

Abstract
Gastric cancer is a complex disease that is affected by multiple genetic and environmental factors. For the precise diagnosis and effective treatment of gastric cancer, the heterogeneity of the disease must be simplified; one way to achieve this is by dividing the disease into subgroups. Toward this effort, recent advances in high-throughput sequencing technology have revealed four molecular subtypes of gastric cancer, which are classified as Epstein-Barr virus-positive, microsatellite instability, genomically stable, and chromosomal instability subtypes. We anticipate that this molecular subtyping will help to extend our knowledge for basic research purposes and will be valuable for clinical use. Here, we review the genomic and epigenomic heterogeneity of the four molecular subtypes of gastric cancer. We also describe a mutational meta-analysis and a reanalysis of DNA methylation that were performed using previously reported gastric cancer datasets.
Keywords: DNA methylation, Gastric cancer, Molecular subtype, Mutation, Next-generation sequencing
Core tip: For the effective diagnosis and treatment of gastric cancer, a recent sequencing study classified gastric cancer into four molecular subtypes, which include Epstein-Barr virus-positive, microsatellite instability, genomically stable, and chromosomal instability subtypes. This molecular subtyping will extend our knowledge for basic research and will be valuable for clinical uses. We herein discuss the genomic and epigenomic heterogeneity of the four molecular subtypes of gastric cancer. We also describe a meta-analysis result that was performed using previously reported sequencing datasets.
INTRODUCTION
Gastric cancer (GC) is a heterogeneous disease that is affected by various genetic and environmental factors. Traditionally, GC has been divided into two histological subtypes, intestinal- and diffuse-type, on the basis of Lauren’s classification[1]. Intestinal-type GC is derived from gastric mucosa cells, characterized by well-differentiated glandular structures, and develops through well-characterized sequential pathological stages, such as chronic gastritis, atrophy, intestinal metaplasia, and dysplasia[2]. Diffuse-type GC is characterized by poorly differentiated infiltrative growth with no definitive premalignant stage and is associated with aggressive behavior and poor prognosis[3]. In addition to the histological subtypes, the clinicopathological characteristics of GCs vary from case to case, making it difficult to identify detailed subtypes and to choose a subtype-optimized therapeutic approach[4].
Over the past decade, advances in sequencing technology and high-throughput analysis have delivered new insights into the genetic and epigenetic heterogeneity that underlies the distinct molecular subtypes of GC[5-14]. Recently, The Cancer Genome Atlas (TCGA) network performed both sequencing-based and array-based approaches to investigate exome sequences, copy-number alterations, gene expression, DNA methylation, and protein activities in GCs, and GC was classified into four subtypes: Epstein-Barr virus (EBV)-positive, microsatellite instability (MSI), genomically stable, and chromosomal instability subtypes[15]. This classification potentially has important biological and clinical implications for basic research, disease diagnosis, and drug treatment.
In this review, we summarize the genomic and epigenomic heterogeneity of the four molecular subtypes of GC. We describe a meta-analysis result that was conducted using the combined data of eight previously reported exome sequencing studies. We also explain a CpG methylation result that was analyzed using TCGA DNA methylation profile data.
SUBTYPE-SPECIFIC GENOMIC ALTERATIONS IN GC
Genomic alterations in EBV-positive subtype
EBV, a gamma-herpes virus containing a 184-kb-long double-stranded DNA genome, was the first virus identified in human malignant cells (Burkitt’s lymphoma)[16]. It was also found in GC epithelial cells in 1990[17]. EBV infection was found in approximately 8.7% of GCs and exhibited a distinct sex and anatomical prevalence[18]: males were predominantly infected, and the proximal stomach such as the gastric cardia and fundus is the major infection site.
There are three latency programs of EBV (Latency I, II, and III) that are defined on the basis of EBV-derived latent gene expression. EBV-positive GC belongs to Latency I or II, which express EBV nuclear antigen I (EBNA1), EBV-encoded small RNA (EBER), BamHI-A rightward transcripts (BARTs), and latent membrane protein 2A (LMP2A)[19]. Latency I neoplasms, including EBV-positive GC, do not express the representative EBV viral oncoproteins, EBNA2 and LMP1, suggesting that EBV contributes to GC development through other mechanisms. As opposed to EBNA2 and LMP1, viral LMP2A expression is one of candidate mechanisms involved in EBV-positive GCs. A previous study demonstrated that two GC cell lines (MKN1 and MKN7), when infected with recombinant LMP2A, recapitulated promoter hypermethylation and the repression of the PTEN tumor suppressor[20], a phenomenon which has been previously observed in EBV-positive GCs.
The first mutation frequently identified from EBV-positive GCs was the ARID1A (AT rich interactive domain 1A) mutation[5,10]. The TCGA project revealed that ARID1A mutations occur in approximately 55% of EBV-positive GCs[15]. Notably, the majority of ARID1A mutations are nonsense mutations that introduce premature stop codons. This result indicates that a loss-of-function of ARID1A may be involved in the tumorigenesis of EBV-positive GCs. Supporting the tumor suppressive role of ARID1A in GC, a loss of ARID1A expression has also been associated with lymphatic invasion, lymph node metastasis, mismatch repair deficiency, and poor prognosis[21,22]. Given that ARID1A is a subunit of the SWI/SNF chromatin remodeling complex[12], it would be interesting to investigate whether ARID1A mutations lead to the extreme hypermethylation phenotype of EBV-positive subtype, referred to as EBV-CIMP (CpG island methylator phenotype, see the section ‘DNA methylation of EBV-positive subtype).
In addition to ARID1A, EBV-positive GCs have frequent mutations in PIK3CA and BCOR (BCL6 corepressor). In a TCGA cohort, approximately 80% of EBV-positive GCs acquired PIK3CA mutations and 23% had BCOR mutations[15]. Interestingly, all of the identified BCOR mutations were nonsense or frameshift mutations, indicating that the inactivation of BCOR is associated with EBV-positive GC. Recurrent mutations of BCOR have also been found in other cancers, including medulloblastoma[23], acute myeloid leukemia[24], and rhabdomyosarcoma[8]. Furthermore, fusion transcripts BCOR-CCNB3 (Cyclin B3) and BCOR-RARA (Retinoic acid receptor alpha) were found in sarcoma[25] and acute myeloid leukemia[26], respectively, suggesting the importance of BCOR in the development of multiple types of tumors. BCOR acts as a transcriptional repressor, and a BCOR complex exhibits ubiquitylation and demethylation activities by recruiting a Polycomb group E3 ubiquitin ligase to histone H2A, a demethylase to histone H3K36, and an SCF E3 ubiquitin ligase[27]. Thus, it is necessary to examine whether BCOR mutations participate in epigenetic chromatin remodeling and the establishment of EBV-CIMP.
