

Abstract
Transendothelial migration (TEM) of leukocytes is the step in leukocyte emigration in which the leukocyte actually leaves the blood vessel to carry out its role in the inflammatory response. It is therefore, arguably the most critical step in emigration. This review focuses on two of the many aspects of this process that have seen important recent developments. The adhesion molecules, PECAM (CD31) and CD99 that regulate two major steps in TEM, do so by regulating specific signals. PECAM initiates the signaling pathway responsible for the calcium flux that is required for TEM. Calcium enters through the cation channel TRPC6 and recruits the first wave of trafficking of membrane from the lateral border recycling compartment (LBRC). CD99 signals through soluble adenylate cyclase to activate protein kinase A to recruit a second wave of LBRC trafficking. Another process that is critical for TEM is transient removal of VE-cadherin from the site of TEM. However, the local signaling pathways that are responsible for this appear to be different from those that open the junctions to increase vascular permeability.
Introduction
The inflammatory response evolved to fight off infectious pathogens and heal wounds. However, the other side of the proverbial double-edged sword of inflammation is that inflammation that is doing more harm than good to the host--inflammation that damages host tissues, that will not resolve, or is self-directed--is involved in most diseases. Thus, there is a significant focus on understanding the molecular regulation of inflammation to develop better anti-inflammatory therapies. Since there is no response in either the innate or adaptive response unless leukocytes cross blood vessels, the mechanisms regulating this process have increasingly been the focus of investigation.
Less than a decade ago, most reviews on transendothelial migration (TEM) of leukocytes would start apologetically with phrases like, “while much has been learned about leukocyte rolling and adhesion, much less is known at the molecular level about transendothelial migration.” Many advances have been made recently in our understanding of the molecules and signaling pathways regulating transendothelial migration. These have been reviewed comprehensively in a number of major reviews including [1–6] to which the reader is referred for a broader discussion. This brief review will summarize two major concepts in the biology and regulation of TEM. First, the “step” of TEM is itself a multistep process regulated by sequential adhesion and signaling interactions between leukocytes and endothelial cells. Second, a topic that is not well appreciated by most scientists—even many of those working in the field: the differences in the regulation of TEM and inflammatory vascular permeability.
TEM itself is a multi-step pathway
It is well-established that extravasation of leukocytes at the site of inflammation involves a sequential series of molecular interactions between leukocytes and endothelial cells. The molecules regulating the steps of capture, fast and slow rolling, activation, adhesion, intraluminal crawling, and transendothelial migration are known [3,7,8]. These steps were determined because selective blocking antibodies, chemical inhibitors, or genetic manipulations of specific molecules were able to inhibit each specific step. TEM is the process by which leukocytes squeeze tightly between (paracellular migration), or in some cases through (transcellular migration) endothelial cells to reach the site of inflammation. However, the process of TEM itself can be dissected into several steps as a result of inhibition or genetic reduction of specific molecules [1,2].
The first step unique to the process of diapedesis is engagement of PECAM on the leukocyte with PECAM on the endothelial cell border [9–11]. Interference with this interaction using blocking antibodies, PECAM-Fc chimeras, or genetic ablation blocks selectively blocks TEM, trapping the leukocyte on the apical surface, where it remains bound to the endothelial cell over the junction. Leukocytes remain activated and crawl along the junction, but cannot get through as long as the block remains [9,12]. In the presence of a PECAM blockade, ICAM-1 clusters under the endothelial cell [13], but this is not sufficient to promote TEM if PECAM is blocked.
Homophilic interactions between leukocyte PECAM and endothelial cell PECAM start signals that recruit membrane from the lateral border recycling compartment (LBRC) to the site of interaction. The LBRC is a reticulum of 50 nm vesicular structures that are connected to each other and to the plasma membrane at intervals along the cell borders [11]. The LBRC contains a representative selection of lateral border membrane with the exception of VE-cadherin [13,14], which is actively excluded [15]. Membrane from the LBRC is moved by kinesin molecular motors along microtubules [16] to surround the leukocyte during its passage across the endothelial border during paracellular migration [11] and through the cell during transcellular migration [13]. This is hypothesized to provide the leukocyte with a VE-cadherin-free passage lined by unligated adhesion molecules with which to interact and increased surface area to permit passage without requiring endothelial cell retraction [1,17]. Any treatment of EC that inhibits targeted recycling of the LBRC (e.g. blocking PECAM, acutely disrupting microtubules, or inhibiting the kinesin molecular motor) inhibits TEM regardless of the leukocyte type, inflammatory stimulus, or path of TEM [11,13,16,18,19].
