

Abstract
Purpose
Prostate cancers incite tremendous morbidity upon metastatic growth. We previously identified Asporin (ASPN) as a potential mediator of metastatic progression found within the tumor microenvironment. ASPN contains an aspartic acid (D)-repeat domain and germline polymorphisms in D-repeat-length have been associated with degenerative diseases. Associations of germline ASPN D polymorphisms with risk of prostate cancer progression to metastatic disease have not been assessed.
Experimental Design
Germline ASPN D-repeat-length was retrospectively analyzed in 1600 men who underwent radical prostatectomy for clinically localized prostate cancer and in 548 non-cancer controls. Multivariable Cox proportional hazards models were used to test the associations of ASPN variations with risk of subsequent oncologic outcomes including metastasis. Orthotopic xenografts were used to establish allele- and stroma-specific roles for ASPN D variants in metastatic prostate cancer.
Results
Variation at the ASPN D locus was differentially associated with poorer oncologic outcomes. ASPN D14 (HR=1.72, 95%CI=1.05–2.81, P=0.032) and heterozygosity for ASPN D13/14 (HR=1.86, 95%CI=1.03–3.35, P=0.040) were significantly associated with metastatic recurrence, while homozygosity for the ASPN D13 variant was significantly associated with a reduced risk of metastatic recurrence (HR=0.44, 95%CI=0.21–0.94, P=0.035) in multivariable analyses. Orthotopic xenografts established biologic roles for ASPN D14 and ASPN D13 variants in metastatic prostate cancer progression that were consistent with patient based data.
Conclusions
We observed associations between ASPN D variants and oncologic outcomes including metastasis. Our data suggest that ASPN expressed in the tumor microenvironment is a heritable modulator of metastatic progression.
Keywords: Asporin, Germline Variants, Metastasis, Tumor Microenvironment, Prostate
Introduction
The burden of prostate cancer-related mortality remains significant due to the subset of prostate cancers that progress to metastatic disease (1). Identification and differentiation of patients with aggressive cancer from those with indolent disease remains a key issue in the management of clinically localized prostate cancer. To date most biomarker studies have focused on carcinoma specific features predictive of aggressive disease. As the tumor microenvironment influences prostate cancer progression (2), we hypothesized that molecules in the primary tumor microenvironment secreted by cancer associated fibroblasts could modulate tumor progression and predict metastatic potential. We previously reported that Asporin (ASPN), a member of a small leucine-rich proteoglycan (SLRP) family of extracellular proteins, was one of the most highly enriched androgen-induced mesenchymal genes during both physiologic prostate development and pathologic growth in the prostate (3). ASPN expression has been shown to be elevated in prostate cancer associated fibroblasts (PCAFs) (4), in blood from men with advanced prostate cancer (5), and in scirrhous gastric cancer associated stroma (6). A recent study supports that ASPN induces coordinated fibroblast and cancer cell invasion through CD44 mediated RAC1 activation (6); however, a biologic role for ASPN in the prostate cancer microenvironment is not well understood.
In addition to expression, a genetic association exists between variation at the ASPN locus and multiple disease states (7–9). ASPN, located at 9q22.31, contains an aspartic acid (D)-repeat domain and over 10 polymorphisms in the length of the D-repeat have been identified with the most common variants ranging from 12–16 aspartate residues (7). Polymorphisms in the length of this allelic repeat have been associated with susceptibility to and severity of several bone and joint diseases including osteoarthritis (7–9). While functional differences among these polymorphisms are not fully known, polymorphisms in the ASPN D-repeat-length have been shown to differentially inhibit Bone Morphogenetic Protein 2 (BMP-2) signaling (10) and Transforming Growth Factor Beta 1 (TGFβ1)-induced gene expression (7). Intriguingly, the loss of TGFβresponsiveness in prostate stromal cells has been shown to promote prostate cancer initiation (11) and the development of bone lesions (12). Due to its stromal specific expression and the differential role of ASPN D variants in TGFβ signaling, we hypothesized that ASPN D-repeat-length variants may differentially modulate metastatic progression in the prostate.
Herein, we genotyped germline ASPN D-repeat-lengths in men with clinically localized prostate cancer and in non-cancer controls to determine if germline ASPN D variants were differentially associated with risk of prostate cancer incidence or progression to metastatic disease. We utilized an orthotopic xenograft model to establish allele- and stroma-specific roles for these variants in facilitating prostate cancer metastasis.
Materials and Methods
JHH Germline Study Population
We retrospectively analyzed a database of 19,142 men who had undergone radical prostatectomy (RP) and pelvic lymphadenectomy at Johns Hopkins Hospital (JHH) since 1992 (PSA era). Germline DNA was available for approximately 10,000 patients. We selected 1672 cases for genomic analysis that were weighted for approximately equal Gleason grades and to generate approximately equal white and non-white race groups, of which 1600 had adequate DNA for germline genotyping. 55.6% men self-reported Caucasian, 43.6% self-reported African American, and 0.8% self-reported other ethnicity or were of unknown ethnicity. We retrospectively analyzed 192 self-reported Caucasian and 370 self-reported African American male controls with no cancer, of which 179 and 369, respectively, had adequate germline DNA for genotyping. All participants provided written informed consent. The protocol and consent documents were approved by the Johns Hopkins University (JHU) Institutional Review Board (IRB).
RP specimens were processed as previously described (13). Each tumor was graded using the Gleason scoring system and staged using the TNM (tumor-node-metastasis) system. Clinical outcome data included biochemical recurrence (BCR) (defined as a post-operative Prostate Specific Antigen [PSA] ≥0.2 ng/ml), distant metastasis (defined as post-operative clinical or radiographic spread of disease to extra-pelvic lymph nodes, bones, or viscera), and prostate-cancer specific mortality (PCSM). Outcome or time to outcome data was not available for all men in the cohort; any resulting differences in sample size number were recorded in the results section.
Genomic DNA isolation
Tissue for genomic DNA isolation was taken from seminal vesicles (SV) at the time of RP (cases) or from blood of men without prostate cancer diagnoses (controls). SV tissue was suspended in 12ml Suspension buffer (20mM Tris; 25mM EDTA; 100mM NaCl) + 1ml 10% SDS + 60µl Proteinase K solution (20mg/mL), inverted twice, and then incubated overnight at 50°C. The next day, RNA was digested by adding 60µl RNase A Solution (Qiagen) and incubating at 37°C for 1 hour. Proteins were precipitated in 4ml Protein Precipitation Solution (Promega) on ice for 20 minutes and then centrifuged at 2,000g for 10 minutes. DNA was extracted from the supernatant with 30ml 100% Isopropanol, centrifuged 2,000g for 5 minutes, and then washed with 70% ethanol followed by centrifugation.
Genotyping of the D-repeat polymorphism
The D-repeat polymorphism located in the N-terminal region of the ASPN gene was PCR amplified using 5’ primer 6-FAM-ATTCCTGGCTTTGTGCTCTG and 3’ primer TGGCTTCTTGGCTCTCTTGT. Primers were designed using Oligo software. Reactions were carried out in 10µL consisting of 30ng DNA, 0.125µM primers, 0.6mM dNTPs (Continental Lab Products), 10mM Tris-HCl pH8.3, 50mM KCL, 1.5mM MgCl, and 0.6units of Taq Gold DNA polymerase (Perkin Elmer). Amplification was performed in a Veriti Thermal Cycler (Applied Biosystems Inc.) for an initial denaturation of 12 minutes at 94°C followed by 40 cycles of 94°C for 20 seconds, 58°C for 20 seconds, 72°C for 30 seconds, and a final 10 minute elongation at 72°C. The PCR products were electrophoresed on an ABI 3730×l DNA Analyzer (Applied Biosystems Inc.). Data was collected and analyzed with GeneMapper software (Applied Biosystems Inc.) that calculates fragment length in reference to an internal lane standard (LIZ500). Three homozygous samples of different repeat-length were confirmed by Sanger sequencing. PCR products were sequenced using fluorescent dideoxy terminator method of cycle sequencing. Reactions were run on a 3730×l DNA Analyzer (Applied Biosystems Division) following Applied Biosystems protocols. Sequence data was analyzed using Sequencher Software (Gene Codes).