Several copy-number alterations, including frequent 18q loss and 9p24.1 gain, were found in EBV-positive GCs[10]. In particular, 9p24.1 amplification correlated with elevated expression levels of JAK2 (Janus kinase 2), CD274, and PDCD1LG2 (Programmed cell death 1 ligand 2)[15]. The elevated expression of PD-L1 and PD-L2, which are encoded by CD274 and PDCD1LG2, was known to mediate tumor evasion from host immune responses[28]. Importantly, antibody-mediated blockade of PD-L1/2 recovers immune function and enhances antitumor activity[29]. Moreover, EBV-positive GCs exhibited dysregulation in immune cell signaling, including IL-12 signaling[15]. Hence, it is required to test the efficacy of therapeutic agents that are used to control immune cell signaling for the treatment of EBV-positive GCs.
Genomic alterations in MSI-high subtype
In the TCGA cohort, MSI-high GCs accounted for approximately 24% of GC patients. MSI-high GCs tended to be diagnosed at relatively older ages and contained a high proportion of intestinal-type GCs. This subtype is characterized by an extensive hypermethylation phenotype referred to as MSI-CIMP that is different from that of EBV-CIMP. A main criterion distinguishing MSI-CIMP from EBV-CIMP is the presence of MLH1 (mutL homolog 1) silencing by promoter hypermethylation[30].
MSI is associated with an absence of DNA mismatch repair activity. DNA mismatch repair genes, including MLH1, MLH3 (mutL homolog 3), PMS1 (PMS1 homolog 1), PMS2, MSH2 (mutS homolog 2), MSH3, and MSH6, maintain genomic integrity by correcting errors (base-base mismatches and insertion/deletions) that are generated during DNA replication and recombination[31]. Due to silencing of DNA mismatch repair genes by promomter hypermethylation, MSI-high GCs exhibit hypermutation; tumors with mutation rates higher than 11-12 mutations per megabase were designated as hypermutated[15,32]. The distinctly high mutational load of MSI-high GCs indicates that this GC subtype may have a unique mutational signature that is different from the other subtypes. Indeed, MSI-high GCs showed a high percentage of a C-to-T substitution signature, whereas the other GC molecular subtypes exhibited the enrichment of an A-to-C substitution signature[33].
In addition to a distinct mutational signature, MSI-high GCs have a different repertoire of mutations compared to non-hypermutated GCs. Liu et al[33] revealed that MSI-high GCs acquired frequent mutations in TP53 (Tumor protein p53), ACVR2A (Activin A receptor, type IIA), PTEN (Phosphatase and tensin homolog ), PIK3CA, KRAS (Kirsten rat sarcoma viral oncogene homolog), ERBB2 (Erb-b2 receptor tyrosine kinase 2), ZBTB1 (Zinc finger and BTB domain containing 1), TRAPPC2L (Trafficking protein particle complex 2-like), GPR39 (G protein-coupled receptor 39), GPR85, and CHRM3 (Cholinergic receptor, muscarinic 3). The TCGA project identified frequent mutations in PIK3CA, ERBB3, RNF43 (Ring finger protein 43), PTEN, TP53, KRAS, ARID1A, HLA-B (Major histocompatibility complex, class I, B), B2M (Beta-2-microglobulin), and NF1 (Neurofibromin 1), in hypermutated GCs[15]. TP53, PIK3CA, and PTEN are the only genes that overlap between the two studies. Hypermutation in this subtype may cause numerous passenger mutations and hinder the detection of driver genes. Thus, rather than therapeutic approaches targeting mutated genes, therapeutic regimens targeting the MSI-CIMP may provide better options for the treatment of MSI-high GCs.
Genomic alterations in genomically stable subtype
Genomically stable GCs are classified according to guidelines from the TCGA network: first, molecular subtypes of EBV-positive and MSI-high tumors are assigned and then the remaining tumors are further divided as being genomically stable or chromosomally unstable based on their degrees of aneuploidy[15]. Genomically stable GC is characterized by enrichment of diffuse-type GCs, a relatively younger patient age at diagnosis, and low mutation rates. Since the diffuse-type GC is an aggressive, invasive, and stem-like histological subtype, its rapid tumor progression may result in a diagnosis at an early age and may not provide enough time to accumulate mutations.
In contrast to intestinal-type GCs that are characterized by a corpus-dominated gastritis with gastric atrophy and intestinal metaplasia, diffuse-type GCs are characterized by gastritis throughout the stomach and a lack of atrophy[34]. Diffuse-type GCs occur more uniformly throughout the world, whereas intestinal-type GCs are predominantly found in specific geographic areas (i.e., Eastern Asia). The histological and genomic alterations of diffuse-type GCs are less recognized compared to those of intestinal-type GCs, which develop through a sequence of events known as the Correa pathway[34]. The highly infiltrative feature of diffuse-type GCs makes it difficult to obtain high purity tumor samples, thereby resulting in a low efficiency of mutation detection. Therefore, as the genomically stable property of diffuse-type GCs could be caused by low purity tumors, the genomic features of diffuse-type GCs should be interpreted with caution. Recent whole-genome or whole-exome sequencing followed by validation with deep sequencing may, in part, overcome this problem and may identify novel mutations of diffuse-type GCs.
Genomically stable GCs have frequent mutations in ARID1A, CDH1 (Cadherin 1), and RHOA (Ras homolog family member A). In the TCGA cohort, mutations in CDH1, which encodes E-cadherin, were found in approximately 37% of genomically stable GCs. In addition to somatic mutations, germ-line mutations have been described as causative variants for hereditary diffuse-type GCs[35-37]. A total of 90 out of 104 known germ-line CDH1 mutations potentially cause a premature translation stop or lack of mRNA expression, thereby affecting the entire coding sequence and all functional domains of a protein[38]. The inactivation of the cell adhesion molecule E-cadherin by mutations may, in part, explain a lack of cellular cohesion of diffuse-type GCs, which is the primarily histological feature.
Recently, three studies sequentially reported recurrent RHOA mutations in diffuse-type GCs[10,14,15]. RHOA mutations were found in approximately 15%-23% of diffuse-type GCs. RHOA mutations occur in highly conserved hotspot sites, including R5W, G17E, L22R, Y34C, F39C/V, E40K/V, Y42C, L57V, and G62E. These RHOA mutations were clustered in two adjacent amino-terminal regions that are known to be functional domains associated with ROCK1 (Rho-associated, coiled-coil containing protein kinase 1) and other effector interaction or GTP binding[39]. The most frequently mutated RhoA Y42 corresponds to HRAS Y40, which is required for the activation of mitogen-activated protein kinase[40].
