

Abstract
Inhibition and induction of drug-metabolizing enzymes are the most frequent and dangerous drug-drug interactions. They are an important cause of serious adverse events that have often resulted in early termination of drug development or withdrawal of drugs from the market. Management of such interactions by dose adjustment in clinical practice is extremely difficult because of the wide interindividual variability in their magnitude. This review examines the genetic, physiological, and environmental factors responsible for this variability, focusing on an important but so far neglected cause of variability, liver functional status. Clinical studies have shown that liver disease causes a reduction in the magnitude of interactions due to enzyme inhibition, which is proportional to the degree of liver function impairment. The effect of liver dysfunction varies quantitatively according to the nature, reversible or irreversible, of the inhibitory interaction. The magnitude of reversible inhibition is more drastically reduced and virtually vanishes in patients with advanced hepatocellular insufficiency. Two mechanisms, in order of importance, are responsible for this reduction: decreased hepatic uptake of the inhibitory drug and reduced enzyme expression. The extent of irreversible inhibitory interactions is only partially reduced, as it is only influenced by the decreased expression of the inhibited enzyme. Thus, for appropriate clinical management of inhibitory drug interactions, both the liver functional status and the mechanism of inhibition must be taken into consideration. Although the inducibility of drug-metabolizing enzymes in liver disease has long been studied, very conflicting results have been obtained, mainly because of methodological differences. Taken together, the results of early animal and human studies indicated that enzyme induction is substantially preserved in compensated liver cirrhosis, whereas no definitive conclusion as to whether it is significantly reduced in the decompensated state of cirrhosis was provided. Since ethical constraints virtually preclude the possibility of performing methodologically rigorous investigations in patients with severe liver dysfunction, studies have recently been performed in animals rigorously stratified according to the severity of liver insufficiency. The results of these studies confirmed that enzyme induction is virtually unaffected in compensated cirrhosis and indicated that the susceptibility of enzyme induction to severe liver dysfunction depends on the type of nuclear receptor involved and also varies among enzyme isoforms under the transcriptional control of the same nuclear receptor. These findings make it clear that no general conclusion can be reached from the study of any particular enzyme and partly explain the conflicting results obtained by previous studies. Since no general guidelines can be provided for the management of drug interactions resulting from enzyme induction, both the effects and the plasma concentration of the induced drug should be strictly monitored. The findings discussed in this review have important methodological implications as they indicate that, contrary to current guidelines, the magnitude of metabolic drug-drug interactions in patients with liver disease cannot be inferred from studies in healthy subjects.
Keywords: Pharmacokinetic interactions, Enzyme inhibition, Enzyme induction, Liver disease, Plasma protein binding
Core tip: The widespread use of polypharmacotherapy makes drug-drug interactions due to inhibition or induction of drug-metabolizing enzymes virtually unavoidable, with consequently high risk of serious or even life-threatening adverse effects. An appropriate management of such interactions by dose adjustment is made difficult by their high interindividual variability. This review, which focuses on the variability associated with liver functional status, presents an updated discussion of the relevant literature, analyzes the mechanisms responsible for the effect of liver dysfunction on the magnitude of these interactions, and outlines the criteria on which their clinical management should be based.
INTRODUCTION
A drug-drug interaction (DDI) may be defined as the modification of a patient’s clinical response to the administered drug by co-administration of another drug. DDIs are termed pharmacodynamic when a pharmacological response is altered through either agonism or antagonism, without affecting the drug kinetics. Pharmacokinetic interactions, i.e., alterations of drug disposition, occur mainly via inhibition or induction of metabolic enzymes or transporters involved in drug absorption, distribution, metabolism, or excretion. DDIs are an important and avoidable cause of serious adverse events and can result in early termination of development or withdrawal of drugs from the market. As polypharmacy is commonplace in many patient populations, the risk of dangerous DDIs is high. For example, in the general population, DDIs have been considered responsible for 20%-30% of all adverse drug reactions[1] and account for about 10% of visits to emergency departments[2]. In hospitalized patients, they represent 3%-5% of medication errors[3] and have been estimated to be the cause of death in 4% of cancer patients[4], to whom drugs are frequently administered at or close to the maximum tolerated dose. Both transporter- and enzyme-mediated DDIs can significantly alter drug pharmacokinetics and have therefore the potential to affect the therapeutic efficacy or toxicity of drugs. However, a very limited number of transporter-mediated DDIs have so far been shown to be clinically relevant, and the effect of environmental variables, such as liver disease, on the extent of these interactions has not yet been investigated[5]. By contrast, metabolism-based DDIs, particularly those due to inhibition and induction of cytochromes P450 (CYPs), have been shown to be rather frequent and the most dangerous ones[6]. The clinical consequences of CYP inhibition or induction depend on the pharmacological and toxic effects of both the parent drug and its metabolite(s) and may be particularly important if the victim drug has a narrow therapeutic index, since metabolism-based DDIs may cause up to 10-fold changes in the concentrations of the drug whose biotransformation is inhibited or induced (see, e.g., Teo et al[7]). Thus, when the parent drug is responsible for the pharmacological effect and the affected metabolic pathway constitutes its main route of elimination, inhibition and induction may cause dangerous toxic effects or complete loss of therapeutic efficacy, respectively. Vice versa, if the parent compound is a pro-drug, inhibition of its metabolic conversion will cause a decrease or loss of therapeutic efficacy, whereas enzyme induction will likely result in toxic effects. Induction may be particularly dangerous when reactive metabolites are generated, as they can frequently cause serious idiosyncratic reactions[8].
Although pharmaceutical companies are devising new approaches to circumvent DDIs mediated by drug-metabolizing enzymes (DMEs), the very broad specificity of CYPs, which results in frequent competition of co-administered drugs for CYP catalytic sites, make these interactions virtually unavoidable. For example, a systematic review[9] of new drug applications approved by the Food and Drug Administration (FDA) in 2013 revealed that, on the basis of in vitro tests performed during drug development, almost all new molecular entities had been found to be perpetrators of metabolic interactions. In particular, 77% inhibited at least one CYP enzyme, and 29% induced CYP enzyme expression to some extent. In addition, 45% of the new molecular entities had been found to be victims of clinically significant metabolic DDIs.
The wide interindividual variability in the magnitude of drug interactions due to enzyme inhibition or induction makes it very difficult to manage such interactions by dose adjustment since, as pointed out by Obach et al[10], there is no proven in vitro method consenting a quantitative prediction of the alteration in exposure to the victim drug. Although more complex and refined predictive models have subsequently been proposed and endorsed by the FDA regulatory guidance[11], it has recently been pointed out that they do not improve predictive capacity[12].
Genetic factors (enzyme polymorphisms), physiological and environmental variables (age, sex, diet, co-medication) as well as various pathological conditions may concur in determining the wide interindividual variability of metabolism-based DDIs. As shown by Christensen et al[13], any displacement from plasma protein-binding sites is a further factor influencing the magnitude of metabolic DDIs. Factors influencing the magnitude of metabolism-based interactions have been reviewed in the past[14-17]. However, only one review focused on the effect of liver disease on both enzyme inhibition and induction[18]. The present review is an update that includes new findings and reinterprets some results in light of newly-acquired knowledge.
The mechanisms underlying altered drug disposition in liver disease are not known with certainty. Therefore, the effect of liver insufficiency on metabolism-based DDIs is difficult to predict. In this review, we interpret the observed liver effects on metabolic DDIs on the basis of the mechanisms proposed to account for altered drug handling in liver disease. Therefore, current theories aiming to explain the influence of liver dysfunction on hepatic drug disposition will be summarized firstly.
MECHANISMS UNDERLYING ALTERED HEPATIC DRUG HANDLING IN LIVER DISEASE
A large number of studies have shown that impairment of drug disposition is strictly correlated with the type and severity of liver disease. In patients with chronic hepatitis without cirrhosis, or primary or secondary liver cancer, hepatic drug metabolism is generally comparable to that of subjects with normal liver function[19]. By contrast, in liver cirrhosis, hepatic drug clearance is in most cases significantly reduced. Therefore, most of the published literature is concerned with the impairment of drug disposition in liver cirrhosis.
Unlike the kidney, for which creatinine clearance is a quantitative measure of renal excretory ability, no single endogenous marker is a reliable index of the degree of liver function impairment and its drug-metabolizing ability. Various classification systems have been developed to characterize the degree of liver injury and predict the prognosis of patients with cirrhosis[20-22]. Because of their fairly good predictive value of hepatic drug-metabolizing ability, the Child[23] and Pugh[24] classifications, which are based on a combination of clinical features (ascites, encephalopathy, and nutritional status) and laboratory variables (serum albumin, bilirubin, and the international normalized ratio of prothrombin time), are currently used in clinical studies aiming to correlate the degree of liver dysfunction with impairment of hepatic drug disposition. According to the Child classification, liver cirrhosis is graded as A, B, or C, in order of increasing severity. Child grade A patients have well preserved liver function (normal or nearly normal values of liver function tests); Child grade B patients have altered values of these tests, but minimal encephalopathy and easily controlled ascites, whereas patients with Child grade C (decompensated) cirrhosis have grossly altered values of liver function tests, and severe encephalopathy and ascites are present. In the Pugh system, a numerical score is given to each clinical feature or biochemical test. Overall scores of 5-6, 7-9, and 10-15 correspond to Child’s grade A, B, and C liver cirrhosis, respectively.
On the basis of experimental observations of the effect of liver cirrhosis on drug disposition and the morphological and functional changes associated with this disease, four mechanisms have been proposed whereby liver cirrhosis can alter hepatic drug handling[19]. The basic tenets of these theories will be briefly summarized, and experimental evidence supporting or disproving each of these theories will be discussed.
Intact hepatocyte theory
According to this theory, the alteration in hepatic drug elimination is the consequence of a reduced mass of hepatocytes, the function and perfusion of which remain normal. Depending on their hepatic elimination characteristics, drugs can be divided into two major groups[25]: (1) drugs with high hepatic extraction ratio (EH > 0.7), which are referred to as flow-limited drugs because their clearance is mainly dependent on the rate of delivery of the drug to the liver, i.e., liver blood flow; and (2) drugs with low hepatic extraction ratio (EH < 0.3), termed capacity-limited drugs because their clearance is primarily dependent on the metabolic capacity of the liver. The intact hepatocyte theory implicitly assumes that the reduction in clearance is the same for drugs with high or low extraction ratio, i.e., irrespective of whether their clearance is limited by blood flow or the intrinsic metabolic ability of the liver, respectively. Experimental evidence both consistent and inconsistent with this theory has been obtained. On the one hand, the total hepatocyte number in liver biopsies of cirrhotic patients was found to be reduced by a mean of 41% with respect to non-cirrhotic subjects, and the clearances of antipyrine and aminopyrine (model compounds with very low extraction ratio (EH about 0.05) used to assess hepatic metabolic efficiency[26]) were found to be proportionally reduced (by about 40%). On the other hand, the hepatic disposition of flow- and capacity-limited drugs was reported to be affected to different extents in the same cirrhotic patients[19]. In addition, this theory cannot explain the preferential reduction of Phase I (oxidative) metabolic reactions compared to Phase II (conjugative) biotransformations.
Sick cell theory
This theory states that liver disease is associated with a decline in hepatocyte function; therefore, it implicitly predicts a reduction in enzyme and/or transporter activity measured in vitro. Although drug transport is a significant contributor to hepatic drug disposition, no information is yet available about the effect of cirrhosis on hepatic drug transport. Accordingly, the following discussion will be necessarily limited to alterations in drug-metabolizing reactions.
Consistent with the sick cell theory, numerous animal and human studies have shown that the hepatic levels of CYP proteins and related enzyme activities are reduced in proportion to the severity of liver cirrhosis. In addition, differential reductions in both the content and activity of CYP enzymes have been reported. For example, protein content and activity of CYPs 1A2, 2C9, and 3A4 decrease to a greater extent and a faster rate than those of CYPs 2D6 and 2E1[25]. As the decline of CYP activities is generally correlated with a decrease in corresponding mRNA levels, inhibition of transcriptional activity by the cirrhosis-associated increase in inflammatory cytokines is considered responsible for CYP downregulation[27].
