

Abstract
The gut microbiota acts as a real organ. The symbiotic interactions between resident micro-organisms and the digestive tract highly contribute to maintain the gut homeostasis. However, alterations to the microbiome caused by environmental changes (e.g., infection, diet and/or lifestyle) can disturb this symbiotic relationship and promote disease, such as inflammatory bowel diseases and cancer. Colorectal cancer is a complex association of tumoral cells, non-neoplastic cells and a large amount of micro-organisms, and the involvement of the microbiota in colorectal carcinogenesis is becoming increasingly clear. Indeed, many changes in the bacterial composition of the gut microbiota have been reported in colorectal cancer, suggesting a major role of dysbiosis in colorectal carcinogenesis. Some bacterial species have been identified and suspected to play a role in colorectal carcinogenesis, such as Streptococcus bovis, Helicobacter pylori, Bacteroides fragilis, Enterococcus faecalis, Clostridium septicum, Fusobacterium spp. and Escherichia coli. The potential pro-carcinogenic effects of these bacteria are now better understood. In this review, we discuss the possible links between the bacterial microbiota and colorectal carcinogenesis, focusing on dysbiosis and the potential pro-carcinogenic properties of bacteria, such as genotoxicity and other virulence factors, inflammation, host defenses modulation, bacterial-derived metabolism, oxidative stress and anti-oxidative defenses modulation. We lastly describe how bacterial microbiota modifications could represent novel prognosis markers and/or targets for innovative therapeutic strategies.
Keywords: Colorectal cancer, Gut microbiota, Dysbiosis, Cyclomodulin, Oxidative stress
Core tip: The gut microbiota acts as a real organ and many changes in its composition have been reported in colorectal cancer. The pro-carcinogenic properties of bacteria are now better understood. In this review, we discuss possible links between the bacterial microbiota and colorectal carcinogenesis, focusing on the dysbiosis-causing and pro-carcinogenic properties of bacteria, such as genotoxicity, inflammation, and oxidative stress. We lastly detail how microbiota modifications may represent novel prognosis markers and/or targets for innovative therapeutic strategies.
INTRODUCTION
Colorectal cancer (CRC) is a complex association of tumoral cells, non-neoplastic cells (i.e., stromal cells) and a large number of micro-organisms. The microbiota may be linked to carcinogenesis via various mechanisms and could lead to the development of novel prognosis markers and/or targets for innovative therapeutic strategies.
GUT MICROBIOTA IN HEALTH
What is the gut microbiota?
Approximately 100 trillion micro-organisms (including bacteria, viruses and fungi) reside in the adult human gut and constitute the microbiota[1,2]. The composition of the microbiota is rather stable along the length of the gut, but the absolute number of micro-organisms varies considerably between the mouth and rectum[3]. The gut microbiota consistently differs among between individuals. It is acquired during the first stages of life via the commensal flora from the mother’s skin, vagina and feces, and matures primarily during the first two years. Microbiota development is the result of interactions between physiological process in the host and micro-organisms that are introduced from the environment[4-6]. After the initial stages, the microbiota stabilizes and maintains a consistent composition, despite some fluctuations throughout adulthood in response to environmental, developmental and pathological events[7,8]. In the elderly, the microbiota composition changes gradually but can maintain similar physiological functions[9-12]. The early acquisition of a diverse and balanced microbiota is likely critical for the development and maturation of a healthy immune system, as suggested by immune abnormalities in germ-free animals raised in bacteria-free conditions[4,13]. The colon is colonized by approximately 103 different microbial species and this colonic microbiota is mostly represented by bacteria[7,9,14]. Indeed, the colon contains approximately 1014 bacteria (70% of the host’s microorganisms)[15,16]. This review will therefore focus on the impact of bacteria in CRC.
Most bacteria cannot be cultured, but modern molecular approaches can be used to identify and classify bacteria such as 16S ribosomal RNA (16S rRNA) sequencing from feces or digestive tissues can be used to identify and classify bacteria. The microbiota can be divided according to location in the gut. Specifically, microbes in the lumen are referred to “luminal flora”, whereas microbes that penetrate the mucosal layer overlying the intestinal epithelium are referred to “mucosa-associated flora”[16]. Indeed, thick mucus layers protect enterocytes from excessive exposure to microorganisms and dietary antigens along the length of the intestine, particularly in the colon, thus preventing hypersensitivity responses[17]. Moreover, the ratio of anaerobes to aerobes is lower at the mucosal surfaces than in the lumen. In addition, the collection of “fecal flora” from the feces is a non-invasive technique that facilitates sampling of colonic microbiota. These bacteria are representative of distal colonic colonization but differ from proximal “associated” flora[18]. It is essential to note that the composition of murine gut microbiota is quite similar to that in humans, lending translational relevance to mouse experimental models of gastrointestinal disease[19,20]. Actually, more than 50 different phyla and 500 bacterial species may comprise the human normal commensal gut microbiota. Although the exact num ber of species and the amount of variability among individuals remain to be characterized[21,22], these factors are likely highly dependent on lifestyle, diet and host genotype[23,24]. Some bacterial species are regularly recovered from different individuals, and the human gut microbiota is dominated by 3 primary phyla: Firmicutes (30%-50%), Bacteroidetes (20%-40%) and Actinobacteria (1%-10%). Strict anaerobes, including Bacteroides, Eubacterium, Bifidobacterium, Fusobacterium, Peptostreptococcus and Atopobium[25], represent a major portion of the gut microbiota, whereas facultative anaerobes, such as Lactobacilli, Enterococci, Streptococci and Enterobacteriaceae, constitute a minority (about 1000-fold lower levels). The fact that composition significantly vary along the gut should also be highlighted, especially given that Bacteroidetes and Actinobacteria represent more than 90% of bacterial phyla in the colon but only 50% in the small intestine, which contains approximately 40% Firmicutes species[26].
Microbiota and gut homeostasis
The gut microbiota constitutes a natural defensive barrier to infection. Moreover, the microbiota is involved in numerous protective, structural and metabolic roles in the intestinal epithelium and plays a large role in maintaining gut homeostasis. The microbiota is involved in several physiological functions[27]. The impact of enteric bacteria on intestinal physiology has been studied primarily in germ-free animals raised in bacteria-free conditions. Such animals are more susceptible to infections and have reduced vascularity, digestive enzyme activity, muscle wall thickness, cytokine production and serum immunoglobulin levels, smaller Peyer’s patches and fewer intraepithelial lymphocytes[28]. The reconstitution of a gut microbiota in germ-free mice is sufficient to restore the mucosal immune system[29] and affects the expression of various host genes that can impact nutrient uptake, metabolism, angiogenesis, mucosal barrier function and development of the intestinal nervous system[30]. Moreover, commensal bacteria influence the normal development and function of the mucosal immune system[31,32], such as the humoral components[33]. These bacteria also modify T-cell repertories and T-helper-cell cytokine profiles[34,35]. Such data support a possible influence of gut microbiota composition on individual variations in immunity.
The structural role of gut microbiota on the intestinal epithelium is increasingly evident and has been studied primarily in germ-free mice. These animals present longer intestinal villi, associated with crypt atrophy, slower renewal of epithelial cells and decrease of angiogenesis phenomenon[3]. Furthermore, it has been reported that mucosa and muscle wall thickness were decreased in these mice[28] and that microbiota enhanced crypt cell turnover in a CRC-predisposed mouse model[36].
Furthermore, the gut microbiota is involved in metabolic functions[27]. For example, the microbiota can participate in (1) anaerobic carbohydrate fermentation through the production of CO2, H2, CH4 and short-chain fatty acids (e.g., butyrate, propionate and acetate); and (2) proteolytic fermentation via metabolites such as phenolic compounds, amines, ammonia, N-nitroso compounds and indoles. These effects can impact on gene expression, intestinal epithelial cell differentiation and proliferation, and can also mediate vitamin synthesis, ion absorption and mucus production[27,35,37,38]. This complex metabolic activity also increases the yield and storage of energy from dietary sources, regulates fat storage, helps to provide absorbable substrates for both the host and the microbiota, and is involved in bacterial growth and proliferation[37-40]. Some of produced metabolites, especially during proteolytic fermentation, can be toxic to the host[27,41,42].
In addition to immune, structural, and metabolic functions, the commensal microbiota inhibits gut colonization of intruding pathogens and ensures “colonization resistance” or “microbial interference”[43]. The involved mechanisms of these effects remain unclear but likely involve competition with adhesion receptors, stabilization of the gut mucosal barrier, competition for nutrients and the production of anti-microbial substances[27]. Indeed, alterations in colonization resistance due to, e.g., pathogens or antibiotics treatment, probably increase the risk of gastrointestinal affections.
MICROBIOTA AND COLORECTAL CANCER
Geographic variability of the incidence of CRC highly suggests the involvement of certain environmental risk factors, such as high-fat diets, obesity or living in a Western country[44,45]. Moreover, Knudson’s two-hit hypothesis suggests that host factors play an important role in the predisposition to carcinogenesis. In this scenario, a second environmental hit can lead to uncontrolled cellular proliferation[46]. Indeed, growing attention has been given to the role of microbial infection in carcinogenesis in recent decades, and microbes are suspected to be involved in approximately 20% of cancers[47], especially CRC[48]. Concerning digestive cancers, even if some pathogens, such as Helicobacter pylori (H. pylori), have been directly and strongly linked to gastric cancer[49], possible infectious cause in CRC remains controversial. It is becoming increasingly clear, however, that pathogens play a role in colorectal carcinogenesis[50]. Interestingly, bacteria levels in the colon are one million-fold higher than in the small intestine, and approximately 12-fold more cancers develop in the former than in the latter, suggesting a potential role of gut microbiota in colorectal carcinogenesis[51]. The first observation linking gut microbiota with CRC was reported in 1975 in germ-free rats that developed less chemically induced colorectal tumor than conventional rats[52]. These results have been reproduced in CRC-predisposed mice[36,53]. Contrary to gastric carcinogenesis, which seems to result from a single pathogen, the following differing hypotheses have emerged to explain the contribution of bacteria to CRC: (1) a dysbiotic microbial community with pro-carcinogenic features are capable of remodeling the microbiome as a whole to drive pro-inflammatory responses and epithelial cell transformation, leading to cancer; and (2) the “driver-passenger” theory, wherein intestinal bacteria, termed “bacteria drivers”, initiate CRC by inducing epithelial DNA damage and tumorigenesis, in turn promoting the proliferation of passenger bacteria that have a growth advantage in the tumoral microenvironment[50,54]. Studies in mouse models of altered immune and inflammatory responses suggest that dysbiosis could be sufficient to promote cancer[55,56]. It is thus likely that the immune system is a key factor in the interactions between the gut microbiota and CRC. In addition to the impact of specific pathogens on carcinogenesis, the high redundancy of gut microbiota at a metagenomic level suggests that dysbiosis could exert pro-carcinogenic effects[57]. These properties could be due to interactions between different emergent bacterial strains activating similar pathways. However, the mechanisms that contribute to dysbiosis and to alterations in microbial richness are not well understood, and it is unknown whether dysbiosis is a cause or a consequence of CRC. Indeed, the CRC microenvironment is characterized by host-derived immune and inflammatory responses that could impact on microbial regulation, alter microbiota composition, and favor the outgrowth of specific bacteria that potentially have carcinogenic effects[58]. Dysbiosis in CRC could thus result in selection of microbiota composition via a tumor-linked microenvironment, with the emergence of “keystone pathogens” that have strong effects on bacterial composition and subsequently amplify dysbiosis[59].