The functional effect of RHOA mutations is not conclusive whether it acts via loss-of-function or gain-of-function: both possibilities were revealed by two different studies. One study emphasized loss-of-function effect of RHOA mutations. Compared to wild-type RhoA, two different mutant RhoA proteins (Y42C and L57V) exhibited a decrease level of its active GTP-bound form. A subsequent functional study demonstrated that the overexpression of RhoA mutants Y42C or L57V in the intestinal organoid resulted in the evasion of cell detachment-induced apoptosis, termed anoikis[10]. Given that lack of cellular cohesion, anchorage-independent growth, and resistance to anoikis may be prerequisites for the development of diffuse-type GCs[13], the inhibition of anoikis by mutant RHOA may provide a selective advantage with respect to tumorigenesis of diffuse-type GC.
Other study revealed a gain-of-function effect of RHOA mutations. siRNA-mediated RHOA knockdown largely decreased the growth rate of tumor cells harboring RHOA mutation; however, when expression was recovered using RhoA Y42C and G17E, tumor growth reinitiated. Conversely, wild type RhoA failed to rescue the growth inhibition affected by siRNA knockdown[10]. In any case, drugs that modulate the RhoA signaling pathway may be valuable to treat diffuse-type GCs harboring RHOA mutations.
CLDN18-ARHGAP fusions were found in 15% of genomically stable GCs and were mutually exclusive from RHOA mutations[15]. As a result, approximately 30% of genomically stable GCs have either RHOA or CLDN18-ARHGAP alterations. However, recent whole-genome sequencing studies revealed that there were no fusions in CLDN18 or ARHGAP6/26[10,11]. Instead, RHOA fusions were found, including RHOA-COL7A1, RHOA-GPX1 by deletion, and RHOA-RBM6 by inversion.
A recently conducted study suggested that diffuse-type GCs could potentially be further divided into two subgroups. One is diffuse-type GC with tubular cell morphology, which presents a mutational signature of NpTpT > NpGpT[11]. The other is genetically quiet diffuse-type GC, which shows infrequent genetic changes and low clonality irrespective of the presence of a TpT dinucleotide mutational signature[11].
Recent studies have revealed several genetic variants in diffuse-type GCs, despite their low tumor purity. However, a high-depth sequencing with high purity tumor samples would increase the likelihood of identifying more significantly mutated driver genes in the genomically stable subtype.
Genomic alterations in chromosomally unstable subtype
According to the TCGA project, chromosomally unstable GCs, which are classified based on degree of aneuploidy, account for approximately 50% of GC patients. Most patients in this subtype are histologically classified as intestinal-type GC[15]. This subtype is characterized as having highly variable chromosomal copy numbers, although it does not exhibit a high mutation rate. This phenomenon may indicate that copy-number alterations and mutations occur through distinct oncogenic processes in different subsets of tumors. Supporting this assertion, a hierarchical classification of 3299 TCGA tumors from 12 cancer types revealed two main pan-cancer classes that are dominated by either mutations or copy-number alterations[41].
Chromosomally unstable GC is primarily characterized by the enrichment of TP53 mutations and recurrent chromosomal amplifications and deletions. This is consistent with the fact that TP53 mutations cause chromosomal instability[9,41,42]. Given that a majority of chromosomally unstable GCs are intestinal-type, it is reasonable to observe increased clonality and ploidy in intestinal-type GCs[11].
Genomic amplification of receptor tyrosine kinases (RTKs) is the most apparent signature of chromosomally unstable GCs. Frequent amplification was found in the genomic regions of RTK-RAS, encompassing EGFR (epidermal growth factor receptor), ERBB2, ERBB3, FGFR2 (fibroblast growth factor receptor 2), MET (MET proto-oncogene), VEGFA (vascular endothelial growth factor A), and KRAS[43-45]. Because of this observation, administering trastuzumab, an anti-HER2 monoclonal antibody[46,47], may be an therapeutic option for GCs harboring ERBB2 amplification[48]. The amplification of RTK-RAS had a mutually exclusive pattern within chromosomally unstable GCs[49].
Other amplified genes are oncogenic transcription factors such as MYC (v-myc avian myelocytomatosis viral oncogene homolog), GATA4 (GATA binding protein 4), and GATA6, and cell cycle regulators including CCNE1 (cyclin E1), CCND1, and CDK6 (Cyclin-dependent kinase 6). Meanwhile, chromosomal deletions have been found in genomic regions containing FHIT (Fragile histidine triad), WWOX (WW domain containing oxidoreductase), STK3 (Serine/threonine kinase 3), CDH1, CTNNA1 (Catenin alpha 1), PARD3 (Par-3 family cell polarity regulator), and RB1 (Retinoblastoma 1).
META-ANALYSIS OF PUBLISHED GC DATASETS
We conducted a meta-analysis to identify significantly mutated genes that have not previously been recognized in GCs. We applied a MutSigCV algorithm to a combined exome data set of 629 GC patients from eight published studies[5,7,11,14,15,33,50,51]. This analysis revealed 20 significantly mutated genes (Q-value < 0.001), including previously identified GC genes such as TP53, RHOA, KRAS, CDH1, GLI3 (GLI family zinc finger 3) and PIK3CA (Table 1 and Figure 1). Additionally, this analysis identified previously unrecognized genes in GC, including DDI1 (DNA-damage inducible 1 homolog 1), DHFR (Dihydrofolate reductase), GHSR (Growth hormone secretagogue receptor), KRT73 (Keratin 73), OR10J3, PCDHGA6 (Protocadherin gamma subfamily A, 6), PREX2 (Phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 2), KIF2B (Kinesin family member 2B), GRM8 (Glutamate receptor, metabotropic 8), RPL22 (Ribosomal protein L22), DNAH5 (Dynein, axonemal, heavy chain 5), EPB41L3 (Erythrocyte membrane protein band 4.1-like 3), DCAF12L1 (DDB1 and CUL4 associated factor 12-like 1), and PLCL1 (Phospholipase C-like 1) (Table 1 and Figure 1).
Table 1.
Significantly mutated genes identified using a combined exome sequencing data from 629 gastric cancer patients
| Gene | Nonsilent mutations from 629 GCs | P value | Q value |
| TP53 | 276 | 0 | 0 |
| KRAS | 41 | 0 | 0 |
| RHOA | 34 | 0 | 0 |
| PCDHGA6 | 34 | 0 | 0 |
| DDI1 | 27 | 0 | 0 |
| KRT73 | 24 | 0 | 0 |
| OR10J3 | 21 | 0 | 0 |
| GHSR | 20 | 0 | 0 |
| DHFR | 4 | 0 | 0 |
| CDH1 | 86 | 6.00 × 10-15 | 1.13 × 10-11 |
| PREX2 | 94 | 2.23 × 10-10 | 3.82 × 10-7 |
| PIK3CA | 100 | 1.75 × 10-9 | 2.75 × 10-6 |
| GLI3 | 81 | 8.46 × 10-9 | 1.23 × 10-5 |
| KIF2B | 46 | 2.62 × 10-8 | 3.53 × 10-5 |
| GRM8 | 47 | 1.73 × 10-7 | 2.18 × 10-4 |
| RPL22 | 16 | 2.92 × 10-7 | 3.44 × 10-4 |
| DNAH5 | 132 | 3.92 × 10-7 | 4.20 × 10-4 |
| EPB41L3 | 48 | 4.07 × 10-7 | 4.20 × 10-4 |
| DCAF12L1 | 29 | 4.23 × 10-7 | 4.20 × 10-4 |
| PLCL1 | 38 | 4.79 × 10-7 | 4.51 × 10-4 |
GC: Gastric cancer.