Pharmacokinetic theory predicts that a decrease in liver metabolic activity reduces hepatic clearance of drugs with low-extraction ratio (capacity-limited drugs) to a significantly greater extent than that of highly extracted (flow-limited) drugs. Thus, if the cirrhosis-associated impairment of hepatic drug disposition is due to reduced enzyme content of hepatocytes (as assumed by the sick cell theory), the clearance of capacity-limited drugs should be more profoundly reduced than that of flow-limited drugs. Contrary to this prediction, one study showed that the clearances of propranolol and antipyrine (flow- and capacity-limited drugs, respectively) were affected in a quantitatively similar fashion in a rat model of cirrhosis[28]. This result has been replicated by clinical studies, which showed that the metabolic clearances of the highly and poorly extracted drugs lidocaine and theophylline, respectively, were reduced in a parallel way and to a virtually identical extent in cirrhotic patients[29,30]. These results indicated that other factors, besides reduced metabolic activity of hepatocytes, must be responsible for the impaired elimination of drugs with flow-dependent clearance.
Impaired drug uptake theory
Because of the very large endothelial fenestrations, the sinusoidal capillaries do not constitute an appreciable barrier to the transfer of drugs and proteins from blood to the space of Disse. Sinusoidal capillarization, i.e., cirrhosis-associated occlusion of fenestrations and formation of a basal lamina, converts the microvascular bed of liver into a two-barrier system, as in the other organs. The impaired drug uptake theory assumes that capillarization of sinusoids limits drug uptake by the hepatic parenchyma, especially of those drugs that are highly bound to plasma proteins. Consistent with this assumption, a study of the highly protein-bound drug propranolol showed that in cirrhotic patients the reduction of the hepatic extraction ratio of this drug could not be ascribed to a decreased diffusion of the free drug but rather to an impeded transfer across the endothelial barrier of protein-bound propranolol[31].
This theory also predicts that the hepatic clearance of a highly diffusible and poorly bound drug, such as antipyrine, should be much less affected by cirrhosis than that of a highly bound drug, such as propranolol. However, contrary to this prediction, the in vivo clearances of these two drugs were found to be reduced to the same extent by cirrhosis[28].
Oxygen limitation theory
This theory assumes that capillarization of the sinusoids reduces oxygen transfer from blood to the hepatocytes. This hypothesis was prompted by two types of experimental evidence: (1) reduced oxygen uptake by the isolated cirrhotic liver and cirrhotic animals[32]; and (2) the generally marked reduction of oxidative Phase I biotransformations, compared with the substantial preservation of Phase II (conjugative) metabolic reactions, which have a lower sensitivity to reduced oxygen concentration and are not significantly decreased until end-stage liver disease is reached[33]. Consistent with this hypothesis, the clearance of theophylline, which undergoes CYP-mediated oxidative metabolism, was shown to be reduced by 37% in cirrhotic rats and restored to normal values by oxygen supplementation[34]. However, these results were not replicated in a similar study of cirrhotic patients[35]. In conclusion, although some in vitro and animal data support this theory, no clear evidence has yet been obtained that sinusoidal capillarization reduces oxygen transfer in humans.
Reduced binding to plasma proteins constitutes an additional mechanism by which liver cirrhosis can modify hepatic drug elimination. The concentrations of both proteins mainly responsible for drug binding [albumin and α1-acid glycoprotein (AAG)] are substantially reduced in severe cirrhosis[36], causing a significant increase in the free level of highly bound drugs. As discussed in detail below, this may result in a paradoxical increase in clearance based on total drug concentration.
Although each of the mechanisms thus far proposed in order to explain the reduction of hepatic drug disposition in liver disease has some experimental support, the above considerations make it clear that no single mechanism can explain the altered disposition of all drugs. A plausible explanation for the observed inconsistencies is that multiple mechanisms generally concur in determining the reduction of drug elimination by the diseased liver, and their relative importance varies according to the physicochemical and pharmacokinetic characteristics of each drug.
ENZYME INHIBITION: MECHANISMS AND BIOLOGICAL VARIABILITY
Information regarding the molecular mechanism of interaction between DMEs and inhibitors has been obtained prevalently from studies involving CYP enzymes. The biochemical mechanisms by which drugs inhibit these enzymes are classically divided into two main classes: reversible and irreversible.
Reversible inhibition
This inhibition results from rapidly reversible binding of a lipophilic drug molecule to a hydrophobic substrate-binding region or to the unoccupied sixth ligand position of the prosthetic heme iron[37]. All the kinetic types of inhibition, i.e., competitive, noncompetitive, and uncompetitive[38], as well as allosteric inhibition[39] have been observed following reversible binding.
Irreversible or mechanism-based inhibition
This type of inhibition can be the result of two distinct chemical mechanisms: formation of a metabolic intermediate (MI) complex and autocatalytic inactivation[37]. In the former case, inhibition is due to the formation of a metabolite that binds tightly to the reduced heme moiety, thereby forming a stable inactive complex with the enzyme, called the MI complex. In the latter case, the enzyme converts the bound drug to a chemically reactive metabolite that binds covalently to the catalytic site and destroys the structure of the hemoprotein. For this reason, this mechanism is also referred to as suicide inactivation. In contrast to suicide inactivation, MI complexation does not cause a permanent alteration of the hemoprotein structure. However, from a clinical point of view, there is no difference between the two mechanisms, because both cause irreversible loss of the inhibited enzyme, and restoration of in vivo enzyme activity requires enzyme re-synthesis. Because of their long-lasting effect, irreversible inhibitors cause generally more dangerous clinical effects than reversible ones[40]. They are also much more selective than reversible inhibitors, as only one or very few CYP can generate chemically reactive metabolites or form MI complexes with a given drug. For example, erythromycin and ethinylestradiol are selective inhibitors of CYP3A4, because only this CYP isoform generates a MI complex with the former drug and a chemically reactive metabolite with the latter[40].
The liver content of the inhibited enzyme is the most important factor determining the magnitude of inhibitory DDIs and, consequently, their clinical relevance. Whenever the inhibited metabolic pathway is not the unique route of elimination, its contribution to the overall elimination of a drug depends on the hepatic content of the relevant enzyme. Hence, the lower the enzyme content, the lesser the fractional inhibition of total drug clearance. Any factor decreasing the expression of a DME is therefore expected to reduce the importance of its inhibition. Marked interindividual variations have been observed in the hepatic content of DMEs, with consequent wide variability in the magnitude of DDIs. The biological factors affecting enzyme expression, which have been investigated mainly by studies of CYP-mediated biotransformations, can be divided in two categories: genetic polymorphism and phenoconversion.
Genetic polymorphism
Various CYP enzymes show genetic polymorphism (defined as the presence of variant alleles in at least 1% of the population), which is the main cause of the wide interindividual variability in metabolic drug disposition. The marked differences in CYP expression levels between extensive (EM) and poor (PM) metabolizers also result in significant variability of the magnitude of inhibitory drug interactions (see Lee et al[15] for a review). Various observations have been made regarding the variability associated with the inhibition of the polymorphic enzymes CYP2C19 and CYP2D6. These studies have consistently shown that the magnitude of metabolic inhibition is markedly greater in EMs than in PMs. For example, co-administration of fluvoxamine, an inhibitor of CYP2C19, with the CYP2C19 substrate lansoprazole, resulted in a 4-fold increase in lansoprazole AUC (corresponding to 75% inhibition of its systemic clearance) in EMs, whereas it had virtually no effect in PMs[41]. An even greater difference was observed when quinidine, a potent CYP2D6 inhibitor, was co-administered with venlafaxine, since the oral clearance of the R enantiomer of venlafaxine, selectively metabolized by CYP2D6, was inhibited by more than 90% in EMs and virtually unaffected in PMs[42]. According to the authors of the above mentioned review[15], genotyping of polymorphic enzymes should become a mandatory aspect of drug development and the extent of inhibitory DDIs should be reported according to the metabolizer genotype. They also pointed out that available results advocate the need for genotyping in the clinical setting, since dose adjustments may be necessary only in EMs.
Phenoconversion
The wide inter-genotype differences in drug metabolism, which account for a substantial fraction of the variability in drug response, have prompted a large number of pharmacogenetic studies trying to establish possible associations between DME genotypes and clinical outcomes. These studies have almost invariably focused on the DME genotypes of the study population, with the assumption that genotypes predict their functional phenotypes. However, this assumption does not always hold since, contrary to the DME genotype that is immutable, DME phenotypes may be changed by environmental and physiological factors. The phenomenon whereby a genotypic EM is converted into a phenotypic PM is called phenoconversion[43]. A large body of evidence, obtained from both non-clinical and clinical studies, indicates that release of proinflammatory cytokines is the main cause of disease-associated phenoconversion, as these mediators downregulate the expression of certain DMEs, especially those of the CYP family[44]. Thus, reduced CYP levels are expected, and have been observed, in patients with infections or other inflammatory conditions, such as rheumatoid arthritis, sepsis, cancer, cardiac or renal failure, and liver disease[27,45]. Besides impairment of drug disposition, these diseases are expected to cause a decrease in the magnitude of inhibitory drug interactions. However, only for liver cirrhosis has this question been addressed by a series of studies carried out in our laboratory[29,30,46,47] (see below).
The expression and activity of various DMEs are also reduced at the extremes of age[48]. It has been observed that phenoconversion associated with aging is “strikingly similar” to that caused by cirrhosis, and the same mechanisms have been proposed to explain reduced drug metabolism[49]. However, only a few and so far inconclusive studies have been devoted to the quantitation of the magnitude of inhibitory DDIs in elderly subjects[49,50]. Ethical concerns generally preclude the analysis of inhibitory DDIs in children by means of prospective pharmacokinetic studies, since such studies entail the administration of a non-therapeutic agent. A recent comprehensive survey of the relevant literature[51], which compared the magnitude of reported metabolic DDIs in pediatric and adult populations, highlighted the paucity of data in children younger than 2 years and noted that magnitudes both somewhat higher and lower than in adults had been reported in children. This study concluded that no age-related trend in the magnitude of inhibitory DDIs could be established in children.
EFFECT OF CIRRHOSIS ON ENZYME INHIBITION
In principle, five types of factors can affect the extent of inhibitory DDIs in liver cirrhosis.
Reduced enzyme content
The expression of various CYP enzymes, particularly that of CYP1A2 and CYP3A4, has been shown to be markedly reduced in cirrhosis[25,52]. As discussed in detail above, reduced enzyme content is expected to result in a decreased inhibitory effect.
Hepatic extraction ratio of the drug with inhibited metabolism
Pharmacokinetic theory predicts that the hepatic clearance of drugs with a low extraction ratio (capacity-limited drugs) is reduced in proportion to the degree of enzyme inhibition, since the clearance of these drugs depends essentially on intrinsic clearance, i.e., the metabolic capacity of the liver. Vice versa, the clearance of drugs with a high extraction ratio (flow-dependent drugs), is almost exclusively determined by liver perfusion and should be virtually unaffected by a decrease in intrinsic clearance caused by enzyme inhibition.
In cirrhosis, intrahepatic shunts and sinusoidal capillarization cause a reduction of effective blood flow through the liver parenchyma. As a consequence, the flow-dependent clearance of drugs with a high extraction ratio is expected to become more and more capacity-limited as liver function worsens. These expectations were confirmed by Huet and Villeneuve[53], who showed that the extraction ratio of lidocaine was reduced from a mean control value of 0.64 to 0.31 in decompensated cirrhotics and that its clearance was no longer related to liver blood flow but rather became capacity-limited, that is dependent on the liver ability to metabolize the drug. These observations predict that the clearance of drugs with high extraction ratio becomes more and more sensitive to the action of metabolic inhibitors as liver dysfunction progresses.
Reduced liver uptake of the inhibitor
Reduced drug uptake by the cirrhotic liver has been observed in vitro for various structurally unrelated basic drugs[54].
Nature (reversible or irreversible) of the inhibitory interaction
The accumulation kinetics in the hepatocyte of reversible and irreversible inhibitors may be differentially affected by liver cirrhosis since reversibly binding molecules rapidly equilibrate between intra- and extracellular spaces, whereas binding of irreversible inhibitors is time-dependent and can proceed up to total enzyme inhibition if the inhibitor concentration exceeds that of the enzymatic protein.
Plasma protein binding of the drug with inhibited biotransformation
When the perpetrator of an interaction both inhibits the metabolism and causes displacement of the victim drug from its plasma protein-binding sites, displacement influences the magnitude of inhibitory DDIs[55].
