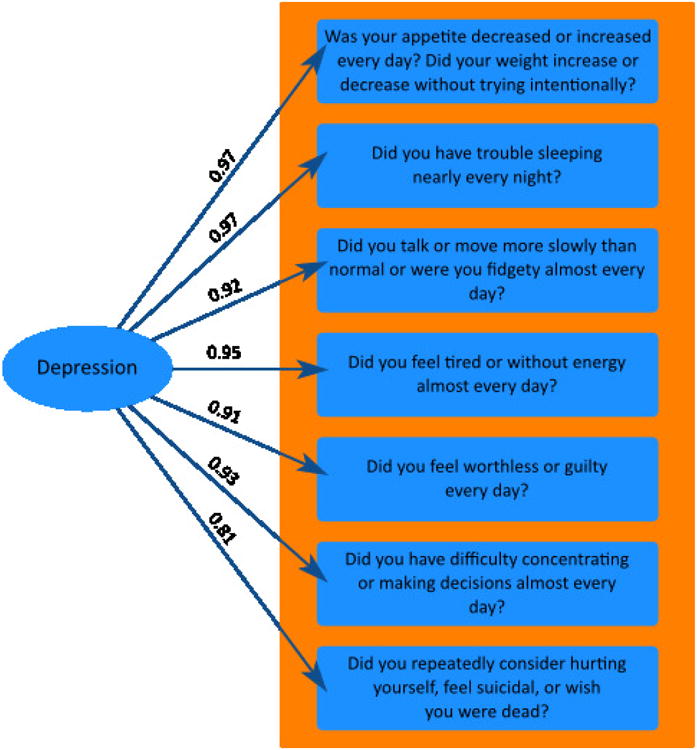
One-factor confirmatory factor model of all items from the past major depressive episode section of the MINI.
Genotyping
Subjects were genotyped for approximately one million SNPs using Illumina HumanHap550v3, HumanExon510Sv1, Human1Mv1 and Human1M-Duov3 BeadChips, according to the Illumina Infinium protocol (Illumina, San Diego, CA). SNP loci were checked for Mendelian consistency utilizing SimWalk2 (Sobel and Lange, 1996). SNPs or samples exhibiting high calling rate failures or requiring excessive blanking (i.e., if <95% of the genotypes are retained) were eliminated from analyses. Missing genotypes were imputed according to Mendelian laws based on available pedigree data using MERLIN (Abecasis et al., 2002). Maximum likelihood techniques, accounting for pedigree structure, were used to estimate allelic frequencies (Boehnke, 1991). For linkage analyses, multipoint identity-by-descent (IBD) matrices were calculated based on 28,387 SNPs selected from the 1M GWAS panel as follows. Using genotypes for 345 founders, SNPs on each chromosome were selected to be at least 1kb apart, MAF >= 5%, and LD within a 100kb sliding window not exceeding |rho| = 0.15. The resulting selection averaged 7-8 SNPs/centimorgan. For each centimorgan location in the genome, multipoint IBD probability matrices were calculated using a stochastic Markov Chain Monte Carlo procedure implemented in the computer package, LOKI (Heath, 1997).
Quantitative Genetic Analyses
All genetic analyses were performed in SOLAR 40. SOLAR implements a maximum likelihood variance decomposition to determine the contribution of genes and environmental influence to a trait by modeling the covariance among family members as a function of expected allele sharing given the pedigree. In the simplest such decomposition, the additive genetic contribution to a trait is represented by the heritability, or h2, index. Univariate variance decomposition analysis was applied to the continuous measure of depression. The trait was normalized using an inverse Gaussian transformation. Age, age2, sex and their interactions were included as covariates.
Linkage and Association Analyses
Quantitative trait linkage analysis was performed to localize specific chromosomal locations influencing MDD 40. Model parameters were estimated using maximum likelihood. The hypothesis of significant linkage was assessed by comparing the likelihood of a classical additive polygenic model with that of a model allowing for both a polygenic component and a variance component due to linkage at a specific chromosomal location (as evidenced by the location-specific identity-by-descent probability matrix). The LOD score, given by the log10 of the ratio of the likelihoods of the linkage and the polygenic null models, served as the test statistic for linkage. Genome-wide thresholds for linkage evidence were computed for this exact pedigree structure and density of markers, using a method derived from 41: a LOD of 1.69 is required for suggestive significance (likely to happen by chance less than once in a genome-wide scan) and a LOD of 2.9 is required for genome-wide significance.
Genomic regions meeting genome-wide significance for linkage were investigated in greater detail using association analysis of the MDD confirmatory factor score and the genetic variants encapsulated by the linkage peak. Statistical significance levels were established according to the effective number of tested variants given the linkage disequilibrium (LD) structure in the region. To this end, the pairwise genotypic correlations were calculated in an effort to establish the effective number of independent tests carried out during association analysis. This method, by Moskvina and Schmidt 42, is considered to be conservative and entails computing the eigenvalues of the genotypic correlation matrix. A corrected P-value is obtained from a Bonferroni correction based on the nominal alpha (=0.05) and the total number of independent tests.