Statistical analysis
Characteristics of patient groups defined by distinct ASPN variations were compared. Exploratory data visualization was used to identify departure from normality. Means of continuous variables were compared by t-tests. Non-normally distributed variables were compared by Wilcoxon-Mann-Whitney rank-sum tests. Chi-squared tests were used to examine the association of ASPN variants with biopsy and pathologic Gleason sum. In lieu of univariate models to test strengths of association, bivariate adjusting for race were performed due to clinical and genomic race based differences in the analyzed cohort. Bivariate and multivariable logistic regression analysis was performed in order to test strengths of association with ASPN variants and pathologic outcomes: extraprostatic extension (EPE), seminal vesicle invasion (SVI), and lymph node involvement (LNI). Similarly, bivariate and multivariable Cox proportional hazards models were used to test the associations of ASPN variations with risk of subsequent oncologic outcomes: BCR and metastasis. This study was under-powered to examine PCSM. For multivariable analyses Gleason sum was categorized as ≤7 or ≥8 and clinical stage was categorized as non-palpable (T1a, T1b, T1c) vs. palpable (T2a, T2b, T2c, T3). Metastasis free survival estimates were derived from Kaplan-Meier life tables with surgery at the zero starting point. Statistics were computed with Stata 11.0 (StataCorp).
Immunohistochemistry, Immunofluorescence and Immunoblotting
For immunohistochemistry (IHC) and immunofluorescence (IF), formalin fixed paraffin embedded (FFPE) tissues were sectioned, deparaffinized, steamed in Target Retrieval Solution Ready to Use (Dako) for 40 minutes, blocked with Protein Block Serum-Free (Dako), incubated with antibodies against ASPN (Sigma Prestige HPA008425; 1:400) and pancytokeratin (Sigma C2562; 1:400) in Antibody Diluent (Invitrogen). Sigma Prestige HPA008425 ASPN antibody was validated for immunohistochemistry and immunoblotting in accordance with JHU pathology TMA criteria (Supplementary Fig. S1A–E). For IHC, primary antibodies were followed by secondary antibodies (Vector Laboratories) and then detected with 3,3’-Diaminobenzidine kit (Vector Laboratories). For IF, primary antibodies were followed by Alexa Fluor Dye secondary antibodies (Invitrogen) and mounted with Vectashield hard-set mounting medium with DAPI counterstain (Vector Laboratories). Images were captured at room temperature on a Nikon E800 fluorescence microscope with 40× Plan Apo objective and a Nikon DS-QiMc camera with Nikon Elements imaging software (vAR 3.0).
For immunoblotting, lysates were fractionated on NuPAGE gels (Invitrogen). Proteins were transferred to poly-vinylidene difluoride membranes, blocked, and then incubated with primary antibodies. Antibodies used included: ASPN (Sigma Prestige HPA008425; 1:1000) and GAPDH (Santa Cruz sc-32233; 1:10,000). Blots were developed using enhanced chemiluminescence (Thermo Fisher Scientific).
Quantitative Real Time PCR
Total RNA was purified using RNeasy Minikit Plus (Qiagen). First-strand cDNA was synthesized using random hexamer primers (Applied Biosystems) and Ready-To-Go You-Prime First-Strand Beads (GE Healthcare), according to the manufacturers’ instructions. Quantitative PCR was performed using TaqMan Universal PCR Master Mix (Applied Biosystems) with TaqMan primers specific to human or to mouse ASPN (Applied Biosystems). Applied Biosystems software was used to calculate threshold cycle values for ASPN and the reference genes HPRT (Applied Biosystems) and GAPDH (Applied Biosystems).
JHU and Harvard University Tissue Microarrays
JHU Gleason grade tissue microarrays (TMAs) were constructed from archival tissue from RP performed at JHU between 2000 and 2001. Cases for the TMAs were reviewed and selected by a genitourinary pathologist. The index lesion was defined as the largest tumor of the highest grade. In each case, both benign adjacent glands and the index tumors of Gleason sum 5, 6, 7, 8, and 9 were spotted in triplicate.
The Harvard Health Professional Follow-Up Study TMAs have been described previously (14). TMAs were constructed from archival FFPE prostate tissue from men treated with RP for prostate cancer who were participants in the HPFS, and included 715 evaluable prostate cancer cases, diagnosed between 1983 and 2004. The mean follow-up time from diagnosis was 13 years. Tumor tissue from RP was reviewed by multiple pathologists to provide uniform evaluation of Gleason score and to identify areas of high-density tumor for construction of TMAs. At least three tumor cores were sampled from each case from the index lesion, which was defined as the largest tumor of the highest grade.
Four µm sections were stained for ASPN by IHC. Cases were scored by urologic pathologists for ASPN expression. Using established scoring schemes, ASPN intensity was evaluated and assigned an incremental score of 0 (negative), 1 (weak), 2 (moderate), or 3 (strong) (15). For the JHU Gleason grade TMAs, the extent of staining was assigned a score for 0–25% (score 0), 26–50% (score 1), 51–75% (score 2) and 76–100% (score 3). For each sample, an ASPN score was calculated by adding the intensity score and the extent score (H-score). For the Harvard HPFS TMAs, the extent of staining was assigned a percentage from 0–100%. For each sample, an ASPN score was calculated by multiplying the intensity score and the extent score (H-score). H-scores were compared using one-way ANOVA with pairwise comparisons. Statistical tests performed using GraphPad Software were two sided and P values less than 0.05 were considered statistically significant.
Mayo Clinic Progression Analyses
Methods including tissue preparation, RNA extraction, microarray hybridization and microarray expression analysis have been previously described (15, 16).
Study Design
Patients were selected from a cohort of high-risk RP patients from the Mayo Clinic with a median follow-up of 8.1 years. The cohort was comprised of 1010 high-risk men that underwent RP between 2000–2006, of which 73 patients developed metastatic disease as evidenced by positive bone or CT scan. High-risk was defined as pre-operative PSA >20ng/ml, pathological Gleason score 8–10, SVI, or GPSM score ≥10. The sub-cohort incorporated all 73 metastatic patients and a 20% random sampling of the entire cohort.
Statistical Analysis
The summarized expression of core transcript cluster, 3214845, was used to represent the expression of ASPN. PAM (Partition Around Medoids) unsupervised clustering method was used on the expression values of all clinical samples to define two groups of high and low expression of ASPN. Statistical analysis on the association of ASPN with clinical outcomes was done using two endpoints: BCR, defined as two consecutive PSA values ≥0.2ng/ml after RP and Metastasis (MET), as defined by a positive bone scan and/or CT/MRI evidence of metastatic disease.
Stable Cell Lines
Quick Change Site directed mutagenesis kit (Agilent Technologies) was used to generate ASPN D13 and ASPN D14 cDNAs from parent human ASPN D17 cDNA (Origene). ASPN D13 and ASPN D14 were subcloned into pIRESNeo3 (Clontech). Stable cell lines were generated through transfection with Fugene (Roche) of cDNAs into WPMY1 (ATCC low passage) according to the manufacturer’s instruction, plating at limiting dilutions, and isolating single clones. WPMY1 cells were grown in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen) and PS (Invitrogen).