In order to assess the influence of both reduced CYP expression and decreased liver perfusion on the magnitude of inhibitory DDIs in liver dysfunction, we examined the effect of cirrhosis on the inhibition of the metabolic disposition of a flow-dependent drug, lidocaine[29], and a capacity-limited drug, theophylline, using the reversible CYP1A2 inhibitor fluvoxamine[30]. Co-administration of therapeutic doses of fluvoxamine reduced the clearance of intravenously administered lidocaine on average by 60% in healthy subjects and caused a proportional prolongation of terminal half-life. Divergent results had been obtained by previous in vitro studies trying to identify the major CYP isoform responsible for lidocaine metabolism, as either CYP3A4[56] or CYP1A2[57] was suggested as the major CYP involved. The above result was the first in vivo demonstration that CYP1A2 is the main CYP enzyme responsible for lidocaine biotransformation. In patients with Child grade A cirrhosis, fluvoxamine inhibited lidocaine clearance to a significantly lower extent (40%), and no statistically significant modification of either clearance or half-life was produced in patients with Child grade C cirrhosis, in spite of the 3-fold higher steady-state plasma levels of fluvoxamine measured in this patient group (Table 1). Thus, contrary to theoretical predictions, the cirrhosis-associated conversion from flow-dependent to capacity-limited clearance does not increase the sensitivity to the action of metabolic inhibitors of drugs with flow-dependent clearance. Probably, the factors that reduce the sensitivity of drug clearance to metabolic inhibition overshadow the effect of clearance conversion. One such factor, the cirrhosis-associated decrease in hepatic CYP1A content (up to 80% in hepatocellular cirrhosis)[52], was proposed as the main determinant of the reduction of clearance inhibition, because a close correlation was observed between the decrease in clearance inhibition and the cirrhosis-associated decline in lidocaine clearance. A large reduction in CYP1A2 content drastically reduced its contribution to the overall lidocaine elimination, thereby minimizing the effect of its inhibition[29].
Table 1.
Differential inhibitory effects of fluvoxamine on systemic lidocaine and theophylline clearances, and on partial metabolic clearances of theophylline in healthy subjects and cirrhotic patients
| Parameter |
Healthy subjects |
Patients with cirrhosis |
||||
|
Child A |
Child C |
|||||
| Placebo | Fluvoxamine | Placebo | Fluvoxamine | Placebo | Fluvoxamine | |
| CL lidocaine | 12.10 ± 3.38 | 4.85 ± 4.85c | 9.83 ± 3.99 | 5.06 ± 1.75c | 4.21 ± 1.56e | 3.65 ± 0.99 |
| Inhibition (%) | 60 (53-77) | 44 (28-59) | 9 (0-19)e | |||
| Fluvoxamine AUC0-14 | 866 ± 331 | 1277 ± 381 | 2316 ± 790d | |||
| CL theophylline | 0.825 ± 0.201 | 0.302 ± 0.080c | 0.660 ± 0.231 | 0.310 ± 0.104a | 0.296 ± 0.124d | 0.251 ± 0.151 |
| Inhibition (%) | 64 (59-69) | 55 (46-66) | 14 (0-29) | |||
| CLf 3-MX | 0.192 ± 0.069 | 0.008 ± 0.007c | 0.124 ± 0.051 | 0.011 ± 0.017c | 0.037 ± 0.012d | 0.023 ± 0.013a |
| Inhibition (%) | 97 (93-100) | 92 (84-100) | 38 (18-58) | |||
| CLf 1-MU | 0.220 ± 0.089 | 0.021 ± 0.019c | 0.178 ± 0.072 | 0.026 ± 0.018c | 0.066 ± 0.025d | 0.039 ± 0.019a |
| Inhibition (%) | 93 (89-98) | 83 (73-93) | 36 (15-57) | |||
| CLf 1,3-DMU | 0.290 ± 0.070 | 0.136 ± 0.024c | 0.222 ± 0.058 | 0.138 ± 0.026c | 0.087 ± 0.040d | 0.079 ± 0.037 |
| Inhibition (%) | 58 (49-67) | 43 (29-58) | 7 (0-22) | |||
| CLR theophylline | 0.123 ± 0.033 | 0.136 ± 0.030 | 0.136 ± 0.050 | 0.135 ± 0.043 | 0.106 ± 0.047 | 0.110 ± 0.062 |
| Inhibition (%) | - | - | - | |||
| Fluvoxamine Css | 75 ± 26 | 95 ± 40 | 130 ± 26b | |||
Data are taken or calculated from Orlando et al[29] 2004 and Orlando et al[30] 2006; all clearance values are expressed as mL/min per kg; inhibition values are given as means with 95% confidence limits.
P < 0.01,
P < 0.001 vs placebo;
P < 0.001 vs healthy subjects;
P < 0.01,
P < 0.001 vs healthy subjects and Child’s grade A patients. CL: Systemic clearance; CLf: Formation clearance; 3-MX: 3-methylxanthine; 1-MU: 1-methyluric acid; 1,3-DMU: 1,3-dimethyluric acid; CLR: Renal clearance; AUC0-14: Area under plasma-concentration-time curve from 0 h to 14 h at steady state (ng/h/mL); Css: Steady-state trough concentration (ng/mL).
In a subsequent study[30], the influence of cirrhosis on the inhibitory effect of fluvoxamine was investigated using theophylline as a victim drug, since this drug is a CYP1A2 substrate with a very low extraction ratio (0.1[58]), i.e., with hepatic elimination characteristics opposite to those of lidocaine, which has an extraction ratio of about 0.7[59]. In this study, an identical protocol was used for fluvoxamine co-administration; whereas, due to safety concerns, theophylline had to be administered as an oral hydro-alcoholic solution. However, its oral bioavailability following administration as a solution is virtually complete (98%-100%[60,61]), which makes it possible to determine real pharmacokinetic parameters. Theophylline was also chosen because, unlike lidocaine whose primary metabolites undergo further biotransformations, its metabolites are directly excreted into the urine. This offers the considerable advantage of evaluating the effect of liver cirrhosis on the inhibition of the formation clearance of each metabolite. In spite of the much lower extraction ratio, the extent of theophylline clearance reduction by fluvoxamine was very similar to that observed with lidocaine in healthy subjects (64% and 60%, respectively). The degree of clearance inhibition also decreased in a quantitatively similar fashion with the progression of liver cirrhosis, since the slopes of the regression lines for the correlation between degree of clearance inhibition and Pugh’s score exhibited very close values: -5.4 (95%CI: -6.8 to -4.1) for lidocaine and -6.2 (95%CI: -7.3 to -5.0) for theophylline. The quantitative similarity of the effects of cirrhosis on the inhibition of lidocaine and theophylline clearances can be further appreciated from Table 1.
As shown in Figure 1, three metabolic pathways concur in theophylline metabolism. Formation of 1-methylxanthine (1-MX) and 3-methylxanthine (3-MX) is catalyzed almost exclusively by CYP1A2, whereas generation of 1,3-dimethyluric acid is also due (by about 40%) to CYPs 2E1 and 3A4. 1-MX is readily oxidized by xanthine oxidase and measured as 1-methyluric acid (1-MU) in urine. A comparison of the inhibitory effects of fluvoxamine on the formation clearance of theophylline metabolites in healthy subjects and cirrhotic patients (Table 1) provides a deeper understanding of the mechanisms whereby cirrhosis decreases the magnitude of inhibitory interactions. The formation clearances of the two metabolites formed exclusively by CYP1A2 (3-MX and 1-MU) were inhibited by almost 100% in healthy subjects. The degree of inhibition decreased drastically in cirrhotic patients, since the formation clearances of 3-MX and 1-MU were only inhibited by about a third in patients with Child grade C cirrhosis. A simple calculation shows that if the residual CYP activity in these patients were inhibited to the same extent as in subjects with normal liver function, their systemic clearance in the presence of fluvoxamine would be 0.150 mL/min per kg instead of 0.251 mL/min per kg. Hence, clearance inhibition would be about 50%, instead of 14% as actually observed. These considerations make it clear that cirrhosis-associated reduction in the extent of inhibition cannot be ascribed only to decreased hepatic content of CYP1A2 but is also due to a “reduced sensitivity” of residual CYP1A2 activity to the inhibitory action of fluvoxamine.
Figure 1.
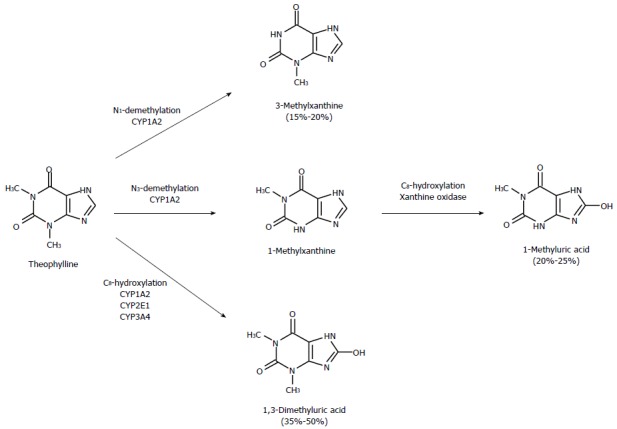
Metabolic pathways of theophylline. Numbers in parentheses indicate percentages of urinary metabolites in adult humans. Theophylline is hydroxylated at the C8 position to form 1,3-dimethyluric acid and demethylated at the N1 and N3 positions, to yield 3-methylxanthine and 1-methylxanthine, respectively. The latter undergoes subsequent oxidation, by xanthine oxidase, to 1-methyluric acid. CYP1A2 is responsible for about 80% of theophylline metabolism[30].
Of the mechanisms thus far proposed to explain impaired drug handling in liver disease (see above), only impaired drug uptake, with consequent reduction in the intracellular concentration of the inhibitory agent, provides a plausible explanation for the decreased inhibition of CYP1A2 activity in cirrhotic patients. In Child C patients, reduced inhibition of residual enzyme activity appears to be the main determinant of the attenuating effect of cirrhosis on the extent of fluvoxamine-theophylline interaction, since the degree of clearance inhibition would not be reduced from 64% to 14% (Table 1) but only to 50% if this mechanism were not operative. Thus, reduced liver content of CYP1A2 and reduced inhibition of its residual activity appear responsible for about 30% and 70%, respectively, of the decrease in the extent of this inhibitory interaction.
The observations that emerged from the analysis of this DDI have both clinical and mechanistic implications, as they indicate that, First, therapeutic doses of fluvoxamine cause almost complete inhibition of CYP1A2, thereby virtually abolishing the CYP1A2-mediated metabolic disposition of any drug in subjects with normal liver function. This may require substitution or substantial dose reduction of the victim drug in healthy subjects, whereas no dose modification may be necessary in patients with severe liver disease, in which the effect of CYP1A2 inhibition is hardly significant. Secondly, although indirect, evidence is provided supporting the hypothesis that reduced drug uptake is the main mechanism affecting hepatic drug handling in cirrhosis since, of the four mechanisms thus far proposed, only reduced drug uptake can explain both reduced hepatic clearance and the decreased inhibitory effect.