With the 16S rRNA sequencing of bacteria from the feces or digestive tissues, numerous studies have reported colonic dysbiosis in patients presenting with CRC[18,60-64]. At present, there is no consensus with respect to the modifications observed in CRC, which are listed in Table 1. However, some bacterial species have been identified and are suspected to play a role in colorectal carcinogenesis[27,54]. These species primarily include Streptococcus bovis (S. bovis)[62,65,66], H. pylori[67-69], Bacteroides fragilis (B. fragilis)[61,62,70-72], Enterococcus faecalis (E. faecalis)[62,73], Clostridium septicum (C. septicum)[74-76], Fusobacterium spp.[77-79] and Escherichia coli (E. coli)[80-82].
Table 1.
Summary of 16S rRNA sequencing and qPCR analyses of colonic microbiota variations in colorectal cancer
| Variation in CRC | Phyla | Genus/species | Population | Ref. |
| Fecal flora | ||||
| ↑ | Enterococcus Faecalis | 20 CRC/17 C | [73] | |
| ↑ | Proteobacteria | Porphyromonas/Escherichia/Shigella Enterococcus/Streptococcus/Peptostreptococcus | 46 CRC/56 C | [62] |
| Bacteroides fragilis | ||||
| ↑ | Bacteroides/Prevotella | 60 CRC/119 C | [61] | |
| ↑ | Peptostreptococcus/Mogibacterium | 21 CRC/22 C | [18] | |
| Anaerococcus/Slakia/Paraprevotella | ||||
| Anaerotruncus/Collinsella/Desulfovibrio | ||||
| Eubacterium/Porphyromonas | ||||
| ↑ | Bacteroidetes | Atopobium/Porphyromonas | 47 CRC/94 C | [63] |
| Fusobacteria | Fusobacterium | |||
| ↑ | Fusobacteria | Fusobacterium/Bacteroides | 19 CRC/20 C | [64] |
| ↑ | Bacteroides/Fusobacterium | 46 CRC/63 C | [214] | |
| Alistipes/Escherichia/Parvimonas/Bilophila | ||||
| ↓ | Faecalibacterium prauznitsii | 20 CRC/17 C | [73] | |
| ↓ | Bacteroidetes | Bacteroides vulgatus/Bacteroides uniformis | 46 CRC/56 C | [62] |
| Roseburia/Butyrate-producing bacteria | ||||
| ↓ | Faecalibacterium prauznitsii/Roseburia | 19 CRC/20 C | [64] | |
| ↓ | Firmicutes (clostridia) | Ruminococcus | 47 CRC/94 C | [63] |
| ↓ | Ruminococcus/Bifidobacterium/Streptococcus | 46 CRC/63 C | [214] | |
| Tumor-associated flora | ||||
| ↑ | Bacteroidetes | Coriobacteriae/Roseburia | 6 CRC/6 AM | [14] |
| Fusobacterium/Faecalibacterium | ||||
| Butyrate-producing bacteria | ||||
| ↑ | Bacteroides | 22 CRC/22 C | [61] | |
| ↑ | Fusobacterium | 95 CRC'95 AM | [106] | |
| ↑ | Bacteroidetes | Bacteroides/Prevotella/Streptococcus | 27 CRC | [18] |
| Fusobacteria | Fusobacterium/Peptostreptococcus | 27 intestinal lumen | ||
| Proteobacteria | Morganella/Porphyromonas | |||
| ↑ | Fusobacterium | 55 CRC/55 AM | [109] | |
| ↓ | Firmicutes | Shigella/Citrobacter/Serratia/Salmonella | 6 CRC/6 AM | [14] |
| ↓ | Firmicutes | Lactobacillus/Roseburia/Pseudobutyrivibrio | 27 CRC | [18] |
| 27 intestinal lumen | ||||
| ↓ | Bacteroidetes | 95 CRC/95 AM | [106] | |
| Firmicutes (clostridia) | ||||
| Mucosa-associated flora | ||||
| ↑ | Fusobacterium/Porphyromonas | 32 CRC | [18] | |
| Peptostreptococcaceae/Gemella | 22 C (swab) | |||
| Mogibacterium/Klebsiella | ||||
| ↓ | Faecalibacterium/Blautia/Anaeroslipes | 32 CRC | [18] | |
| Lachospira/Bifidobacterium | 22 C (swab) | |||
CRC: Colorectal cancer; C: Control patients; AM: Adjacent tumor, normal mucosa of patients with CRC.
S. bovis/gallolyticus
S. bovis was the first bacteria indirectly associated with CRC. Indeed, McCoy et al[83] reported the first recognized case of enterococcal endocarditis associated with CRC in 1951. This association was later confirmed by Hoppes et al[84], who reported that approximately two-thirds of S. bovis endocarditis cases were associated with gastrointestinal disease. In addition, Klein et al[85] reported a 5-fold increased incidence of CRC in patients presenting such endocarditis. Interestingly, in this study, most of patients had asymptomatic colorectal tumors that were occasionally benign adenomas, suggesting the involvement of S. bovis at an early step of CRC development. However, there are contrasting results in studies of the association between S. bovis and CRC; such differences could be due to variations in the collection of feces, sample processing, bacterial culturing or the molecular analysis of fecal samples[66,86,87].
Studies that confirmed the association between S. bovis infection and CRC reported prevalences from 33% to 100% of S. bovis with underlying CRC[88]. It was shown that the mucosal detection of S. bovis could be a better tool than fecal level to assess its presence in patients with CRC[27,89]. More recently, Abdulamir et al[90] used molecular techniques to show increased S. bovis in colorectal adenomas and CRC tissues, strengthening the possible involvement of this pathogen in colorectal carcinogenesis. The mechanisms behind this link remain unclear, but these three hypotheses have been reported: (1) S. bovis adheres to both normal epithelial and neoplastic cells; (2) this species attains a competitive growth advantage in a tumor microenvironment by foraging on tumor metabolites; and (3) S. bovis induces inflammation and/or pro-carcinogenic pathways, leading to tumor progression, especially from pre-malignant tumors[27]. Thus, the fact that increase in the numbers of S. bovis probably occurs at an early step in colorectal carcinogenesis could lead its use for early detection of CRC[91]. Moreover, these results directly impact clinical practice, leading to recommended colorectal endoscopic exploration in all patients presenting with S. bovis infection.
H. pylori
H. pylori has been directly and strongly linked to gastric cancer[49] and has recently been classified as a carcinogen of the gastrointestinal tract by the International Agency for Research on Cancer. The role of H. pylori in colorectal carcinogenesis remains controversial due to differences between studies with respect to the H. pylori strains and their specific virulence factors. A meta-analysis of 11 studies conducted between 1991 and 2002 was published by Zumkeller et al[69] and reported a 1.4-fold increased risk of CRC in patients presenting with a H. pylori infection. More recently, Guo et al[92] reported a statistical association between H. pylori and colorectal adenomas in a meta-analysis of 7679 Asian patients. This result suggested a carcinogenic role of H. pylori at an early stage of carcinogenesis. The presence of high levels of H. pylori has been reported in CRC tissue using a specific 16S rDNA polymerase chain reaction (PCR) assay and pyrosequencing[67]. Jones et al[68] analyzed paraffin-embedded tissue blocks of normal colonic mucosa, adenomas and adenocarcinomas from 180 patients. The results indicated a significant increase in colonic mucosa-associated colonization by H. pylori in adenomas and adenocarcinomas compared to normal mucosa[68].
Although the role of H. pylori in gastric carcinogenesis has been better studied and described than its involvement in colorectal carcinogenesis, some hypotheses can be extrapolated from the pathophysiology of bacteria-linked gastric cancer. Bacterial cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA) are encoded by pathogenicity islands in some H. pylori strains and may induce the activation of inflammation pathways and cellular proliferation in gastric cancer[93]. Shmuely et al[94] reported that patients presenting with seropositivity for CagA-positive H. pylori strains had a significantly increased risk of CRC. Another hypothesis is the direct and indirect production of pro-oxidative reactive oxygen and nitrogen species by some H. pylori strains that could participate to colorectal carcinogenesis[49].
B. fragilis
B. fragilis are common anaerobic bacteria that are detected in up to 80% of children and adults but which represent less than 1% of gut microbiota[95,96]. There are two molecular subtypes of B. fragilis, nontoxigenic or enterotoxigenic, with the latter known to be responsible in diarrheal illnesses in humans, especially children[97]. Enterotoxigenic B. fragilis is easily detected in fecal samples of up to 40% of healthy adults, facilitating studies on the role of these bacteria in gastrointestinal diseases[98]. Indeed, some studies have reported increased colonic colonization by B. fragilis in patients presenting with CRC[61,62]. For example, Sobhani et al[61] performed pyrosequencing on stools from 179 patients with CRC or controls who had a normal colposcopy. The results indicated more colonization by B. fragilis in patients presenting with CRC than in control patients[61]. Most of enterotoxigenic B. fragilis strains detected in mucosal samples from patients with CRC harbored the bft gene, which encodes the bacterial toxin B. fragilis toxin (BFT). This toxin is involved in the pathogenicity of enterotoxigenic B. fragilis[99,100]. Indeed, the involvement of BFT in colorectal carcinogenesis is becoming increasingly evident as this toxin directly affects pathways that lead to increased cell proliferation, the epithelial release of pro-inflammatory effectors, and DNA damage in in vitro and CRC-predisposed mouse models[70-72].
E. faecalis
E. faecalis is a facultative anaerobic commensal bacterium of the oral cavity and the gastrointestinal tract. Recently, this species has emerged as a human pathogen[101]. Balamurugan et al[73] performed 16S rRNA real-time PCR from the feces of patients with CRC and healthy volunteers. The authors reported significantly higher E. faecalis fecal populations in patients with CRC compared to healthy controls[62,73]. These results were confirmed more recently by Wang et al[62]. The mechanisms linking E. faecalis to colorectal carcinogenesis remain unclear, but the production of pro-oxidative reactive oxygen species (ROS) by E. faecalis has been described in cellular and animal models[102,103]. Moreover, E. faecalis can trigger colitis, dysplasia and CRC in a susceptible interleukin (IL)-10-/- mouse model[104].
C. septicum
C. septicum is a rare cause of bacteremia (less than 1%). As for S. bovis endocarditis, C. septicum infections have been clinically linked to CRC[76]. Indeed, it has been reported that gastrointestinal disease and/or colorectal malignancies can be found in up to 40% of patients presenting with C. septicum infections[74,75]. Hermsen et al[75] reported an analysis of 320 cases of C. septicum infections, of which more than 40% had a gastrointestinal origin, primarily malignant. The underlying mechanisms of this association remain unknown, but one hypothesis is that the hypoxic and acidic tumor microenvironment favors the germination of C. septicum spores via ingestion of contaminated food[27,105]. However, no direct involvement of C. septicum in colorectal carcinogenesis has been well defined.
Fusobacterium nucleatum
Fusobacterium nucleatum (F. nucleatum) is an anaerobic Gram-negative pathogenic bacterium that recently emerged as a potential candidate for CRC susceptibility. Indeed, recent metagenomic analyses using whole-genome and bacterial 16S rRNA sequencing revealed an enrichment of F. nucleatum in colonic tumor-associated microbiota compared to adjacent normal mucosa in patients with CRC[106,107]. Moreover, other studies found that luminal F. nucleatum colonization was higher in patients presenting with colorectal adenomas compared to healthy patients. An increase of F. nucleatum colonization in adenomas compared to adjacent colonic normal mucosa has also been reported[77,78]. These data strongly suggest that, more than simply being associated with CRC, F. nucleatum likely acts at the early steps of colorectal carcinogenesis promotion. This effect may be mediated via its FadA adhesion and activates the Wnt/β-catenin pathway[79]. This hypothesis has been verified in a CRC-predisposed adenomatous polyposis coli (APC)min/+ mouse model. In this animal model, infection with F. nucleatum increased tumor multiplicity and selectively recruited tumor-infiltrating myeloid cells, which can promote tumor progression[77]. Interestingly, F. nucleatum is detected in up to 80% of tumor samples. The increased colonic colonization of this species has been correlated with CRC stage, as has been described for other pathogens[108,109]. Indeed, Viljoen et al[109] identified significant associations between high-level colonization by F. nucleatum and advanced stage III-IV CRC, as well as with microsatellite instability tumor phenotype. These results could lead to further studies that explore (1) the role of F. nucleatum in modulating the DNA repair systems involved in colorectal carcinogenesis; and (2) the potential pathogenic interactions between different pathogenic bacterial species.