An increase in cytosolic free calcium ion (↑[Ca+2]i) was shown in 1993 to be required for TEM to take place [20]. The transient receptor potential canonical family member 6 (TRPC6) cation channel has recently been shown to be the channel responsible for the ↑[Ca+2]i [18]. PECAM signaling triggers activation of TRPC6 during TEM. Exogenous activation of endothelial cell TRPC6 using the selective activator Hyp9 can overcome a block to TEM mediated by anti-PECAM antibody by directly activating the signaling pathway downstream of PECAM and recruiting the LBRC. Knockdown in vitro or knockout in vivo of TRPC6 only in EC produces the same phenotype as blocking PECAM [18].
Real-time observation of TEM events across EC monolayers expressing VE-cadherin-GFP revealed that PMN transmigrate at regions of relatively low VE-cadherin expression and/or that VE-cadherin moved out of the way during TEM [21]. This is not an epiphenomenon, but apparently a requirement for TEM [22–24]. There is evidence both for [23] and against [24] internalization of VE-cadherin during this process. Regardless, the phosphorylation state of VE-cadherin tyrosine 731 (and perhaps tyrosines) is involved [22–24].
After the LBRC has been recruited to the site of TEM via PECAM-PECAM interactions, and VE-cadherin has left the site of TEM, migration still requires additional molecular interactions. This is demonstrable because one can selectively and reversibly block the transmigration step using antibodies against specific molecules. In a sequential transmigration assay, one can block TEM using monoclonal antibody (mAb) against molecule A for an appropriate time, then wash out the block, add a mAb against molecule B, and resume the TEM assay. If molecule B is involved in a process downstream of A, mAb B will block any further TEM. If molecule B is involved in a step of TEM before molecule A, it will not [17,25,26].
This was first shown for the relationship of PECAM and CD99 [26]. CD99 is expressed at the borders of EC and is in the LBRC [13]. It is a fairly unique molecule with only one paralog in the genome [27] and no signaling motifs on its cytoplasmic tail. Inhibition of homophilic interaction between CD99 on the leukocyte and CD99 on the endothelial cell arrests TEM with the leukocyte partway across the endothelial junction with its leading lamellipodium under the EC and a trailing uropod on the apical surface [26,28], in contrast to the appearance of leukocytes blocked at the PECAM-dependent step, which appear entirely on the apical surface hovering over the junctions [9]. The sequential transmigration assay demonstrated that the site of blockade was functionally (as well as morphologically) downstream of the step controlled by PECAM [26].
We recently demonstrated that that homophilic CD99 interaction stimulates another wave of targeted recycling of the LBRC to promote the completion of TEM [19]. CD99 is constitutively in a complex with the A-kinase anchoring protein ezrin, the unique soluble adenylate cyclase (sAC), and protein kinase A (PKA). Engagement of CD99 activates sAC to produce cAMP locally to activate PKA and orchestrate the next wave of LBRC recruitment [19].
The fact that two distinct molecules regulate different steps in TEM begs the question of whether there are other molecules that mediate additional steps in the process. In fact, poliovirus receptor (CD155, PVR), expressed at EC borders, is involved in TEM of monocytes [29]. Using the sequential TEM assay, we found that PVR, interacting with the leukocyte ligand DNAX accessory molecule-1, controls a step in TEM that is between those regulated by PECAM and CD99 [25]. PVR is also in the LBRC and, as expected, PVR mAb delivered to the LBRC blocked TEM [25]. How and why these molecules, all of which are together in the LBRC [15], regulate TEM sequentially is not known and is currently the subject of investigation.
Vascular Permeability and TEM are Distinct Phenomena
Increased vascular permeability occurs at sites of inflammation, resulting in edema that provides the “tumor” in Aulus Cornelius Celsus’ classic description of inflammation: tumor, rubor, calor, dolor (swelling, redness, heat, and pain). Vascular leakage and leukocyte extravasation both take place in postcapillary venules. For many years, the common assumption was that these processes were directly related, with leaky vessels, which preceded leukocyte extravasation, providing an easy pathway for the leukocytes.