Orthotopic Xenografts
All animal experiments were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University. 500,000 WPMY1, WPMY1 ASPN D13 (A), and WPMY1 ASPN D14 (A) cells were mixed with 500,000 PC3 cells (authenticated at JHU in September, 2014) in Matrigel (BD Biosciences) at a 1:1 ratio. Cells were grafted into the prostate of male NOD scid gamma (NSG) mice (JHU) that were 10 weeks in age. 7 mice were used for each group. An isoflurane gas anesthetic system (Caliper Life Sciences) was used for surgical anesthesia. Mice were induced by inhalation of 2% isoflurane (Oncology Animal Resources) in oxygen and then maintained on 1.5% isoflurane with adjustment to maintain a deep plane of anesthesia. A midline incision was made in the lower abdomen to externalize the seminal vesicles, bladder, and prostate. Following injection of 20µl of cells 1:1 in Matrigel into the anterior lobe of the prostate, the wound was closed with surgical sutures and surgical metal clips. Postoperatively, mice were administered 5mg/kg Ketoprofen subcutaneously and then monitored daily for distress. One mouse with a WPMY1 ASPN D13/PC3 orthotopic xenograft was euthanized at day 19 due to an unrelated condition, and was excluded from the final analyses. The experiment was terminated on day 59 due to jaundice in three of the mice with WPMY1 ASPN D14/PC3 orthotopic xenografts. Jaundice was not detected in the other mice. Mice were euthanized, necropsied, and the following organs including any associated mesentery and lymph nodes were removed from each mouse and inspected for gross evidence of metastatic disease in a blinded fashion: salivary gland, thymus, heart, lung, liver, gall bladder, stomach, pancreas, intestine, spleen, kidneys, prostate, seminal vesicles, testes, and bladder. Metastases were quantified by counting the number of visible metastases for each mouse in the above organs and associated distant lymph nodes. The prostate regional lymph nodes were not included in the analyses examining distant metastases (distant lymph nodes and other organs). Following visual quantification of evidence of metastatic disease, organs were formalin-fixed, sectioned, paraffin embedded, and then sectioned using standard methods. One H&E stained slide of every dissected organ was examined for confirmation of metastatic disease. Micrometastatic disease observed on H&E was not include in the analyses. Orthotopic xenografts were repeated with independent WPMY1 ASPN D13 (B) and WPMY1 ASPN D14 (B) clones. The experiment was terminated on day 61 due to jaundice in one mouse with a WPMY1 ASPN D14/PC3 orthotopic xenograft. One mouse with a WPMY1/PC3 orthotopic xenograft from experiment B was excluded from final analyses due to cell leakage into the body cavity as evidenced by over 30 metastatic lesions on the body cavity wall.
Results
Germline ASPN D-Repeat-Length Variants and Adverse Prostate Cancer Clinical Outcomes
To determine if ASPN D-repeat-length correlates with prostate cancer progression to metastasis, ASPN D-repeat-length was examined in 1600 men who underwent RP for clinically localized prostate cancer at JHH (Table 1). Men in this study had ASPN D-repeat-lengths ranging from 10 to 19 aspartate residues with most (approximately 90%) of men having alleles containing 13, 14, 15, or 16 residues (Supplementary Fig. S2). The most common ASPN D-length genotypes in JHH men with prostate cancer were (where the integer corresponds to the number of aspartate-coding repeats in a single allele of ASPN): 13/15, 13/13, 15/15, 14/15 and 13/14 with the remaining genotypes between 0.1% and 5.8% of the study population (Supplementary Table S1). To allow for adequately-powered comparative tests, genotypes present in greater than 10% of the population were selected for comparison to all other genotypes in the study population. Furthermore, single ASPN D-repeat-length alleles (either homozygous or heterozygous) present in greater than 10% of the population were also analyzed: Any 13, Any 14, and Any 15. Similar to prior reports (7, 9, 17, 18), the distribution of ASPN D-repeat-length varied with ethnicity (Supplementary Fig. S2 and Supplementary Table S1), and thus all analyses were either matched or controlled for race.
Table 1.
Baseline Characteristics and Cancer Outcomes of JHH Prostate Cancer Cases.
| N | 1600 |
| Median age (years) (IQR) | 58 (53, 63) |
| Caucasian | 889 (55.6%) |
| African-American | 697 (43.6%) |
| Other or Unknown | 14 (0.8%) |
| Median PSA (ng/ml) (IQR) | 5.59 (4.2, 8.3) |
| Clinical stage | |
| T1 | 1118 (69.9%) |
| T2 | 427 (26.7%) |
| T3 | 13 (0.8%) |
| unknown | 42 (2.6%) |
| Biopsy Gleason | |
| ≤6 | 814 (51.0%) |
| 7 (3+4) | 305 (19.1%) |
| 7 (4+3) | 215 (13.5%) |
| 8 | 171 (10.7%) |
| 9–10 | 91 (5.7%) |
| Pathologic Stage | |
| pT2N0 | 921 (57.8%) |
| pT3aN0 | 461 (28.9%) |
| pT3bN0 | 137 (8.6%) |
| pN1 | 75 (4.7%) |
| Pathologic Gleason | |
| ≤6 | 556 (34.9%) |
| 7 (3+4) | 366 (22.9%) |
| 7 (4+3) | 153 (9.6%) |
| 8 | 273 (17.1%) |
| 9–10 | 248 (15.5%) |
| Positive surgical margin | 294 (18.5%) |
| Median Follow-up (years) (IQR) | 3.0 (2.0, 6.0) |
| Biochemical Recurrence (BCR) | 269/1115 (24.1%) |
| Median time to BCR (years) (IQR) | 3.0 (1.0, 5.0) |
| Metastasis (Met) | 83/1093 (7.6%) |
| Median time to Met (years) (IQR) | 3.0 (2.0, 6.0) |
| Prostate cancer specific mortality (PCSM) | 37/1133 (3.3%) |
| Median time to PCSM (years) (IQR) | 6.0 (3.0, 10.0) |
Germline ASPN D-repeat-length variants and adverse prostate cancer clinical outcomes
We examined the relationship between ASPN-D-repeat length and metastatic disease progression following local therapy for prostate cancer. Through Cox regression analyses, we found that germline ASPN D13/14 was significantly associated with metastatic recurrence following surgery (HR=2.02, 95%CI=1.16–3.53, P=0.013) even when adjusted for pre-operative variables and race (HR=1.86, 95%CI=1.03–3.35, P=0.040) (Tables 2 and 3). Consistent with this, Kaplan-Meier survival estimates demonstrated that heterozygous ASPN D13/14 was a significant germline prognostic marker for worse metastasis free survival (Fig. 1). In addition, germline carriers of the ASPN D14 allele were significantly more likely to progress to metastatic disease by multivariable analyses compared to all other alleles (HR=1.72, 95%CI=1.05–2.81, P=0.032; Tables 2 and 3). These data suggest that germline ASPN D13/14 or any ASPN D14 is a risk factor for metastatic recurrence following surgery.
Table 2.
Predictors of Metastatic Recurrence: Bivariate Cox Regression Analyses Adjusted for Race of the Most Common Germline ASPN D Genotypes/Alleles Compared to All Other Genotypes/Alleles.
| Genotype/Allele | Metastatic Recurrence (n=1082) |
||
|---|---|---|---|
| HR | 95% Cl | P | |
| 13/13 | 0.51 | 0.24 to 1.07 | 0.073 |
| 13/14 | 2.02 | 1.16 to 3.53 | 0.013 |
| 13/15 | 0.99 | 0.55 to 1.78 | 0.982 |
| 14/15 | 1.20 | 0.54 to 2.65 | 0.656 |
| 15/15 | 0.45 | 0.14 to 1.46 | 0.186 |
| Any 13 | 1.28 | 0.76 to 2.16 | 0.344 |
| Any 14 | 1.61 | 1.00 to 2.58 | 0.049 |
| Any 15 | 0.82 | 0.51 to 1.32 | 0.417 |
NOTE. Boldfaced values represent statistically significant HRs.
Abbreviations: HR, Hazard Ratio; Cl, Confidence Interval.
Table 3.