Results
Confirmatory Factor Analysis
All MINI items were shown to be significantly heritabile (Table 1). The bivariate tetrachoric correlations (see Table S1) were uniformly moderate to high with little discriminability between items, suggesting a single underlying dimension. A one-factor model fit the data excellently (χ212 = 13.82 p = 0.129, RMSEA = .020 (.000 - .040) p = 0.995, CFI = 1.000, TLI = 1.000, WRMR = .617).
Heritability and Linkage Analysis
The score derived from the factor model was deemed to be significantly heritable (h2 = 0.21, p = 2.3×10-05). Significant univariate linkage was detected for the depression trait on chromosome 10 at 153cM (LOD = 3.43; Figure 2). The majority of this linkage signal originated from a single multiplex MDD pedigree within the data (h2 = 0.33, p = 1.7×10-02, LOD = 1.54) and the top LOD for the multiplex pedigree within the region encapsulated by the linkage peak met the criteria for suggestive significance (LOD = 1.84, 152cM).
Figure 2.
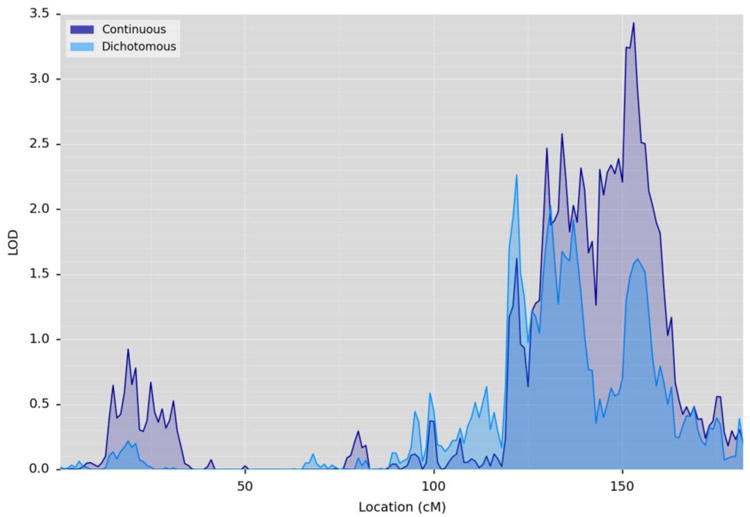
Chromosome 10 multipoint plot for the univariate linkages of the continuous depression factor score (dark blue) and also the dichotomous measure of depression derived from precisely the same items (light blue).
Association Analysis
Association analysis was conducted using all variants within the linkage peak (defined as 150-154cM) and the continuous factor score of (Table 2 and Figure 3), the peak-wide (LD-adjusted Bonferroni-corrected) significance level = 8.7×10-05 (975 SNPs, 590.69 effective SNPs). For association analysis run in the entire sample, the top-ranked variant was suggestively significant (rs11245316, χ2 = 11.28, p = 7.8×10-04) and located within an intron of the gene LHPP (phospholysine phosphohistidine inorganic pyrophosphate phosphatase). Association analysis run only in the multiplex MDD pedigree from which the majority of the linkage signal originated revealed a variant that met peak-wide significance (rs7913161, χ2 = 17.84, p = 2.4×10-05) within an intron of the gene CPXM2 (carboxypeptidase X (M14 family), member 2). When the SNP rs7913161 was included as a covariate in the linkage analysis of the continuous depression factor score in the multiplex pedigree, the LOD score observed without the covariate (LOD = 1.84) was reduced substantially (LOD = 0.46). This linkage conditional on association test gives additional support for the involvement of rs7913161 in depression risk within the multiplex MDD as it implicates the variant in the linkage model for MDD within the pedigree. Association for this variant was not significant in either the entire sample (χ2 = 1.68, p = 0.19) or in any other individual pedigree (the next best association for rs7913161 in any other pedigree did not reach suggestive significance, χ2 = 3.97, p = 0.04), suggesting that the variant is likely marking a functional and rare variant present only in the multiplex pedigree.
Table 2.
Estimates for the Top Five SNPs from the QTL-specific Association Analysis in (a) the Entire Sample and (b) the Multiplex MDD Pedigree for the Continuous Depression Score.
| Entire Sample | ||||||
|---|---|---|---|---|---|---|
| SNP | χ2 | p-value | β | Variance Explained | MAF | HWE p-value |
| rs11245316 | 11.28 | 0.000783 | 0.15 | 0.009 | 0.213 | 0.88 |
| rs3884528 | 9.09 | 0.002565 | -0.11 | 0.008 | 0.466 | 0.18 |
| rs4578341 | 7.51 | 0.006127 | 0.12 | 0.008 | 0.219 | 0.98 |
| rs1123988 | 7.38 | 0.00658 | 0.11 | 0.007 | 0.006 | 0.86 |
| rs859556 | 7.30 | 0.00688 | -0.58 | 0.006 | 0.006 | 0.96 |
| Multiplex Pedigree | ||||||
| rs7913161 | 17.84 | 0.000024 | 0.69 | 0.150 | 0.163 | 0.38 |
| rs4995180 | 16.19 | 0.000057 | 1.35 | 0.139 | 0.025 | 0.45 |
| rs7906808 | 15.35 | 0.000089 | 1.95 | 0.133 | 0.006 | 0.66 |
| rs7906939 | 14.28 | 0.000158 | 1.52 | 0.131 | 0.010 | 0.69 |
| rs7095366 | 14.17 | 0.000167 | 1.18 | 0.116 | 0.021 | 0.51 |
Figure 3.