This concept was supported by studies showing that molecules known to be involved in leukocyte adhesion and transmigration sent signals that activated pathways known to be involved in breakdown of endothelial junctions. The pathways for endothelial permeability had been worked out for mediators of permeability such as thrombin, bradykinin, and histamine (extensively reviewed in [30] and see Fig. 1). These agonists activated G-protein coupled receptors, opening of calcium store operated channels, activation of small GTPases, activation of myosin light chain kinase (MLCK), cytoskeletal rearrangements, and endothelial cell shape changes. The study showing that ↑[Ca2+]i was required for neutrophil TEM [20] was quickly followed by a study demonstrating that FMLF-activated neutrophils or neutrophils transmigrating across IL-1β activated EC induce phosphorylation of MLCK and isometric contraction in the endothelial monolayers [31]. The presumption was that the activated PMN were simulating the ↑[Ca2+]i and that was responsible for the activation of MLCK, which induced the contraction, loosening the junctions and allowing PMN to transmigrate. Antibody-mediated cross-linking of ICAM-1 [32] and VCAM-1 [33], simulating interactions with leukocytes, stimulates an increase in cytosolic free calcium ions. Similarly, clustering of ICAM-1 activates RhoA through Rho GEF 12; RhoA activates Rho kinase (ROCK) (reviewed in [34]). This signaling is particularly enhanced by mechanical forces exerted on ICAM-1 by leukocytes engaging it [35]. ROCK phosphorylates and inactivates the myosin phosphatase targeting subunit (MYPT1) of the trimeric myosin phosphatase, the major phosphatase inactivating MLCK. The end result is potentiation of actin-myosin contraction. Activation of RhoA also induces the phosphorylation myosin light chain through the activation myosin light chain kinase (MLCK) [36,37].
Figure 1. Global signals open junctions for vascular permeability.
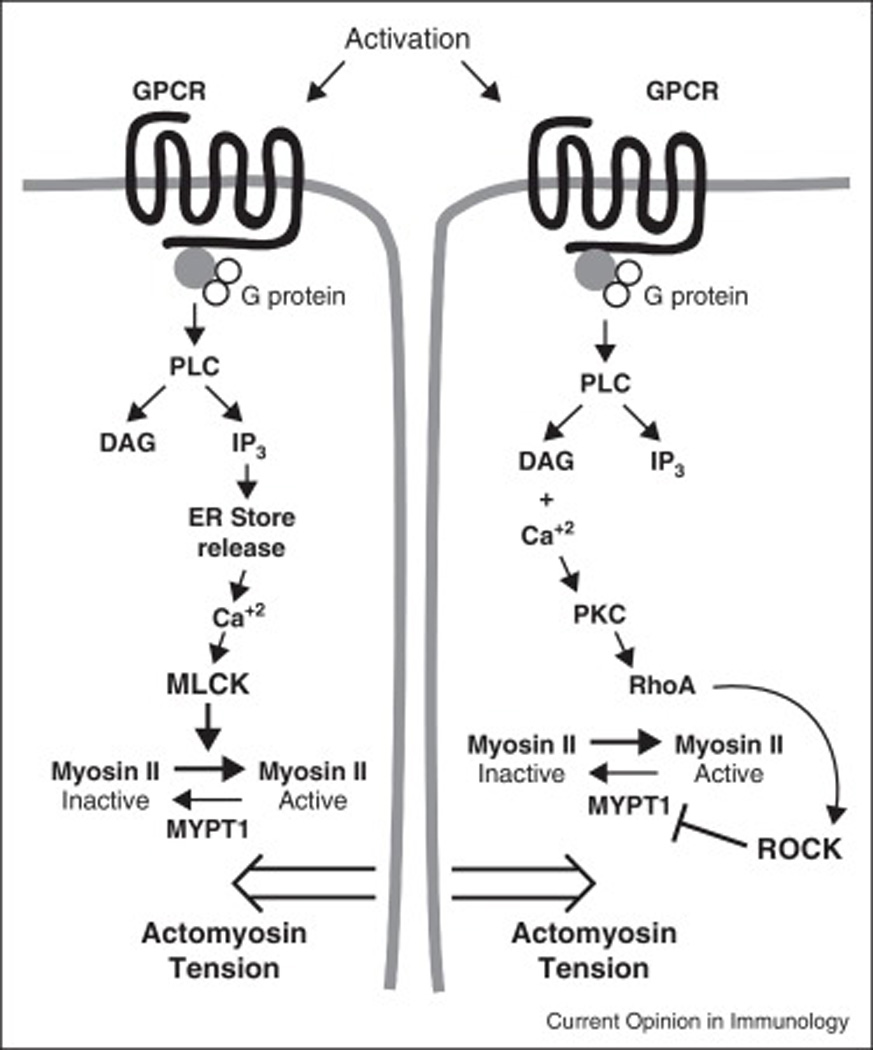
The diagram depicts stimulation of G protein coupled receptors (GPCR) by a prototype soluble agonist. The activation of phospholipase C (PLC) results in the conversion of phosphatidyl inositol in the plasma membrane to the second messengers diacyl glycerol (DAG) and inositol triphosphate (IP3). (Left pathway) IP3 stimulates calcium release from stores in the endoplasmic reticulum. The calcium activates myosin light chain kinase (MLCK) to phosphorylate myosin light chain promoting activation of myosin II and actomyosin tension. This is believed to cause contraction of the endothelial cells weakening the junction and promoting vascular permeability. (Right pathway) In a parallel pathway DAG and calcium activate protein kinase C (PKC), which activates the GTPase RhoA, which in turn activates Rho-associated coiled coil kinase (ROCK), which phosphorylates and inactivates myosin phosphatase target subunit 1 (MYPT1) to inactivate it. Since MYPT1 serves to dephosphorylate and hence activate Myosin II light chain, this further stabilizes the actomyosin tension in the cells.