Predictors of Metastatic Recurrence: Multivariable Cox Regression Analysis of Germline ASPN D13/14, ASPN D14, and ASPN D13/13 Compared to All Other Genotypes/Alleles.
| Characteristic | Metastatic Recurrence | |||
|---|---|---|---|---|
| Multivariable§ (n=1032) | ||||
| HR | 95% Cl | P | RAF | |
| 13/14 | 1.86 | 1.03 to 3.35 | 0.040 | 0.10 |
| Any 14 | 1.72 | 1.05 to 2.81 | 0.032 | 0.29 |
| 13/13 | 0.44 | 0.21 to 0.94 | 0.035 | 0.14 |
| Biopsy Gleason Sum | 3.47 | 2.07 to 5.81 | <0.001 | -- |
| Clinical Stage | 4.02 | 2.35 to 6.90 | <0.001 | -- |
| PSA | 1.05 | 1.03 to 1.08 | <0.001 | -- |
| Age | 1.01 | 0.97 to 1.05 | 0.528 | -- |
| Gland Volume | 1.01 | 1.00 to 1.02 | 0.249 | -- |
| Year of Surgery | 1.03 | 0.97 to 1.10 | 0.319 | -- |
| Race | 0.74 | 0.38 to 1.44 | 0.375 | -- |
NOTE. Boldfaced values represent statistically significant HRs.
Abbreviations: HR, Hazard Ratio; Cl, Confidence Interval; RAF, RiskAllele Frequency; PSA, Prostate Specific Antigen.
Multivariable adjusted for Biopsy Gleason Sum, Clinical Stage, PSA, Age, Gland Volume, Year of Surgery and Race.
Figure 1.
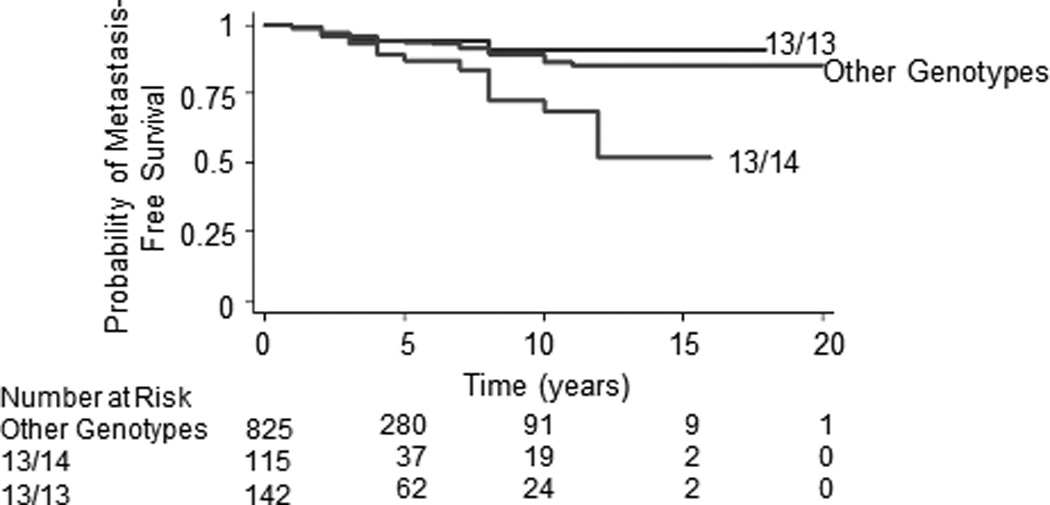
Kaplan-Meier metastasis-free survival curves for germline ASPN D 13/14 compared to ASPN D13/13 (P=0.012) or to all other germline ASPN D genotypes (P=0.003).
In contrast to germline carriers of ASPN D13/14 or any ASPN D14, multivariable Cox regression analyses demonstrated that homozygous ASPN D13 (ASPN D13/13) was significantly associated with a reduced risk of metastatic progression following surgery (HR=0.44, 95%CI=0.21–0.94, P=0.035; Table 3). Kaplan-Meier survival estimates demonstrated that germline carriers of ASPN D13/13 had better metastasis-free survival than men with germline ASPN D13/14 (Fig. 1). When compared to germline ASPN D13/13, men with germline ASPN D13/14 had over a four-fold higher hazard of metastatic progression (HR=4.53, 95%CI=1.65–12.42, P=0.003; Supplementary Table S2). While ASPN D13/13 remained significantly protective when controlled for race, Chi-Square analysis demonstrated that significantly fewer African American men carried the ASPN D13/13 genotype (P<0.0001; Supplementary Table S1).
Given the association of BCR with metastatic progression (19), we evaluated the relationship between ASPN D-repeat-length and BCR. When compared to all other genotypes, bivariate analyses controlled for race demonstrated that ASPN D13/14 was significantly prognostic of BCR following surgery (HR=1.43, 95%CI=1.01–2.03, P=0.042; Supplementary Table S3); however when controlled for pre-surgical variables in multivariable Cox regression analyses, the hazard ratio associated with ASPN D13/14 only approached significance (Supplementary Table S4).
Germline ASPN D-repeat-length variants and adverse prostate cancer pathology
Both pre- and post-treatment clinical findings are used by the NCCN to stratify risk of disease recurrence and to guide treatment recommendations (20). Logistic regression analyses demonstrated that none of the genotypes examined were significantly associated with biopsy or pathologic Gleason grade, PSA, local invasion beyond the prostate (EPE) or invasion into the seminal vesicles (SVI) (Supplementary Table S5). However, logistic regression analyses demonstrated that ASPN D13/14 was significantly associated with prostate cancer involving the lymph nodes at surgery (OR=2.52, 95%CI=1.44–4.42, P=0.001; Supplementary Table S6). Multivariable logistic regression analyses adjusted for pre-operative variables and race demonstrated that men with ASPN D13/14 (OR=2.42, 95%CI=1.29–4.56, P=0.006) or carriers of any ASPN D14 (homozygous or heterozygous) (OR=1.74, 95%CI=1.02–2.96, P=0.041) were significantly more likely to have lymph node involvement at surgery than men with other ASPN D genotypes or alleles (Supplementary Table S7).
Germline ASPN D-repeat-length variants in men with prostate cancer and non-cancer diagnoses
Polymorphisms in ASPN D-repeat-length have reported associations with susceptibility to diseases such as osteoarthritis and lumbar-disc degeneration (7, 9). To determine if ASPN D-repeat length was associated with prostate cancer incidence, we compared ASPN D-repeat-length in prostate cancer cases and non-cancer male controls matched for race. The distribution of ASPN D-repeat-length in JHH race-matched prostate cancer cases and controls was nearly identical with none of the alleles or genotypes being statistically different by Chi-square analyses (Supplementary Fig. S2 and Supplementary Table S1) suggesting that ASPN D allelic repeat-lengths are not associated with prostate cancer incidence.
ASPN Expression in Cancer Associated Fibroblasts
Our data suggest that germline ASPN-repeat-length variants are differentially associated with metastatic progression; however, little is known about the biologic role of ASPN in cancer progression. To better understand this, we first analyzed human TMAs and demonstrated that ASPN expression was not elevated in tumor cells but rather was significantly elevated in PCAFs (Fig. 2A, B, D). Both mRNA and protein expression of ASPN were elevated in primary PCAFs isolated from human prostates compared to primary benign associated fibroblasts (BAF) and an immortalized benign associated fibroblast cell line (WPMY1) (Fig. 2C and Supplementary Fig. S1E). ASPN expression was not detected in primary human prostate epithelial cells (PrEC) or in prostate cancer cell lines (Fig. 1C and Supplementary Fig. S1E). Interestingly, elevated levels of ASPN mRNA were also detected in multiple prostate cancer cohorts (Supplementary Fig. S3A, B); however, ASPN induction was not noted in benign prostatic hyperplasia (BPH) or in prostatic intraepithelial neoplasia (PIN) (Supplementary Fig. S3B). Similar to prostate cancer, ASPN expression was also significantly elevated in multiple other cancers including breast, colon, and pancreatic compared to normal tissue (Supplementary Fig. S3C), suggesting that ASPN may have a conserved role in cancer development.
Figure 2.