Most clinically relevant inhibitory interactions are the consequence of mechanism-based CYP inactivation, especially of CYP3A4[62,63], which metabolizes about 50% of drugs presently on the market. Since an early in vitro study had identified CYP3A4 as the enzyme mainly responsible for lidocaine metabolism[56] and the expression level of this CYP isoform had been reported to be markedly reduced in hepatocellular cirrhosis[52], our first study examining the cirrhosis-associated variability of inhibitory DDIs investigated the inhibition of lidocaine disposition by erythromycin[46]. As mentioned above, this antibiotic causes selective irreversible inhibition of CYP3A4 due to its conversion to a nitroso-alkane metabolite that forms a stable MI complex with this CYP enzyme. Contrary to expectations, pretreatment of subjects with repeated doses of erythromycin until steady state was attained, reduced lidocaine clearance by only 18% in healthy volunteers. As subsequently shown[29], this was because CYP3A4 plays a minor role in lidocaine metabolism. A very similar (about 20%) degree of inhibition was recorded in patients with either Child grade A or grade C liver cirrhosis. Although these findings indicated that cirrhosis does not affect the extent of irreversible inhibitory effects, the baseline low degree of clearance inhibition was such as to preclude the observation of any substantial reduction in the magnitude of this inhibitory interaction in cirrhotic patients. Therefore, a study was planned[47] that examined the inhibitory effect of erythromycin on the metabolic disposition of quinine, a validated probe of CYP3A4, since its major metabolite, 3-hydroquinone, is generated exclusively by this CYP isoform[64,65]. Quinine was also selected because, like theophylline, it has an absolute oral availability close to 100%[66], which makes it possible to determine the true values of its pharmacokinetic parameters. This drug has a low extraction ratio and is extensively bound in plasma (about 90%), mostly to AAG[67]. Therefore, it has a capacity-limited, binding-sensitive hepatic elimination, i.e., strictly dependent on the degree of plasma protein binding. In consideration of the possible decrease in the extent of quinine binding in plasma due to the cirrhosis-associated decrease of AAG concentration[68], and of the expected competition for protein binding sites with erythromycin, a drug exclusively bound to AAG[69], we determined both total and free quinine concentrations. This allowed us to evaluate the effect of erythromycin on both the free plasma concentration and unbound clearance of quinine which, for a capacity-limited drug, is equal to intrinsic clearance[70]. The data reported in Table 2 show that the inhibitory effect of erythromycin on the total plasma clearance of quinine decreased with the progression of liver cirrhosis (from 33% in healthy subjects to a paradoxical 7% increase in Child grade C patients). Thus, they seem to indicate that liver dysfunction exerts similar effects on the magnitude of irreversible (erythromycin) and reversible (fluvoxamine; Table 1) inhibitory interactions. However, in apparent contrast with these findings, the formation clearance of 3-hydroquinone was reduced to similar extents (by approximately 60%) in controls and cirrhotic patients. These apparently discrepant observations are explained by considering the effects of erythromycin on protein binding and unbound (intrinsic) clearance of quinine. As also shown in Table 2, the degree of quinine plasma protein binding decreased and the extent of its displacement by erythromycin increased significantly (from 41% to 76%) with the severity of liver cirrhosis. A close correlation (r = 0.87, P < 0.001) was found between the increase in the displacing effect and the cirrhosis-associated increase of quinine free fraction. This observation is fully consistent with theoretical considerations predicting that the lesser the plasma-binding capacity, the greater the extent of displacement by a competitor of the bound drug[13]. The erythromycin effect on unbound (intrinsic) clearances, which is the only true measure of inhibition of quinine metabolism in the presence of changes in plasma protein binding, clearly indicated that irreversible inhibition of CYP-mediated metabolism is independent of liver functional status, since the unbound formation clearance of 3-hydroxyquinine was inhibited to virtually identical extents (by about 75%) in subjects with normal and impaired liver function. This indicated that, contrary to reversible binding, irreversible inhibitor binding to the target enzyme was not reduced by the morphological and functional changes caused by liver cirrhosis. The limited cirrhosis-associated decrease in the magnitude of the inhibition of unbound systemic clearance of quinine (about 30%) was consistent with the decreased liver content of CYP3A4 which, like CYP1A2, is markedly reduced in hepatocellular cirrhosis[52].
Table 2.
Effect of erythromycin on the pharmacokinetic parameters of quinine and 3-hydroxyquinine in healthy subjects and cirrhotic patients
| Parameter |
Healthy subjects |
Patients with cirrhosis |
||||
|
Child A |
Child C |
|||||
| Placebo | Erythromycin | Placebo | Erythromycin | Placebo | Erythromycin | |
| CL quinine | 1.775 ± 0.310 | 1.161 ± 0.309a | 1.504 ± 0.362 | 1.040 ± 0.303a | 1.281 ± 0.394b | 1.401 ± 0.534 |
| Inhibition (%) | 33 (27-38) | 30 (26-34) | -7 (-16-2)f | |||
| CLf 3-hydroxiquinine | 0.174 ± 0.051 | 0.059 ± 0.017c | 0.140 ± 0.077 | 0.056 ± 0.032c | 0.085 ± 0.028d | 0.037 ± 0.023c |
| Inhibition (%) | 65 (61-70) | 61 (56-67) | 59 (46-72) | |||
| Unbound quinine (%) | 6.7 ± 0.8 | 9.4 ± 1.0c | 7.1 ± 1.6 | 10.3 ± 2.8c | 11.9 ± 3.7f | 19.9 ± 5.5cf |
| Increase (%) | 41 (28-55) | 45 (33-55) | 76 (42-111)b | |||
| CLu quinine | 26.81 ± 11.49 | 12.30 ± 3.16c | 21.18 ± 8.45 | 10.10 ± 3.88c | 10.95 ± 2.19f | 7.24 ± 2.77bc |
| Inhibition (%) | 51 (44-59) | 50 (45-55) | 35 (21-48)b | |||
| CLuf 3-hydroxiquinine | 2.63 ± 0.88 | 0.64 ± 0.20c | 1.97 ± 1.09 | 0.55 ± 0.31c | 7.01 ± 3.33f | 4.06 ± 2.58ad |
| Inhibition (%) | 75 (70-80) | 73 (70-77) | 74 (64-84) | |||
| Erythromycin Css | 1.33 ± 0.25 | 1.18 ± 0.44 | 1.85 ± 1.00 | |||
Data are taken or calculated from Orlando et al[47]; all clearance values are expressed as mL/min per kg; inhibition or increase values are given as means with 95% confidence limits.
P < 0.01,
P < 0.001 vs placebo;
P < 0.05 vs healthy subjects and patients with Child grade A cirrhosis;
P < 0.001 vs healthy subjects and P < 0.05 vs patients with Child grade A cirrhosis;
P < 0.001 vs healthy subjects and patients with Child grade A cirrhosis. CL: Systemic clearance; CLf: Formation clearance; CLu: Unbound systemic clearance; CLuf: Unbound formation clearance; Css: Steady-state trough concentration (μg/mL).
The total plasma clearance of a capacity-limited, binding-sensitive drug such as quinine is directly proportional to its free fraction[70]. Therefore, any factor causing an increase in unbound drug concentration (displacement interaction, decreased albumin or AAG plasma level) also causes an increase in clearance calculated on the basis of total plasma concentration. Thus, the cirrhosis-associated increase in the effectiveness of erythromycin as a displacer of quinine from plasma protein-binding sites (Table 2) causes an increase in clearance that masks the decrease in quinine clearance caused by inhibition of its metabolism. This explains the apparently paradoxical observation that the inhibitory effect of erythromycin on total plasma clearance of quinine progressively disappears as liver function worsens, despite its inhibitory effect on the metabolic disposition of quinine (unbound formation clearance of 3-hydroxyquinine) remains unaltered.
The results of the studies thus far performed indicate that reversible and irreversible enzyme inhibitions are differentially affected by liver cirrhosis. The magnitude of DDIs caused by reversible CYP inhibition decreases progressively to clinically negligible levels as liver function worsens. This is a general phenomenon, independent of the pharmacokinetic characteristics of the victim drug, since it is observed with drugs with both flow-dependent (i.e., lidocaine) and capacity-limited (i.e., theophylline) hepatic clearance. Two of the four possible mechanisms by which liver cirrhosis alters hepatic drug handling appear responsible for the decreased extent of the reversible inhibitory effect: (1) reduced hepatic uptake of the inhibitory drug, which is the quantitatively more important mechanism, and is consistent with a previous observation that various structurally unrelated drugs are taken up to a reduced extent by the isolated cirrhotic liver[54]; and (2) reduced expression of the inhibited CYP enzyme, since the hepatic content of various CYP isoforms is markedly decreased in cirrhosis[25,52]. In contrast with reversible inhibition, the degree of irreversible inhibition is not decreased by liver dysfunction. As observed with reversible inhibition, this conclusion holds irrespective of the hepatic elimination characteristics of the victim drug, because it is based on observations with both a flow-dependent (lidocaine) and a capacity-limited drug (quinine)[46,47]. Thus, only the less important mechanism, namely reduced hepatic content of the inhibited enzyme, may be operant in decreasing the magnitude of DDIs due to irreversible enzyme inhibition. It is worth noting that, in the two cases in which the contribution of this mechanism could be quantified[30,47], it appeared responsible for a similar (about 30%), limited reduction in the magnitude of the inhibitory interaction.
The analysis of the erythromycin-quinine interaction has also provided the first experimental demonstration that the decrease in plasma protein concentration associated with cirrhosis increases significantly the magnitude of drug interactions consequent to plasma protein-binding displacement. As displacement masks the consequences of enzyme inhibition, the effect on the total plasma clearance of the victim drug by any perpetrator causing both metabolic inhibition and plasma protein-binding displacement, will decrease with the increase in liver dysfunction. However, the free concentration of the victim drug will increase to hardly predictable levels. This has important clinical consequences when a drug causing plasma protein-binding displacement and metabolic inhibition is co-administered to a patient already stabilized with an appropriate drug dose, since the perpetrator will produce a dramatic increase in the free concentration and, consequently, the effect of the victim drug, in proportion to the degree of liver dysfunction. In such cases, only measurement of steady-state free concentration will allow an appropriate dose adjustment.
ENZYME INDUCTION
Induction is an adaptive response of mammalian organisms to the exposure to lipophilic substances, which results in increased disposition of xenobiotics by upregulation of drug-metabolizing systems and transporters. Enzyme induction is often defined as enhanced transcription and increased synthesis of drug metabolizing enzymes. Although prevalent, this is not the only mechanism responsible for the increased metabolic disposition of xenobiotics. Therefore, enzyme induction may be more generally defined as the increased amount or activity of a drug metabolizing enzyme following exposure to foreign substances, irrespective of the underlying mechanism, which may be of three types: pre-translational (enhanced transcription), translational (stabilization of the RNA transcript), and post-translational (stabilization of the enzymatic protein)[71]. Since the relevance of enzyme induction to xenobiotic metabolism has been recognized for more than 50 years, long before the underlying molecular mechanisms were unraveled, inducers have been traditionally classified on the basis of the spectrum of enzyme activities they increased. Starting from 1998, various experimental observations showed that enzyme induction is mainly the result of enhanced transcription due to the activation of transcription factors belonging to the superfamily of nuclear receptors. Following ligand binding, the ligand-receptor complex, which is usually localized to the cytoplasm, translocates to the nucleus, where it dimerizes with other transcription factors and interacts with coactivator proteins, thereby inducing the transcription of a battery of target genes[72-74]. Detailed knowledge of the properties and functions of these nuclear receptors now consents a classification of enzyme induction based on the type of receptor involved in the induction process. Seven nuclear receptors, described below, have been found to be involved in enzyme induction.
The aryl hydrocarbon receptor (AhR) is a member of the bHLH-PAS family of transcription factors[16]. Once activated, typically by pollutants such as dioxin and polycyclic aromatic hydrocarbons, this cytosolic receptor translocates into the nucleus where it dimerizes with another bHLH-PAS factor, the Ah receptor nuclear translocator (Arnt), and primarily induces the expression of genes encoding the CYP1A and 1B subfamilies of CYP enzymes.
The constitutive androstane receptor (CAR) is often referred to as an orphan receptor, since no endogenous high-affinity ligand is yet known. Only very few ligands, such as the antimycotic drug clotrimazole, bind directly to human CAR. Classical CAR activators, such as phenobarbital, do not bind to CAR, but stimulate indirectly its translocation from the cytoplasm to the nucleus, where CAR dimerizes with the retinoid X receptor. More than 70 enzymatic and non-enzymatic proteins have been reported to be induced via CAR activation, including CYP2B and CYP3A subfamilies and various phase II enzymes such as glutathione-, glucuronyl- and sulfo-transferases[75].
The pregnane X receptor, which has approximately 40% identity with CAR in the ligand-binding domain, is activated by a great variety of structurally unrelated xenobiotics and endobiotics. Unlike CAR, a direct correlation between ligand binding and receptor activation has been shown for PXR. This nuclear receptor exhibits species-specific activation due to sequence differences in its ligand-binding domain. For example, pregnenolone 16-αcarbonitrile, which is the prototypical agent causing PXR-mediated induction of rat CYP3A, does not induce this CYP subfamily in rabbit or humans. Conversely, rifampicin, the most potent inducer of rabbit and human CYP3A, does not activate PXR and, consequently, does not induce CYP3A in rats. PXR activates the transcription of various genes encoding DMEs such as the CYP3A subfamily, CYP2B6, CYPs 2C8 and 2C9, some Phase II enzymes such as glutathione-S-transferases, and transporters, such as multidrug resistance protein 1, multidrug resistance-associated protein 2, and organic anion transporting polypeptide 2 (OATP2), which are also under CAR regulation. Thus, CAR and PXR exhibit considerable cross-regulation of their target genes. In addition, several substances can activate both receptors. These largely overlapping specificities make it difficult to identify the precise mechanism of any inducer acting through activation of these nuclear receptors[75].