Many studies provided data to reinforce the idea that TEM and vascular permeability were regulated by the same signaling pathways, and as a corollary, that TEM required an increase in vascular permeability. This, in spite of both older [38] and newer data that efficient TEM does not require an increase in endothelial cell junctional permeability [18,39]. Mice in which VE-cadherin was replaced with a chimera encoding a VE-cadherin/α-catenin fusion protein that kept VE-cadherin tethered to the actin cytoskeleton, were virtually completely resistant to the effects of VEGF and histamine on permeability and had a very significant reduction in leukocyte extravasation in several vascular beds [40]. The authors correctly concluded that plasticity of the VE-cadherin junction is critical for TEM {Schulte, 2011 #5960. However, the data were interpreted by some others as evidence that they were controlled by the same mechanism.
Several studies used the C3 exotoxin to block RhoA activation or the compound Y-27632 to inhibit Rho kinase activity in endothelial cells, and studied the effects on transmigration {Barreiro, 2002 #4608;Carman, 2003 #4631;Carman, 2004 #4433;Strey, 2002 #5890}. Often these groups obtained discrepant results, but these could potentially be explained by differences in cell type and assay conditions. It should be noted that this toxin is not specific for RhoA, but also blocks RhoB and RhoC, and at high concentrations may inhibit Rac1, as well [41]. Neither could it always be excluded that the effect of the toxin did not spill over to affect the leukocytes.
Nonetheless, growing appreciation that intracellular signaling is a localized event [42], and recently published data are resulting in a re-evaluation of these studies. The McDonald group published studies of inflammation in the rat tracheal vasculature showing that smaller postcapillary venules (20 –40 µm) were the sites of vascular leakage while most PMN exited from more distal venules (40 –60 µm) in the same animal [43,44]. Thus, these processes do not have to occur together.
There are several examples of localized signaling between leukocyte and endothelial cell resulting in outcomes different from when the same molecules are activated by global mediators [2]. In the local environment of clustered ICAM-1 in TEM, src-mediated phosphorylation of cortactin enhances transmigration, whereas in response to global stimulation by sphingosine-1 phosphate (S1P), a rapid increase in cortactin phosphorylation is associated with a Rac1-dependent association of cortactin with myosin light chain kinase and a tightening of the endothelial barrier. [45,46]. Furthermore, interaction of lymphocyte VLA-4 with endothelial cell VCAM-1 led to activation of Rac1, which activated NADPH oxidase to produce ROS [47]. This was recently shown to activate Pyk2 to promote dissociation of vascular endothelial protein tyrosine phosphatase (VE-PTP) from VE-cadherin to favor its phosphorylation [48]. Through local activation of VE-PTP, local VE-cadherin phosphorylation may be accomplished (Fig. 2).
Figure 2. Localized signals open junctions for transmigration.