ASPN expression in prostate cancer associated fibroblasts is associated with Gleason grade and oncologic outcomes. A, ASPN expression as determined by IHC in prostate tissue following radical prostatectomy. B, ASPN expression in prostate cancer associated fibroblasts in prostate tissue following radical prostatectomy as determined by IF of ASPN and pancytokeratin at 400× magnification. C, ASPN expression in human primary prostate cancer associated fibroblasts (PCAF), human primary benign prostate associated fibroblasts (BAF), an immortalized human benign prostate fibroblast cell line (WPMY1), primary human benign prostate epithelial cells (PreC), and human prostate cancer cell lines (LNCaP, 22RV1, PC3) as determined by qRT-PCR (top) and immunoblotting (bottom). D, ASPN expression as determined by IHC in JHU TMAs containing stroma adjacent to benign (n=48), Gleason sum ≤6 (n=31), Gleason sum 7 (n=6), and Gleason sum 8–9 (n=27) prostate tissue. Statistical analysis performed by one-way ANOVA with Tukey’s multiple comparison test (mean ± SEM; **P≤ 0.001, ***P ≤ 0.0001; upper). ASPN staining in human prostate cancer sections at 200× magnification (lower). E, ASPN expression is associated with biochemical recurrence (BCR). Kaplan–Meier survival curves of ASPN in MSKCC and in Mayo High Risk Cohort for BCR-free survival. F, ASPN expression is associated with metastatic recurrence. Kaplan–Meier survival curves of ASPN in Mayo High Risk Cohort for metastatic recurrence.
ASPN Expression and Prostate Cancer Gleason Grade
Gleason grade of the primary tumor is the strongest single prognostic marker of prostate cancer aggressiveness (21). While low Gleason grade tumors (sum ≤6) rarely progress to metastasis, high Gleason grade tumors (sum 8–10) can have poor oncologic outcomes, even after RP (21). IHC analysis of ASPN protein expression in TMAs from JHU (15) and Harvard (22) demonstrated that ASPN was associated with increasing Gleason grade with the highest expression seen in PCAFs surrounding Gleason sum 8–9 tumors (Fig. 2D; Supplementary Fig. S3D). Consistent with protein expression, analysis of ASPN mRNA expression in a cohort from Memorial Sloan-Kettering Cancer Center (MSKCC) (23) demonstrated that ASPN was significantly elevated in samples from Gleason sum 8–9 tumors (Supplementary Fig. S3E).
ASPN Expression and Prostate Cancer Outcomes
To determine if ASPN expression in the primary tumor microenvironment was associated with disease progression, we examined ASPN mRNA expression in prostate cancer cohorts from MSKCC (23) and the Mayo Clinic (15, 24) and ASPN protein expression in a cohort from Harvard (22). ASPN expression was significantly associated with BCR in all three cohorts as demonstrated by Kaplan-Meir survival curves and/or Cox regression analyses (Fig. 2E and Supplementary Fig. S4A, B). While Kaplan-Meir analysis was not available for the MSKCC cohort due to metastatic event time being unavailable, Kaplan-Meir survival curves demonstrated that ASPN expression was significantly associated with metastatic recurrence in the Mayo Clinic cohort (Fig. 2F). Univariate analyses also demonstrated a significant association between ASPN expression and metastatic recurrence in both the MSKCC and Mayo Clinic cohorts; however, this was not significant in the Harvard cohort (Supplementary Fig. 4C, D).
ASPN D-Repeat Domain Variants in Animal Models of Metastatic Progression
Germline ASPN D14 was associated with metastatic recurrence, while homozygous germline ASPN D13 was associated with a reduced risk of metastases; yet, the functional significance of these variants in metastatic progression is not known. To begin to investigate this, we examined tumor progression in an in vivo model incorporating tumor/fibroblast interactions. We stably overexpressed ASPN D14 and ASPN D13 in an immortalized human prostate fibroblast cell line derived from stroma adjacent to benign glands (WPMY1) (Fig. 3A). While WPMY1 are germline homozygous for ASPN D13, they did not express ASPN in vitro (Fig. 3A). Two independent stable clones of both WPMY1 over-expressing ASPN D13 [WPMY1 ASPN D13 (A) and (B)] and WPMY1 over-expressing ASPN D14 [WPMY1 ASPN D14 (A) and (B)] were generated. Parental WPMY1, WPMY1 ASPN D13, and WPMY1 ASPN D14 were mixed with PC3 cells and then grafted into murine prostates. Expression of ASPN polymorphisms did not affect average orthotopic xenograft size as measured by GU weight (Fig. 3B, C); however, ASPN polymorphisms significantly affected the number of metastases (Fig. 3D, E). Due to differences in 5-year survival rates between regional and distant prostate cancer in humans (1), we quantified the number of metastases to distant lymph nodes and to other organs in the orthotopic xenografts. WPMY1 ASPN D13 significantly decreased the number of metastases compared to parental WPMY1, while conversely WPMY1 ASPN D14 significantly increased the number of metastases to distant lymph nodes and other organs including lung, liver, and pancreas (Fig. 3D, E). Both gross inspection and pathological analyses supported that metastatic obstruction of the bile duct led to jaundice in approximately 33% of the WPMY1 ASPN D14 mice, while, conversely, jaundice was not detected in WPMY1 or WPMY1 ASPN D13 mice. Histological examination demonstrated micro-metastatic disease in WPMY1 ASPN D13 animals compared to the much larger visual metastases in WPMY1 ASPN D14 animals (Fig. 3E). Collectively, these studies suggest that ASPN polymorphisms may have both prognostic and biologic roles in disease progression.
Figure 3.
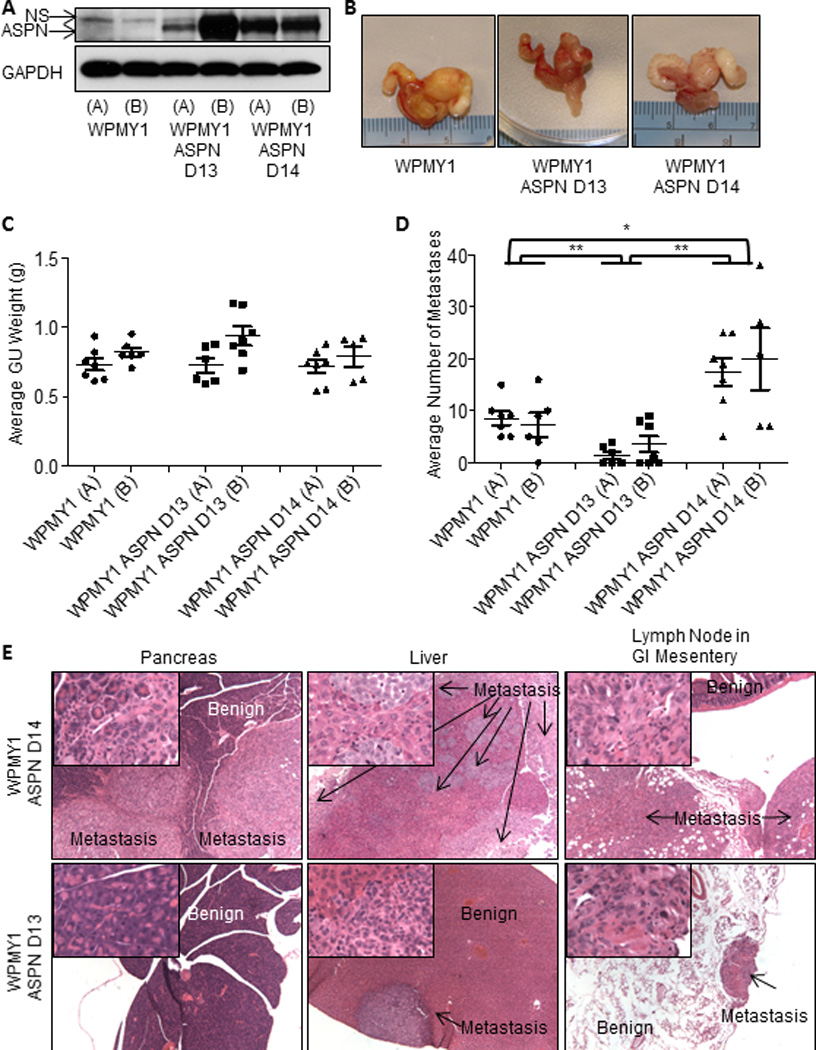
ASPN D13 is protective of metastatic progression while ASPN D14 promotes metastatic progression. A, ASPN expression in WPMY1 (A and B), WPMY1 ASPN D13 (A and B), and ASPN D14 (A and B) as measured by immunoblotting. B and C, Photomicrographs and GU weight of WPMY1 (A) + PC3 (n=7), WPMY1 (B) + PC3 (n=5), WPMY1 ASPN D13 (A) + PC3 (n=6), WPMY1 ASPN D13 (B) + PC3 (n=7), WPMY1 ASPN D14 (A) + PC3 (n=7), and WPMY1 ASPN D14 (B) + PC3 (n=5) prostate orthotopic xenografts (mean ± SEM). D, Average number of visible metastases to other organs and distant lymph nodes in WPMY1 (A) + PC3 (n=7), WPMY1 (B) + PC3 (n=5), WPMY1 ASPN D13 (A) + PC3 (n=6), WPMY1 ASPN D13 (B) + PC3 (n=7), WPMY1 ASPN D14 (A) + PC3 (n=7), and WPMY1 ASPN D14 (B) + PC3 (n=5) prostate orthotopic xenografts (mean ± SEM). E, Representative images of metastases taken at 40× magnification from WPMY1 ASPN D13 + PC3 and WPMY1 ASPN D14 + PC3 prostate orthotopic xenografts. Inset is a 400× magnification of the tumor region. Statistical analyses performed by One-way ANOVA with Tukey’s multiple comparison test to compare WPMY1 (A+B), WPMY1 ASPN D13 (A+B), and WPMY1 ASPN D14 (A+B) (mean ± SEM; *P≤0.004 and **P≤0.0003).