Figure 1.
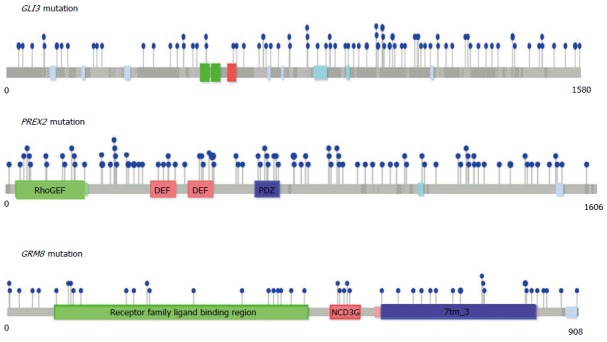
Mutational positions in three significantly mutated genes, GLI3, PREX2, and GRM8.
Among these genes, GLI3, a downstream component of the hedgehog pathway, was found to be a significantly mutated GC driver gene in two independent studies, our current meta-analysis and a previous whole-genome sequencing study by Wang et al[10]. Our results also identified PREX2 as a significantly mutated gene. Supporting this, a recent whole-genome sequencing study identified PREX2, a negative regulator of PTEN, as a new candidate driver of melanoma[52] and pancreatic cancer[53].
GRM8 was identified as a new cancer driver gene in three studies, including our meta-analysis, a study conducted on 441 tumor samples encompassing breast, lung, ovarian, and prostate cancer[54], and a whole-exome sequencing study of endometrial cancer[55]. Thus, GRM8 may be a promising therapeutic target for multiple types of cancer. Additionally, RPL22 was found to be significantly mutated both in colorectal cancer with MSI[56] and in GC. Other significantly mutated genes from our meta-analysis included GHSR, KIF2B, and EPB41L3, which have been shown to play crucial roles in tumorigenesis[57-59]. Thus, further studies are required to evaluate the functional roles of these genes and their mutations during tumorigenesis. Moreover, our meta-analysis suggests that increasing the sample sizes still provides a chance to detect previously unrecognized significantly mutated genes.
SUBTYPE-SPECIFIC DNA METHYLATION IN GC
Thus far, over one hundred genes have been reported to be hypermethylated and downregulated in GC. To elucidate the subtype-specific methylation status of these reported genes, we analyzed CpG methylation levels of 86 genes and 14 microRNAs using the 295 GC DNA methylation data that have been provided by TCGA[15]. Supporting the extensiveness of EBV-CIMP, the majority of the hypermethylated genes were found in EBV-positive subtype (Figure 2). Using K-means clustering, we clustered the hypermethylated genes into three groups: hypermethylated in EBV-positive subtype (Figure 2A), hypermethylated in both EBV-positive and MSI-high subtypes (Figure 2B), and other hypermethylated genes (Figure 2C). In the remainder of this review, we will summarize the methylation patterns of these three groups.
Figure 2.
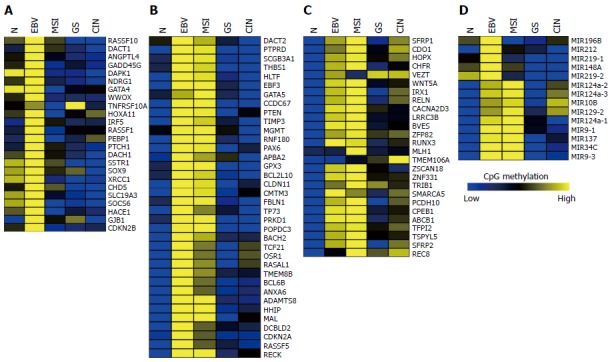
Methylation profiles of the previously reported 86 genes and 14 miRNAs analyzed using the Cancer Genome Atlas methylation data. A: Genes hypermethylated in Epstein-Barr virus (EBV)-positive gastric cancers (GCs); B: Genes hypermethylated in both EBV-positive and MSI-high GCs; C: Genes hypermethylated in more than two GC subtypes; D: Methylation profiles of miRNAs in four GC subtypes.
DNA methylation of EBV-positive subtype
As stated above, EBV has been identified in epithelial malignancies including GC, and nearly 9% of GCs are EBV-positive[60]. Hypermethylation of tumor suppressor genes is a key abnormality in EBV-positive GCs[61].
Unsupervised clustering of CpG methylation clearly revealed that EBV-positivity is the major GC molecular subtype[15]. The most representative feature of EBV-positive GCs is an extensive hypermethylation phenotype EBV-CIMP, which includes CDKN2A promoter hypermethylation. EBV-positive GCs exhibit a global, non-random CpG island hypermethylation phenotype in promoter regions of many cancer-related genes, including p14ARF, p15, p16INK4A, p73, TIMP3, E-cadherin, DAPK, and GSTP1[62]. This CpG island hypermethylation leads to downregulation of the expression level of many tumor suppressor genes that are responsible for GC tumorigenesis.
Interestingly, three DNA methyltransferases, DNMT1, DNMT3A, and DNMT3B, are overexpressed in EBV-positive GC compared to other subtypes (Figure 3). Although the precise molecular mechanism that leads to an increase in the expression of DNMTs during EBV infection is not fully understood, the expression of EBV genes such as LMP2A has been reported to activate DNMT1 transcription by inducing the phosphorylation of STAT3 (Signal transducer and activator of transcription 3)[20]. As DNMT1 plays an important role in the establishment, maintenance, and regulation of tissue specific global methylation patterns, the upregulation of DNMT1 by viral LMP2A might drive the extensive EBV-CIMP. Further studies are required to determine whether EBV viral proteins affect the expression level of other DNA methylation regulators, including DNMTs, HDACs (Histone deacetylases), and TETs (Tet methylcytosine dioxygenases).
Figure 3.
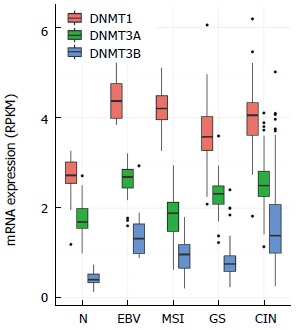
Expression levels of DNMT1, DNMT3A, and DNMT3B analyzed using the Cancer Genome Atlas RNA sequencing data. EBV: Epstein-Barr virus.