The peroxisome proliferator activated receptor (PPAR) family is comprised of three members: α, β, and γ. PPARα, which is localized to the nucleus, is the receptor of the fibrate class of the hypolipidemic drugs. In addition to fatty acid-metabolizing enzymes, PPARα induces the CYP4A subfamily of enzymes[76].
The glucocorticoid receptor (GR) regulates hundreds of genes, either directly by binding to the glucocorticoid response element of target genes, or indirectly, through an interaction with other transcription factors. As far as drug metabolism is concerned, GR positively regulates PXR expression, which plays an important role in the induction of CYP3A enzymes[77,78]. Recently, several CYPs as well as P-glycoprotein, have also been found to be directly regulated by GR[79].
The farnesoid X receptor (FXR) and the vitamin D receptor (VDR) are activated by bile acids and vitamin D, respectively. The former is considered a master regulator of bile acid homeostasis[80]; the latter, in addition to bone metabolism and calcium homeostasis, regulates cell growth and differentiation, immunomodulation, and other hormonal systems[81]. A limited involvement in CYP induction has been thus far demonstrated for these nuclear receptors; specifically, a binding site for FXR has been shown in the regulatory region of the CYP3A4 gene[82], and a VDR-mediated induction of biotransformations catalyzed by CYP3A has been observed[83].
The “ethanol-type” of induction of CYP2E1 is an exception to the transcriptional mechanism, as it is not mediated by the activation of an intracellular receptor but is due to both translational and post-translational mechanisms, i.e., stabilization of CYP2E1 mRNA and protein. This stabilization is the result of the binding of this type of inducers (ethanol, isoniazide, acetone) to both mRNA and enzymatic protein with consequent prevention of their catabolism[75].
Like enzyme inhibition, the induction potential of new molecular entities and the susceptibility of their metabolism to prototypical inducers are assessed in early stages of drug development in order to predict dose adjustment. However, the extremely wide variability (up to hundreds of times in healthy subjects[84]) of inductive effects makes any quantitative prediction particularly difficult. The biological factors contributing to inter- and intra-individual variability in enzyme induction, which have emerged from human in vitro and in vivo studies, are briefly outlined below:
Genetic factors
Polymorphism of the genes coding for the transporters that mediate the influx or efflux of inducing agents. As with drug-metabolizing enzymes, this can result in altered expression or functionality of the encoded transporters and, consequently, in different intracellular levels of inducers transported by polymorphic carriers. For example, CYP3A4 induction by rifampicin has been found to be reduced in cells overexpressing the drug-efflux transporter P-glycoprotein[85].
Polymorphism of the genes encoding the induced enzyme, since it has been clearly shown that induction by rifampicin of the metabolism mediated by the polymorphic enzyme CYP2C19 is observable only in EMs, not in PMs[86]. This apparently indicates that the presence of a fully functional allele is a necessary condition for inducibility.
Genetic polymorphism of nuclear receptors. In consideration of the central role that nuclear receptors and coregulators play in enzyme induction, genetic mutations resulting in altered expression or function of these proteins are expected to exert a marked influence on inducibility. These expectations have been confirmed by studies correlating AhR phenotypes with CYP1A1 induction. The inducibility of this CYP isoform in human lymphocytes has been found to be positively correlated with the expression of AhR and its heterodimerization partner Arnt[87]. In addition, two phenotypes, high and low inducibility, have been observed regarding CYP1A1 in human placenta. This polymorphic induction was due to the presence of two distinct AhRs displaying more than 20-fold differences in their ligand binding affinity[88]. In contrast to AhR, limited and inconclusive information is available about PXR and CAR genetic polymorphisms and their influence on the inducibility of the numerous enzymes regulated by these two nuclear receptors[17].
Physiological and environmental factors
Although age-related induction of DMEs has long been studied, the question of whether aging reduces inducibility remains controversial. Reduced enzyme induction in the elderly was reported in an early study[89], but more recent investigations could find no difference in the extent of induction between old and young individuals (see, e.g., Dilger et al[90] and Fromm et al[91]). Conflicting results have also been reported regarding sex-related differences. For example, induction of midazolam metabolism, a measure of CYP3A4 activity, by rifampicin appeared greater in men if oral clearance was measured, whereas induction was greater in women when systemic clearance was taken as a measure of midazolam biotransformation[92]. This and other observations suggest that sex-related differences in induction may depend on the tissue (intestinal or hepatic) and the CYP isoform involved[17]. Thus, no generalization can as yet be made regarding the effect of sex on the induction response.
Many dietary compounds act on nuclear receptors as agonists or antagonists. For example, resveratrol, which is present in red wines and various fruits and vegetables, prevents induction of CYP1A enzymes by inhibiting the binding of activated AhR to the promoter sequences of CYP1A genes and/or by acting as a competitive antagonist of ligands that activate AhR[16]. Antagonism at the AhR ligand-binding site is also the mechanism by which dietary flavones and flavonols inhibit, at low concentrations, CYP1A induction[93]. Therefore, the type of diet is another contributing factor to the variability of enzyme induction.
Like enzyme inhibition, induction has been shown to be profoundly altered in disease states, especially in inflammatory conditions and liver insufficiency. As discussed earlier in relation to enzyme inhibition, increased cytokine levels associated with inflammation suppress the basal expression of most CYP enzymes. Evidence showing that cytokines reduce CYP inducibility has also accumulated. For instance, a study with human hepatocytes showed that interleukin-6 and tumor necrosis factor α inhibit the induction of CYP1A enzymes by β-naphthoflavone. Interleukin-6 also represses the induction of the CYP2B, CYP2C, and CYP3A subfamilies in consequence of a marked reduction in the expression of PXR and CAR[43].
Unlike enzyme inhibition, which has only recently been shown to be reduced in liver disease (see preceding section), the influence of liver functional status on enzyme induction has long been investigated. However, conflicting results have been reported, and no coherent picture has emerged. Thus, it is not surprising that two previous reviews came to the opposite conclusions that enzyme induction is greatly curtailed[94] or not impaired in severe liver cirrhosis[95]. The purpose of the present review is to re-analyze the data so far obtained and to identify the factors (biological and/or methodological) responsible for the observed differential alterations of DME inducibility caused by liver dysfunction.
EFFECT OF CIRRHOSIS ON ENZYME INDUCTION
AhR-mediated induction
Few studies investigated the effect of liver disease on AhR-mediated enzyme induction. The first animal study to address this question[96] examined the inducing effect of the AhR activator β-naphthoflavone in the classical rat model of experimental cirrhosis produced by prolonged administration of carbon tetrachloride (CCl4). Although cirrhosis was confirmed histologically in CCl4-treated rats, biochemical tests of liver function “were only marginally abnormal”, indicating mild liver dysfunction. Treatment with β-naphthoflavone increased the CYP1A1-mediated aryl hydrocarbon (benzo-α-pyrene, BP) hydroxylase activity of liver microsomes to similar extents in control and cirrhotic rats. A second study, which examined AhR-mediated induction in CCl4-treated rats with “compensated micronodular cirrhosis” found that 3-methylcholantrene induced 7-ethoxycoumarin-O-deethylase activity to similar levels in normal and cirrhotic rats[97]. Thus, the results of both studies indicated that AhR-mediated induction is well preserved in mild cirrhosis.
The effect of liver disease on AhR-mediated induction was subsequently investigated in two human studies[98,99] by examining the inducing effect of smoking, a known AhR activator[75]. Because of the difficulty of obtaining truly abstinent smokers, both studies had to evaluate the effect of smoking by means of between-group comparisons (smokers vs non-smokers), rather than by intra-individual comparisons. One study[98], which used the CYP1A2 probe substrate caffeine, found that caffeine clearance was 40% lower in a group of patients with mild-to-moderate alcoholic cirrhosis, compared with healthy subjects. Among both healthy and cirrhotic subjects, caffeine clearance was almost twice as high in smokers than non-smokers. The other study[99] evaluated the clearance of antipyrine, a non-specific CYP probe, since it was principally designed to assess the overall drug metabolizing capacity of the liver. This study, which examined a mixed patient population with chronic active hepatitis C, with or without cirrhosis, noted that antipyrine clearance was significantly higher (about 80%) in smoking than non-smoking patients. These results are difficult to interpret as the biochemical indices of liver function were not reported, and a control group was not included.
As exemplified by the studies reviewed above, induction is assessed in animals by determining the effect of the administered inducer on the activity of the liver enzymes of sacrificed animals. Moreover, by measuring the mRNA and protein expressions of the induced enzymes and the relevant nuclear receptors, animal studies consent an identification of the mechanism(s) of induction (transcriptional, translational, and/or post-translational) and of the possible cirrhosis-associated alterations. Because of ethical considerations and practical limitations, such determinations (in tissues obtained at biopsy) can rarely be performed in human studies, and the inductive effect is generally inferred from changes in the pharmacokinetic parameters of a metabolic probe. Therefore, human studies provide clinically relevant information but no insight into the mechanisms underlying the effect of liver disease on induction.
Regarding AhR-mediated induction, the mechanism of cirrhosis-associated alterations was not investigated by the two animal studies described above[96,97], because the techniques for measuring mRNA and protein expression were not yet currently available. Thus, the mechanism by which cirrhosis may alter AhR-mediated induction remain to be elucidated. Moreover, both animal and human studies evaluated AhR-mediated induction in mild or moderate liver cirrhosis, and their observation that induction was substantially preserved was to some extent predictable, as significant alterations in the expression and/or activity of hepatic DMEs have consistently been observed only in animals or human beings with severe liver dysfunction[25,52]. Therefore, these studies left unresolved the question of whether this type of induction is compromised in the decompensated state of cirrhosis. To clarify these questions, a study was undertaken in our laboratory[100], which assessed the effect of liver dysfunction on AhR-mediated induction by measuring the mRNA and protein expressions of AhR and CYP1A enzymes, as well as CYP1A activity in rats rigorously stratified according to the severity of liver cirrhosis. To this purpose, rats were treated with CCl4, a method which produces animals with either compensated (corresponding to Child grades A and B) or decompensated (corresponding to Child grade C) liver cirrhosis, depending on the length of exposure to CCl4. Induction was obtained by administration of the prototypical AhR ligand BP. BP treatment caused marked proportional increases in mRNA and protein expressions as well as in the enzymatic activities of CYP1A1 and CYP1A2 in rats with normal and mild to moderate liver cirrhosis. In contrast, induced mRNA and protein levels as well as enzymatic activities of both CYPs were significantly lower in rats with decompensated cirrhosis; moreover, both constitutive and induced protein expressions of AhR were markedly lower in this animal group. These results indicated that the AhR-mediated inducibility of CYP1A enzymes is strictly related to the degree of liver dysfunction as it is still preserved in compensated cirrhosis, whereas it is markedly reduced when decompensation occurs. These findings have also clarified the mechanism by which cirrhosis reduces AhR-mediated inducibility, since the observation that induced mRNA level, protein expression, and enzymatic activity are proportionally decreased in rats with severe cirrhosis indicates that induction is compromised at the transcriptional level due to reduced expression of AhR.
CAR- and PXR-mediated induction
In the following discussion, CAR- and PXR-mediated inductions are grouped together because the most frequently used inducer, phenobarbital, is an activator of both nuclear receptors[75]. The pioneering work of Marshall and McLean[101] evaluated the inducing effect of phenobarbital by comparing hepatic microsomal CYP content and pyramidon demethylation (a non-specific marker reaction) in five healthy rats and five rats with varying degrees of liver cirrhosis, as assessed by histological examination. Although the authors observed that inducibility decreased as the severity of cirrhosis increased, this investigation provided only qualitative results, since differences were not statistically significant. The inducing effect of phenobarbital was also assessed by the two animal studies described above that evaluated the AhR-mediated induction in rats with mild liver cirrhosis[96,97]. The effect of the specific PXR ligand pregnenolone 16-αcarbonitrile was also assessed in one of these studies[96]. Like AhR-mediated induction, CAR- and PXR-mediated inductive effects remained quantitatively unaltered in animals with compensated cirrhosis.