E. coli
Although E. coli is a commensal bacteria of the human microbiota and represents the most common cultivable, Gram-negative, aero-anaerobic bacteria in the gut, various studies have demonstrated a clear link between mucosa-adherent E. coli and CRC[80-82]. E. coli belonging to phylogroups B2 and D comprise most of the pathogenic strains that express virulence factors, and some of these species are involved in chronic inflammatory bowel diseases, which are known risk factors for CRC[110,111].
Some studies have reported higher levels of colonic colonization by mucosa-associated E. coli in patients with CRC compared to healthy patients[80,81]. For example, Swidsinski et al[81] performed 16S PCR using colonic mucosa tissue samples and detected E. coli in only 3% of healthy patient biopsies. However, E. coli was detected and predominant in 62% and 77% of patients presenting with adenomas and carcinomas, respectively[81]. A few years later, Martin et al[80] reported that more than 70% of mucosa samples from patients with CRC were colonized by bacteria, generally E. coli. Increased mucosa- and tumor-associated E. coli colonization in CRC patients has been confirmed by several studies, strongly implicating E. coli in colorectal carcinogenesis[82,108,112,113].
CRC samples commonly exhibit colonic mucosal E. coli that could express genes (afa, lpfA, eae and/or cyclomodulin toxins) that confer characteristics that are relevant to pathogenesis, including M-cell translocation, angiogenesis and genotoxicity[112,114-117]. Cyclomodulins such as colibactin, are toxins that interfere with the eukaryotic cell cycle and induce DNA damage and genomic instability, which are involved in colorectal carcinogenesis[118,119]. A pro-carcinogenic effect of B2 phylogroup colibactin-producing E. coli has been confirmed in mouse models. Specifically, these bacteria are involved in inflammation pathways and cell proliferation fueling[82,108,120]. Moreover, possible interactions between E. coli and DNA repair system involved in colorectal carcinogenesis, such as the DNA mismatch repair (MMR) system, have been described[112,121]. Interestingly, these B2 colibactin-producing E. coli are preferentially detected in patients with CRC[82,108,115,120] and were more prevalent in the mucosa of patients presenting with advanced stage III/IV CRC than in those with stage I CRC[108]. These results suggest that E. coli could serve as a prognostic factor in CRC.
SUSPECTED INVOLVED MECHANISMS
As described above, some bacterial species may be involved in colorectal carcinogenesis. Many suspected involved mechanisms, occasionally shared by different species, have been described. These mechanisms include bacterial-derived genotoxins, microbial-derived metabolism, the modulation of host defenses and inflammation pathways, oxidative stress induction, and anti-oxidative defense regulation (Figure 1).
Figure 1.
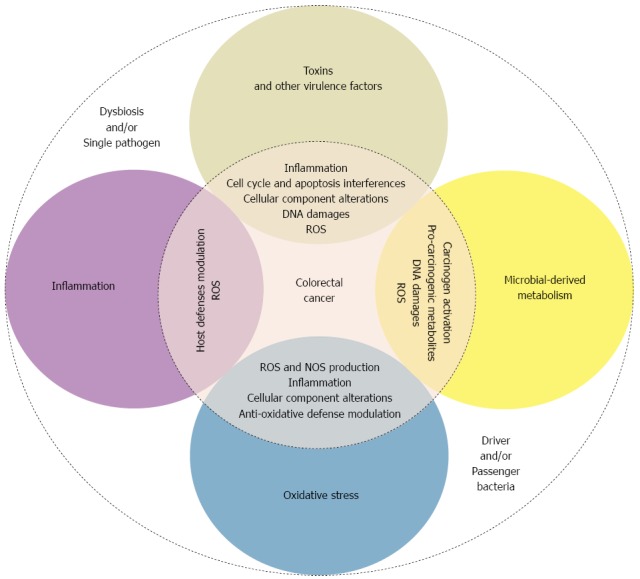
Suspected mechanisms by which the gut bacterial microbiota participates in colorectal carcinogenesis. The bacterial microbiota induces colorectal carcinogenesis through several mechanisms. The primary suspected mechanisms are bacteria-derived genotoxin production, microbial-derived metabolism, the modulation of host defenses and inflammation pathways, oxidative stress induction, and anti-oxidative defense regulation. These mechanisms result in various cellular effects and alterations of host defenses that lead to genomic instability and epithelial cell proliferation, which are involved in colorectal carcinogenesis. ROS: Reactive oxygen species; NOS: Nitrogen species.
Bacterial-derived genotoxins and other bacterial virulence factors
During their phylogenetic evolution, bacteria progressively acquired virulence factors that conferred pathogenicity. For example, bacteria developed the ability to penetrate the gut mucosal barrier, as well as to adhere to and invade intestinal epithelial cells, specifically through the use of flagellum, pili, and adhesins[110,122,123]. Most of the disease-promoting and pro-carcinogenic effects of pathogens depend on these virulence factors[58]. Pathogens can interact with adhesion molecules. For example, F. nucleatum uses the FadA virulence factor to adhere to and invade cells[124], thereby activating β-catenin signaling pathway and promoting CRC[79]. In the same way, certain CRC-associated E. coli strains have acquired virulence factors, such as the afa and eae adhesins, which confer the ability to adhere to and invade the intestinal epithelium[112,116].
Toxins may be involved in colorectal carcinogenesis by modulating certain host-derived signaling pathways, resulting in the activation of carcinogenesis-promoting pathways. For example, CagA or VacA are produced by some H. pylori strains and increase inflammation and cancer rates[125,126]. B. fragilis has been associated with CRC[71,127], and BFT is another well-described example of a bacteria-derived toxin involved in colorectal carcinogenesis. BFT is a 20 kDa zinc-dependent metalloprotease toxin that is grouped into three isotypes, namely, BFT-1, BFT-2 and BFT-3[100]. These toxins are encoded by a unique B. fragilis-specific gene, which was described by Moncrief et al[128] in 1995. A recent study reported that mucosa-associated BFT-producing B. fragilis were more prevalent in late-stage CRC, suggesting possible role of BFT in CRC promotion and progression[99]. At the molecular level, BFT binds to a specific colonic epithelial receptor, activating the Wnt and nuclear factor-kappa B (NF-κB) pathways. These effects lead to increased cell proliferation, the epithelial release of pro-inflammatory mediators, and the induction of DNA damage[97,129-132]. Furthermore, in the CRC-predisposed multiple intestinal neoplasia APCmin+/- mouse model, BFT promotes IL-17-dependent carcinogenesis[127].
Other toxins, named cyclomodulins [cytolethal distending toxin (CDT), colibactin, cytotoxic necrotizing factor and cycle inhibiting factor], can induce DNA damage, interfere with the cell cycle and/or apoptosis[82,118,127,133-136]. Only CDT and colibactin can directly damage DNA and are linked genomic instability; these proteins are therefore considered true genotoxins[118,133]. Both CDT and colibactin induce double-strand DNA breaks, activate the ataxia telangiectasia mutated (ATM)-checkpoint kinase 2 signaling pathway, and lead to H2AX histone phosphorylation. These effects result in transient G2/M cell cycle arrest and cell swelling.
Most of the Gram-negative bacteria that are involved in CRC produce CDT, which is by far the most well-characterized bacterial-derived genotoxin[137]. The CdtA and CdtC subunits permit the interaction between pathogens and host cells; subsequently, the cytoplasmic CdtB subunit can translocate to the nucleus, act as a DNase and damage DNA[133]. CDT also favors persistent gut colonization and induces the production of pro-inflammatory molecules, such as NF-κB, tumor necrosis factor (TNF)-α, IL-6 and cyclooxygenase (COX) 2. These factors are involved in many carcinogenic processes. CDT has also been reported to induce dysplasia in a Helicobacter hepaticus-linked hepatocarcinoma mouse model[138,139]. When combined, DNA damage, interference with the cell cycle and the modulation of pro-inflammatory pathways can lead to mutations that are involved in genomic instability in CRC.
Colibactin is another bacterial-derived genotoxin that has recently attracted attention. It was first described in 2006 by Nougayrède et al[119] but has not yet been isolated and purified. Colibactin synthesis and activation requires complex machinery that involves three non-ribosomal peptide megasynthases (NRPS), three polyketide megasynthases (PKS), two NRPS/PKS hybrids and at least eight of nine accessory enzymes encoded in the pks island[119]. The pks island is found primarily in the Enterobacteriaceae family, especially in the E. coli B2 phylogroup[115,119]. Interestingly, other micro-organisms, such as Proteus mirabilis and Klebsiella pneumoniae, which are associated with bacterial-induced colitis and an immunodeficient-CRC-mouse model, also carry the pks island[140-142]. Colibactin induces ROS formation, double-strand DNA breaks, and the associated ATM-mediated DNA damage response, cell cycle arrest and genomic instability[82,118]. Furthermore, colibactin-producing E. coli can enhance tumor growth by inducing the emergence of senescent epithelial cells secreting growth factors and/or by the secretion of pro-tumoral molecules by infiltrating cells[58,114,120]. Interestingly, even though toxin-producing bacteria constitute a minority of the colonic microbiota, metatranscriptomic analyses performed on human CRC tissues samples reveal a high expression of these toxins in the colon[143].
Microbial-derived metabolism affecting carcinogenesis
In addition to the involvement of bacterial virulence factors, such as genotoxin production, it has become increasingly clear that microbial-derived metabolism strongly impacts CRC development[144]. These metabolic activities may affect colorectal carcinogenesis via the following several processes: regulating the generation of CRC-promoting secondary bile acids; the metabolic activation or inactivation of pro-carcinogenic compounds, dietary phytochemicals and xenobiotics; hormone metabolism; and the modification of inflammation pathways[58].
The interplay between diet, bile acids and gut microbiota is complex. Indeed, high-fat diets are correlated with increased bile secretion and an increased risk of CRC[145-147]. Primary bile acids excreted into the gut are converted through microbial derived-metabolism, including hydrolase activities, into secondary bile acids. These acids are used by microorganisms as an energy source[148] but are also known to be involved in many colorectal carcinogenesis-linked process, such as apoptosis, cell proliferation, DNA damage induction and tumor promotion[27]. Bernstein et al[146] reported a higher incidence of tumors in bile acid-exposed gut in a mouse model. Indeed, bacteria-transformed bile acids can result in DNA damage by the production of pro-oxidative molecules, such as ROS and nitrogen species (NOS)[149,150]. Therefore, chronic exposure to increased levels of secondary bile acids may favor the induction of DNA damage, leading to genomic instability, which is involved in CRC. Furthermore, strong antimicrobial bile acid activities lead to significant changes in the gut microbiota composition, with a relative increase in some Gammaproteobacteria and Bacteroidetes species that are associated with CRC[151].
Some carcinogens are inactivated in the liver by glucuronic acid-mediated conjugation and are excreted via bile in the digestive tract. In the gut, and particularly in the colon, this process may be reversed by bacterial β-glucuronidase activity, which can lead to CRC. Indeed, Takada et al[152] reported a decrease in tumor number with the inhibition of bacterial β-glucuronidase activity in a CRC rat model. Moreover, fecal β-glucuronidase activity is increased in patients with CRC compared to healthy controls[153]. These results strongly support the involvement of bacterial β-glucuronidase activity in the initiation and progression of CRC via the reactivation of toxic components. Moreover, bacterial β-glucuronidase activity plays a major role in the metabolism of xenobiotics and affects the activity and side effects of certain antitumor drugs[58,154]. For example, irinotecan, a commonly used chemotherapy for CRC that is inactivated in the liver, is locally modified in the gut, generating intermediate molecules without systemic anti-tumor effect and inducing major treatment-limiting side effects (e.g., severe diarrhea). These undesired effects can be prevented with the use of antibiotics or bacterial β-glucuronidase inhibitors[155].