Apoptosis-related genes such as RASSF1 (Ras association domain family member 1), DAPK1 (Death-associated protein kinase 1), and GADD45G (Growth arrest and DNA-damage-inducible gamma) exhibit EBV-positive subtype-specific methylation (Figure 2A). RASSF1A is hypermethylated and inactivated in lung, breast, ovarian, kidney, prostate, thyroid, and other cancers[63]. RASSF1A possesses tumor suppressor function through its modulation of apoptosis via the Hippo and Bax pathways and by controlling the cell cycle[64]. Figure 4A illustrates the high methylation level of RASSF1A in EBV-positive GCs of the TCGA cohort. RASSF1A hypermethylation has been detected in 43% of primary GCs and 60% of GC cell lines[65], and significant RASSF1A silencing was found in advanced GC[65]. Therefore, it is important to elucidate a relationship between EBV infection and progressive methylation of RASSF1A during tumorigenesis of GC. Interestingly, aberrant methylation of RASSF1A was detected in 67% of EBV-positive GCs but only in 4% of EBV-negative GCs[66].
Figure 4.
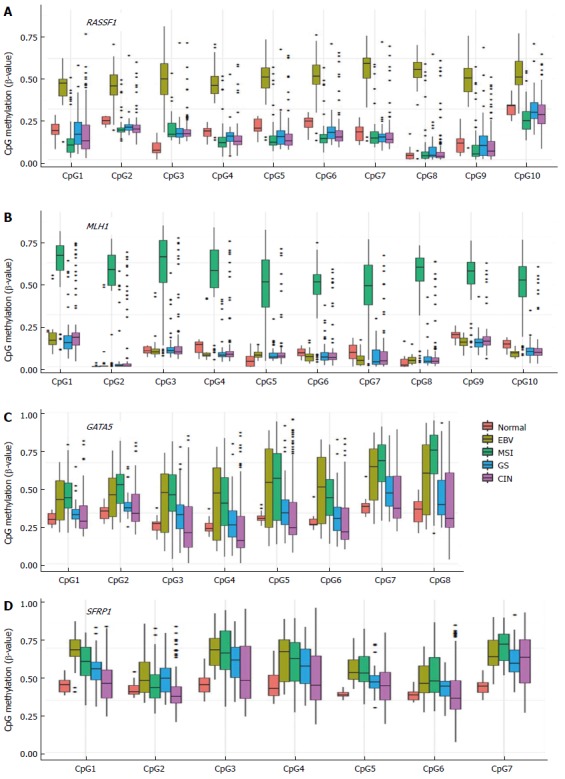
CpG methylation level in promoter regions of RASSF1 (A), MLH1 (B), GATA5 (C), and SFRP1 (D) analyzed using the Cancer Genome Atlas methylation data.
DNA methylation of MSI-high subtype
The most common feature of MSI-high subtype is the hypermethylation of MLH1 promoter. In addition to MLH1, many other tumor suppressive genes are frequently hypermethylated in MSI-high subtype[67], exhibiting MSI-CIMP. Interestingly, we found high mutation rates in DNA methylation regulators, DNMTs and TETs, in MSI-high GC (Supplementary Figure 1)[68], although we cannot rule out the possibility that these high mutation rates are caused by hypermutation in MSI-high GCs. Fifty-two percent (33 of 64) of MSI-high GCs exhibited truncating or missense mutation in TETs (Supplementary Figure 1).
As shown in Figure 4B, MLH1 is hypermethylated only in MSI-high subtype, whereas numerous tumor suppressor genes exhibited hypermethylation patterns in both the EBV and MSI GC subtypes (Figure 2B). This group includes many development-related genes, such as GATA5 (GATA binding protein 5), HHIP (Hedgehog interacting protein), OSR1 (Odd-skipped related 1), PAX6 (Paired box 6), and POPDC3 (Figure 2B). GATA factors are zinc finger DNA binding proteins that control the development of diverse tissues, including the gastrointestinal tract[69,70], and epigenetic inactivation of GATA4 and GATA5 has been reported in GC[71,72]. Using TCGA data, we found that GATA4 is hypermethylated in the EBV-positive subtype of GC, whereas GATA5 is hypermethylated in both EBV and MSI subtypes (Figure 4C).
Hypermethylated genes across more than two subtypes
A subset of genes was found to be hypermethylated in more than two subtypes of GC (Figure 2C). Genes such as SFRP1 (Secreted Frizzled-related protein 1), BVES, IRX1 (Iroquois homeobox 1), RUNX3 (Runt-related transcription factor 3), and WNT5A (Wingless-type MMTV integration site family member 5A) belong to this subset (Figure 4D). The Wnt signaling is important for cell proliferation during development of the gut, and activation of the signaling pathway has been implicated in gastric carcinogenesis[2]. SFRP proteins are secreted glycoproteins that inhibit the Wnt signaling either by competing with Wnt ligands to bind to Fz receptors or by binding directly to Fz[73]. SFRP1 hypermethylation has been detected in 91% of primary GCs and 100% of GC cell lines[74]. This hypermethylation of SFRP was found to occur during an early stage of GC[74]. Hypermethylation of WNT5A, a non-transforming WNT family member that antagonizes the Wnt signaling[75], has also been frequently detected in early GC[76]. Thus, the aberrant methylation patterns of these genes during early GC may serve as useful markers for the early detection of GC.
HYPERMETHYLATED MICRORNAS IN GC
miRNAs are single-stranded, non-coding, small RNAs (18-22 nucleotides in length), which are involved in various biological processes. The aberrant expression of miRNAs and their target genes has a critical role in cancer initiation, progression, and metastasis[77]. The aberrant DNA methylation of miRNAs has frequently been reported in GC[78]. The TCGA miRNA-seq data revealed a subtype-specific aberrant DNA methylation pattern of miRNAs (Figure 2D). miR-196B, miR-212, miR-148A, miR-219-1, and miR-219-2 are hypermethylated in the EBV-positive subtype of GC, whereas miR-9-1, miR-137, miR-34C, and mir-9-3 are hypermethylated in both the EBV and MSI subtypes. Finally, mir-10B, miR-129-2, miR-124a-1, miR-124a-2, and miR-124a-3 are hypermethylated in all of the GC subtypes. Further studies on subtype-specific epigenetic regulation of miRNAs will enable to understand the regulation mechanism of miRNA-driven target genes for GC development.
CONCLUSION
We herein discussed four molecular subtypes of GCs. Each subtype has unique characteristics that facilitate the effective diagnosis and treatment of GCs. The EBV-positive subtype has EBV-driven extensive CpG hypermethylation. The MSI subtype has hypermutation and extreme CpG hypermethylation along with MLH1 silencing. The genomically stable GC subtype exhibits diffuse-type histology harboring frequent RHOA or CDH1 mutations. The chromosomally unstable subtype has the RTK-RAS activation caused by copy-number amplification. This classification simplifies and clarifies the heterogeneous characteristics of GC, thus serving as a foundation for future research, diagnosis, and treatment of GC. Nonetheless, there are many obstacles that must still be overcome. For instance, defining only four molecular subtypes of GC may oversimplify the complexity of the disease. Therefore, further classification of each molecular subtype may be required. Additionally, causative genetic variants that drive the genomically stable GC subtype are still largely unknown. Finally, since a primary purpose of genomic studies is to discover therapeutic targets for GC treatment, the classification scheme may eventually be utilized to facilitate personalized medicine. In that sense, the druggable targets that underlie each GC subtype should be further investigated.