Discussion
Significant progress has been made in the past several years to identify genetic risk factors for prostate cancer. Genome wide association studies (GWAS) have identified many single nucleotide polymorphisms associated with prostate cancer incidence (25, 26); yet few inherited determinants of aggressive prostate cancer have been identified (27, 28). Furthermore, associations of germline variants expressed by non-tumor cells in the tumor microenvironment with metastatic prostate cancer have not been described. Based upon its association with other diseases, we report a hypothesis-driven study to determine if germline variants in ASPN D-repeat-length were differentially associated with metastatic prostate cancer. The distribution of ASPN D-repeat-length variants was similar between prostate cancer cases and race matched controls, supporting the absence of an association between ASPN D variants and prostate cancer incidence. This is consistent with prior GWAS or family-based linkage studies that have not found a strong association between this genomic region and prostate cancer risk (29–31). However, two common variants in ASPN D-repeat-length affected the risk of having metastatic disease. Multivariable analyses demonstrated that homozygous ASPN D13/13 was significantly protective of metastatic recurrence following surgery. Conversely, germline genotype ASPN D13/14 and the ASPN D14 allele were significantly prognostic of lymph node involvement and metastatic recurrence following surgery. While previous studies have not reported an association of this genomic region with metastatic prostate cancer, this may be due to limitations of prior studies that examined candidate genes (27) or genes associated with incidence (32). Consistent with our patient-based findings, an animal model of prostate cancer metastasis supports that ASPN D14 specifically expressed in PCAFs promotes metastatic prostate cancer progression while ASPN D13 hinders metastatic progression.
Development of a minimally invasive and cost-effective genomic test for the early identification of aggressive prostate cancer has the potential to impact therapeutic decisions in clinical settings. For instance, controversy exists over the optimal treatment volumes and the use of hormonal suppression for men with NCCN intermediate risk prostate cancer receiving radiation therapy (20, 33–36). Additionally there is an increasing emphasis on the use of surveillance for men with low-grade cancers. Incorporation of germline markers of aggressive prostate cancer at cancer diagnosis may better identify patients in need of more aggressive therapies while also preventing the overtreatment of patients with indolent tumors. Additional studies including prospective trials are needed; however, this initial large study suggests that germline ASPN D-repeat-length is associated with clinical outcome.
Prostate cancer incidence and mortality have been shown to differ by race (1). While the etiology driving racial disparities is multi-factorial, differences in biologic and genetic factors most likely play contributing roles (37–40). The observation that African American men were approximately one eighth as likely to be germline for ASPN D13/13 is intriguing and the first time that germline factors in the tumor microenvironment have been described to vary by race.
The molecular roles of ASPN D variants in metastatic prostate cancer progression are not well understood. It has been postulated that ASPN and in particular ASPN D14 inhibits TGFβ family members including TGFβ1 (7) and BMP2 (10, 41). Two recent reports support that ASPN induces CD44 (7) and EGFR (42) mediated signaling pathways in gastric cancer cells. It is plausible that ASPN variants differentially regulate multiple signaling pathways; however, a role for ASPN variants in CD44 or EGFR signaling in the prostate has not been reported.
The mesenchymal/fibroblast specific expression of ASPN in prostate development and cancer and its extracellular localization suggest that ASPN variants may have differential roles in modifying the extracellular matrix (ECM). Dynamic regulation of the ECM has been shown to influence disease progression (43, 44). ASPN may modify the ECM through direct collagen interactions. ASPN has recently been shown to regulate osteoblast-driven collagen mineralization and to induce osteogenic factors (45). While the ASPN D-repeat domain has been shown to bind calcium (45), the C-terminal domain of ASPN containing 10 Leucine Rich Repeats has been shown to bind to Type I (45) and to Type II (46) collagens. ASPN competes with Decorin, an inhibitor of prostate tumor growth (47), for collagen binding (45, 48, 49). In contrast to ASPN expression, which is increased (4), Decorin expression is decreased in PCAFs (50). ASPN and Decorin may work antagonistically to regulate the ECM environment. The D-repeat domain has been shown to directly regulate collagen mineralization; however, the differential roles ASPN D-repeat-length variants play in these processes have not been examined.
While the function of the ASPN D motif is not known, it is possible that it regulates protein aggregation or functions as a scaffold. Interestingly, heterozygous carriers of ASPN D13 (D13/14 or D13/15) were not protected from prostate cancer metastases suggesting that potential protective functions of ASPN D13 are recessive to other alleles. As ASPN D13/14 and the D14 allele were associated with metastatic recurrence, the function of ASPN D14 may be dominant. Unfortunately, the number of germline carriers of homozygous ASPN D14 were not powered for adequate comparisons. It is possible that ASPN D13 and ASPN D14 monomers or ASPN D14 monomers have a better binding affinity than two ASPN D13 monomers. In support of this, ASPN D14 has been shown to have better binding affinity to BMP2 than ASPN D13 (10).
In sum, this study suggests that ASPN is a key regulator in the microenvironment and may promote metastatic progression of prostate cancer. Men with the ASPN D13/14 genotype or carriers of the ASPN D14 allele may be at higher risk of disease progression and thus genotyping ASPN has the potential of improving risk stratification and early therapeutic decision making in localized prostate cancer. Further understanding of the biologic role of ASPN in the stroma has the potential to impact both prognostic and therapeutic development.
Supplementary Material
Statement of Translational Relevance.
In this study, we report that germline ASPN D-repeat-length variants expressed in the tumor microenvironment are differentially associated with progression to metastatic prostate cancer. To the best of our knowledge, this is the first report of an association of a germline variant expressed by non-tumor cells in the tumor microenvironment with metastatic prostate cancer progression. We additionally show that ASPN expression is associated with local prostate cancer aggressiveness as measured by Gleason grade and poorer oncologic outcomes including metastasis. We begin to establish biologic roles for ASPN D-repeat-length variants in progression to metastatic prostate cancer. This study highlights the potential clinical utility of using germline ASPN D-repeat-length to improve early therapeutic decisions for men who have localized cancer with lethal potential, as well as providing novel heritable and mechanistic insights into progression to lethal disease.