Contrary to microbial carbohydrate fermentation, which can benefit the host through the generation of short chain fatty acids (e.g., butyrate, acetate, propionate)[156], microbial protein fermentation generates potentially toxic and pro-carcinogenic metabolites involved in CRC, such as phenols, sulfides, ammonia and nitrosamines[58]. It has been reported that protein-rich and low-carbohydrate diets can lead to the increased microbial production of toxic metabolites to the detriment of cancer-protective metabolites[157], increasing the risk for CRC[158]. However, concentration-dependent effects on CRC have been described for butyrate. At lower concentrations, butyrate appears to stimulate epithelial cell proliferation[159], while other studies have demonstrated anti-proliferative and anti-cancer properties of this compound[160]. A subset of bacteria, including Bacteroidetes and Firmicutes species, produce potentially bioactive substances via the degradation of amino acids, especially nitrogenous compounds, in the gut[161-163]. These compounds can exert carcinogenic effects through DNA alkylation, leading to mutations that have been reported in Western diet-linked CRC[161-163]. Moreover, sulfides produced in the gut by the bacterial reduction of dietary sulphate, as well as the metabolism of other compounds[164], are enterotoxic[165]. These sulfides have genotoxic effects on human cell lines at physiological concentrations[166]. These effects occur primarily via the induction of ROS formation and DNA damage[167].
Chronic and/or excessive consumption of alcohol has been found to be an important risk factor for many cancers, including CRC[168]. Microbial metabolism may contribute to the toxicity of alcohol, especially in the gastrointestinal tract, where aerobic and facultative anaerobic bacteria convert ethanol to acetaldehyde[169]. Indeed, acetaldehyde is known to be a highly toxic and pro-carcinogenic compound with various negative effects, ranging from DNA damage and impaired DNA excision repair to the degradation of folate. All of these effects have been implicated in colorectal carcinogenesis[169-171]. The role of microbiota in this process has also been reinforced by Homann et al[172], who reported that the conversion of ethanol to acetaldehyde was inhibited by the use of antibiotics, such as ciprofloxacin. This drug kills primarily aerobic and facultative anaerobic bacterial populations[172].
Host defense modulation and inflammation
As previously mentioned, the intestinal mucosa constitute the first line of defense against gut commensal or pathogen bacteria and related microbial molecules. Intestinal epithelial cells need to rapidly detect the presence of pathogens in order to mount a suitable immune response. However, these cells also must maintain a moderate immune response against or tolerance for non-pathogenic bacteria[173]. The maintenance of gut homeostasis and these interactions between the host and microbiota involve innate immunity receptors, such as Toll Like Receptors (TLRs) and Nod Like Receptors (NLRs), which recognize particular molecular motifs associated with pathogens. The activation of these receptors leads to a cellular response, including the activation of MAPK, NF-κB or PI3K/AKT signaling pathways[174]. The activation of these pathways can induce the expression of pro-inflammatory cytokines (e.g., TNF-α, IL-6, IL-8) and/or antimicrobial peptides, all of which are involved in the development of an inflammatory response. Indeed, a decrease in intestinal tumor number has been reported in azoxymethane (AOM)-treated APCMin/+ or IL-10-/- mice invalidated for MyD88 gene, which encodes a TLR signaling adaptor[175,176]. However, these results remain controversial given that another recent study reported that MyD88-/- mice treated with AOM/dextran sulfate sodium (DSS) exhibit increased susceptibility to colorectal tumors. These results suggest that the role of TLR signaling and the host response in CRC differ between colitis-associated and chemically induced CRC[177]. Moreover, Nod1- or Nod2-deficient APCMin/+ and AOM/DSS-treated mice exhibit an increase in the number of colorectal tumors compared to control animals, suggesting the involvement of NLRs in colorectal carcinogenesis[55,178]. These results highlight the major role of the host immune response to gut microbiota in CRC development.
It is now well established that inflammatory bowel disease patients, who are known to have an increased risk for developing CRC, present many changes in their microbiota composition[179-181]. For these reasons, the involvement of inflammation in the establishment of dysbiosis-related CRC is increasingly evident. Some in vivo studies have shown that the gut microbiota composition differs between AOM-treated and untreated IL-10-/- mice. Moreover the emergence of dysbiosis after each cycle of DSS treatment in mice treated with AOM has been observed[82,182]. Taken together, these data constitute strong evidence that inflammation plays an important role in the modulation of the microbiota and dysbiosis emergence during colorectal carcinogenesis.
However, inflammation could also be linked to the host response induce by bacteria during CRC development. It has been previously reported in a CRC-predisposed APCMin/+ mouse model that BFT induces a Th17 pro-inflammatory response, which is involved in the development of early-stage tumors[127]. Furthermore, the contribution of S. bovis to colorectal carcinogenesis is associated with the increased expression of pro-inflammatory genes, such as IL-1, IL-8 and COX-2[90]. Moreover, APCMin/+ and IL-10-/- mice infected with F. nucleatum and E. faecalis, respectively, exhibit increased immune cell infiltration in tumors and colonic mucosa and heightened expression of pro-inflammatory cytokines, such as TNF-α, IL-1β, IL-6 and IL-8[77,104,183]. E. coli is one of the best-characterized bacteria associated with inflammatory bowel disease. An abnormal colonization of the gut mucosa by adherent and invasive E. coli in inflammatory bowel disease patients has been reported[110,111]. Moreover, Raisch et al[184] showed that CRC-associated E. coli can induce the expression of the pro-inflammatory gene COX-2 in macrophages, supporting the bacterial modulation of inflammation in colorectal carcinogenesis.
Oxidative stress and anti-oxidative defenses modulation
ROS induction appears to have a major and central role in microbiota-linked CRC via the previously described mechanisms. ROS can be generated by cells during infection and inflammation, as previously discussed, or directly by gut microbiota[177]. The induction of ROS is known to be a major defense mechanism of infected cells, contributing to the elimination of bacteria[48,119]. It has been reported in both in vitro and in vivo studies that some Enterococci species, especially E. faecalis, generate hydroxyl radicals[102,103]. These radicals are powerful mutagens that can cause DNA breaks, point mutations and protein-DNA crosslinking, all contributing to genomic instability in CRC[185-187]. Furthermore, this bacterium can induce aneuploidy in colonic epithelial cells, and the use of inhibitors of ROS and NOS can prevent this effect, supporting the role of bacterial-induced oxidative stress in colorectal carcinogenesis[82]. Moreover, the role of H. pylori in gastric carcinogenesis via the induction of oxidative stress has been clearly demonstrated[49], and this species is able to both produce and induce the production of ROS by immune cells. In this manner, H. pylori affects many signal transduction pathways in gastric cells and thereby promotes gastric cancer. It could thus extrapolate the fact that existence of colorectal chemical-, bacterial- and/or immune-induced inflammation also induces the recruitment of neutrophils and macrophages, which are major sources of ROS, leading to the genetic and epigenetic alterations involved in CRC[188-190]. The gut microbiota also promote host-derived production of nitric oxide and its secondary NOS, especially through the activation of macrophages in the inflammation response, which can induce DNA damage. Some bacterial species can directly generate NOS[191]. Sobko et al[192,193] reported that Lactobacilli and Bifidobacteria generate significant levels of NOS in germ-free and monoassociated mice and that a nitrate-enriched diet increased NOS production. However, it is not clear whether in ROS and NOS produced in vivo are sufficiently long lived to diffuse from immune cells to the extracellular matrix and subsequently enter the nucleus of epithelial cells to induce damage DNA[194].
Oxidative stress is defined by an imbalance between the levels of pro-oxidative molecules (e.g., ROS and NOS) and the effectiveness of anti-oxidative defenses[195]. Oxidative stress results in primarily irreversible direct or organic substrate-mediated cell damage, including DNA breaks and damage, protein aggregation or fragmentation, and cellular membranes dysfunction[185,195]. The toxic effects of ROS and NOS are thus balanced by various enzymatic and non-enzymatic anti-oxidative defenses, which help to regulating ROS/NOS production and repair mechanisms[196,197]. DNA repair mechanisms are altered in CRC[198,199]. Moreover, under specific conditions, the balance between pro- and anti-oxidative compounds is lost, especially in cases of bacterial infection. The bacterial modulation of anti-oxidative defenses, especially DNA repair systems, is becoming increasingly clear. Mangerich et al[200] reported decreased expression of DNA repair and oxidative response genes in the colons of colitis-induced CRC mice model that were infected with H. hepaticus. Moreover, Maddocks et al[112,121] reported in vitro results demonstrating the ability of some enteropathogenic E. coli strains to downregulate the DNA MMR system in infected HT-29 intestinal epithelial cells via post-transcriptional effect of a secreted effector protein. This downregulation of the MMR system led to the accumulation of mutations involved in colorectal carcinogenesis[112,121]. Furthermore, a study on APCMin/+ MMR-deficient mice strongly supports a possible role of an interaction between gut microbiota and MMR deficiency in CRC induction[159]. Indeed, Belcheva et al[159] reported that (1) altering the microbiota composition reduces tumorigenesis; (2) gut microbes can fuel the hyperproliferation of MMR-altered intestinal epithelial cells; and (3) the MMR pathway has a role in regulating APC/β-catenin activity and modulating the differentiation of transit-amplifying cells in the colon[159]. Moreover, Viljoen et al[109] reported increased colonic colonization by F. nucleatum in CRC patients with an MMR deficiency-linked microsatellite instability phenotype. All of these data strengthen the hypothesis of an interaction between the gut microbiota and the DNA repair system in colorectal carcinogenesis.
MICROBIOTA IMBALANCE AND CLINICAL IMPLICATIONS
As previously mentioned, the gut microbiota likely plays a major role in the promotion and progression of CRC via several mechanisms, including inflammation, metabolism and genotoxicity. There are therefore many possible ways by which to target the microbiota in terms of CRC prevention strategies. Indeed, the use of probiotics or fecal transplantation protocols could combat CRC-associated dysbiosis and thus restore eubiosis in chronic diseases, helping to reduce microbiota-induced genotoxicity and activation of inflammatory, proliferative and pro-carcinogenic pathways[58]. However, this microbiota-targeting therapy approach has not been well studied in CRC.
Genotoxins are a target of interest in the context of CRC treatment. For example, supportive evidence has recently been provided that colibactin-producing E. coli could be major actors in CRC-related genomic instability[58,114,115,119,120]. Colibactin synthesis requires the serine enzyme ClbP, which acts as a peptidase to produce colibactin NRPS compounds[201-203]. On this basis, it has been reported that boronic acid compounds, which are potent inhibitors of active serine enzymes, suppress the genotoxic activity of colibactin-producing E. coli in vitro and in vivo[204]. In CRC mice model, treatment with such compounds was shown to prevent cell proliferation and genotoxin-induced tumorigenesis compared to water-treated mice[204].