Footnotes
Supported by Grants from the genomics program of the National Research Foundation of Korea funded by the Ministry of Science, ICT, and Future Planning, NRF-2012M3A9D1054670 and NRF-2014M3C9A3068554 (to Kim SY); and Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education, NRF-2013R1A1A2006621 (to Kim M); and the Korea Research Institute of Bioscience and Biotechnology research initiative grant.
Conflict-of-interest statement: Authors declare no conflict of interests for this article.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: April 22, 2015
First decision: August 26, 2015
Article in press: October 13, 2015
P- Reviewer: Bhak J, Guo ZS, Liang H, Lin YW S- Editor: Yu J L- Editor: A E- Editor: Zhang DN
References
- 1.Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49. doi: 10.1111/apm.1965.64.1.31. [DOI] [PubMed] [Google Scholar]
- 2.Yuasa Y. Control of gut differentiation and intestinal-type gastric carcinogenesis. Nat Rev Cancer. 2003;3:592–600. doi: 10.1038/nrc1141. [DOI] [PubMed] [Google Scholar]
- 3.Chiaravalli AM, Klersy C, Vanoli A, Ferretti A, Capella C, Solcia E. Histotype-based prognostic classification of gastric cancer. World J Gastroenterol. 2012;18:896–904. doi: 10.3748/wjg.v18.i9.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grabsch HI, Tan P. Gastric cancer pathology and underlying molecular mechanisms. Dig Surg. 2013;30:150–158. doi: 10.1159/000350876. [DOI] [PubMed] [Google Scholar]
- 5.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, Chan TL, Kan Z, Chan AS, Tsui WY, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat Genet. 2011;43:1219–1223. doi: 10.1038/ng.982. [DOI] [PubMed] [Google Scholar]
- 6.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, Rajasegaran V, Heng HL, Deng N, Gan A, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–574. doi: 10.1038/ng.2246. [DOI] [PubMed] [Google Scholar]
- 7.Lee YS, Cho YS, Lee GK, Lee S, Kim YW, Jho S, Kim HM, Hong SH, Hwang JA, Kim SY, et al. Genomic profile analysis of diffuse-type gastric cancers. Genome Biol. 2014;15:R55. doi: 10.1186/gb-2014-15-4-r55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, Ambrogio L, Auclair D, Wang J, Song YK, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer Discov. 2014;4:216–231. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sieren JC, Meyerholz DK, Wang XJ, Davis BT, Newell JD, Hammond E, Rohret JA, Rohret FA, Struzynski JT, Goeken JA, et al. Development and translational imaging of a TP53 porcine tumorigenesis model. J Clin Invest. 2014;124:4052–4066. doi: 10.1172/JCI75447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014;46:573–582. doi: 10.1038/ng.2983. [DOI] [PubMed] [Google Scholar]
- 11.Wong SS, Kim KM, Ting JC, Yu K, Fu J, Liu S, Cristescu R, Nebozhyn M, Gong L, Yue YG, et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat Commun. 2014;5:5477. doi: 10.1038/ncomms6477. [DOI] [PubMed] [Google Scholar]
- 12.Wu RC, Wang TL, Shih IeM. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15:655–664. doi: 10.4161/cbt.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou J, Hayakawa Y, Wang TC, Bass AJ. RhoA mutations identified in diffuse gastric cancer. Cancer Cell. 2014;26:9–11. doi: 10.1016/j.ccr.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kakiuchi M, Nishizawa T, Ueda H, Gotoh K, Tanaka A, Hayashi A, Yamamoto S, Tatsuno K, Katoh H, Watanabe Y, et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat Genet. 2014;46:583–587. doi: 10.1038/ng.2984. [DOI] [PubMed] [Google Scholar]
- 15.Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet. 1964;1:702–703. doi: 10.1016/s0140-6736(64)91524-7. [DOI] [PubMed] [Google Scholar]
- 17.Burke AP, Yen TS, Shekitka KM, Sobin LH. Lymphoepithelial carcinoma of the stomach with Epstein-Barr virus demonstrated by polymerase chain reaction. Mod Pathol. 1990;3:377–380. [PubMed] [Google Scholar]
- 18.Murphy G, Pfeiffer R, Camargo MC, Rabkin CS. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology. 2009;137:824–833. doi: 10.1053/j.gastro.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young LS, Rickinson AB. Epstein-Barr virus: 40 years on. Nat Rev Cancer. 2004;4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 20.Hino R, Uozaki H, Murakami N, Ushiku T, Shinozaki A, Ishikawa S, Morikawa T, Nakaya T, Sakatani T, Takada K, et al. Activation of DNA methyltransferase 1 by EBV latent membrane protein 2A leads to promoter hypermethylation of PTEN gene in gastric carcinoma. Cancer Res. 2009;69:2766–2774. doi: 10.1158/0008-5472.CAN-08-3070. [DOI] [PubMed] [Google Scholar]
- 21.Inada R, Sekine S, Taniguchi H, Tsuda H, Katai H, Fujiwara T, Kushima R. ARID1A expression in gastric adenocarcinoma: clinicopathological significance and correlation with DNA mismatch repair status. World J Gastroenterol. 2015;21:2159–2168. doi: 10.3748/wjg.v21.i7.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang DD, Chen YB, Pan K, Wang W, Chen SP, Chen JG, Zhao JJ, Lv L, Pan QZ, Li YQ, et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS One. 2012;7:e40364. doi: 10.1371/journal.pone.0040364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pugh TJ, Weeraratne SD, Archer TC, Pomeranz Krummel DA, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grossmann V, Tiacci E, Holmes AB, Kohlmann A, Martelli MP, Kern W, Spanhol-Rosseto A, Klein HU, Dugas M, Schindela S, et al. Whole-exome sequencing identifies somatic mutations of BCOR in acute myeloid leukemia with normal karyotype. Blood. 2011;118:6153–6163. doi: 10.1182/blood-2011-07-365320. [DOI] [PubMed] [Google Scholar]
- 25.Puls F, Niblett A, Marland G, Gaston CL, Douis H, Mangham DC, Sumathi VP, Kindblom LG. BCOR-CCNB3 (Ewing-like) sarcoma: a clinicopathologic analysis of 10 cases, in comparison with conventional Ewing sarcoma. Am J Surg Pathol. 2014;38:1307–1318. doi: 10.1097/PAS.0000000000000223. [DOI] [PubMed] [Google Scholar]