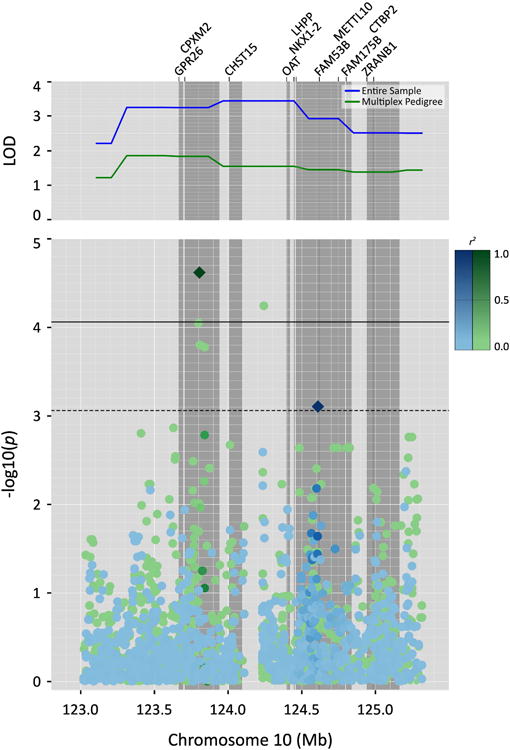
QTL-specific association analysis within the QTL on chromosome 10 for the continuous depression factor score in the entire sample (blue) and the multiplex MDD pedigree (green). The top plot shows the linkage signal in the entire sample and the multiplex pedigree. The plot below shows the results of association analysis in the same region. Intergenic regions are pale gray and genes are represented by the dark gray bars. The top ranked variant in each subject group is represented by a diamond and the degree of linkage disequilibrium is represented by the color scale.
Validation of the Continuous Factor Score Measure of Depression
The factor score derived from the one-factor model of depression correlates highly with the analogous dichotomous diagnosis of depression (indicating the presence or absence of a depressive episode over the lifetime of an individual), derived from the same section of the MINI (rphenotypic = 0.87 (p = 1.22×10-217), rgenetic = 1.00 (p = 2.46×10-06)). This high correlation is unsurprising given that the two traits are derived from precisely the same items. However, to provide further validation of the factor model, we ran linkage analysis for the dichotomous diagnostic trait within the region of chromosome 10 where we observed a genome-wide significant peak for the continuous factor score. For this analysis, the dichotomous diagnostic trait was transformed into a normally-distributed liability trait based on disease prevalence following standard quantitative genetic practice 43 (pp. 299-309). The analogous liability measure from the same items in the entire sample did not exhibit genome-wide significant linkage but neared suggestive significance (h2 = 0.38, p = 4.7×10-06, LOD = 1.58 at 153cM; Figure 2). Moreover, association analysis with the top ranked variant (for the continuous factor score) and the dichotomous measure exhibited some signal without reaching suggestive significance (χ2= 3.93, p = 4.7×10-02), while the top-ranked variant (in the multiplex pedigree) also reached significance for the dichotomous measure of depression (χ2= 16.23, p = 5.6×10-05). Thus, while the continuous measure of depression derived from the single-factor model overlaps almost completely with the dichotomous measure derived from the same items; the use of a continuous measure was shown to be more successful for gene-finding efforts. It is of note that the distribution of our continuous factor score is bimodal which is in line with a number of unaffected individuals within the data. Indeed, the kurtosis score (-0.5887) indicates a platykurtic distribution. However, previous work indicates that positive kurtosis, not negative, may inflate the false-positive rate for linkage 44, 45. Nonetheless, we ran an emprical LOD adjustment routine in SOLAR which calculates an adjustment factor by which to multiply the peak LOD from linkage analysis; 10,000 simulations run on the inverse normalized factor score calculated an adjustment factor of 1.08 where an adjustment factor > 1 means that our LOD of 3.43 is in fact somewhat conservative. Thus, we are satisfied that our trait, despite being negatively skewed, has not resulted in an inflated LOD score in the present paper.