Numerous clinical studies have evaluated the effect of liver dysfunction on enzyme induction mediated by CAR and PXR. However, no clear picture has emerged from these studies, since they yielded very conflicting results. As most of these investigations date back to the 1960s and 1970s, they do not comply with the methodological standards now required for pharmacokinetic studies in patients with liver disease[102], and their discrepant conclusions can often be ascribed to methodological differences. For example, they either included or did not include a control group of healthy subjects. Moreover, few studies made intra-individual comparisons, i.e., before and after inducer treatment. Because of ethical concerns relating to the administration of repeated doses of a non-therapeutic inducing agent to patients with severe liver dysfunction, most studies had to rely on hepatopathic patients taking an inducer for therapeutic purposes and, consequently, made interindividual comparisons, i.e., between patient groups taking and not taking inducing drugs. In this type of studies, the generally large variation in individual metabolic clearances may mask the effect of the inducer. An additional reason why the complex of these studies did not provide clear indications about the effect of liver disease on induction is due to the lack of rigorous criteria of patient selection, as they often examined pathologically heterogeneous populations, including patients with both compensated and decompensated liver cirrhosis or with unspecified degree of liver dysfunction. These studies have been analyzed individually in our previous review[18] and will not be further discussed. Somewhat clearer indications have been provided by two studies that examined either a patient population with sufficiently homogeneous liver pathology[103] or patient groups stratified according to the severity of liver dysfunction[104]. In the former study, a considerable difference was observed in rifampicin autoinduction between healthy subjects and patients with severe cirrhosis, since rifampicin through concentration decreased on average by 60% in healthy subjects following a 17-d rifampicin course, whereas it remained unaltered in cirrhotic patients. However, the statistical power of this study was limited, since it evaluated only four subjects per group. The latter study, in which patients were stratified in three groups according to the severity of liver disease (assessed from histological findings), evaluated enzyme induction by measuring morphine demethylase activity (a CYP3A4 marker reaction) in hepatic tissue obtained at biopsy and antipyrine half-life (a non-specific metabolic probe). Within each group, these parameters were compared between patients taking or not taking enzyme-inducing drugs (phenobarbital, phenytoin, or glutethimide) for therapeutic purposes. Morphine demethylase activity was significantly greater and antipyrine half-life significantly shorter in smokers belonging to a control group of healthy subjects and the two patient groups with mild-to-moderate liver dysfunction, whereas no significant difference between smokers and non-smokers was observed in the group with more severe liver disease.
The above considerations make it clear that the studies performed thus far could not provide an exhaustive and clear picture of the effect of liver dysfunction on CAR- and PXR-mediated induction. The three studies using animal models of cirrhosis[96,97,101] provided limited information because they examined animals with essentially mild liver dysfunction. Although the results of the two human studies described above[103,104] suggested that inducibility is preserved in mild to moderate liver dysfunction, whereas it is compromised in severe hepatic insufficiency, definitive conclusions can only be provided by studies making intra-individual comparisons in patient and control groups of adequate sizes. As ethical and practical constrains virtually preclude the possibility of performing human studies complying with such methodological standards, we recently re-evaluated the effect of liver cirrhosis on the PXR-mediated induction of CYP3A enzymes in control and cirrhotic rats obtained by treatment with CCl4, according to an experimental protocol identical to that previously used to assess the effect of cirrhosis on AhR-mediated induction (see above). To this purpose, control and cirrhotic rats, stratified according to the severity of liver insufficiency, were treated with the PXR activator dexamethasone (DEX), which had been shown to be the most effective inducer of rat CYP3A enzymes[105]. Unlike AhR, for which transcription was found to be reduced in the presence of liver cirrhosis[100], PXR mRNA and protein expressions were not reduced by liver function impairment. Consistent with this observation, DEX significantly increased mRNA level, protein content, and enzymatic activity of CYP3A1 in healthy and cirrhotic rats, irrespective of the degree of liver dysfunction. However, CYP3A2 induction proved to be susceptible to liver dysfunction, since the inducing effect of DEX on CYP3A2 mRNA and protein expressions, and its enzymatic activity were greatly curtailed when liver insufficiency became severe. A plausible explanation for this apparent discrepancy may be provided by the observation that the mRNA expressions of CYP3A1 and CYP3A2 are not coordinately regulated in response to DEX, since the mRNA level of CYP3A1 is increased by DEX to a far greater extent than that of CYP3A2[105,106]. Thus, it is conceivable that the gene transcription of CYP3A2 involves PXR co-activators that are more sensitive to the alterations associated with liver cirrhosis. Although the precise mechanism responsible for the selective impairment of CYP3A2 transcription remains to be elucidated, these results make it clear that no general conclusion can be drawn from the study of any particular enzyme, because even the induction of CYP isoforms under the transcriptional regulation of the same nuclear receptor may be differentially affected by liver dysfunction.
CONCLUSION
The experimental observations discussed in this review have important clinical implications. In patients with mild to moderate liver dysfunction, dose adjustment of drugs coadministered with an inhibitor or an inducer of their metabolism should be similar to that adopted for healthy subjects.
Management of inhibitory drug interactions in patients with severe liver dysfunction depends on the mechanism of the enzyme inhibition, since reversible and irreversible inhibition are differentially affected by liver disease. Virtually no dose adjustment is needed following coadministration of a reversible inhibitor, as the inhibitory effect becomes negligible in advanced hepatocellular insufficiency. The magnitude of irreversible inhibition is only partially decreased in severe liver dysfunction and the extent of such a reduction is expected to vary in proportion to the reduction in the expression of the inhibited enzyme, which cannot be measured in clinical practice. Therefore, individual dose adjustments are necessary, ideally based on therapeutic drug monitoring. When inhibition of metabolism is accompanied by displacement from plasma protein-binding sites, the free concentration of the victim drug increases in proportion to the degree of liver dysfunction. Therefore, free plasma drug concentration should be measured in order to manage appropriately such an interaction.
No general guidelines can as yet be proposed for the management of drug interactions resulting from enzyme induction in patients with decompensated cirrhosis, since definitive clinical studies have not yet been performed and animal studies have indicated that the effect of severe liver dysfunction on inducibility depends on both the type of nuclear receptor and the enzyme isoform involved. Thus, when such an interaction cannot be avoided, both the effects and the plasma concentration of the induced drug should be strictly monitored.
The observation that liver cirrhosis drastically modifies the magnitude of DDIs consequent to enzyme inhibition or induction also has important methodological implications. In vitro[10-12] and in vivo[107] methods used to predict the extent of DDIs caused by changes in CYP activity apply only to healthy subjects. Furthermore, the last FDA draft guidance to Drug Interaction Studies[108] states that clinical studies “can be performed using healthy volunteers, and findings in healthy volunteers will predict findings in the patient population for which the drug is intended.” Only phenotype or genotype determinations are recommended for the selection of the study population, if the relevant enzyme or transporter is polymorphically expressed. However, the data discussed in this review make it clear that, in addition to genetic polymorphisms, liver functional status is also an important determinant of the extent of pharmacokinetic interactions due to enzyme inhibition or induction. Therefore, results obtained in healthy subjects cannot be extended a priori to patients with liver disease.
Footnotes
Conflict-of-interest statement: The authors have no conflict of interest to report.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: June 4, 2015
First decision: July 14, 2015
Article in press: November 19, 2015
P- Reviewer: Barau C S- Editor: Yu J L- Editor: A E- Editor: Liu XM
References
- 1.Köhler GI, Bode-Böger SM, Busse R, Hoopmann M, Welte T, Böger RH. Drug-drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38:504–513. doi: 10.5414/cpp38504. [DOI] [PubMed] [Google Scholar]
- 2.Hohl CM, Dankoff J, Colacone A, Afilalo M. Polypharmacy, adverse drug-related events, and potential adverse drug interactions in elderly patients presenting to an emergency department. Ann Emerg Med. 2001;38:666–671. doi: 10.1067/mem.2001.119456. [DOI] [PubMed] [Google Scholar]
- 3.Leape LL, Bates DW, Cullen DJ, Cooper J, Demonaco HJ, Gallivan T, Hallisey R, Ives J, Laird N, Laffel G. Systems analysis of adverse drug events. ADE Prevention Study Group. JAMA. 1995;274:35–43. [PubMed] [Google Scholar]
- 4.Buajordet I, Ebbesen J, Erikssen J, Brørs O, Hilberg T. Fatal adverse drug events: the paradox of drug treatment. J Intern Med. 2001;250:327–341. doi: 10.1046/j.1365-2796.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- 5.Müller F, Fromm MF. Transporter-mediated drug-drug interactions. Pharmacogenomics. 2011;12:1017–1037. doi: 10.2217/pgs.11.44. [DOI] [PubMed] [Google Scholar]
- 6.Pirmohamed M, Park BK. Cytochrome P450 enzyme polymorphisms and adverse drug reactions. Toxicology. 2003;192:23–32. doi: 10.1016/s0300-483x(03)00247-6. [DOI] [PubMed] [Google Scholar]
- 7.Teo YL, Ho HK, Chan A. Metabolism-related pharmacokinetic drug-drug interactions with tyrosine kinase inhibitors: current understanding, challenges and recommendations. Br J Clin Pharmacol. 2015;79:241–253. doi: 10.1111/bcp.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams DH, Ju C, Ramaiah SK, Uetrecht J, Jaeschke H. Mechanisms of immune-mediated liver injury. Toxicol Sci. 2010;115:307–321. doi: 10.1093/toxsci/kfq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu J, Ritchie TK, Mulgaonkar A, Ragueneau-Majlessi I. Drug disposition and drug-drug interaction data in 2013 FDA new drug applications: a systematic review. Drug Metab Dispos. 2014;42:1991–2001. doi: 10.1124/dmd.114.060392. [DOI] [PubMed] [Google Scholar]