- 26.Ichikawa S, Ichikawa S, Ishikawa I, Takahashi T, Fujiwara T, Harigae H. Successful treatment of acute promyelocytic leukemia with a t(X; 17)(p11.4; q21) and BCOR-RARA fusion gene. Cancer Genet. 2015;208:162–163. doi: 10.1016/j.cancergen.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Gearhart MD, Corcoran CM, Wamstad JA, Bardwell VJ. Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol Cell Biol. 2006;26:6880–6889. doi: 10.1128/MCB.00630-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, Nakamura S, Enomoto K, Yagita H, Azuma M, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13:2151–2157. doi: 10.1158/1078-0432.CCR-06-2746. [DOI] [PubMed] [Google Scholar]
- 29.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geddert H, Zur Hausen A, Gabbert HE, Sarbia M. EBV-infection in cardiac and non-cardiac gastric adenocarcinomas is associated with promoter methylation of p16, p14 and APC, but not hMLH1. Anal Cell Pathol (Amst) 2010;33:143–149. doi: 10.3233/ACP-CLO-2010-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 32.Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, McCleland M, Stawiski EW, Gnad F, Mayba O, Haverty PM, Durinck S, Chen YJ, Klijn C, Jhunjhunwala S, et al. Integrated exome and transcriptome sequencing reveals ZAK isoform usage in gastric cancer. Nat Commun. 2014;5:3830. doi: 10.1038/ncomms4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richards FM, McKee SA, Rajpar MH, Cole TR, Evans DG, Jankowski JA, McKeown C, Sanders DS, Maher ER. Germline E-cadherin gene (CDH1) mutations predispose to familial gastric cancer and colorectal cancer. Hum Mol Genet. 1999;8:607–610. doi: 10.1093/hmg/8.4.607. [DOI] [PubMed] [Google Scholar]
- 36.Huntsman DG, Carneiro F, Lewis FR, MacLeod PM, Hayashi A, Monaghan KG, Maung R, Seruca R, Jackson CE, Caldas C. Early gastric cancer in young, asymptomatic carriers of germ-line E-cadherin mutations. N Engl J Med. 2001;344:1904–1909. doi: 10.1056/NEJM200106213442504. [DOI] [PubMed] [Google Scholar]
- 37.Humar B, Toro T, Graziano F, Müller H, Dobbie Z, Kwang-Yang H, Eng C, Hampel H, Gilbert D, Winship I, et al. Novel germline CDH1 mutations in hereditary diffuse gastric cancer families. Hum Mutat. 2002;19:518–525. doi: 10.1002/humu.10067. [DOI] [PubMed] [Google Scholar]
- 38.Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015;16:e60–e70. doi: 10.1016/S1470-2045(14)71016-2. [DOI] [PubMed] [Google Scholar]
- 39.Dvorsky R, Blumenstein L, Vetter IR, Ahmadian MR. Structural insights into the interaction of ROCKI with the switch regions of RhoA. J Biol Chem. 2004;279:7098–7104. doi: 10.1074/jbc.M311911200. [DOI] [PubMed] [Google Scholar]
- 40.Joneson T, White MA, Wigler MH, Bar-Sagi D. Stimulation of membrane ruffling and MAP kinase activation by distinct effectors of RAS. Science. 1996;271:810–812. doi: 10.1126/science.271.5250.810. [DOI] [PubMed] [Google Scholar]
- 41.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–1133. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwartz JL, Jordan R, Liber H, Murnane JP, Evans HH. TP53-dependent chromosome instability is associated with transient reductions in telomere length in immortal telomerase-positive cell lines. Genes Chromosomes Cancer. 2001;30:236–244. [PubMed] [Google Scholar]
- 43.Thiel A, Ristimäki A. Targeted therapy in gastric cancer. APMIS. 2015;123:365–372. doi: 10.1111/apm.12359. [DOI] [PubMed] [Google Scholar]
- 44.Janbabai G, Oladi Z, Farazmandfar T, Taghvaei T, Naghshvar F. The prognostic impact of EGFR, ErbB2 and MET gene amplification in human gastric carcinomas as measured by quantitative Real-Time PCR. J Cancer Res Clin Oncol. 2015;141:1945–1952. doi: 10.1007/s00432-015-1965-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su X, Zhan P, Gavine PR, Morgan S, Womack C, Ni X, Shen D, Bang YJ, Im SA, Ho Kim W, et al. FGFR2 amplification has prognostic significance in gastric cancer: results from a large international multicentre study. Br J Cancer. 2014;110:967–975. doi: 10.1038/bjc.2013.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Mello RA, Marques AM, Araújo A. HER2 therapies and gastric cancer: a step forward. World J Gastroenterol. 2013;19:6165–6169. doi: 10.3748/wjg.v19.i37.6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luis M, Tavares A, Carvalho LS, Lara-Santos L, Araújo A, de Mello RA. Personalizing therapies for gastric cancer: molecular mechanisms and novel targeted therapies. World J Gastroenterol. 2013;19:6383–6397. doi: 10.3748/wjg.v19.i38.6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, Lordick F, Ohtsu A, Omuro Y, Satoh T, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–697. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 49.Das K, Gunasegaran B, Tan IB, Deng N, Lim KH, Tan P. Mutually exclusive FGFR2, HER2, and KRAS gene amplifications in gastric cancer revealed by multicolour FISH. Cancer Lett. 2014;353:167–175. doi: 10.1016/j.canlet.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 50.Kim TM, Jung SH, Kim MS, Baek IP, Park SW, Lee SH, Lee HH, Kim SS, Chung YJ, Lee SH. The mutational burdens and evolutionary ages of early gastric cancers are comparable to those of advanced gastric cancers. J Pathol. 2014;234:365–374. doi: 10.1002/path.4401. [DOI] [PubMed] [Google Scholar]
- 51.Chen K, Yang D, Li X, Sun B, Song F, Cao W, Brat DJ, Gao Z, Li H, Liang H, et al. Mutational landscape of gastric adenocarcinoma in Chinese: implications for prognosis and therapy. Proc Natl Acad Sci USA. 2015;112:1107–1112. doi: 10.1073/pnas.1422640112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berger MF, Hodis E, Heffernan TP, Deribe YL, Lawrence MS, Protopopov A, Ivanova E, Watson IR, Nickerson E, Ghosh P, et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature. 2012;485:502–506. doi: 10.1038/nature11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518:495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 55.Liang H, Cheung LW, Li J, Ju Z, Yu S, Stemke-Hale K, Dogruluk T, Lu Y, Liu X, Gu C, et al. Whole-exome sequencing combined with functional genomics reveals novel candidate driver cancer genes in endometrial cancer. Genome Res. 2012;22:2120–2129. doi: 10.1101/gr.137596.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreira AM, Tuominen I, van Dijk-Bos K, Sanjabi B, van der Sluis T, van der Zee AG, Hollema H, Zazula M, Sijmons RH, Aaltonen LA, et al. High frequency of RPL22 mutations in microsatellite-unstable colorectal and endometrial tumors. Hum Mutat. 2014;35:1442–1445. doi: 10.1002/humu.22686. [DOI] [PubMed] [Google Scholar]