Changes in gut microbiota composition could also lead to an altered host immune response. On this basis, some authors have studied the impact of the oral administration of probiotics on immunologic signaling. These studies have provided supportive evidence that the gut microbiota plays an essential role in intestinal epigenomic mechanisms of the host[205,206]. Moreover, it was reported that the deletion of lipoteichoic acid (LTA), a TLR2 ligand, normalizes innate and adaptive pathogenic immune responses and decreases the number of tumors in a CRC-predisposed murine model. It was also reported that LTA-deficient Lactobacillus acidophilus (1) decreased inflammation and protected against CRC[207]; (2) prevented or induced the regression of established colitis and polyposis[207,208]; and (3) downregulated downstream signaling[209], and stimulated tumor suppressor gene expression in CRC cell lines[210]. Furthermore, some commensal bacteria, such as Bifidobacterium breve and Lactobacillus rhamnosus, inhibit the production of pro-inflammatory cytokines and decrease host DNA methylation and histone acetylation events that are involved in colorectal carcinogenesis[211-213]. All of these results underline a feasible microbiota-targeting therapy approach in CRC through the use of probiotics or genetically modified bacteria. Even if fecal transplantation has not been well studied in CRC, future transplantation studies using germ-free CRC animal models likely represent an important next step in this line of inquiry.
To provide adequate treatments as part of modern personalized medicine, accurate prognosis factor have yet to be establish. Prognostic factors in CRC primarily depend on morphologic and histologic results. Recently, there has been a significant body of work involving the gut microbiota as a prognosis factor in CRC. Bonnet et al[108] reported increased colonic colonization by cyclomodulin-producing E. coli in advanced stage CRC, that have been ever described by Viljoen et al[109] for enterotoxigenic B. fragilis and F. nucleatum. These results support a possible use of microbiota CRC prognosis markers that may be used to improve patient selection for aggressive, suitable treatments.
CONCLUSION
The advent of modern molecular microbiota sequencing techniques has strongly improved the characterization of microbiota variations in CRC. However, a better understanding of the interactions between the host and pathogens in colorectal carcinogenesis requires further microbiota functional studies, especially with respect to metabolomics and RNA sequencing approaches. All of the studies published in this regard have been performed without classifying tumors according to their molecular phenotype. Investigations should also consider the heterogeneity of CRC tumors by studying microbiota imbalances in relation to molecular pathways involved in colorectal carcinogenesis, such as chromosomal and microsatellite instabilities or CpG island methylator phenotypes. In summary, the role of the microbiota in CRC is increasingly evident and perhaps represents a new approach towards the improved therapeutic management of patients with CRC.
Footnotes
Supported by Inserm and Université d’Auvergne (UMR 1071), INRA (USC-2018); and grants from “Conseil regional d’Auvergne”, “Nuovo Soldati Foundation for Cancer Research” and “Fondation pour la recherche médicale”.
Conflict-of-interest statement: None of the authors have any conflicts of interest to declare.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 6, 2015
First decision: August 25, 2015
Article in press: October 20, 2015
P- Reviewer: Altomare DF S- Editor: Gong ZM L- Editor: A E- Editor: Wang CH
References
- 1.Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol. 1977;31:107–133. doi: 10.1146/annurev.mi.31.100177.000543. [DOI] [PubMed] [Google Scholar]
- 2.Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Doré J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Neish AS. Microbes in gastrointestinal health and disease. Gastroenterology. 2009;136:65–80. doi: 10.1053/j.gastro.2008.10.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goncharova GI, Dorofeĭchuk VG, Smolianskaia AZ, Sokolova KIa. [Microbial ecology of the intestines in health and in pathology] Antibiot Khimioter. 1989;34:462–466. [PubMed] [Google Scholar]
- 5.Dominguez-Bello MG, Blaser MJ, Ley RE, Knight R. Development of the human gastrointestinal microbiota and insights from high-throughput sequencing. Gastroenterology. 2011;140:1713–1719. doi: 10.1053/j.gastro.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulder IE, Schmidt B, Lewis M, Delday M, Stokes CR, Bailey M, Aminov RI, Gill BP, Pluske JR, Mayer CD, et al. Restricting microbial exposure in early life negates the immune benefits associated with gut colonization in environments of high microbial diversity. PLoS One. 2011;6:e28279. doi: 10.1371/journal.pone.0028279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dethlefsen L, Eckburg PB, Bik EM, Relman DA. Assembly of the human intestinal microbiota. Trends Ecol Evol. 2006;21:517–523. doi: 10.1016/j.tree.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 8.Stanghellini V, Barbara G, Cremon C, Cogliandro R, Antonucci A, Gabusi V, Frisoni C, De Giorgio R, Grasso V, Serra M, et al. Gut microbiota and related diseases: clinical features. Intern Emerg Med. 2010;5 Suppl 1:S57–S63. doi: 10.1007/s11739-010-0451-0. [DOI] [PubMed] [Google Scholar]
- 9.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, et al. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rajilić-Stojanović M, Heilig HG, Molenaar D, Kajander K, Surakka A, Smidt H, de Vos WM. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol. 2009;11:1736–1751. doi: 10.1111/j.1462-2920.2009.01900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3:213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zwielehner J, Liszt K, Handschur M, Lassl C, Lapin A, Haslberger AG. Combined PCR-DGGE fingerprinting and quantitative-PCR indicates shifts in fecal population sizes and diversity of Bacteroides, bifidobacteria and Clostridium cluster IV in institutionalized elderly. Exp Gerontol. 2009;44:440–446. doi: 10.1016/j.exger.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchesi JR. Human distal gut microbiome. Environ Microbiol. 2011;13:3088–3102. doi: 10.1111/j.1462-2920.2011.02574.x. [DOI] [PubMed] [Google Scholar]
- 15.Hakansson A, Molin G. Gut microbiota and inflammation. Nutrients. 2011;3:637–682. doi: 10.3390/nu3060637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 17.Kelly D, Mulder IE. Microbiome and immunological interactions. Nutr Rev. 2012;70 Suppl 1:S18–S30. doi: 10.1111/j.1753-4887.2012.00498.x. [DOI] [PubMed] [Google Scholar]
- 18.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 2012;7:e39743. doi: 10.1371/journal.pone.0039743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arthur JC, Jobin C. The struggle within: microbial influences on colorectal cancer. Inflamm Bowel Dis. 2011;17:396–409. doi: 10.1002/ibd.21354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mai V. Dietary modification of the intestinal microbiota. Nutr Rev. 2004;62:235–242. doi: 10.1301/nr2004.jun235-242. [DOI] [PubMed] [Google Scholar]
- 22.Rastall RA. Bacteria in the gut: friends and foes and how to alter the balance. J Nutr. 2004;134:2022S–2026S. doi: 10.1093/jn/134.8.2022S. [DOI] [PubMed] [Google Scholar]
- 23.Hopkins MJ, Sharp R, Macfarlane GT. Age and disease related changes in intestinal bacterial populations assessed by cell culture, 16S rRNA abundance, and community cellular fatty acid profiles. Gut. 2001;48:198–205. doi: 10.1136/gut.48.2.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zoetendal EG, Akkermans ADL, Akkermans-van Vilet WM, de Visser JAGM, de Vos WM. The host genotype affects the bacterial community in the human gastrointestinal tract. Microb Ecol Health Dis. 2001;13:129–134. [Google Scholar]
- 25.Tlaskalová-Hogenová H, Stepánková R, Hudcovic T, Tucková L, Cukrowska B, Lodinová-Zádníková R, Kozáková H, Rossmann P, Bártová J, Sokol D, et al. Commensal bacteria (normal microflora), mucosal immunity and chronic inflammatory and autoimmune diseases. Immunol Lett. 2004;93:97–108. doi: 10.1016/j.imlet.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 26.Peterson DA, Frank DN, Pace NR, Gordon JI. Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe. 2008;3:417–427. doi: 10.1016/j.chom.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boleij A, Tjalsma H. Gut bacteria in health and disease: a survey on the interface between intestinal microbiology and colorectal cancer. Biol Rev Camb Philos Soc. 2012;87:701–730. doi: 10.1111/j.1469-185X.2012.00218.x. [DOI] [PubMed] [Google Scholar]
- 28.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Umesaki Y, Okada Y, Matsumoto S, Imaoka A, Setoyama H. Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol Immunol. 1995;39:555–562. doi: 10.1111/j.1348-0421.1995.tb02242.x. [DOI] [PubMed] [Google Scholar]
- 30.Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci USA. 2003;100:10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mazmanian SK, Liu CH, Tzianabos AO, Kasper DL. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell. 2005;122:107–118. doi: 10.1016/j.cell.2005.05.007. [DOI] [PubMed] [Google Scholar]
- 32.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 33.Weinstein PD, Cebra JJ. The preference for switching to IgA expression by Peyer’s patch germinal center B cells is likely due to the intrinsic influence of their microenvironment. J Immunol. 1991;147:4126–4135. [PubMed] [Google Scholar]
- 34.Cebra JJ. Influences of microbiota on intestinal immune system development. Am J Clin Nutr. 1999;69:1046S–1051S. doi: 10.1093/ajcn/69.5.1046s. [DOI] [PubMed] [Google Scholar]
- 35.Shanahan F. The host-microbe interface within the gut. Best Pract Res Clin Gastroenterol. 2002;16:915–931. doi: 10.1053/bega.2002.0342. [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Kundu P, Seow SW, de Matos CT, Aronsson L, Chin KC, Kärre K, Pettersson S, Greicius G. Gut microbiota accelerate tumor growth via c-jun and STAT3 phosphorylation in APCMin/+ mice. Carcinogenesis. 2012;33:1231–1238. doi: 10.1093/carcin/bgs137. [DOI] [PubMed] [Google Scholar]
- 37.Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307:1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 39.Laparra JM, Sanz Y. Interactions of gut microbiota with functional food components and nutraceuticals. Pharmacol Res. 2010;61:219–225. doi: 10.1016/j.phrs.2009.11.001. [DOI] [PubMed] [Google Scholar]
- 40.Wall R, Ross RP, Shanahan F, O’Mahony L, O’Mahony C, Coakley M, Hart O, Lawlor P, Quigley EM, Kiely B, et al. Metabolic activity of the enteric microbiota influences the fatty acid composition of murine and porcine liver and adipose tissues. Am J Clin Nutr. 2009;89:1393–1401. doi: 10.3945/ajcn.2008.27023. [DOI] [PubMed] [Google Scholar]
- 41.Manning TS, Gibson GR. Microbial-gut interactions in health and disease. Prebiotics. Best Pract Res Clin Gastroenterol. 2004;18:287–298. doi: 10.1016/j.bpg.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 42.Simmering R, Pforte H, Jacobasch G, Blaut M. The growth of the flavonoid-degrading intestinal bacterium, Eubacterium ramulus, is stimulated by dietary flavonoids in vivo. FEMS Microbiol Ecol. 2002;40:243–248. doi: 10.1111/j.1574-6941.2002.tb00957.x. [DOI] [PubMed] [Google Scholar]