- 10.Obach RS, Walsky RL, Venkatakrishnan K, Houston JB, Tremaine LM. In vitro cytochrome P450 inhibition data and the prediction of drug-drug interactions: qualitative relationships, quantitative predictions, and the rank-order approach. Clin Pharmacol Ther. 2005;78:582–592. doi: 10.1016/j.clpt.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Zhang YD, Zhao P, Huang SM. Predicting drug-drug interactions: an FDA perspective. AAPS J. 2009;11:300–306. doi: 10.1208/s12248-009-9106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenblatt DJ. In vitro prediction of clinical drug interactions with CYP3A substrates: we are not there yet. Clin Pharmacol Ther. 2014;95:133–135. doi: 10.1038/clpt.2013.230. [DOI] [PubMed] [Google Scholar]
- 13.Christensen H, Baker M, Tucker GT, Rostami-Hodjegan A. Prediction of plasma protein binding displacement and its implications for quantitative assessment of metabolic drug-drug interactions from in vitro data. J Pharm Sci. 2006;95:2778–2787. doi: 10.1002/jps.20733. [DOI] [PubMed] [Google Scholar]
- 14.Cheng PY, Morgan ET. Hepatic cytochrome P450 regulation in disease states. Curr Drug Metab. 2001;2:165–183. doi: 10.2174/1389200013338676. [DOI] [PubMed] [Google Scholar]
- 15.Lee LS, Nafziger AN, Bertino JS. Evaluation of inhibitory drug interactions during drug development: genetic polymorphisms must be considered. Clin Pharmacol Ther. 2005;78:1–6. doi: 10.1016/j.clpt.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 16.Ma Q, Lu AY. Origins of individual variability in P4501A induction. Chem Res Toxicol. 2003;16:249–260. doi: 10.1021/tx0200919. [DOI] [PubMed] [Google Scholar]
- 17.Tang C, Lin JH, Lu AY. Metabolism-based drug-drug interactions: what determines individual variability in cytochrome P450 induction? Drug Metab Dispos. 2005;33:603–613. doi: 10.1124/dmd.104.003236. [DOI] [PubMed] [Google Scholar]
- 18.Palatini P, De Martin S, Pegoraro P, Orlando R. Enzyme inhibition and induction in liver disease. Curr Clin Pharmacol. 2008;3:56–69. doi: 10.2174/157488408783329896. [DOI] [PubMed] [Google Scholar]
- 19.Morgan DJ, McLean AJ. Clinical pharmacokinetic and pharmacodynamic considerations in patients with liver disease. An update. Clin Pharmacokinet. 1995;29:370–391. doi: 10.2165/00003088-199529050-00005. [DOI] [PubMed] [Google Scholar]
- 20.Schuppan D, Afdhal NH. Liver cirrhosis. Lancet. 2008;371:838–851. doi: 10.1016/S0140-6736(08)60383-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet. 2014;383:1749–1761. doi: 10.1016/S0140-6736(14)60121-5. [DOI] [PubMed] [Google Scholar]
- 22.Gaba RC, Couture PM, Bui JT, Knuttinen MG, Walzer NM, Kallwitz ER, Berkes JL, Cotler SJ. Prognostic capability of different liver disease scoring systems for prediction of early mortality after transjugular intrahepatic portosystemic shunt creation. J Vasc Interv Radiol. 2013;24:411–420, 420.e1-e4; quiz 421. doi: 10.1016/j.jvir.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 23.Conn HO. A peek at the Child-Turcotte classification. Hepatology. 1981;1:673–676. doi: 10.1002/hep.1840010617. [DOI] [PubMed] [Google Scholar]
- 24.Pugh RN, Murray-Lyon IM, Dawson JL, Pietroni MC, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 25.Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147–1161. doi: 10.1007/s00228-008-0553-z. [DOI] [PubMed] [Google Scholar]
- 26.Sinha V, Brendel K, Mayersohn M. A simplified isolated perfused rat liver apparatus: characterization and measurement of extraction ratios of selected compounds. Life Sci. 2000;66:1795–1804. doi: 10.1016/s0024-3205(00)00503-8. [DOI] [PubMed] [Google Scholar]
- 27.Morgan ET. Impact of infectious and inflammatory disease on cytochrome P450-mediated drug metabolism and pharmacokinetics. Clin Pharmacol Ther. 2009;85:434–438. doi: 10.1038/clpt.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huet PM, Villeneuve JP, Fenyves D. Drug elimination in chronic liver diseases. J Hepatol. 1997;26 Suppl 2:63–72. doi: 10.1016/s0168-8278(97)80498-9. [DOI] [PubMed] [Google Scholar]
- 29.Orlando R, Piccoli P, De Martin S, Padrini R, Floreani M, Palatini P. Cytochrome P450 1A2 is a major determinant of lidocaine metabolism in vivo: effects of liver function. Clin Pharmacol Ther. 2004;75:80–88. doi: 10.1016/j.clpt.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 30.Orlando R, Padrini R, Perazzi M, De Martin S, Piccoli P, Palatini P. Liver dysfunction markedly decreases the inhibition of cytochrome P450 1A2-mediated theophylline metabolism by fluvoxamine. Clin Pharmacol Ther. 2006;79:489–499. doi: 10.1016/j.clpt.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 31.Gariépy L, Fenyves D, Kassissia I, Villeneuve JP. Clearance by the liver in cirrhosis. II. Characterization of propranolol uptake with the multiple-indicator dilution technique. Hepatology. 1993;18:823–831. doi: 10.1002/hep.1840180412. [DOI] [PubMed] [Google Scholar]
- 32.Le Couteur DG, Fraser R, Hilmer S, Rivory LP, McLean AJ. The hepatic sinusoid in aging and cirrhosis: effects on hepatic substrate disposition and drug clearance. Clin Pharmacokinet. 2005;44:187–200. doi: 10.2165/00003088-200544020-00004. [DOI] [PubMed] [Google Scholar]
- 33.McLean AJ, Morgan DJ. Clinical pharmacokinetics in patients with liver disease. Clin Pharmacokinet. 1991;21:42–69. doi: 10.2165/00003088-199121010-00004. [DOI] [PubMed] [Google Scholar]
- 34.Hickey PL, Angus PW, McLean AJ, Morgan DJ. Oxygen supplementation restores theophylline clearance to normal in cirrhotic rats. Gastroenterology. 1995;108:1504–1509. doi: 10.1016/0016-5085(95)90700-9. [DOI] [PubMed] [Google Scholar]
- 35.Froomes PR, Morgan DJ, Smallwood RA, Angus PW. Comparative effects of oxygen supplementation on theophylline and acetaminophen clearance in human cirrhosis. Gastroenterology. 1999;116:915–920. doi: 10.1016/s0016-5085(99)70075-2. [DOI] [PubMed] [Google Scholar]
- 36.Howden CW, Birnie GG, Brodie MJ. Drug metabolism in liver disease. Pharmacol Ther. 1989;40:439–474. doi: 10.1016/0163-7258(89)90088-0. [DOI] [PubMed] [Google Scholar]
- 37.Murray M. Mechanisms and significance of inhibitory drug interactions involving cytochrome P450 enzymes (review) Int J Mol Med. 1999;3:227–238. [PubMed] [Google Scholar]
- 38.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Annu Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 39.Koley AP, Robinson RC, Markowitz A, Friedman FK. Drug-drug interactions: effect of quinidine on nifedipine binding to human cytochrome P450 3A4. Biochem Pharmacol. 1997;53:455–460. doi: 10.1016/s0006-2952(96)00836-2. [DOI] [PubMed] [Google Scholar]
- 40.Zhou S, Yung Chan S, Cher Goh B, Chan E, Duan W, Huang M, McLeod HL. Mechanism-based inhibition of cytochrome P450 3A4 by therapeutic drugs. Clin Pharmacokinet. 2005;44:279–304. doi: 10.2165/00003088-200544030-00005. [DOI] [PubMed] [Google Scholar]
- 41.Yasui-Furukori N, Saito M, Uno T, Takahata T, Sugawara K, Tateishi T. Effects of fluvoxamine on lansoprazole pharmacokinetics in relation to CYP2C19 genotypes. J Clin Pharmacol. 2004;44:1223–1229. doi: 10.1177/0091270004269015. [DOI] [PubMed] [Google Scholar]
- 42.Eap CB, Lessard E, Baumann P, Brawand-Amey M, Yessine MA, O’Hara G, Turgeon J. Role of CYP2D6 in the stereoselective disposition of venlafaxine in humans. Pharmacogenetics. 2003;13:39–47. doi: 10.1097/00008571-200301000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Shah RR, Smith RL. Addressing phenoconversion: the Achilles’ heel of personalized medicine. Br J Clin Pharmacol. 2015;79:222–240. doi: 10.1111/bcp.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan ET. Regulation of cytochrome P450 by inflammatory mediators: why and how? Drug Metab Dispos. 2001;29:207–212. [PubMed] [Google Scholar]
- 45.Shah RR, Smith RL. Inflammation-induced phenoconversion of polymorphic drug metabolizing enzymes: hypothesis with implications for personalized medicine. Drug Metab Dispos. 2015;43:400–410. doi: 10.1124/dmd.114.061093. [DOI] [PubMed] [Google Scholar]
- 46.Orlando R, Piccoli P, De Martin S, Padrini R, Palatini P. Effect of the CYP3A4 inhibitor erythromycin on the pharmacokinetics of lignocaine and its pharmacologically active metabolites in subjects with normal and impaired liver function. Br J Clin Pharmacol. 2003;55:86–93. doi: 10.1046/j.1365-2125.2003.01718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orlando R, De Martin S, Pegoraro P, Quintieri L, Palatini P. Irreversible CYP3A inhibition accompanied by plasma protein-binding displacement: a comparative analysis in subjects with normal and impaired liver function. Clin Pharmacol Ther. 2009;85:319–326. doi: 10.1038/clpt.2008.216. [DOI] [PubMed] [Google Scholar]
- 48.Benedetti MS, Whomsley R, Canning M. Drug metabolism in the paediatric population and in the elderly. Drug Discov Today. 2007;12:599–610. doi: 10.1016/j.drudis.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 49.Le Couteur DG, McLean AJ. The aging liver. Drug clearance and an oxygen diffusion barrier hypothesis. Clin Pharmacokinet. 1998;34:359–373. doi: 10.2165/00003088-199834050-00003. [DOI] [PubMed] [Google Scholar]
- 50.Parkinson A, Mudra DR, Johnson C, Dwyer A, Carroll KM. The effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme activity in human liver microsomes and inducibility in cultured human hepatocytes. Toxicol Appl Pharmacol. 2004;199:193–209. doi: 10.1016/j.taap.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 51.Salem F, Rostami-Hodjegan A, Johnson TN. Do children have the same vulnerability to metabolic drug-drug interactions as adults? A critical analysis of the literature. J Clin Pharmacol. 2013;53:559–566. doi: 10.1002/jcph.13. [DOI] [PubMed] [Google Scholar]
- 52.Farrell G. Effects of disease on expression and regulation of CYPs. Mol Aspects Med. 1999;20:55–70, 137. [PubMed] [Google Scholar]
- 53.Huet PM, Villeneuve JP. Determinants of drug disposition in patients with cirrhosis. Hepatology. 1983;3:913–918. doi: 10.1002/hep.1840030604. [DOI] [PubMed] [Google Scholar]
- 54.Hung DY, Chang P, Cheung K, McWhinney B, Masci PP, Weiss M, Roberts MS. Cationic drug pharmacokinetics in diseased livers determined by fibrosis index, hepatic protein content, microsomal activity, and nature of drug. J Pharmacol Exp Ther. 2002;301:1079–1087. doi: 10.1124/jpet.301.3.1079. [DOI] [PubMed] [Google Scholar]
- 55.Sandson NB, Marcucci C, Bourke DL, Smith-Lamacchia R. An interaction between aspirin and valproate: the relevance of plasma protein displacement drug-drug interactions. Am J Psychiatry. 2006;163:1891–1896. doi: 10.1176/ajp.2006.163.11.1891. [DOI] [PubMed] [Google Scholar]
- 56.Bargetzi MJ, Aoyama T, Gonzalez FJ, Meyer UA. Lidocaine metabolism in human liver microsomes by cytochrome P450IIIA4. Clin Pharmacol Ther. 1989;46:521–527. doi: 10.1038/clpt.1989.180. [DOI] [PubMed] [Google Scholar]
- 57.Wang JS, Backman JT, Taavitsainen P, Neuvonen PJ, Kivistö KT. Involvement of CYP1A2 and CYP3A4 in lidocaine N-deethylation and 3-hydroxylation in humans. Drug Metab Dispos. 2000;28:959–965. [PubMed] [Google Scholar]
- 58.Piafsky KM, Sitar DS, Rangno RE, Ogilvie RI. Theophylline disposition in patients with hepatic cirrhosis. N Engl J Med. 1977;296:1495–1497. doi: 10.1056/NEJM197706302962603. [DOI] [PubMed] [Google Scholar]
- 59.Pieper JA, Johnson KE. Lidocaine. In: Evans WE, Schentag JJ, Jusko WJ, editors. Applied Pharmacokinetics. In: Principles of Therapeutic Drug Monitoring. Vancouver (WA): Applied Therapeutics; 1992. p. chapter 21. [Google Scholar]
- 60.Fixley M, Shen DD, Azarnoff DL. Theophylline bioavailability: a comparison of the oral absorption of a theophylline elixir and two combination theophylline tablets to intravenous aminophylline. Am Rev Respir Dis. 1977;115:955–962. doi: 10.1164/arrd.1977.115.6.955. [DOI] [PubMed] [Google Scholar]