Acknowledgments
We thank Phuoc Tran, Ballentine Carter, Ken Pienta, Patrick Walsh, John Isaacs and Mike Brown for helpful discussions and/or critical reading of this manuscript. We thank John Isaacs for reagents. We thank Laura Kasch-Semenza at the Johns Hopkins GRCF Fragment Analysis Facility and the Prostate Cancer Biorepository Network (PCBN), supported by the Department of Defense Prostate Cancer Research Program, DOD Award No W81XWH-10-2-0056 and W81XWH-10-2-0046.
Grant Support: This work was supported by The Department of Defense Prostate Cancer Research Program Idea Award, DOD Award No PC110902 (PJH, LM, EMS); Flight Attendant Medical Research Institute Young Clinical Scientist Award (PJH); The Patrick C. Walsh Prostate Cancer Fund Beth W. and A. Ross Myer Scholar (PJH); The Prostate Cancer Foundation Hagen Challenge Award (EMS, PJH); National Institutes of Health Grant T32DK007552 (DS); and National Institutes of Health Grant P30CA006973 (LM); The Johns Hopkins Clinician Scientist Award (AER).
PJH and EMS have submitted a provisional patent through the Johns Hopkins Technology Transfer office.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors do not have financial interests that are a conflict of interest. We have the following disclosures. ED, IAV, and NE are employees of GenomeDx and have stock in the company. BHP is a consultant for LOXO Oncology and Horizon Discovery. AER and EMS are consultants for GenomeDx.
Authors’ Contributions
Conception and design: PJH and EMS.
Acquisition of data: PJH, BWS, RMH, BS, RMM, BB, IAV, NE, ED, RJK, GY, CE, SDI, KMJ, JRR, LAM, WBI, AER, and EMS.
Analysis and interpretation of the data: PJH, DS, BS, BWS, SFF, GN, IAV, NE, ED, RJK, DMB, JRR, LAM, KMJ, SSA, JH, WBI, LM, BHP, AER, and EMS.
Writing, review and/or revision of the manuscript: PJH, DS, BHP, DMB, ED, AER, SSA, JRR, WBI, LAM, LM, and EMS.
References
- 1.National Cancer Institute Surveillance E, and End Results Program. seer.cancer.gov/statfacts/html/prost.html.
- 2.Barron DA, Rowley DR. The reactive stroma microenvironment and prostate cancer progression. Endocrine-related cancer. 2012;19:R187–R204. doi: 10.1530/ERC-12-0085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaeffer EM, Marchionni L, Huang Z, Simons B, Blackman A, Yu W, et al. Androgen-induced programs for prostate epithelial growth and invasion arise in embryogenesis and are reactivated in cancer. Oncogene. 2008;27:7180–7191. doi: 10.1038/onc.2008.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Orr B, Riddick AC, Stewart GD, Anderson RA, Franco OE, Hayward SW, et al. Identification of stromally expressed molecules in the prostate by tag-profiling of cancer-associated fibroblasts, normal fibroblasts and fetal prostate. Oncogene. 2012;31:1130–1142. doi: 10.1038/onc.2011.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klee EW, Bondar OP, Goodmanson MK, Dyer RB, Erdogan S, Bergstralh EJ, et al. Candidate serum biomarkers for prostate adenocarcinoma identified by mRNA differences in prostate tissue and verified with protein measurements in tissue and blood. Clin Chem. 2012;58:599–609. doi: 10.1373/clinchem.2011.171637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Satoyoshi R, Kuriyama S, Aiba N, Yashiro M, Tanaka M. Asporin activates coordinated invasion of scirrhous gastric cancer and cancer-associated fibroblasts. Oncogene. 2015;34:650–660. doi: 10.1038/onc.2013.584. [DOI] [PubMed] [Google Scholar]
- 7.Kizawa H, Kou I, Iida A, Sudo A, Miyamoto Y, Fukuda A, et al. An aspartic acid repeat polymorphism in asporin inhibits chondrogenesis and increases susceptibility to osteoarthritis. Nat Genet. 2005;37:138–144. doi: 10.1038/ng1496. [DOI] [PubMed] [Google Scholar]
- 8.Liu D, Yang Q, Li M, Mu K, Zhang Y. Association of an asporin repeat polymorphism with ankylosing spondylitis in Han Chinese population: a case-control study. Clin Invest Med. 2010;33:E63–E68. doi: 10.25011/cim.v33i1.11839. [DOI] [PubMed] [Google Scholar]
- 9.Song YQ, Cheung KM, Ho DW, Poon SC, Chiba K, Kawaguchi Y, et al. Association of the asporin D14 allele with lumbar-disc degeneration in Asians. Am J Hum Genet. 2008;82:744–747. doi: 10.1016/j.ajhg.2007.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kajikawa T, Yamada S, Tauchi T, Awata T, Yamaba S, Fujihara C, et al. Inhibitory effects of PLAP-1/asporin on periodontal ligament cells. Journal of dental research. 2014;93:400–405. doi: 10.1177/0022034513520549. [DOI] [PubMed] [Google Scholar]
- 11.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–851. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Sterling JA, Fan KH, Vessella RL, Shyr Y, Hayward SW, et al. Loss of TGF-beta responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Molecular cancer research: MCR. 2012;10:494–503. doi: 10.1158/1541-7786.MCR-11-0506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Epstein JI, Pizov G, Walsh PC. Correlation of pathologic findings with progression after radical retropubic prostatectomy. Cancer. 1993;71:3582–3593. doi: 10.1002/1097-0142(19930601)71:11<3582::aid-cncr2820711120>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 14.Flavin R, Pettersson A, Hendrickson WK, Fiorentino M, Finn S, Kunz L, et al. SPINK1 protein expression and prostate cancer progression. Clin Cancer Res. 2014;20:4904–4911. doi: 10.1158/1078-0432.CCR-13-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hurley PJ, Marchionni L, Simons BW, Ross AE, Peskoe SB, Miller RM, et al. Secreted protein, acidic and rich in cysteine-like 1 (SPARCL1) is down regulated in aggressive prostate cancers and is prognostic for poor clinical outcome. Proc Natl Acad Sci U S A. 2012;109:14977–14982. doi: 10.1073/pnas.1203525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karnes RJ, Bergstralh EJ, Davicioni E, Ghadessi M, Buerki C, Mitra AP, et al. Validation of a genomic classifier that predicts metastasis following radical prostatectomy in an at risk patient population. J Urol. 2013;190:2047–2053. doi: 10.1016/j.juro.2013.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mustafa Z, Dowling B, Chapman K, Sinsheimer JS, Carr A, Loughlin J. Investigating the aspartic acid (D) repeat of asporin as a risk factor for osteoarthritis in a UK Caucasian population. Arthritis Rheum. 2005;52:3502–3506. doi: 10.1002/art.21399. [DOI] [PubMed] [Google Scholar]
- 18.Atif U, Philip A, Aponte J, Woldu EM, Brady S, Kraus VB, et al. Absence of association of asporin polymorphisms and osteoarthritis susceptibility in US Caucasians. Osteoarthritis Cartilage. 2008;16:1174–1177. doi: 10.1016/j.joca.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. Natural history of progression after PSA elevation following radical prostatectomy. Jama. 1999;281:1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 20.Mohler JL, Armstrong AJ, Bahnson RR, Boston B, Busby JE, D'Amico AV, et al. Prostate cancer, Version 3.2012: featured updates to the NCCN guidelines. Journal of the National Comprehensive Cancer Network : JNCCN. 2012;10:1081–1087. doi: 10.6004/jnccn.2012.0114. [DOI] [PubMed] [Google Scholar]