- 43.Stecher B, Hardt WD. The role of microbiota in infectious disease. Trends Microbiol. 2008;16:107–114. doi: 10.1016/j.tim.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 44.Alexander DD, Cushing CA, Lowe KA, Sceurman B, Roberts MA. Meta-analysis of animal fat or animal protein intake and colorectal cancer. Am J Clin Nutr. 2009;89:1402–1409. doi: 10.3945/ajcn.2008.26838. [DOI] [PubMed] [Google Scholar]
- 45.Sandler RS. Epidemiology and risk factors for colorectal cancer. Gastroenterol Clin North Am. 1996;25:717–735. doi: 10.1016/s0889-8553(05)70271-5. [DOI] [PubMed] [Google Scholar]
- 46.Knudson A. Alfred Knudson and his two-hit hypothesis. (Interview by Ezzie Hutchinson) Lancet Oncol. 2001;2:642–645. doi: 10.1016/s1470-2045(01)00524-1. [DOI] [PubMed] [Google Scholar]
- 47.Zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. 2009;392:1–10. doi: 10.1016/j.virol.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 48.Collins D, Hogan AM, Winter DC. Microbial and viral pathogens in colorectal cancer. Lancet Oncol. 2011;12:504–512. doi: 10.1016/S1470-2045(10)70186-8. [DOI] [PubMed] [Google Scholar]
- 49.Handa O, Naito Y, Yoshikawa T. Helicobacter pylori: a ROS-inducing bacterial species in the stomach. Inflamm Res. 2010;59:997–1003. doi: 10.1007/s00011-010-0245-x. [DOI] [PubMed] [Google Scholar]
- 50.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe. 2014;15:317–328. doi: 10.1016/j.chom.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Proctor LM. The Human Microbiome Project in 2011 and beyond. Cell Host Microbe. 2011;10:287–291. doi: 10.1016/j.chom.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 52.Weisburger JH, Reddy BS, Narisawa T, Wynder EL. Germ-free status and colon tumor induction by N-methyl-N’-nitro-N-nitrosoguanidine. Proc Soc Exp Biol Med. 1975;148:1119–1121. doi: 10.3181/00379727-148-38700. [DOI] [PubMed] [Google Scholar]
- 53.Vannucci L, Stepankova R, Kozakova H, Fiserova A, Rossmann P, Tlaskalova-Hogenova H. Colorectal carcinogenesis in germ-free and conventionally reared rats: different intestinal environments affect the systemic immunity. Int J Oncol. 2008;32:609–617. [PubMed] [Google Scholar]
- 54.Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol. 2012;10:575–582. doi: 10.1038/nrmicro2819. [DOI] [PubMed] [Google Scholar]
- 55.Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123:700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu B, Elinav E, Huber S, Strowig T, Hao L, Hafemann A, Jin C, Wunderlich C, Wunderlich T, Eisenbarth SC, et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci USA. 2013;110:9862–9867. doi: 10.1073/pnas.1307575110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schwabe RF, Jobin C. The microbiome and cancer. Nat Rev Cancer. 2013;13:800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. 2012;10:717–725. doi: 10.1038/nrmicro2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, et al. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 2012;6:1858–1868. doi: 10.1038/ismej.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 2011;6:e16393. doi: 10.1371/journal.pone.0016393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012;6:320–329. doi: 10.1038/ismej.2011.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 2013;105:1907–1911. doi: 10.1093/jnci/djt300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, Chen Y, Yang F, Lu N, Wang Z, et al. Dysbiosis signature of fecal microbiota in colorectal cancer patients. Microb Ecol. 2013;66:462–470. doi: 10.1007/s00248-013-0245-9. [DOI] [PubMed] [Google Scholar]
- 65.Abdulamir AS, Hafidh RR, Abu Bakar F. The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res. 2011;30:11. doi: 10.1186/1756-9966-30-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klein RS, Recco RA, Catalano MT, Edberg SC, Casey JI, Steigbigel NH. Association of Streptococcus bovis with carcinoma of the colon. N Engl J Med. 1977;297:800–802. doi: 10.1056/NEJM197710132971503. [DOI] [PubMed] [Google Scholar]
- 67.Grahn N, Hmani-Aifa M, Fransén K, Söderkvist P, Monstein HJ. Molecular identification of Helicobacter DNA present in human colorectal adenocarcinomas by 16S rDNA PCR amplification and pyrosequencing analysis. J Med Microbiol. 2005;54:1031–1035. doi: 10.1099/jmm.0.46122-0. [DOI] [PubMed] [Google Scholar]
- 68.Jones M, Helliwell P, Pritchard C, Tharakan J, Mathew J. Helicobacter pylori in colorectal neoplasms: is there an aetiological relationship? World J Surg Oncol. 2007;5:51. doi: 10.1186/1477-7819-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zumkeller N, Brenner H, Zwahlen M, Rothenbacher D. Helicobacter pylori infection and colorectal cancer risk: a meta-analysis. Helicobacter. 2006;11:75–80. doi: 10.1111/j.1523-5378.2006.00381.x. [DOI] [PubMed] [Google Scholar]
- 70.Housseau F, Sears CL. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/-) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle. 2010;9:3–5. doi: 10.4161/cc.9.1.10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Toprak NU, Yagci A, Gulluoglu BM, Akin ML, Demirkalem P, Celenk T, Soyletir G. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect. 2006;12:782–786. doi: 10.1111/j.1469-0691.2006.01494.x. [DOI] [PubMed] [Google Scholar]
- 72.Wu S, Morin PJ, Maouyo D, Sears CL. Bacteroides fragilis enterotoxin induces c-Myc expression and cellular proliferation. Gastroenterology. 2003;124:392–400. doi: 10.1053/gast.2003.50047. [DOI] [PubMed] [Google Scholar]
- 73.Balamurugan R, Rajendiran E, George S, Samuel GV, Ramakrishna BS. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J Gastroenterol Hepatol. 2008;23:1298–1303. doi: 10.1111/j.1440-1746.2008.05490.x. [DOI] [PubMed] [Google Scholar]
- 74.Chew SS, Lubowski DZ. Clostridium septicum and malignancy. ANZ J Surg. 2001;71:647–649. doi: 10.1046/j.1445-1433.2001.02231.x. [DOI] [PubMed] [Google Scholar]
- 75.Hermsen JL, Schurr MJ, Kudsk KA, Faucher LD. Phenotyping Clostridium septicum infection: a surgeon’s infectious disease. J Surg Res. 2008;148:67–76. doi: 10.1016/j.jss.2008.02.027. [DOI] [PubMed] [Google Scholar]
- 76.Mirza NN, McCloud JM, Cheetham MJ. Clostridium septicum sepsis and colorectal cancer - a reminder. World J Surg Oncol. 2009;7:73. doi: 10.1186/1477-7819-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe. 2013;14:207–215. doi: 10.1016/j.chom.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One. 2013;8:e53653. doi: 10.1371/journal.pone.0053653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 2013;14:195–206. doi: 10.1016/j.chom.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 81.Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology. 1998;115:281–286. doi: 10.1016/s0016-5085(98)70194-5. [DOI] [PubMed] [Google Scholar]
- 82.Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB, et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 2012;338:120–123. doi: 10.1126/science.1224820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McCoy WC, Mason JM 3rd. Enterococcal endocarditis associated with carcinoma of the sigmoid; report of a case. J Med Assoc State Ala. 1951;21:162–166. [PubMed] [Google Scholar]
- 84.Hoppes WL, Lerner PI. Nonenterococcal group-D streptococcal endocarditis caused by Streptococcus bovis. Ann Intern Med. 1974;81:588–593. doi: 10.7326/0003-4819-81-5-588. [DOI] [PubMed] [Google Scholar]
- 85.Klein RS, Catalano MT, Edberg SC, Casey JI, Steigbigel NH. Streptococcus bovis septicemia and carcinoma of the colon. Ann Intern Med. 1979;91:560–562. doi: 10.7326/0003-4819-91-4-560. [DOI] [PubMed] [Google Scholar]
- 86.Dubrow R, Edberg S, Wikfors E, Callan D, Troncale F, Vender R, Brand M, Yapp R. Fecal carriage of Streptococcus bovis and colorectal adenomas. Gastroenterology. 1991;101:721–725. doi: 10.1016/0016-5085(91)90531-o. [DOI] [PubMed] [Google Scholar]
- 87.Potter MA, Cunliffe NA, Smith M, Miles RS, Flapan AD, Dunlop MG. A prospective controlled study of the association of Streptococcus bovis with colorectal carcinoma. J Clin Pathol. 1998;51:473–474. doi: 10.1136/jcp.51.6.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Boleij A, van Gelder MM, Swinkels DW, Tjalsma H. Clinical Importance of Streptococcus gallolyticus infection among colorectal cancer patients: systematic review and meta-analysis. Clin Infect Dis. 2011;53:870–878. doi: 10.1093/cid/cir609. [DOI] [PubMed] [Google Scholar]
- 89.Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol. 2002;68:3401–3407. doi: 10.1128/AEM.68.7.3401-3407.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Abdulamir AS, Hafidh RR, Bakar FA. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol Cancer. 2010;9:249. doi: 10.1186/1476-4598-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boleij A, Tjalsma H. The itinerary of Streptococcus gallolyticus infection in patients with colonic malignant disease. Lancet Infect Dis. 2013;13:719–724. doi: 10.1016/S1473-3099(13)70107-5. [DOI] [PubMed] [Google Scholar]
- 92.Guo Y, Li HY. Association between Helicobacter pylori infection and colorectal neoplasm risk: a meta-analysis based on East Asian population. J Cancer Res Ther. 2014;10 Suppl:263–266. doi: 10.4103/0973-1482.151482. [DOI] [PubMed] [Google Scholar]
- 93.Higashi H, Tsutsumi R, Fujita A, Yamazaki S, Asaka M, Azuma T, Hatakeyama M. Biological activity of the Helicobacter pylori virulence factor CagA is determined by variation in the tyrosine phosphorylation sites. Proc Natl Acad Sci USA. 2002;99:14428–14433. doi: 10.1073/pnas.222375399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shmuely H, Passaro D, Figer A, Niv Y, Pitlik S, Samra Z, Koren R, Yahav J. Relationship between Helicobacter pylori CagA status and colorectal cancer. Am J Gastroenterol. 2001;96:3406–3410. doi: 10.1111/j.1572-0241.2001.05342.x. [DOI] [PubMed] [Google Scholar]
- 95.Huang JY, Lee SM, Mazmanian SK. The human commensal Bacteroides fragilis binds intestinal mucin. Anaerobe. 2011;17:137–141. doi: 10.1016/j.anaerobe.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Macfarlane S, Woodmansey EJ, Macfarlane GT. Colonization of mucin by human intestinal bacteria and establishment of biofilm communities in a two-stage continuous culture system. Appl Environ Microbiol. 2005;71:7483–7492. doi: 10.1128/AEM.71.11.7483-7492.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sears CL. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev. 2009;22:349–69, Table of Contents. doi: 10.1128/CMR.00053-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zitomersky NL, Coyne MJ, Comstock LE. Longitudinal analysis of the prevalence, maintenance, and IgA response to species of the order Bacteroidales in the human gut. Infect Immun. 2011;79:2012–2020. doi: 10.1128/IAI.01348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boleij A, Hechenbleikner EM, Goodwin AC, Badani R, Stein EM, Lazarev MG, Ellis B, Carroll KC, Albesiano E, Wick EC, et al. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin Infect Dis. 2015;60:208–215. doi: 10.1093/cid/ciu787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rhee KJ, Wu S, Wu X, Huso DL, Karim B, Franco AA, Rabizadeh S, Golub JE, Mathews LE, Shin J, et al. Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect Immun. 2009;77:1708–1718. doi: 10.1128/IAI.00814-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pillar CM, Gilmore MS. Enterococcal virulence--pathogenicity island of E. Faecalis. Front Biosci. 2004;9:2335–2346. doi: 10.2741/1400. [DOI] [PubMed] [Google Scholar]
- 102.Huycke MM, Moore D, Joyce W, Wise P, Shepard L, Kotake Y, Gilmore MS. Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol Microbiol. 2001;42:729–740. doi: 10.1046/j.1365-2958.2001.02638.x. [DOI] [PubMed] [Google Scholar]
- 103.Huycke MM, Moore DR. In vivo production of hydroxyl radical by Enterococcus faecalis colonizing the intestinal tract using aromatic hydroxylation. Free Radic Biol Med. 2002;33:818–826. doi: 10.1016/s0891-5849(02)00977-2. [DOI] [PubMed] [Google Scholar]