The diagram depicts two processes critical for TEM: targeted recycling of the LBRC and disruption of VE-cadherin at the site of TEM. PECAM-PECAM interactions between leukocyte and endothelial cell trigger signals that activate ↑[Ca+2]i through TRPC6 to recruit the LBRC to the site of TEM. VLA-4 on leukocytes interacts with VCAM-1 on EC to activate Rac1, which activates NADPH oxidase (NOX) to generate reactive oxygen species (ROS) that activate the kinase Pyk2. This phosphorylates a putative substrate for vascular endothelial receptor protein tyrosine phosphatase (VE-PTP), which reduces the affinity of VE-PTP for VE-cadherin, favoring phosphorylation (circled P) of VE-cadherin and its removal from the adherens junction.
The Vestweber group showed that vascular permeability and neutrophil TEM could be molecularly uncoupled in vivo: Mice genetically deficient in the actin bundling protein cortactin demonstrated an increase in baseline and histamine-induced permeability, while leukocyte extravasation was reduced [49]. The increase in permeability was due to reduced levels of Rap1, while the extravasation defect was due to impaired ICAM-1 clustering (hence reduced PMN adhesion to endothelium) in the cortactin-deficient mice [49]. Different tyrosine residues on the cytoplasmic tail of VE-cadherin are responsible for its role in preventing vascular permeability and TEM [23]. VE-cadherin mutant mice were knocked in to a VE-cadherin-deficient mice. Those expressing the Y685F mutation were defective in the induction of vascular permeability, but displayed normal leukocyte extravasation; those expressing the Y731F mutation were defective in neutrophil extravasation but not in their response to permeability-inducing agents [23].
Re-evaluation of some previously published papers may shed some light on this. Most studies of TEM in vitro use leukocyte:EC ratios of 1–3:1. Under these conditions, there is efficient TEM without a change in endothelial barrier function [38,39]. The experiments that demonstrated activation of MLCK and increase in isometric tension in endothelial cells [31] used PMN: endothelial cell ratios of 10–12:1. These ratios may have been necessary to stimulate a large enough phosphorylation of MLCK to measure; however, they may have transformed a local signal into a global one [38] (and Muller, unpublished data). The study by Schulte, et al. [40] show that opening of the adherens junctions is required for response to soluble permeabilizing agents, and for efficient TEM, but not for all TEM. Modulation of the adherens junction involving VE-cadherin tyrosine residue 731 is necessary for efficient TEM to occur in vitro [22] and in vivo [23]. However, the mechanisms for breaking down adherens junctions for increasing permeability and modulating VE-cadherin permitting TEM may be different, even if they involved some of the same molecules. In retrospect, this should not come as a surprise. TEM is a local phenomenon involving multiple signals between leukocytes and endothelial cells [1,2]. Permeability in response to most soluble mediators is a global response via GPCRs [30].
Conclusions
Our understanding of the process of TEM is rapidly evolving. We know several molecules involved in this process and how they signal to carry out their roles. There may well be other steps in the diapedesis process regulated by yet-to-be-discovered mechanisms. Following TEM, the leukocyte crawls between the abluminal side of the endothelial cell and the basal lamina, often along pericytes [50] seeking areas of low expression of select matrix molecules across which to migrate into the interstitium [51]. The ventral side of the endothelium remodels after the leukocyte has finished TEM [52]. These steps are equally important and complex in their own right, but must be left for the subjects of other reviews.
There is a growing appreciation that signaling during TEM is local. Opening of junctions during TEM uses different mechanisms than global stimulation of the endothelial permeability in response to soluble mediators. We still know very little about local bidirectional signals between leukocyte and endothelium during TEM. These intimate interactions may play a key role in regulating TEM, and represent a challenge for future work in this field.
Highlights.
Transendothelial migration (TEM) itself is a multi-step process.
TEM requires trafficking of the LBRC to, and removal of VE-cadherin from, the site
PECAM signals via TRPC6 to generate the ↑[Ca+2]i required for TEM.
CD99 in a complex with ezrin, sAC, and PKA signals a subsequent step in TEM
Junctional modeling during TEM is regulated differently than vascular permeability
Acknowledgements
I wish to thank Drs. Jaap van Buul, Ronen Alon, and Dietmar Vestweber for helpful and insightful discussions; and Dr. David Sullivan for making the figures. This work was supported by NIH grants R01 HL046849 and R37 HL064774.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests Statement
The author has no conflicts of interest to declare.