Discussion
Repeated attempts to identify genetic influences on MDD using genome-wide association have been met with difficulty. Conversely, several genome-wide significant loci have been identified using linkage analysis 32, 33, including in the same region of chromosome 10, and more specifically in the same gene LHPP, as identified here 31. Moreover, a recent whole genome sequence study has also highlighted the role of the gene LHPP in MDD risk 46. The present study extends the previous literature by supporting the role of 10q26.13 (and possibly LHPP) in risk for MDD. Moreover, the present study identifies a novel and interesting gene, CPXM2, in a newly identified large multiplex MDD pedigree from whom the majority of the linkage signal originates – though this finding needs replication before it can be considered a risk gene for MDD. The linkage conditional on association test for the variant within CPXM2 suggests that it is partially responsible (either directly or via LD with another variant) for the linkage signal within the multiplex pedigree 47. Indeed, it is likely that the variant identified in the pedigree is marking a functional and rare variant that exists only in this pedigree. Thus, the present study, through a combination of linkage in extended pedigrees and a dimensional index of depression, highlights a two interesting genes for MDD risk and, potentially, the role of rare variation in risk for the illness.
LHPP encodes the protein phospholysine phosphohistidine inorganic pyrophosphate phosphatase (Lhpp) 48 and is highly expressed in brain 31, 49. Neff and colleagues have previously implicated the gene LHPP in MDD risk using a combination of linkage and association analysis 31. However, the LHPP associations were dependent on HTR1A genotype, which is not a finding that we were able to replicate in the present paper. First, many of the LHPP variants identified by Neff and colleagues as being associated with MDD risk are not present in our sample, although those that are present are in partial LD with our top-ranked variant (rs12265012, r2 = 0.21; rs10794134, r2 = 0.17) 31. Second, while Neff and colleagues showed an interaction between HTR1A (and specifically the 1019C>G genotype, rs2495, which is not present in out sample) and LHPP, we did not. Three HTR1A variants are present in our sample, and an interaction term between our top-ranked LHPP variant and any of the HTR1A variants was not significant when included as covariates in a polygenic model of the depression factor score (rs10052087, χ2= 0.75, p = 0.39; rs6449693, χ2= 0.01, p = 0.93; rs6294, χ2= 0.75, p = 0.39); and none of the HTR1A variants were significantly associated with the continuous depression score in isolation (rs10052087, χ2= 0.46, p = 0.50; rs6449693, χ2= 2.16, p = 0.14; rs6294, χ2= 0.46, p = 0.50). Relatively little is known about the function of LHPP. A single study implicates the role of Lhpp in thyroid function 50, which could be interesting given that thyroid function is thought to mediate the function of certain anti-depressants 51. However, an explicit relationship between thyroid function, Lhpp and MDD is not apparent based on current research.
The gene CPXM2 is a member of the metallocarboxypeptidase A family of digestive enzymes and is highly expressed in brain, particularly in the hippocampus, hypothalamus, choroid plexus and throughout the cerebral cortex 52. CPXM2 is distinct from other gene-family members as it lacks the active site residues necessary for enzyme function and as a consequence it may fulfill an alternative role as a phospholipid binding protein 53. In rats CPE, a paralog of CPXM1 (which is highly similar to CPXM2 in that it also lacks the catalytic activity found in other carboxypetidases 54), mediates dopamine transporter (DAT) expression such that co-expression of CPE and DAT results in increased dopamine reuptake in brain 55. Also, a variant of CPXM2 is suggestively associated with cognitive decline in schizophrenia 56, where cognitive ability, and more specifically cognitive impairment in schizophrenia, is thought to be modulated by dopaminergic signaling 57-59. Insofar as the role of dopamine is well established in MDD 60-62 and that the gene CPXM2, or at least very similar genes in the same family, appears to influence dopamine functioning in the brain, the present paper highlights a new candidate gene for MDD in a newly established large multiplex MDD pedigree that warrants further investigation.
The association for the variant rs7913161 in CPXM2 in the larger sample was low and not significant, which could suggests that the variant is marking a rare and functional variant that exists only within the multiplex MDD pedigree, one which likely makes up a haplotype of many variants. The authors of the most recent mega-analysis GWA study (GWAS) from the PGC, which comprised approximately seventy thousand subjects, cite the need for even greater sample sizes and increased power in order to detect genetic variants for depression 30. However, it is also possible that genetic heterogeneity for MDD is greater than for other disorders, meaning that it will be necessary to reduce genetic heterogeneity (for example, by studying a group of genetically-homogenous kindred) in order to find risk genes. The present study makes a case for the latter approach; namely, the use of whole genome sequence data in extended pedigrees. The common disease-rare variant hypothesis states that the genetic causes of common, polygenic disorders such as depression are likely to be rare in the population. Clearly the use of rare variation alone will not solve the power issues highlighted by the PGC. However, identifying a rare functional variant (with a large effect size) in only a handful of affected individuals can be sufficient to verify that a given gene is involved in an illness. Data from the 1000 Genomes Project confirm that rare (<1%) variants constitute the vast majority (73%) of polymorphic sites in humans 63. A key factor for identification of specific rare functional variants is detecting sufficient copies of that variant for statistical inference 64, 65. Pedigree-based studies represent an implicit enrichment strategy for identifying rare variants as Mendelian transmissions from parents to offspring maximize the chance that multiple copies of rare variants exist in the pedigree. Family-based cohorts have substantially greater power than unrelated cases to detect rare genetic effects given an equivalent number of sampled individuals 66, 67. For example, genes for hypertension have been identified for blood pressure in the general population by focusing research efforts on an extended pedigree with a rare form of hypertension 68. Indeed, rare deleterious mutations are known to occur in genes that also harbor common variants with modest effects on disease risk 69. For example, 11 of 30 genes with common variants associated with lipid levels also carry known rare alleles of large effect in Mendelian dyslipidemias 70, 71. Furthermore, rare variants may contribute to loci identified through common variation 72.