- 104.Balish E, Warner T. Enterococcus faecalis induces inflammatory bowel disease in interleukin-10 knockout mice. Am J Pathol. 2002;160:2253–2257. doi: 10.1016/S0002-9440(10)61172-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dylewski J, Luterman L. Septic arthritis and Clostridium septicum: a clue to colon cancer. CMAJ. 2010;182:1446–1447. doi: 10.1503/cmaj.091946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012;22:292–298. doi: 10.1101/gr.126573.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H. Towards the human colorectal cancer microbiome. PLoS One. 2011;6:e20447. doi: 10.1371/journal.pone.0020447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, Déchelotte P, Bonnet R, Pezet D, Darfeuille-Michaud A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20:859–867. doi: 10.1158/1078-0432.CCR-13-1343. [DOI] [PubMed] [Google Scholar]
- 109.Viljoen KS, Dakshinamurthy A, Goldberg P, Blackburn JM. Quantitative profiling of colorectal cancer-associated bacteria reveals associations between fusobacterium spp., enterotoxigenic Bacteroides fragilis (ETBF) and clinicopathological features of colorectal cancer. PLoS One. 2015;10:e0119462. doi: 10.1371/journal.pone.0119462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. doi: 10.1053/j.gastro.2004.04.061. [DOI] [PubMed] [Google Scholar]
- 111.Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 112.Maddocks OD, Short AJ, Donnenberg MS, Bader S, Harrison DJ. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS One. 2009;4:e5517. doi: 10.1371/journal.pone.0005517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martins SA, Prazeres DM, Cabral JM, Monteiro GA. Comparison of real-time polymerase chain reaction and hybridization assays for the detection of Escherichia coli genomic DNA in process samples and pharmaceutical-grade plasmid DNA products. Anal Biochem. 2003;322:127–129. doi: 10.1016/j.ab.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 114.Arthur JC, Jobin C. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes. 2013;4:253–258. doi: 10.4161/gmic.24220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One. 2013;8:e56964. doi: 10.1371/journal.pone.0056964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, Knight P, Codling C, Marchesi JR, Winstanley C, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 2014;63:761–770. doi: 10.1136/gutjnl-2013-304739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallée A, Déchelotte P, Darcha C, Pezet D, Bonnet R, et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. 2014;20:6560–6572. doi: 10.3748/wjg.v20.i21.6560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 2010;107:11537–11542. doi: 10.1073/pnas.1001261107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 2006;313:848–851. doi: 10.1126/science.1127059. [DOI] [PubMed] [Google Scholar]
- 120.Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, Sauvanet P, Darcha C, Déchelotte P, Bonnet M, et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut. 2014;63:1932–1942. doi: 10.1136/gutjnl-2013-305257. [DOI] [PubMed] [Google Scholar]
- 121.Maddocks OD, Scanlon KM, Donnenberg MS. An Escherichia coli effector protein promotes host mutation via depletion of DNA mismatch repair proteins. MBio. 2013;4:e00152–e00113. doi: 10.1128/mBio.00152-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Escobar-Páramo P, Grenet K, Le Menac’h A, Rode L, Salgado E, Amorin C, Gouriou S, Picard B, Rahimy MC, Andremont A, et al. Large-scale population structure of human commensal Escherichia coli isolates. Appl Environ Microbiol. 2004;70:5698–5700. doi: 10.1128/AEM.70.9.5698-5700.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Le Gall T, Clermont O, Gouriou S, Picard B, Nassif X, Denamur E, Tenaillon O. Extraintestinal virulence is a coincidental by-product of commensalism in B2 phylogenetic group Escherichia coli strains. Mol Biol Evol. 2007;24:2373–2384. doi: 10.1093/molbev/msm172. [DOI] [PubMed] [Google Scholar]
- 124.Han YW, Ikegami A, Rajanna C, Kawsar HI, Zhou Y, Li M, Sojar HT, Genco RJ, Kuramitsu HK, Deng CX. Identification and characterization of a novel adhesin unique to oral fusobacteria. J Bacteriol. 2005;187:5330–5340. doi: 10.1128/JB.187.15.5330-5340.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, Higashi H, Musashi M, Iwabuchi K, Suzuki M, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natl Acad Sci USA. 2008;105:1003–1008. doi: 10.1073/pnas.0711183105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moncrief JS, Obiso R, Barroso LA, Kling JJ, Wright RL, Van Tassell RL, Lyerly DM, Wilkins TD. The enterotoxin of Bacteroides fragilis is a metalloprotease. Infect Immun. 1995;63:175–181. doi: 10.1128/iai.63.1.175-181.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Goodwin AC, Destefano Shields CE, Wu S, Huso DL, Wu X, Murray-Stewart TR, Hacker-Prietz A, Rabizadeh S, Woster PM, Sears CL, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci USA. 2011;108:15354–15359. doi: 10.1073/pnas.1010203108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wu S, Lim KC, Huang J, Saidi RF, Sears CL. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA. 1998;95:14979–14984. doi: 10.1073/pnas.95.25.14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu S, Powell J, Mathioudakis N, Kane S, Fernandez E, Sears CL. Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-kappaB pathway. Infect Immun. 2004;72:5832–5839. doi: 10.1128/IAI.72.10.5832-5839.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wu S, Shin J, Zhang G, Cohen M, Franco A, Sears CL. The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect Immun. 2006;74:5382–5390. doi: 10.1128/IAI.00060-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nesić D, Hsu Y, Stebbins CE. Assembly and function of a bacterial genotoxin. Nature. 2004;429:429–433. doi: 10.1038/nature02532. [DOI] [PubMed] [Google Scholar]
- 134.Nougayrède JP, Taieb F, De Rycke J, Oswald E. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005;13:103–110. doi: 10.1016/j.tim.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 135.Oswald E, Nougayrède JP, Taieb F, Sugai M. Bacterial toxins that modulate host cell-cycle progression. Curr Opin Microbiol. 2005;8:83–91. doi: 10.1016/j.mib.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 136.Travaglione S, Fabbri A, Fiorentini C. The Rho-activating CNF1 toxin from pathogenic E. coli: a risk factor for human cancer development? Infect Agent Cancer. 2008;3:4. doi: 10.1186/1750-9378-3-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Smith JL, Bayles DO. The contribution of cytolethal distending toxin to bacterial pathogenesis. Crit Rev Microbiol. 2006;32:227–248. doi: 10.1080/10408410601023557. [DOI] [PubMed] [Google Scholar]
- 138.Ge Z, Rogers AB, Feng Y, Lee A, Xu S, Taylor NS, Fox JG. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell Microbiol. 2007;9:2070–2080. doi: 10.1111/j.1462-5822.2007.00939.x. [DOI] [PubMed] [Google Scholar]
- 139.Ge Z, Schauer DB, Fox JG. In vivo virulence properties of bacterial cytolethal-distending toxin. Cell Microbiol. 2008;10:1599–1607. doi: 10.1111/j.1462-5822.2008.01173.x. [DOI] [PubMed] [Google Scholar]
- 140.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Garrett WS, Punit S, Gallini CA, Michaud M, Zhang D, Sigrist KS, Lord GM, Glickman JN, Glimcher LH. Colitis-associated colorectal cancer driven by T-bet deficiency in dendritic cells. Cancer Cell. 2009;16:208–219. doi: 10.1016/j.ccr.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Putze J, Hennequin C, Nougayrède JP, Zhang W, Homburg S, Karch H, Bringer MA, Fayolle C, Carniel E, Rabsch W, et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun. 2009;77:4696–4703. doi: 10.1128/IAI.00522-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dutilh BE, Backus L, van Hijum SA, Tjalsma H. Screening metatranscriptomes for toxin genes as functional drivers of human colorectal cancer. Best Pract Res Clin Gastroenterol. 2013;27:85–99. doi: 10.1016/j.bpg.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 144.Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12:661–672. doi: 10.1038/nrmicro3344. [DOI] [PubMed] [Google Scholar]
- 145.Barrasa JI, Olmo N, Lizarbe MA, Turnay J. Bile acids in the colon, from healthy to cytotoxic molecules. Toxicol In Vitro. 2013;27:964–977. doi: 10.1016/j.tiv.2012.12.020. [DOI] [PubMed] [Google Scholar]
- 146.Bernstein H, Bernstein C, Payne CM, Dvorak K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J Gastroenterol. 2009;15:3329–3340. doi: 10.3748/wjg.15.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ou J, DeLany JP, Zhang M, Sharma S, O’Keefe SJ. Association between low colonic short-chain fatty acids and high bile acids in high colon cancer risk populations. Nutr Cancer. 2012;64:34–40. doi: 10.1080/01635581.2012.630164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Philipp B. Bacterial degradation of bile salts. Appl Microbiol Biotechnol. 2011;89:903–915. doi: 10.1007/s00253-010-2998-0. [DOI] [PubMed] [Google Scholar]
- 149.Bernstein H, Bernstein C, Payne CM, Dvorakova K, Garewal H. Bile acids as carcinogens in human gastrointestinal cancers. Mutat Res. 2005;589:47–65. doi: 10.1016/j.mrrev.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 150.Dvorak K, Payne CM, Chavarria M, Ramsey L, Dvorakova B, Bernstein H, Holubec H, Sampliner RE, Guy N, Condon A, et al. Bile acids in combination with low pH induce oxidative stress and oxidative DNA damage: relevance to the pathogenesis of Barrett’s oesophagus. Gut. 2007;56:763–771. doi: 10.1136/gut.2006.103697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499:97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 152.Takada H, Hirooka T, Hiramatsu Y, Yamamoto M. Effect of beta-glucuronidase inhibitor on azoxymethane-induced colonic carcinogenesis in rats. Cancer Res. 1982;42:331–334. [PubMed] [Google Scholar]
- 153.Kim DH, Jin YH. Intestinal bacterial beta-glucuronidase activity of patients with colon cancer. Arch Pharm Res. 2001;24:564–567. doi: 10.1007/BF02975166. [DOI] [PubMed] [Google Scholar]
- 154.Haiser HJ, Turnbaugh PJ. Is it time for a metagenomic basis of therapeutics? Science. 2012;336:1253–1255. doi: 10.1126/science.1224396. [DOI] [PubMed] [Google Scholar]
- 155.Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh LA, Mani S, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 2010;330:831–835. doi: 10.1126/science.1191175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nyangale EP, Mottram DS, Gibson GR. Gut microbial activity, implications for health and disease: the potential role of metabolite analysis. J Proteome Res. 2012;11:5573–5585. doi: 10.1021/pr300637d. [DOI] [PubMed] [Google Scholar]
- 157.Russell WR, Gratz SW, Duncan SH, Holtrop G, Ince J, Scobbie L, Duncan G, Johnstone AM, Lobley GE, Wallace RJ, et al. High-protein, reduced-carbohydrate weight-loss diets promote metabolite profiles likely to be detrimental to colonic health. Am J Clin Nutr. 2011;93:1062–1072. doi: 10.3945/ajcn.110.002188. [DOI] [PubMed] [Google Scholar]
- 158.Hughes R, Cross AJ, Pollock JR, Bingham S. Dose-dependent effect of dietary meat on endogenous colonic N-nitrosation. Carcinogenesis. 2001;22:199–202. doi: 10.1093/carcin/22.1.199. [DOI] [PubMed] [Google Scholar]
- 159.Belcheva A, Irrazabal T, Robertson SJ, Streutker C, Maughan H, Rubino S, Moriyama EH, Copeland JK, Kumar S, Green B, et al. Gut microbial metabolism drives transformation of MSH2-deficient colon epithelial cells. Cell. 2014;158:288–299. doi: 10.1016/j.cell.2014.04.051. [DOI] [PubMed] [Google Scholar]