- 57.Moskalev EA, Jandaghi P, Fallah M, Manoochehri M, Botla SK, Kolychev OV, Nikitin EA, Bubnov VV, von Knebel Doeberitz M, Strobel O, et al. GHSR DNA hypermethylation is a common epigenetic alteration of high diagnostic value in a broad spectrum of cancers. Oncotarget. 2015;6:4418–4427. doi: 10.18632/oncotarget.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Welburn JP, Cheeseman IM. The microtubule-binding protein Cep170 promotes the targeting of the kinesin-13 depolymerase Kif2b to the mitotic spindle. Mol Biol Cell. 2012;23:4786–4795. doi: 10.1091/mbc.E12-03-0214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perez-Janices N, Blanco-Luquin I, Tuñón MT, Barba-Ramos E, Ibáñez B, Zazpe-Cenoz I, Martinez-Aguillo M, Hernandez B, Martínez-Lopez E, Fernández AF, et al. EPB41L3, TSP-1 and RASSF2 as new clinically relevant prognostic biomarkers in diffuse gliomas. Oncotarget. 2015;6:368–380. doi: 10.18632/oncotarget.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fukayama M, Hino R, Uozaki H. Epstein-Barr virus and gastric carcinoma: virus-host interactions leading to carcinoma. Cancer Sci. 2008;99:1726–1733. doi: 10.1111/j.1349-7006.2008.00888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kaneda A, Matsusaka K, Aburatani H, Fukayama M. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012;72:3445–3450. doi: 10.1158/0008-5472.CAN-11-3919. [DOI] [PubMed] [Google Scholar]
- 62.Fukayama M, Ushiku T. Epstein-Barr virus-associated gastric carcinoma. Pathol Res Pract. 2011;207:529–537. doi: 10.1016/j.prp.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 63.Dammann R, Schagdarsurengin U, Strunnikova M, Rastetter M, Seidel C, Liu L, Tommasi S, Pfeifer GP. Epigenetic inactivation of the Ras-association domain family 1 (RASSF1A) gene and its function in human carcinogenesis. Histol Histopathol. 2003;18:665–677. doi: 10.14670/HH-18.665. [DOI] [PubMed] [Google Scholar]
- 64.Donninger H, Clark J, Rinaldo F, Nelson N, Barnoud T, Schmidt ML, Hobbing KR, Vos MD, Sils B, Clark GJ. The RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol Cell Biol. 2015;35:277–287. doi: 10.1128/MCB.00202-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Byun DS, Lee MG, Chae KS, Ryu BG, Chi SG. Frequent epigenetic inactivation of RASSF1A by aberrant promoter hypermethylation in human gastric adenocarcinoma. Cancer Res. 2001;61:7034–7038. [PubMed] [Google Scholar]
- 66.Kang GH, Lee S, Kim WH, Lee HW, Kim JC, Rhyu MG, Ro JY. Epstein-barr virus-positive gastric carcinoma demonstrates frequent aberrant methylation of multiple genes and constitutes CpG island methylator phenotype-positive gastric carcinoma. Am J Pathol. 2002;160:787–794. doi: 10.1016/S0002-9440(10)64901-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamamoto H, Watanabe Y, Maehata T, Morita R, Yoshida Y, Oikawa R, Ishigooka S, Ozawa S, Matsuo Y, Hosoya K, et al. An updated review of gastric cancer in the next-generation sequencing era: insights from bench to bedside and vice versa. World J Gastroenterol. 2014;20:3927–3937. doi: 10.3748/wjg.v20.i14.3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zheng R, Blobel GA. GATA Transcription Factors and Cancer. Genes Cancer. 2010;1:1178–1188. doi: 10.1177/1947601911404223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laverriere AC, MacNeill C, Mueller C, Poelmann RE, Burch JB, Evans T. GATA-4/5/6, a subfamily of three transcription factors transcribed in developing heart and gut. J Biol Chem. 1994;269:23177–23184. [PubMed] [Google Scholar]
- 71.Akiyama Y, Watkins N, Suzuki H, Jair KW, van Engeland M, Esteller M, Sakai H, Ren CY, Yuasa Y, Herman JG, et al. GATA-4 and GATA-5 transcription factor genes and potential downstream antitumor target genes are epigenetically silenced in colorectal and gastric cancer. Mol Cell Biol. 2003;23:8429–8439. doi: 10.1128/MCB.23.23.8429-8439.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wen XZ, Akiyama Y, Pan KF, Liu ZJ, Lu ZM, Zhou J, Gu LK, Dong CX, Zhu BD, Ji JF, et al. Methylation of GATA-4 and GATA-5 and development of sporadic gastric carcinomas. World J Gastroenterol. 2010;16:1201–1208. doi: 10.3748/wjg.v16.i10.1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jones SE, Jomary C. Secreted Frizzled-related proteins: searching for relationships and patterns. Bioessays. 2002;24:811–820. doi: 10.1002/bies.10136. [DOI] [PubMed] [Google Scholar]
- 74.Nojima M, Suzuki H, Toyota M, Watanabe Y, Maruyama R, Sasaki S, Sasaki Y, Mita H, Nishikawa N, Yamaguchi K, et al. Frequent epigenetic inactivation of SFRP genes and constitutive activation of Wnt signaling in gastric cancer. Oncogene. 2007;26:4699–4713. doi: 10.1038/sj.onc.1210259. [DOI] [PubMed] [Google Scholar]
- 75.Li J, Ying J, Fan Y, Wu L, Ying Y, Chan AT, Srivastava G, Tao Q. WNT5A antagonizes WNT/β-catenin signaling and is frequently silenced by promoter CpG methylation in esophageal squamous cell carcinoma. Cancer Biol Ther. 2010;10:617–624. doi: 10.4161/cbt.10.6.12609. [DOI] [PubMed] [Google Scholar]
- 76.Hibi K, Sakata M, Yokomizi K, Kitamura YH, Sakuraba K, Shirahata R, Goto T, Mizukami H, Saito M, Ishibashi K, et al. Methylation of the WNT5A gene is frequently detected in early gastric carcinoma. Hepatogastroenterology. 2012;59:2661–2663. doi: 10.5754/hge09664. [DOI] [PubMed] [Google Scholar]
- 77.Chan SH, Wang LH. Regulation of cancer metastasis by microRNAs. J Biomed Sci. 2015;22:9. doi: 10.1186/s12929-015-0113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guo X, Xia J, Yan J. Promoter methylated microRNAs: potential therapeutic targets in gastric cancer. Mol Med Rep. 2015;11:759–765. doi: 10.3892/mmr.2014.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]