Another issue the PGC highlights in the hunt for depression genes is the possibility that the depression phenotypes used in genetic studies are ‘suboptimal’ 30 (p. 9). This observation dovetails with the Research Domain Criteria (RDoC) strategy that was recently proposed by the NIMH. This strategy encourages researchers to focus their efforts on developing new ways of classifying psychopathology by developing a dimension-based taxonomy of functioning that encompasses behavior, neuroscience and genetics 73, 74. MDD is typically treated as a categorical trait it is assumed that MDD reflects the tail end of an underlying normal distribution of mood, and that diagnosis occurs when a threshold for liability is crossed. It seems plausible that the genes which moderate behavior at the tail end of the distribution are the same as those that underlie the regulation of normal mood 37 and by dichotomizing the MDD distribution, we ignore a substantial proportion of variance that would contribute to gene-finding efforts. Thus the present paper is in line with the RDoC strategy whereby depression is represented as a continuum or dimension and moreover the use of a continuous measure of depression derived from a single-factor model of interview items versus the analogous dichotomous measure from the same items was shown to be more successful in the present study. The notion that continuous models of complex disorders derived from commonly used questionnaires can be used in association studies represents a significant advancement over studies that rely entirely on a diagnostic endpoint. Other studies have examined this notion in detail, and developed multidimensional models of depressive symptomatology 75-77. The present work focussed specifically on the MINI and as such fewer dimensions were derived (indeed, inspection of the correlations between items strongly supports the existence of a single dimension in the data used in the present study; Table S1), however the utility of the present study over those published previously is the inclusion of genetic data which allowed the identification of possible candidate genes (LHPP and CPXM2).
In summary, the present study represents advancement in our understanding in the genetics of depression in two ways. First, it confirms the probable involvement of a gene previously implicated in illness risk (LHPP) and, through the use of a multiplex MDD pedigree, it highlights a novel risk gene (CPXM2), which warrants further investigation. Second, it draws attention to an alternative methodology for the hunt for depression genes, which is focusing on rare variation in a multiplex MDD pedigree combined with the use of dimensional indices of MDD symptomatology.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: Review and meta-analysis. Am J Psychiatry. 2000;157:1552–62. doi: 10.1176/appi.ajp.157.10.1552. [DOI] [PubMed] [Google Scholar]
- 2.Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. The epidemiology of major depressive disorder: Results from the national comorbidity survey replication (NCS-R) JAMA. 2003;289:3095–105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- 3.Greenberg PE, Kessler RC, Birnbaum HG, Leong SA, Lowe SW, Berglund PA, et al. The economic burden of depression in the united states: How did it change between 1990 and 2000? J Clin Psychiatry. 2003;64:1465–75. doi: 10.4088/jcp.v64n1211. [DOI] [PubMed] [Google Scholar]
- 4.Wells KB, Stewart A, Hays RD, Burnam MA, Rogers W, Daniels M, et al. The functioning and well-being of depressed patients. results from the medical outcomes study. JAMA. 1989;262:914–9. [PubMed] [Google Scholar]
- 5.Hays RD, Wells KB, Sherbourne CD, Rogers W, Spritzer K. Functioning and well-being outcomes of patients with depression compared with chronic general medical illnesses. Arch Gen Psychiatry. 1995;52:11–9. doi: 10.1001/archpsyc.1995.03950130011002. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization. Depression fact sheet number 369. 2012; 2012(11/06)Web Page.