REFERENCES
- 1.Muller WA. Mechanisms of leukocyte transendothelial migration. Annu. Rev. Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller WA. The regulation of transendothelial migration: new knowledge and new questions. Cardiovasc Res. 2015 doi: 10.1093/cvr/cvv145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 4.van Buul JD, Kanters E, Hordijk PL. Endothelial signaling by Ig-like cell adhesion molecules. Arterioscler Thromb Vasc Biol. 2007;27:1870–1876. doi: 10.1161/ATVBAHA.107.145821. [DOI] [PubMed] [Google Scholar]
- 5.Fernandez-Borja M, van Buul JD, Hordijk PL. The regulation of leucocyte transendothelial migration by endothelial signalling events. Cardiovasc Res. 2010;86:202–210. doi: 10.1093/cvr/cvq003. [DOI] [PubMed] [Google Scholar]
- 6.Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. 2010;11:366–378. doi: 10.1038/nrm2889. [DOI] [PubMed] [Google Scholar]
- 7.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 8.Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 9.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;178:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Muller WA. PECAM: Regulating the start of diapedesis. In: Ley K, editor. Adhesion Molecules: Function and Inhibition. Birkhauser Verlag AG; 2007. pp. 201–220. [Parnham MJ (Series Editor): Progress in Inflammation Research. [Google Scholar]
- 11.Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA. Targeted recycling of PECAM from endothelial cell surface-connected compartments during diapedesis. Nature. 2003;421:748–753. doi: 10.1038/nature01300. [DOI] [PubMed] [Google Scholar]
- 12.Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. 2004;5:393–400. doi: 10.1038/ni1051. [DOI] [PubMed] [Google Scholar]
- 13.Mamdouh Z, Mikhailov A, Muller WA. Transcellular migration of leukocytes is mediated by the endothelial lateral border recycling compartment. J. Exp. Med. 2009;206:2795–2808. doi: 10.1084/jem.20082745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sullivan DP, Ruffer C, Muller WA. Isolation of the lateral border recycling compartment using a diaminobenzidine-induced density shift. Traffic. 2014;15:1016–1029. doi: 10.1111/tra.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng G, Sullivan DP, Han F, Muller WA. Segregation of VE-cadherin from the LBRC depends on the ectodomain sequence required for homophilic adhesion. Journal of cell science. 2015;128:576–588. doi: 10.1242/jcs.159053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mamdouh Z, Kreitzer GE, Muller WA. Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J. Exp. Med. 2008;205:951–966. doi: 10.1084/jem.20072328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan DP, Muller WA. Neutrophil and monocyte recruitment by PECAM, CD99, and other molecules via the LBRC. Seminars in immunopathology. 2014;36:193–209. doi: 10.1007/s00281-013-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18••. Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the Endothelial Calcium Channel That Regulates Leukocyte Transendothelial Migration During the Inflammatory Response. J. Exp. Med. 2015 Oct;212 doi: 10.1084/jem.20150353. This paper shows that the long-sought source of endothelial cell cytosolic free calcium required for transmigration is TRPC6, activated downstream of PECAM.
- 19••. Watson RL, Buck J, Levin LR, Winger RC, Wang J, Arase H, Muller WA. Endothelial CD99 signals through soluble adenylyl cyclase and PKA to regulate leukocyte transendothelial migration. The Journal of experimental medicine. 2015;212:1021–1041. doi: 10.1084/jem.20150354. This paper identifies how a complex of CD99, ezrin, soluble adenylate cyclase and PKA provide the signal that is critical for continued recruitment of the LBRC.
- 20.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J.Cell Biol. 1993;120:1371–1380. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaw SK, Bamba PS, Perkins BN, Luscinskas FW. Real-time imaging of vascular endothelial-cadherin during leukocyte transmigration across endothelium. J Immunol. 2001;167:2323–2330. doi: 10.4049/jimmunol.167.4.2323. [DOI] [PubMed] [Google Scholar]
- 22.Allingham MJ, van Buul JD, Burridge K. ICAM-1-mediated, Src- and Pyk2-dependent vascular endothelial cadherin tyrosine phosphorylation is required for leukocyte transendothelial migration. J Immunol. 2007;179:4053–4064. doi: 10.4049/jimmunol.179.6.4053. [DOI] [PubMed] [Google Scholar]
- 23••. Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nature immunology. 2014;15:223–230. doi: 10.1038/ni.2824. This paper uses mice in which specific tyrosine residuse of VE-cadherin are mutated to phenylalanine to show that Y685 is required for vascular permeability while Y731 is required for efficient TEM.