- 21.Eggener SE, Scardino PT, Walsh PC, Han M, Partin AW, Trock BJ, et al. Predicting 15-year prostate cancer specific mortality after radical prostatectomy. J Urol. 2011;185:869–875. doi: 10.1016/j.juro.2010.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Flavin RJ, Pettersson A, Hendrickson WK, Fiorentino M, Finn SP, Kunz L, et al. SPINK1 Protein Expression and Prostate Cancer Progression. Clin Cancer Res. 2014;20:4904–4911. doi: 10.1158/1078-0432.CCR-13-1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erho N, Crisan A, Vergara IA, Mitra AP, Ghadessi M, Buerki C, et al. Discovery and validation of a prostate cancer genomic classifier that predicts early metastasis following radical prostatectomy. PloS one. 2013;8:e66855. doi: 10.1371/journal.pone.0066855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eeles RA, Kote-Jarai Z, Giles GG, Olama AA, Guy M, Jugurnauth SK, et al. Multiple newly identified loci associated with prostate cancer susceptibility. Nat Genet. 2008;40:316–321. doi: 10.1038/ng.90. [DOI] [PubMed] [Google Scholar]
- 26.Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet. 2008;40:310–315. doi: 10.1038/ng.91. [DOI] [PubMed] [Google Scholar]
- 27.Lin DW, FitzGerald LM, Fu R, Kwon EM, Zheng SL, Kolb S, et al. Genetic variants in the LEPR, CRY1, RNASEL, IL4, and ARVCF genes are prognostic markers of prostate cancer-specific mortality. Cancer Epidemiol Biomarkers Prev. 2011;20:1928–1936. doi: 10.1158/1055-9965.EPI-11-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stark JR, Wiklund F, Gronberg H, Schumacher F, Sinnott JA, Stampfer MJ, et al. Toll-like receptor signaling pathway variants and prostate cancer mortality. Cancer Epidemiol Biomarkers Prev. 2009;18:1859–1863. doi: 10.1158/1055-9965.EPI-08-0981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al Olama AA, Kote-Jarai Z, Berndt SI, Conti DV, Schumacher F, Han Y, et al. A meta-analysis of 87,040 individuals identifies 23 new susceptibility loci for prostate cancer. Nat Genet. 2014;46:1103–1109. doi: 10.1038/ng.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu L, Cancel-Tassin G, Valeri A, Cussenot O, Lange EM, Cooney KA, et al. Chromosomes 4 and 8 implicated in a genome wide SNP linkage scan of 762 prostate cancer families collected by the ICPCG. Prostate. 2012;72:410–426. doi: 10.1002/pros.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu J, Dimitrov L, Chang BL, Adams TS, Turner AR, Meyers DA, et al. A combined genomewide linkage scan of 1,233 families for prostate cancer-susceptibility genes conducted by the international consortium for prostate cancer genetics. Am J Hum Genet. 2005;77:219–229. doi: 10.1086/432377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helfand BT, Roehl KA, Cooper PR, McGuire BB, Fitzgerald LM, Cancel-Tassin G, et al. Associations of prostate cancer risk variants with disease aggressiveness: results of the NCI-SPORE Genetics Working Group analysis of 18,343 cases. Hum Genet. 2015;134:439–450. doi: 10.1007/s00439-015-1534-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran PT, Hales RK, Zeng J, Aziz K, Salih T, Gajula RP, et al. Tissue biomarkers for prostate cancer radiation therapy. Current molecular medicine. 2012;12:772–787. doi: 10.2174/156652412800792589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nguyen PL, D'Amico AV. Targeting pelvic lymph nodes in men with intermediate- and high-risk prostate cancer despite two negative randomized trials. J Clin Oncol. 2008;26:2055–2056. doi: 10.1200/JCO.2007.15.9939. author reply 6–7. [DOI] [PubMed] [Google Scholar]
- 35.Morikawa LK, Roach M., 3rd Pelvic nodal radiotherapy in patients with unfavorable intermediate and high-risk prostate cancer: evidence, rationale, and future directions. International journal of radiation oncology, biology, physics. 2011;80:6–16. doi: 10.1016/j.ijrobp.2010.11.074. [DOI] [PubMed] [Google Scholar]
- 36.Bastian PJ, Boorjian SA, Bossi A, Briganti A, Heidenreich A, Freedland SJ, et al. High-risk prostate cancer: from definition to contemporary management. Eur Urol. 2012;61:1096–1106. doi: 10.1016/j.eururo.2012.02.031. [DOI] [PubMed] [Google Scholar]
- 37.Powell IJ, Bock CH, Ruterbusch JJ, Sakr W. Evidence supports a faster growth rate and/or earlier transformation to clinically significant prostate cancer in black than in white American men, influences racial progression and mortality disparity. J Urol. 2010;183:1792–1796. doi: 10.1016/j.juro.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khani F, Mosquera JM, Park K, Blattner M, O'Reilly C, MacDonald TY, et al. Evidence for Molecular Differences in Prostate Cancer between African American and Caucasian Men. Clin Cancer Res. 2014;20:4925–4934. doi: 10.1158/1078-0432.CCR-13-2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sundi D, Schaeffer EM. Active surveillance for African-American men with prostate cancer: proceed with caution. Con. Oncology (Williston Park) 2014;28:83–85. [PubMed] [Google Scholar]
- 40.Freedland SJ, Isaacs WB. Explaining racial differences in prostate cancer in the United States: sociology or biology? Prostate. 2005;62:243–252. doi: 10.1002/pros.20052. [DOI] [PubMed] [Google Scholar]
- 41.Tomoeda M, Yamada S, Shirai H, Ozawa Y, Yanagita M, Murakami S. PLAP-1/asporin inhibits activation of BMP receptor via its leucine-rich repeat motif. Biochem Biophys Res Commun. 2008;371:191–196. doi: 10.1016/j.bbrc.2008.03.158. [DOI] [PubMed] [Google Scholar]
- 42.Ding Q, Zhang M, Liu C. Asporin participates in gastric cancer cell growth and migration by influencing EGF receptor signaling. Oncology reports. 2015;33:1783–1790. doi: 10.3892/or.2015.3791. [DOI] [PubMed] [Google Scholar]
- 43.Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. The Journal of cell biology. 2012;196:395–406. doi: 10.1083/jcb.201102147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harbor perspectives in biology. 2011;3:1–24. doi: 10.1101/cshperspect.a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kalamajski S, Aspberg A, Lindblom K, Heinegard D, Oldberg A. Asporin competes with decorin for collagen binding, binds calcium and promotes osteoblast collagen mineralization. Biochem J. 2009;423:53–59. doi: 10.1042/BJ20090542. [DOI] [PubMed] [Google Scholar]
- 46.Kou I, Nakajima M, Ikegawa S. Binding characteristics of the osteoarthritis-associated protein asporin. J Bone Miner Metab. 2010;28:395–402. doi: 10.1007/s00774-009-0145-8. [DOI] [PubMed] [Google Scholar]
- 47.Hu Y, Sun H, Owens RT, Wu J, Chen YQ, Berquin IM, et al. Decorin suppresses prostate tumor growth through inhibition of epidermal growth factor and androgen receptor pathways. Neoplasia. 2009;11:1042–1053. doi: 10.1593/neo.09760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lorenzo P, Aspberg A, Onnerfjord P, Bayliss MT, Neame PJ, Heinegard D. Identification and characterization of asporin. a novel member of the leucine-rich repeat protein family closely related to decorin and biglycan. J Biol Chem. 2001;276:12201–12211. doi: 10.1074/jbc.M010932200. [DOI] [PubMed] [Google Scholar]
- 49.Henry SP, Takanosu M, Boyd TC, Mayne PM, Eberspaecher H, Zhou W, et al. Expression pattern and gene characterization of asporin. a newly discovered member of the leucine-rich repeat protein family. J Biol Chem. 2001;276:12212–12221. doi: 10.1074/jbc.M011290200. [DOI] [PubMed] [Google Scholar]
- 50.Henke A, Grace OC, Ashley GR, Stewart GD, Riddick AC, Yeun H, et al. Stromal expression of decorin, Semaphorin6D, SPARC, Sprouty1 and Tsukushi in developing prostate and decreased levels of decorin in prostate cancer. PLoS One. 7:e42516. doi: 10.1371/journal.pone.0042516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.