- 7.American Psychiatric Association. Diagnostic and statistical manual of mental disorders : DSM-IV. American Psychiatric Association; Washington, D.C.: 1994. [Google Scholar]
- 8.Binder EB, Holsboer F. Pharmacogenomics. Handb Exp Pharmacol. 2005;(169):527–46. doi: 10.1007/3-540-28082-0_19. [DOI] [PubMed] [Google Scholar]
- 9.Bromet EJ, Dunn LO, Connell MM, Dew MA, Schulberg HC. Long-term reliability of diagnosing lifetime major depression in a community sample. Arch Gen Psychiatry. 1986;43:435–40. doi: 10.1001/archpsyc.1986.01800050033004. [DOI] [PubMed] [Google Scholar]
- 10.Kendler KS, Neale MC, Kessler RC, Heath AC, Eaves LJ. The lifetime history of major depression in women. reliability of diagnosis and heritability. Arch Gen Psychiatry. 1993;50:863–70. doi: 10.1001/archpsyc.1993.01820230054003. [DOI] [PubMed] [Google Scholar]
- 11.Keller MB, Hanks DL, Klein DN. Summary of the DSM-IV mood disorders field trial and issue overview. Psychiatr Clin North Am. 1996;19:1–28. doi: 10.1016/s0193-953x(05)70270-7. [DOI] [PubMed] [Google Scholar]
- 12.U.S. Department of Health and Human Services, Public Health Service Agency for Healthcare Policy and Research. treatment of major depression clinical practice guideline, number 5. Vol. 2. rockville, MD: AHCPR publication 93-0551; Depression guideline panel. depression in primary care. april 1993. 1993Laws/Statutes. [Google Scholar]
- 13.Lopez MF, Compton WM, Grant BF, Breiling JP. Dimensional approaches in diagnostic classification: A critical appraisal. Int J Methods Psychiatr Res. 2007;16 Suppl 1:S6–7. doi: 10.1002/mpr.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. Am J Psychiatry. 2006;163:1905–17. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 15.Miller DB, O'Callaghan JP. Personalized medicine in major depressive disorder --opportunities and pitfalls. Metabolism. 2013;62 Suppl 1:S34–9. doi: 10.1016/j.metabol.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flint J, Kendler KS. The genetics of major depression. Neuron. 2014;81:484–503. doi: 10.1016/j.neuron.2014.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kendler KS, Gatz M, Gardner CO, Pedersen NL. A swedish national twin study of lifetime major depression. Am J Psychiatry. 2006;163:109–14. doi: 10.1176/appi.ajp.163.1.109. [DOI] [PubMed] [Google Scholar]
- 18.Cohen-Woods S, Craig IW, McGuffin P. The current state of play on the molecular genetics of depression. Psychol Med. 2013;43:673–87. doi: 10.1017/S0033291712001286. [DOI] [PubMed] [Google Scholar]
- 19.Bosker FJ, Hartman CA, Nolte IM, Prins BP, Terpstra P, Posthuma D, et al. Poor replication of candidate genes for major depressive disorder using genome-wide association data. Mol Psychiatry. 2011;16:516–32. doi: 10.1038/mp.2010.38. [DOI] [PubMed] [Google Scholar]
- 20.Wray NR, Pergadia ML, Blackwood DH, Penninx BW, Gordon SD, Nyholt DR, et al. Genome-wide association study of major depressive disorder: New results, meta-analysis, and lessons learned. Mol Psychiatry. 2012;17:36–48. doi: 10.1038/mp.2010.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sullivan PF, de Geus EJ, Willemsen G, James MR, Smit JH, Zandbelt T, et al. Genome-wide association for major depressive disorder: A possible role for the presynaptic protein piccolo. Mol Psychiatry. 2009;14:359–75. doi: 10.1038/mp.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rietschel M, Mattheisen M, Frank J, Treutlein J, Degenhardt F, Breuer R, et al. Genome-wide association-, replication-, and neuroimaging study implicates HOMER1 in the etiology of major depression. Biol Psychiatry. 2010;68:578–85. doi: 10.1016/j.biopsych.2010.05.038. [DOI] [PubMed] [Google Scholar]
- 23.Lewis CM, Ng MY, Butler AW, Cohen-Woods S, Uher R, Pirlo K, et al. Genome-wide association study of major recurrent depression in the U.K. population Am J Psychiatry. 2010;167:949–57. doi: 10.1176/appi.ajp.2010.09091380. [DOI] [PubMed] [Google Scholar]
- 24.Muglia P, Tozzi F, Galwey NW, Francks C, Upmanyu R, Kong XQ, et al. Genome-wide association study of recurrent major depressive disorder in two european case-control cohorts. Mol Psychiatry. 2010;15:589–601. doi: 10.1038/mp.2008.131. [DOI] [PubMed] [Google Scholar]
- 25.Kohli MA, Lucae S, Saemann PG, Schmidt MV, Demirkan A, Hek K, et al. The neuronal transporter gene SLC6A15 confers risk to major depression. Neuron. 2011;70:252–65. doi: 10.1016/j.neuron.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shyn SI, Shi J, Kraft JB, Potash JB, Knowles JA, Weissman MM, et al. Novel loci for major depression identified by genome-wide association study of sequenced treatment alternatives to relieve depression and meta-analysis of three studies. Mol Psychiatry. 2011;16:202–15. doi: 10.1038/mp.2009.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi J, Potash JB, Knowles JA, Weissman MM, Coryell W, Scheftner WA, et al. Genome-wide association study of recurrent early-onset major depressive disorder. Mol Psychiatry. 2011;16:193–201. doi: 10.1038/mp.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hek K, Demirkan A, Lahti J, Terracciano A, Teumer A, Cornelis MC, et al. A genome-wide association study of depressive symptoms. Biol Psychiatry. 2013;73:667–78. doi: 10.1016/j.biopsych.2012.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Neff CD, Abkevich V, Packer JC, Chen Y, Potter J, Riley R, et al. Evidence for HTR1A and LHPP as interacting genetic risk factors in major depression. Mol Psychiatry. 2009;14:621–30. doi: 10.1038/mp.2008.8. [DOI] [PubMed] [Google Scholar]