- 24.Alcaide P, Newton G, Auerbach S, Sehrawat S, Mayadas TN, Golan DE, Yacono P, Vincent P, Kowalczyk A, Luscinskas FW. p120-Catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood. 2008;112:2770–2779. doi: 10.1182/blood-2008-03-147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sullivan DP, Seidman MA, Muller WA. Poliovirus receptor (CD155) regulates a step in transendothelial migration between PECAM and CD99. Am. J. Pathol. 2013;182:1031–1042. doi: 10.1016/j.ajpath.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat. Immunol. 2002;3:143–150. doi: 10.1038/ni749. [DOI] [PubMed] [Google Scholar]
- 27.Suh YH, Shin YK, Kook MC, Oh KI, Park WS, Kim SH, Lee IS, Park HJ, Huh TL, Park SH. Cloning, genomic organization, alternative transcripts and expression analysis of CD99L2, a novel paralog of human CD99, and identification of evolutionary conserved motifs. Gene. 2003;307:63–76. doi: 10.1016/s0378-1119(03)00401-3. [DOI] [PubMed] [Google Scholar]
- 28.Lou O, Alcaide P, Luscinskas FW, Muller WA. CD99 is a key mediator of the transendothelial migration of neutrophils. J. Immunol. 2007;178:1136–1143. doi: 10.4049/jimmunol.178.2.1136. [DOI] [PubMed] [Google Scholar]
- 29.Reymond N, Imbert AM, Devilard E, Fabre S, Chabannon C, Xerri L, Farnarier C, Cantoni C, Bottino C, Moretta A, et al. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J Exp Med. 2004;199:1331–1341. doi: 10.1084/jem.20032206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 31.Hixenbaugh EA, Goeckeler ZM, Papaiya NN, Wysolmerski RB, Silverstein SC, Huang AJ. Chemoattractant-stimulated neutrophils induce regulatory myosin light chain phosphorylation and isometric tension development in endothelial cells. American Journal of Physiology. 1997;273:H981–H988. doi: 10.1152/ajpheart.1997.273.2.H981. [DOI] [PubMed] [Google Scholar]
- 32.Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO. ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol. 2000;165:3375–3383. doi: 10.4049/jimmunol.165.6.3375. [DOI] [PubMed] [Google Scholar]
- 33.Lorenzon P, Vecile E, Nardon E, Ferrero E, Harlan JM, Tedesco F, Dobrina A. Endothelial cell E-and P-selectin and vascular cell adhesion molecule-1 function as signaling receptors. J. Cell Biol. 1998;142:1381–1391. doi: 10.1083/jcb.142.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cernuda-Morollon E, Ridley AJ. Rho GTPases and leukocyte adhesion receptor expression and function in endothelial cells. Circ Res. 2006;98:757–767. doi: 10.1161/01.RES.0000210579.35304.d3. [DOI] [PubMed] [Google Scholar]
- 35.Lessey-Morillon EC, Osborne LD, Monaghan-Benson E, Guilluy C, O'Brien ET, Superfine R, Burridge K. The RhoA guanine nucleotide exchange factor, LARG, mediates ICAM-1-dependent mechanotransduction in endothelial cells to stimulate transendothelial migration. Journal of immunology. 2014;192:3390–3398. doi: 10.4049/jimmunol.1302525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Nieuw Amerongen GP, van Delft S, Vermeer MA, Collard JG, van Hinsbergh VW. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 37.Saito H, Minamiya Y, Kitamura M, Saito S, Enomoto K, Terada K, Ogawa J. Endothelial myosin light chain kinase regulates neutrophil migration across human umbilical vein endothelial cell monolayer. J. Immunol. 1998;161:1533–1540. [PubMed] [Google Scholar]
- 38.Huang AJ, Furie MB, Nicholson SC, Fischbarg J, Liebovitch LS, Silverstein SC. Effects of human neutrophil chemotaxis across human endothelial cell monolayers on the permeability of these monolayers to ions and macromolecules. J. Cell. Physiol. 1988;135:355–366. doi: 10.1002/jcp.1041350302. [DOI] [PubMed] [Google Scholar]
- 39.Winger RC, Koblinski JE, Kanda T, Ransohoff RM, Muller WA. Rapid Remodeling of Tight Junctions during Paracellular Diapedesis in a Human Model of the Blood-Brain Barrier. Journal of immunology. 2014;193:2427–2437. doi: 10.4049/jimmunol.1400700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•. Schulte D, Kuppers V, Dartsch N, Broermann A, Li H, Zarbock A, Kamenyeva O, Kiefer F, Khandoga A, Massberg S, et al. Stabilizing the VE-cadherin-catenin complex blocks leukocyte extravasation and vascular permeability. EMBO J. 2011;30:4157–4170. doi: 10.1038/emboj.2011.304. Mice with a chimeric VE-cadherin/α-catenin that remains bound to the actin cytoskeletin show a dramatic reduction in vascular permeability and leukocyte extravasation.