- 160.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40:128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gill CI, Rowland IR. Diet and cancer: assessing the risk. Br J Nutr. 2002;88 Suppl 1:S73–S87. doi: 10.1079/BJN2002632. [DOI] [PubMed] [Google Scholar]
- 162.Russell WR, Duncan SH, Scobbie L, Duncan G, Cantlay L, Calder AG, Anderson SE, Flint HJ. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol Nutr Food Res. 2013;57:523–535. doi: 10.1002/mnfr.201200594. [DOI] [PubMed] [Google Scholar]
- 163.Loh YH, Jakszyn P, Luben RN, Mulligan AA, Mitrou PN, Khaw KT. N-Nitroso compounds and cancer incidence: the European Prospective Investigation into Cancer and Nutrition (EPIC)-Norfolk Study. Am J Clin Nutr. 2011;93:1053–1061. doi: 10.3945/ajcn.111.012377. [DOI] [PubMed] [Google Scholar]
- 164.Magee EA, Richardson CJ, Hughes R, Cummings JH. Contribution of dietary protein to sulfide production in the large intestine: an in vitro and a controlled feeding study in humans. Am J Clin Nutr. 2000;72:1488–1494. doi: 10.1093/ajcn/72.6.1488. [DOI] [PubMed] [Google Scholar]
- 165.Roediger WE, Moore J, Babidge W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig Dis Sci. 1997;42:1571–1579. doi: 10.1023/a:1018851723920. [DOI] [PubMed] [Google Scholar]
- 166.Attene-Ramos MS, Nava GM, Muellner MG, Wagner ED, Plewa MJ, Gaskins HR. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ Mol Mutagen. 2010;51:304–314. doi: 10.1002/em.20546. [DOI] [PubMed] [Google Scholar]
- 167.Attene-Ramos MS, Wagner ED, Gaskins HR, Plewa MJ. Hydrogen sulfide induces direct radical-associated DNA damage. Mol Cancer Res. 2007;5:455–459. doi: 10.1158/1541-7786.MCR-06-0439. [DOI] [PubMed] [Google Scholar]
- 168.Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- 169.Homann N. Alcohol and upper gastrointestinal tract cancer: the role of local acetaldehyde production. Addict Biol. 2001;6:309–323. doi: 10.1080/13556210020077028. [DOI] [PubMed] [Google Scholar]
- 170.Hooper SJ, Wilson MJ, Crean SJ. Exploring the link between microorganisms and oral cancer: a systematic review of the literature. Head Neck. 2009;31:1228–1239. doi: 10.1002/hed.21140. [DOI] [PubMed] [Google Scholar]
- 171.Choi SW, Kim YI, Weitzel JN, Mason JB. Folate depletion impairs DNA excision repair in the colon of the rat. Gut. 1998;43:93–99. doi: 10.1136/gut.43.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Homann N, Tillonen J, Salaspuro M. Microbially produced acetaldehyde from ethanol may increase the risk of colon cancer via folate deficiency. Int J Cancer. 2000;86:169–173. doi: 10.1002/(sici)1097-0215(20000415)86:2<169::aid-ijc4>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 173.Cario E. Bacterial interactions with cells of the intestinal mucosa: Toll-like receptors and NOD2. Gut. 2005;54:1182–1193. doi: 10.1136/gut.2004.062794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5:749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- 175.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 176.Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 2009;4:e6026. doi: 10.1371/journal.pone.0006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Irrazábal T, Belcheva A, Girardin SE, Martin A, Philpott DJ. The multifaceted role of the intestinal microbiota in colon cancer. Mol Cell. 2014;54:309–320. doi: 10.1016/j.molcel.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 178.Chen GY, Shaw MH, Redondo G, Núñez G. The innate immune receptor Nod1 protects the intestine from inflammation-induced tumorigenesis. Cancer Res. 2008;68:10060–10067. doi: 10.1158/0008-5472.CAN-08-2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–1728. doi: 10.1053/j.gastro.2011.01.054. [DOI] [PubMed] [Google Scholar]
- 180.Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, Starling N. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- 181.Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9:599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 182.Zackular JP, Baxter NT, Iverson KD, Sadler WD, Petrosino JF, Chen GY, Schloss PD. The gut microbiome modulates colon tumorigenesis. MBio. 2013;4:e00692–e00613. doi: 10.1128/mBio.00692-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Yang Y, Wang X, Moore DR, Lightfoot SA, Huycke MM. TNF-α mediates macrophage-induced bystander effects through Netrin-1. Cancer Res. 2012;72:5219–5229. doi: 10.1158/0008-5472.CAN-12-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Raisch J, Rolhion N, Dubois A, Darfeuille-Michaud A, Bringer MA. Intracellular colon cancer-associated Escherichia coli promote protumoral activities of human macrophages by inducing sustained COX-2 expression. Lab Invest. 2015;95:296–307. doi: 10.1038/labinvest.2014.161. [DOI] [PubMed] [Google Scholar]
- 185.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17:1195–1214. doi: 10.1096/fj.02-0752rev. [DOI] [PubMed] [Google Scholar]
- 186.Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004;567:1–61. doi: 10.1016/j.mrrev.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 187.Wang D, Kreutzer DA, Essigmann JM. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat Res. 1998;400:99–115. doi: 10.1016/s0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 188.Bartsch H, Nair J. Potential role of lipid peroxidation derived DNA damage in human colon carcinogenesis: studies on exocyclic base adducts as stable oxidative stress markers. Cancer Detect Prev. 2002;26:308–312. doi: 10.1016/s0361-090x(02)00093-4. [DOI] [PubMed] [Google Scholar]
- 189.Keshavarzian A, Zapeda D, List T, Mobarhan S. High levels of reactive oxygen metabolites in colon cancer tissue: analysis by chemiluminescence probe. Nutr Cancer. 1992;17:243–249. doi: 10.1080/01635589209514193. [DOI] [PubMed] [Google Scholar]
- 190.Roessner A, Kuester D, Malfertheiner P, Schneider-Stock R. Oxidative stress in ulcerative colitis-associated carcinogenesis. Pathol Res Pract. 2008;204:511–524. doi: 10.1016/j.prp.2008.04.011. [DOI] [PubMed] [Google Scholar]
- 191.Lundberg JO, Weitzberg E, Cole JA, Benjamin N. Nitrate, bacteria and human health. Nat Rev Microbiol. 2004;2:593–602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 192.Sobko T, Huang L, Midtvedt T, Norin E, Gustafsson LE, Norman M, Jansson EA, Lundberg JO. Generation of NO by probiotic bacteria in the gastrointestinal tract. Free Radic Biol Med. 2006;41:985–991. doi: 10.1016/j.freeradbiomed.2006.06.020. [DOI] [PubMed] [Google Scholar]
- 193.Sobko T, Reinders CI, Jansson E, Norin E, Midtvedt T, Lundberg JO. Gastrointestinal bacteria generate nitric oxide from nitrate and nitrite. Nitric Oxide. 2005;13:272–278. doi: 10.1016/j.niox.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 194.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Klaunig JE, Kamendulis LM, Hocevar BA. Oxidative stress and oxidative damage in carcinogenesis. Toxicol Pathol. 2010;38:96–109. doi: 10.1177/0192623309356453. [DOI] [PubMed] [Google Scholar]
- 196.Dröge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 197.Hazane-Puch F, Bonnet M, Valenti K, Schnebert S, Kurfurst R, Favier A, Sauvaigo S. Study of fibroblast gene expression in response to oxidative stress induced by hydrogen peroxide or UVA with skin aging. Eur J Dermatol. 2010;20:308–320. doi: 10.1684/ejd.2010.0934. [DOI] [PubMed] [Google Scholar]
- 198.Moreno V, Gemignani F, Landi S, Gioia-Patricola L, Chabrier A, Blanco I, González S, Guino E, Capellà G, Canzian F. Polymorphisms in genes of nucleotide and base excision repair: risk and prognosis of colorectal cancer. Clin Cancer Res. 2006;12:2101–2108. doi: 10.1158/1078-0432.CCR-05-1363. [DOI] [PubMed] [Google Scholar]
- 199.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21:1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 200.Mangerich A, Knutson CG, Parry NM, Muthupalani S, Ye W, Prestwich E, Cui L, McFaline JL, Mobley M, Ge Z, et al. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc Natl Acad Sci USA. 2012;109:E1820–E1829. doi: 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Cougnoux A, Gibold L, Robin F, Dubois D, Pradel N, Darfeuille-Michaud A, Dalmasso G, Delmas J, Bonnet R. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J Mol Biol. 2012;424:203–214. doi: 10.1016/j.jmb.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 202.Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, Bouchon B, Bringer MA, Nougayrède JP, Oswald E, et al. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J Biol Chem. 2011;286:35562–35570. doi: 10.1074/jbc.M111.221960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Reimer D, Pos KM, Thines M, Grün P, Bode HB. A natural prodrug activation mechanism in nonribosomal peptide synthesis. Nat Chem Biol. 2011;7:888–890. doi: 10.1038/nchembio.688. [DOI] [PubMed] [Google Scholar]
- 204.Cougnoux A, Delmas J, Gibold L, Faïs T, Romagnoli C, Robin F, Cuevas-Ramos G, Oswald E, Darfeuille-Michaud A, Prati F, et al. Small-molecule inhibitors prevent the genotoxic and protumoural effects induced by colibactin-producing bacteria. Gut. 2015:Epub ahead of print. doi: 10.1136/gutjnl-2014-307241. [DOI] [PubMed] [Google Scholar]
- 205.Licciardi PV, Wong SS, Tang ML, Karagiannis TC. Epigenome targeting by probiotic metabolites. Gut Pathog. 2010;2:24. doi: 10.1186/1757-4749-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ritchie ML, Romanuk TN. A meta-analysis of probiotic efficacy for gastrointestinal diseases. PLoS One. 2012;7:e34938. doi: 10.1371/journal.pone.0034938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Khazaie K, Zadeh M, Khan MW, Bere P, Gounari F, Dennis K, Blatner NR, Owen JL, Klaenhammer TR, Mohamadzadeh M. Abating colon cancer polyposis by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci USA. 2012;109:10462–10467. doi: 10.1073/pnas.1207230109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Mohamadzadeh M, Pfeiler EA, Brown JB, Zadeh M, Gramarossa M, Managlia E, Bere P, Sarraj B, Khan MW, Pakanati KC, et al. Regulation of induced colonic inflammation by Lactobacillus acidophilus deficient in lipoteichoic acid. Proc Natl Acad Sci USA. 2011;108 Suppl 1:4623–4630. doi: 10.1073/pnas.1005066107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Saber R, Zadeh M, Pakanati KC, Bere P, Klaenhammer T, Mohamadzadeh M. Lipoteichoic acid-deficient Lactobacillus acidophilus regulates downstream signals. Immunotherapy. 2011;3:337–347. doi: 10.2217/imt.10.119. [DOI] [PubMed] [Google Scholar]
- 210.Lightfoot YL, Yang T, Sahay B, Mohamadzadeh M. Targeting aberrant colon cancer-specific DNA methylation with lipoteichoic acid-deficient Lactobacillus acidophilus. Gut Microbes. 2013;4:84–88. doi: 10.4161/gmic.22822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ghadimi D, Helwig U, Schrezenmeir J, Heller KJ, de Vrese M. Epigenetic imprinting by commensal probiotics inhibits the IL-23/IL-17 axis in an in vitro model of the intestinal mucosal immune system. J Leukoc Biol. 2012;92:895–911. doi: 10.1189/jlb.0611286. [DOI] [PubMed] [Google Scholar]
- 212.Hedin C, Whelan K, Lindsay JO. Evidence for the use of probiotics and prebiotics in inflammatory bowel disease: a review of clinical trials. Proc Nutr Soc. 2007;66:307–315. doi: 10.1017/S0029665107005563. [DOI] [PubMed] [Google Scholar]
- 213.Marteau PR, de Vrese M, Cellier CJ, Schrezenmeir J. Protection from gastrointestinal diseases with the use of probiotics. Am J Clin Nutr. 2001;73:430S–436S. doi: 10.1093/ajcn/73.2.430s. [DOI] [PubMed] [Google Scholar]
- 214.Feng Q, Liang S, Jia H, Stadlmayr A, Tang L, Lan Z, Zhang D, Xia H, Xu X, Jie Z, et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun. 2015;6:6528. doi: 10.1038/ncomms7528. [DOI] [PubMed] [Google Scholar]