- 32.Breen G, Webb BT, Butler AW, van den Oord EJ, Tozzi F, Craddock N, et al. A genome-wide significant linkage for severe depression on chromosome 3: The depression network study. Am J Psychiatry. 2011;168:840–7. doi: 10.1176/appi.ajp.2011.10091342. [DOI] [PubMed] [Google Scholar]
- 33.Pergadia ML, Glowinski AL, Wray NR, Agrawal A, Saccone SF, Loukola A, et al. A 3p26-3p25 genetic linkage finding for DSM-IV major depression in heavy smoking families. Am J Psychiatry. 2011;168:848–52. doi: 10.1176/appi.ajp.2011.10091319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glahn DC, Curran JE, Winkler AM, Carless MA, Kent JW, Jr, Charlesworth JC, et al. High dimensional endophenotype ranking in the search for major depression risk genes. Biol Psychiatry. 2012;71:6–14. doi: 10.1016/j.biopsych.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ripke S, O'Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–9. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collins AL, Sullivan PF. Genome-wide association studies in psychiatry: What have we learned? Br J Psychiatry. 2013;202:1–4. doi: 10.1192/bjp.bp.112.117002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luft FC. Hypertension as a complex genetic trait. Semin Nephrol. 2002;22:115–26. doi: 10.1053/snep.2002.30211. [DOI] [PubMed] [Google Scholar]
- 38.Duggirala R, Williams JT, Williams-Blangero S, Blangero J. A variance component approach to dichotomous trait linkage analysis using a threshold model. Genet Epidemiol. 1997;14:987–92. doi: 10.1002/(SICI)1098-2272(1997)14:6<987::AID-GEPI71>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 39.Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The mini-international neuropsychiatric interview (M.I.N.I.): The development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59 Suppl 20:22–33. quiz 34-57. [PubMed] [Google Scholar]
- 40.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feingold E, Brown PO, Siegmund D. Gaussian models for genetic linkage analysis using complete high-resolution maps of identity by descent. Am J Hum Genet. 1993;53:234–51. [PMC free article] [PubMed] [Google Scholar]
- 42.Moskvina V, Schmidt KM. On multiple-testing correction in genome-wide association studies. Genet Epidemiol. 2008;32:567–73. doi: 10.1002/gepi.20331. [DOI] [PubMed] [Google Scholar]
- 43.Falconer DS. Introduction to quantitative genetics. Longman; Harlow: 1996. [Google Scholar]
- 44.Blangero J, Williams JT, Almasy L. Variance component methods for detecting complex trait loci. In: Rao DC, Province MA, editors. Genetic Dissection of Complex Traits. Academic Press; San Diego: 2001. pp. 151–81. [DOI] [PubMed] [Google Scholar]
- 45.Blangero J, Williams JT, Almasy L, Williams-Blangero S. Chapter 11. mapping genes influencing human quantitative trait variation. In: Crawford Michael H., editor. Anthropological Genetics: Theory, Methods and Applications. Cambridge University Press; New York, NY: 2007. pp. 306–33. [Google Scholar]
- 46.CONVERGE consortium. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature. 2015;523:588–91. doi: 10.1038/nature14659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biernacka JM, Cordell HJ. Exploring causality via identification of SNPs or haplotypes responsible for a linkage signal. Genet Epidemiol. 2007;31:727–40. doi: 10.1002/gepi.20236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seal US, Binkley F. An inorganic pyrophosphatase of swine brain. J Biol Chem. 1957;228:193–9. [PubMed] [Google Scholar]
- 49.Yokoi F, Hiraishi H, Izuhara K. Molecular cloning of a cDNA for the human phospholysine phosphohistidine inorganic pyrophosphate phosphatase. J Biochem. 2003;133:607–14. doi: 10.1093/jb/mvg078. [DOI] [PubMed] [Google Scholar]
- 50.Koike E, Toda S, Yokoi F, Izuhara K, Koike N, Itoh K, et al. Expression of new human inorganic pyrophosphatase in thyroid diseases: Its intimate association with hyperthyroidism. Biochem Biophys Res Commun. 2006;341:691–6. doi: 10.1016/j.bbrc.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Altshuler LL, Bauer M, Frye MA, Gitlin MJ, Mintz J, Szuba MP, et al. Does thyroid supplementation accelerate tricyclic antidepressant response? A review and meta-analysis of the literature. Am J Psychiatry. 2001;158:1617–22. doi: 10.1176/appi.ajp.158.10.1617. [DOI] [PubMed] [Google Scholar]
- 52.Xin X, Day R, Dong W, Lei Y, Fricker LD. Identification of mouse CPX-2, a novel member of the metallocarboxypeptidase gene family: CDNA cloning, mRNA distribution, and protein expression and characterization. DNA Cell Biol. 1998;17:897–909. doi: 10.1089/dna.1998.17.897. [DOI] [PubMed] [Google Scholar]
- 53.Xin X, Varlamov O, Day R, Dong W, Bridgett MM, Leiter EH, et al. Cloning and sequence analysis of cDNA encoding rat carboxypeptidase D. DNA Cell Biol. 1997;16:897–909. doi: 10.1089/dna.1997.16.897. [DOI] [PubMed] [Google Scholar]