- 61.Hendeles L, Weinberger M, Bighley L. Absolute bioavailability of oral theophylline. Am J Hosp Pharm. 1977;34:525–527. [PubMed] [Google Scholar]
- 62.Zhou SF, Xue CC, Yu XQ, Li C, Wang G. Clinically important drug interactions potentially involving mechanism-based inhibition of cytochrome P450 3A4 and the role of therapeutic drug monitoring. Ther Drug Monit. 2007;29:687–710. doi: 10.1097/FTD.0b013e31815c16f5. [DOI] [PubMed] [Google Scholar]
- 63.Venkatakrishnan K, Obach RS, Rostami-Hodjegan A. Mechanism-based inactivation of human cytochrome P450 enzymes: strategies for diagnosis and drug-drug interaction risk assessment. Xenobiotica. 2007;37:1225–1256. doi: 10.1080/00498250701670945. [DOI] [PubMed] [Google Scholar]
- 64.Christensen M, Andersson K, Dalén P, Mirghani RA, Muirhead GJ, Nordmark A, Tybring G, Wahlberg A, Yaşar U, Bertilsson L. The Karolinska cocktail for phenotyping of five human cytochrome P450 enzymes. Clin Pharmacol Ther. 2003;73:517–528. doi: 10.1016/S0009-9236(03)00050-X. [DOI] [PubMed] [Google Scholar]
- 65.Mirghani RA, Ericsson O, Tybring G, Gustafsson LL, Bertilsson L. Quinine 3-hydroxylation as a biomarker reaction for the activity of CYP3A4 in man. Eur J Clin Pharmacol. 2003;59:23–28. doi: 10.1007/s00228-003-0575-5. [DOI] [PubMed] [Google Scholar]
- 66.Wanwimolruk S, Wong SM, Zhang H, Coville PF. Simultaneous determination of quinine and a major metabolite, 3-hydroxyquinine, in biological fluids by HPLC without extraction. J Liq Chromatog Rel Technol. 1996;19:293–305. [Google Scholar]
- 67.Krishna S, White NJ. Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications. Clin Pharmacokinet. 1996;30:263–299. doi: 10.2165/00003088-199630040-00002. [DOI] [PubMed] [Google Scholar]
- 68.Barry M, Keeling PW, Weir D, Feely J. Severity of cirrhosis and the relationship of alpha 1-acid glycoprotein concentration to plasma protein binding of lidocaine. Clin Pharmacol Ther. 1990;47:366–370. doi: 10.1038/clpt.1990.41. [DOI] [PubMed] [Google Scholar]
- 69.Routledge PA. The plasma protein binding of basic drugs. Br J Clin Pharmacol. 1986;22:499–506. doi: 10.1111/j.1365-2125.1986.tb02927.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Toutain PL, Bousquet-Melou A. Free drug fraction vs free drug concentration: a matter of frequent confusion. J Vet Pharmacol Ther. 2002;25:460–463. doi: 10.1046/j.1365-2885.2002.00442.x. [DOI] [PubMed] [Google Scholar]
- 71.Park BK, Kitteringham NR, Pirmohamed M, Tucker GT. Relevance of induction of human drug-metabolizing enzymes: pharmacological and toxicological implications. Br J Clin Pharmacol. 1996;41:477–491. doi: 10.1046/j.1365-2125.1996.03482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mankowski DC, Ekins S. Prediction of human drug metabolizing enzyme induction. Curr Drug Metab. 2003;4:381–391. doi: 10.2174/1389200033489352. [DOI] [PubMed] [Google Scholar]
- 73.Tirona RG, Kim RB. Nuclear receptors and drug disposition gene regulation. J Pharm Sci. 2005;94:1169–1186. doi: 10.1002/jps.20324. [DOI] [PubMed] [Google Scholar]
- 74.Lonard DM, O’Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell. 2006;125:411–414. doi: 10.1016/j.cell.2006.04.021. [DOI] [PubMed] [Google Scholar]
- 75.Pelkonen O, Turpeinen M, Hakkola J, Honkakoski P, Hukkanen J, Raunio H. Inhibition and induction of human cytochrome P450 enzymes: current status. Arch Toxicol. 2008;82:667–715. doi: 10.1007/s00204-008-0332-8. [DOI] [PubMed] [Google Scholar]
- 76.Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- 77.Huss JM, Kasper CB. Two-stage glucocorticoid induction of CYP3A23 through both the glucocorticoid and pregnane X receptors. Mol Pharmacol. 2000;58:48–57. doi: 10.1124/mol.58.1.48. [DOI] [PubMed] [Google Scholar]
- 78.Pascussi JM, Drocourt L, Fabre JM, Maurel P, Vilarem MJ. Dexamethasone induces pregnane X receptor and retinoid X receptor-alpha expression in human hepatocytes: synergistic increase of CYP3A4 induction by pregnane X receptor activators. Mol Pharmacol. 2000;58:361–372. doi: 10.1124/mol.58.2.361. [DOI] [PubMed] [Google Scholar]
- 79.Dvorak Z, Pavek P. Regulation of drug-metabolizing cytochrome P450 enzymes by glucocorticoids. Drug Metab Rev. 2010;42:621–635. doi: 10.3109/03602532.2010.484462. [DOI] [PubMed] [Google Scholar]
- 80.Gadaleta RM, Cariello M, Sabbà C, Moschetta A. Tissue-specific actions of FXR in metabolism and cancer. Biochim Biophys Acta. 2015;1851:30–39. doi: 10.1016/j.bbalip.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 81.Dusso AS, Brown AJ, Slatopolsky E. Vitamin D. Am J Physiol Renal Physiol. 2005;289:F8–28. doi: 10.1152/ajprenal.00336.2004. [DOI] [PubMed] [Google Scholar]
- 82.Gnerre C, Blättler S, Kaufmann MR, Looser R, Meyer UA. Regulation of CYP3A4 by the bile acid receptor FXR: evidence for functional binding sites in the CYP3A4 gene. Pharmacogenetics. 2004;14:635–645. doi: 10.1097/00008571-200410000-00001. [DOI] [PubMed] [Google Scholar]
- 83.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 84.Lin JH, Lu AY. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol. 2001;41:535–567. doi: 10.1146/annurev.pharmtox.41.1.535. [DOI] [PubMed] [Google Scholar]
- 85.Schuetz EG, Schinkel AH, Relling MV, Schuetz JD. P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc Natl Acad Sci USA. 1996;93:4001–4005. doi: 10.1073/pnas.93.9.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.de Morais SM, Goldstein JA, Xie HG, Huang SL, Lu YQ, Xia H, Xiao ZS, Ile N, Zhou HH. Genetic analysis of the S-mephenytoin polymorphism in a Chinese population. Clin Pharmacol Ther. 1995;58:404–411. doi: 10.1016/0009-9236(95)90053-5. [DOI] [PubMed] [Google Scholar]
- 87.Lin P, Hu SW, Chang TH. Correlation between gene expression of aryl hydrocarbon receptor (AhR), hydrocarbon receptor nuclear translocator (Arnt), cytochromes P4501A1 (CYP1A1) and 1B1 (CYP1B1), and inducibility of CYP1A1 and CYP1B1 in human lymphocytes. Toxicol Sci. 2003;71:20–26. doi: 10.1093/toxsci/71.1.20. [DOI] [PubMed] [Google Scholar]
- 88.Micka J, Milatovich A, Menon A, Grabowski GA, Puga A, Nebert DW. Human Ah receptor (AHR) gene: localization to 7p15 and suggestive correlation of polymorphism with CYP1A1 inducibility. Pharmacogenetics. 1997;7:95–101. doi: 10.1097/00008571-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 89.Salem SA, Rajjayabun P, Shepherd AM, Stevenson IH. Reduced induction of drug metabolism in the elderly. Age Ageing. 1978;7:68–73. doi: 10.1093/ageing/7.2.68. [DOI] [PubMed] [Google Scholar]
- 90.Dilger K, Hofmann U, Klotz U. Enzyme induction in the elderly: effect of rifampin on the pharmacokinetics and pharmacodynamics of propafenone. Clin Pharmacol Ther. 2000;67:512–520. doi: 10.1067/mcp.2000.106872. [DOI] [PubMed] [Google Scholar]
- 91.Fromm MF, Dilger K, Busse D, Kroemer HK, Eichelbaum M, Klotz U. Gut wall metabolism of verapamil in older people: effects of rifampicin-mediated enzyme induction. Br J Clin Pharmacol. 1998;45:247–255. doi: 10.1046/j.1365-2125.1998.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gorski JC, Vannaprasaht S, Hamman MA, Ambrosius WT, Bruce MA, Haehner-Daniels B, Hall SD. The effect of age, sex, and rifampin administration on intestinal and hepatic cytochrome P450 3A activity. Clin Pharmacol Ther. 2003;74:275–287. doi: 10.1016/S0009-9236(03)00187-5. [DOI] [PubMed] [Google Scholar]
- 93.Hodek P, Trefil P, Stiborová M. Flavonoids-potent and versatile biologically active compounds interacting with cytochromes P450. Chem Biol Interact. 2002;139:1–21. doi: 10.1016/s0009-2797(01)00285-x. [DOI] [PubMed] [Google Scholar]
- 94.Hoyumpa AM, Schenker S. Major drug interactions: effect of liver disease, alcohol, and malnutrition. Annu Rev Med. 1982;33:113–149. doi: 10.1146/annurev.me.33.020182.000553. [DOI] [PubMed] [Google Scholar]
- 95.Elbekai RH, Korashy HM, El-Kadi AO. The effect of liver cirrhosis on the regulation and expression of drug metabolizing enzymes. Curr Drug Metab. 2004;5:157–167. doi: 10.2174/1389200043489054. [DOI] [PubMed] [Google Scholar]
- 96.Farrell GC, Zaluzny L. Microsomal protein synthesis and induction of cytochrome P-450 in cirrhotic rat liver. Aust J Exp Biol Med Sci. 1984;62(Pt 3):291–301. doi: 10.1038/icb.1984.29. [DOI] [PubMed] [Google Scholar]
- 97.Wu ZQ, Piché D, Vallières S, Huet PM, Gascon-Barré M. Unimpaired induction of drug-metabolizing enzymes in hepatocytes isolated from rats with micronodular cirrhosis. Can J Physiol Pharmacol. 1991;69:426–436. doi: 10.1139/y91-065. [DOI] [PubMed] [Google Scholar]
- 98.Joeres R, Klinker H, Heusler H, Epping J, Zilly W, Richter E. Influence of smoking on caffeine elimination in healthy volunteers and in patients with alcoholic liver cirrhosis. Hepatology. 1988;8:575–579. doi: 10.1002/hep.1840080323. [DOI] [PubMed] [Google Scholar]
- 99.Coverdale S, Byth K, Field J, Liddle C, Lin R, Farrell GC. Antipyrine clearance and response to interferon treatment in patients with chronic active hepatitis C. Hepatology. 1995;22:1065–1071. doi: 10.1016/0270-9139(95)90610-x. [DOI] [PubMed] [Google Scholar]
- 100.Floreani M, De Martin S, Gabbia D, Barbierato M, Nassi A, Mescoli C, Orlando R, Bova S, Angeli P, Gola E, et al. Severe liver cirrhosis markedly reduces AhR-mediated induction of cytochrome P450 in rats by decreasing the transcription of target genes. PLoS One. 2013;8:e61983. doi: 10.1371/journal.pone.0061983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marshall WJ, McLean AE. The effect of cirrhosis of the liver on microsomal detoxications and cytochrome P-450. Br J Exp Pathol. 1969;50:578–583. [PMC free article] [PubMed] [Google Scholar]
- 102.FDA guidance for industry. Pharmacokinetics in patients with impaired hepatic function - study design, data analysis, and impact on dosing and labeling. Rockville (MD): Food and Drug Administration; 1999. [Google Scholar]
- 103.Capelle P, Dhumeaux D, Mora M, Feldmann G, Berthelot P. Effect of rifampicin on liver function in man. Gut. 1972;13:366–371. doi: 10.1136/gut.13.5.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Farrell GC, Cooksley WG, Powell LW. Drug metabolism in liver disease: activity of hepatic microsomal metabolizing enzymes. Clin Pharmacol Ther. 1979;26:483–492. doi: 10.1002/cpt1979264483. [DOI] [PubMed] [Google Scholar]
- 105.De Martin S, Gabbia D, Albertin G, Sfriso MM, Mescoli C, Albertoni L, Paliuri G, Bova S, Palatini P. Differential effect of liver cirrhosis on the pregnane X receptor-mediated induction of CYP3A1 and 3A2 in the rat. Drug Metab Dispos. 2014;42:1617–1626. doi: 10.1124/dmd.114.058511. [DOI] [PubMed] [Google Scholar]
- 106.Choudhuri S, Zhang XJ, Waskiewicz MJ, Thomas PE. Differential regulation of cytochrome P450 3A1 and P450 3A2 in rat liver following dexamethasone treatment. J Biochem Toxicol. 1995;10:299–307. doi: 10.1002/jbt.2570100604. [DOI] [PubMed] [Google Scholar]
- 107.Hisaka A, Ohno Y, Yamamoto T, Suzuki H. Prediction of pharmacokinetic drug-drug interactions caused by changes in cytochrome P450 activity using in vivo information. Pharmacol Ther. 2010;125:230–248. doi: 10.1016/j.pharmthera.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 108.FDA Center for Drug Evaluation and Research. Guidance for Industry: Drug Interaction Studies - Study Design, Data Analysis, Implications for Dosing, and Labeling Reccomendations. Rockville (MD): CDER; 2012. p. Feb [online]. Available from: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. [Google Scholar]