- 41.Vogelsgesang M, Pautsch A, Aktories K. C3 exoenzymes, novel insights into structure and action of Rho-ADP-ribosylating toxins. Naunyn Schmiedebergs Arch Pharmacol. 2007;374:347–360. doi: 10.1007/s00210-006-0113-y. [DOI] [PubMed] [Google Scholar]
- 42.Zaccolo M. cAMP signal transudction in the heart: understanding spatial control for the development of novel therapeutic strategies. Br J Pharmacol. 2009;158:50–60. doi: 10.1111/j.1476-5381.2009.00185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baluk P, Bolton P, Hirata A, Thurston G, McDonald DM. Endothelial gaps and adherent leukocytes in allergen-induced early- and late-phase plasma leakage in rat airways. Am J Pathol. 1998;152:1463–1476. [PMC free article] [PubMed] [Google Scholar]
- 44.McDonald DM. Endothelial gaps and permeability of venules in rat tracheas exposed to inflammatory stimuli. Am J Physiol. 1994;266:L61–L83. doi: 10.1152/ajplung.1994.266.1.L61. [DOI] [PubMed] [Google Scholar]
- 45.Brown M, Adyshev D, Bindokas V, Moitra J, Garcia JG, Dudek SM. Quantitative distribution and colocalization of non-muscle myosin light chain kinase isoforms and cortactin in human lung endothelium. Microvasc Res. 2010;80:75–88. doi: 10.1016/j.mvr.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dudek SM, Jacobson JR, Chiang ET, Birukov KG, Wang P, Zhan X, Garcia JG. Pulmonary endothelial cell barrier enhancement by sphingosine 1-phosphate: roles for cortactin and myosin light chain kinase. J Biol Chem. 2004;279:24692–24700. doi: 10.1074/jbc.M313969200. [DOI] [PubMed] [Google Scholar]
- 47.Cook-Mills JM, Johnson JD, Deem TL, Ochi A, Wang L, Zheng Y. Calcium mobilization and Rac1 activation are required for VCAM-1 (vascular cell adhesion molecule-1) stimulation of NADPH oxidase activity. Biochem J. 2004;378:539–547. doi: 10.1042/BJ20030794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48•. Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood. 2013;122:2512–2522. doi: 10.1182/blood-2013-04-499228. This study extends the work of ref. 47 and shows that signals downstream of VE-cadherin lead to dissociation of VE-PTP from VE-cadherin, locally destabilizing the junction.
- 49••. Schnoor M, Lai FP, Zarbock A, Klaver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–1735. doi: 10.1084/jem.20101920. This study clearly shows that vascular permeability and transendothelial migration, although sharing common molecular elements, are distinct and potentially independent phenomena.
- 50.Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–1234. doi: 10.1084/jem.20111622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang S, Voisin MB, Larbi KY, Dangerfield J, Scheiermann C, Tran M, Maxwell PH, Sorokin L, Nourshargh S. Venular basement membranes contain specific matrix protein low expression regions that act as exit points for emigrating neutrophils. J Exp Med. 2006;203:1519–1532. doi: 10.1084/jem.20051210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, Toporsian M, Dvorak AM, Kirchhausen T, Springer TA, et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. The Journal of cell biology. 2013;201:449–465. doi: 10.1083/jcb.201209077. [DOI] [PMC free article] [PubMed] [Google Scholar]