- 54.Lei Y, Xin X, Morgan D, Pintar JE, Fricker LD. Identification of mouse CPX-1, a novel member of the metallocarboxypeptidase gene family with highest similarity to CPX-2. DNA Cell Biol. 1999;18:175–85. doi: 10.1089/104454999315565. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H, Li S, Wang M, Vukusic B, Pristupa ZB, Liu F. Regulation of dopamine transporter activity by carboxypeptidase E. Mol Brain. 2009;2:10–6606-2-10. doi: 10.1186/1756-6606-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hashimoto R, Ikeda M, Ohi K, Yasuda Y, Yamamori H, Fukumoto M, et al. Genome-wide association study of cognitive decline in schizophrenia. Am J Psychiatry. 2013;170:683–4. doi: 10.1176/appi.ajp.2013.12091228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nieoullon A. Dopamine and the regulation of cognition and attention. Prog Neurobiol. 2002;67:53–83. doi: 10.1016/s0301-0082(02)00011-4. [DOI] [PubMed] [Google Scholar]
- 58.Goldman-Rakic PS, Castner SA, Svensson TH, Siever LJ, Williams GV. Targeting the dopamine D1 receptor in schizophrenia: Insights for cognitive dysfunction. Psychopharmacology (Berl) 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- 59.Knowles EEM, Mathias SR, McKay DR, Sprooten E, Blangero J, Almasy L, et al. Genome-wide analyses of working memory: A review. Current Behavioral Neuroscience Reports. 2014 doi: 10.1007/s40473-014-0028-8. Journal, Electronic. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–9. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- 61.Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch Gen Psychiatry. 2007;64:327–37. doi: 10.1001/archpsyc.64.3.327. [DOI] [PubMed] [Google Scholar]
- 62.Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–41. doi: 10.1038/nature11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, et al. The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. 2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kent JW, Jr, Dyer TD, Goring HH, Blangero J. Type I error rates in association versus joint linkage/association tests in related individuals. Genet Epidemiol. 2007;31:173–7. doi: 10.1002/gepi.20200. [DOI] [PubMed] [Google Scholar]
- 65.Blangero J, Goring HH, Kent JW, Williams JT, Peterson CP, Almasy L, et al. Quantitative trait nucleotide analysis using bayesian model selection 2005. Hum Biol. 2009;81:829–47. doi: 10.3378/027.081.0625. [DOI] [PubMed] [Google Scholar]
- 66.Li M, Boehnke M, Abecasis GR. Efficient study designs for test of genetic association using sibship data and unrelated cases and controls. Am J Hum Genet. 2006;78:778–92. doi: 10.1086/503711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saad M, Wijsman EM. Power of family-based association designs to detect rare variants in large pedigrees using imputed genotypes. Genet Epidemiol. 2014;38:1–9. doi: 10.1002/gepi.21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tobin MD, Tomaszewski M, Braund PS, Hajat C, Raleigh SM, Palmer TM, et al. Common variants in genes underlying monogenic hypertension and hypotension and blood pressure in the general population. Hypertension. 2008;51:1658–64. doi: 10.1161/HYPERTENSIONAHA.108.112664. [DOI] [PubMed] [Google Scholar]
- 69.Bodmer W, Bonilla C. Common and rare variants in multifactorial susceptibility to common diseases. Nat Genet. 2008;40:695–701. doi: 10.1038/ng.f.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cohen JC, Pertsemlidis A, Fahmi S, Esmail S, Vega GL, Grundy SM, et al. Multiple rare variants in NPC1L1 associated with reduced sterol absorption and plasma low-density lipoprotein levels. Proc Natl Acad Sci U S A. 2006;103:1810–5. doi: 10.1073/pnas.0508483103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Romeo S, Pennacchio LA, Fu Y, Boerwinkle E, Tybjaerg-Hansen A, Hobbs HH, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–6. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–9. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cuthbert BN, Insel TR. Toward new approaches to psychotic disorders: The NIMH research domain criteria project. Schizophr Bull. 2010;36:1061–2. doi: 10.1093/schbul/sbq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): Toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010;167:748–51. doi: 10.1176/appi.ajp.2010.09091379. [DOI] [PubMed] [Google Scholar]
- 75.Uher R, Farmer A, Maier W, Rietschel M, Hauser J, Marusic A, et al. Measuring depression: Comparison and integration of three scales in the GENDEP study. Psychol Med. 2008;38:289–300. doi: 10.1017/S0033291707001730. [DOI] [PubMed] [Google Scholar]
- 76.Korszun A, Moskvina V, Brewster S, Craddock N, Ferrero F, Gill M, et al. Familiality of symptom dimensions in depression. Arch Gen Psychiatry. 2004;61:468–74. doi: 10.1001/archpsyc.61.5.468. [DOI] [PubMed] [Google Scholar]
- 77.Kendler KS, Aggen SH, Neale MC. Evidence for multiple genetic factors underlying DSM-IV criteria for major depression. JAMA Psychiatry. 2013;70:599–607. doi: 10.1001/jamapsychiatry.2013.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.